

Abstract
Selective serotonin reuptake inhibitors are clinically prescribed antidepressants that act by increasing the local concentration of neurotransmitter at synapses and in extracellular spaces via blockade of the serotonin transporter. Here we report x-ray structures of engineered thermostable variants of the human serotonin transporter bound to the antidepressants sertraline, fluvoxamine, and paroxetine. The drugs prevent serotonin binding by occupying the central substrate binding site and stabilizing the transporter in an outward-open conformation. These structures explain how residues within the central site orchestrate binding of chemically diverse inhibitors and mediate transporter-drug selectivity.
INTRODUCTION
In the central nervous system the serotonin transporter (SERT) modulates serotoninergic signaling by carrying out the uptake of serotonin (5-HT) from the synaptic space into presynaptic neurons1. 5-HT influences many aspects of behavior including memory, learning, sleep, hunger, pain, sexual function, and mood2. A substantial number of nonsynonymous single nucleotide polymorphisms are known to affect 5-HT transport and are enriched in families with psychiatric disorders including obsessive-compulsive disorder and Asperger’s syndrome3. Moreover, SERT is the primary target of therapeutic drugs used in the treatment of major depression, anxiety disorders, and attention-deficient hyperactivity disorder1. 5-HT is also found in the circulatory system where it acts as a vasoconstrictor, and altered plasma concentrations of 5-HT have been implicated in several pathologies, including hypertension4. The plasma levels of 5-HT are regulated by SERT, which stores 5-HT in platelets, ensuring a stable blood flow5. Transporters for dopamine (DAT) and norepinephrine (NET) are related to SERT in amino acid sequence and in biological function and are also well-established targets for pharmacological agents that influence brain chemistry6. These monoamine transporters exploit the ion gradients of Na+ and Cl− in order to render neurotransmitter (NT) uptake thermodynamically favorable7. Monoamine transporters have twelve transmembrane helices (TM) with TMs 1–5 and TMs 6–10 forming an inverted-topological repeat within which is a central binding site for substrate and ions approximately halfway across the membrane8–10. An extracellular vestibule in the outward-facing conformation8,10,11 forms a secondary, allosteric site which, when occupied by ligands, can result in modulation of transporter activity by altering the kinetics of ligand dissociation from the central site12–14. Addictive substances such as cocaine and amphetamine bind to monoamine transporters and can either inhibit NT transport or promote NT efflux, respectively15,16. Selective serotonin reuptake inhibitors (SSRIs) are a class of small molecules that are highly selective for SERT over DAT and NET and that inhibit 5-HT reuptake with nanomolar potency17. SSRIs are typically used as antidepressants in the treatment of major depressive disorder and anxiety disorder1 and are the most widely prescribed antidepressants18.
SSRIs have a diverse chemical structure and, in many instances, they do not share common structural motifs. The diversity of SSRI chemical structures, in turn, results in compounds with substantial pharmacological differences19. Sertraline and fluvoxamine, as examples, differ in chemical structure from other SSRIs such as paroxetine, citalopram, and fluoxetine (Fig. 1a), and as a consequence these compounds bind with a range of affinities to SERT. Sertraline contains a tetrahydronaphthalene ring system linked to a secondary amine together with a meta and para substituted dichlorophenyl group. Fluvoxamine, by contrast, consists of a 2-aminoethyloxime moiety attached to a methyoxybutyl group, and a phenyl group containing a trifluoronated methyl at the para positon. Fluoxetine also contains a trifluoronated aromatic group but is instead coupled to a phenylpropylamine moiety. Paroxetine and citalopram also differ substantially in the structures of their aromatic, amine, and halogenated substitutents.
Figure 1. Saturation and competition binding experiments.

a, Chemical structures of SSRIs. b, Shown are the saturation binding of 3H-paroxetine to ts3 (red, squares) and to ts2 (blue, circles) variants. Panels c–g show competition experiments between the binding of 3H-paroxetine and cold SSRIs, also on the ts3 and ts2 SERT variants, as described in panel b. Graphs depict the average of triplicate measurements from a representative experiment, repeated three independent times (error bars represent s.e.m.).
Recently, we solved x-ray structures of a thermostable, transport-inactive construct of human SERT, deemed the ts3 construct, in complex with the SSRIs paroxetine and citalopram9. We employed the thermostable variant of SERT to facilitate purification and crystallization20,21. One of the thermostabilizing mutations, however, involves a residue (Thr439Ser) that is directly positioned within the central binding site, in close proximity to the bound SSRIs. Moreover, recent computational modeling of a wild-type SERT-paroxetine complex, in which the structure of the Drosophila DAT was used as a template for the SERT structure, yielded a pose for paroxetine in the SERT binding site that is different from that found in the SERT ts3 crystal structure22. In light of the computational study and because residue 439 is near the central binding site, we were motivated to determine crystal structures of SERT bound with SSRIs where residue 439 is the wild-type threonine amino acid.
Despite the importance of SSRIs in medicine and in the biophysical study of SERT, there is little structural explanation for how these diverse ligands bind to SERT 23. Until recently, the structural understanding of how SSRIs bind to SERT has been largely guided by studies of the bacterial homolog LeuT24,25, computational modeling26–30, and more recently, the structures of SERT in complex with paroxetine and citalopram9. In order to accommodate various SSRIs within the central binding site, we reasoned that residues lining the central binding site might adopt different conformations. Thus we determined x-ray structures of Thr439 ts2 SERT in complex with paroxetine and the structure of the ts3 transporter bound to sertraline and fluvoxamine. These structures provide insight into how different SSRIs are bound within the central binding site of SERT.
RESULTS
Binding of antidepressants to ts2 and ts3 transporters
To probe the capacity of the ts3 and ts2 transporters to bind antidepressants we first carried out binding studies using 3H-paroxetine (Fig. 1b). Paroxetine binds with a dissociation constant (Kd) value of 1.09 ± 0.08 and 0.63 ± 0.07 nM to the ts3 and ts2 variants (student’s t-test, p-value less than 0.05). Next, we measured the Ki values for various SSRIs by competition with 3H-paroxetine (Fig. 1c–g). With the exception of sertraline, SSRIs bind with slightly lower affinity to ts3 vs. ts2. S-citalopram exhibited a Ki of 10 ± 1 and 7 ± 1 nM (Fig. 1c, p-value greater than 0.05); sertraline, 2.0 ± 0.2 and 7 ± 1 nM (Fig. 1d, p-value greater than 0.05); S-fluoxetine, 35 ± 3 and 10 ± 1 nM (Fig. 1e, p-value less than 0.05); R-fluoxetine, 41 ± 4 and 9 ± 1 nM (Fig. 1f, p-value less than 0.05); and fluvoxamine, 69 ± 9 and 10 ± 2 nM (Fig. 1g, p-value less than 0.05). These dissociation constants and the relative differences of the dissociation constants between the SSRIs agree well with previous reports31. The presence of the Y110A mutant in both the ts3 and the ts2 variants renders them inactive in 5-HT transport20.
Transporter - antidepressant interactions at the central site
The structure of paroxetine bound to ts2 was determined using diffraction data that extend to Bragg spacing of 3.6 Å. Electron density for several of the side chains for residues in the intracellular gate and within the C-terminal hinge and helix were also better resolved in the ts2 structure reported here in comparison to the previously reported ts3 structure, thus allowing for more complete modeling of these regions (Supplementary Fig. 1), including part of the recognition sequence for SEC24C32,33. Despite the medium resolution of the diffraction data, the electron density features in the central site are of sufficient quality to allow us to position paroxetine. We find that the piperidine ring is best accommodated in subsite A, the benzodioxol in subsite B, and the fluorophenyl group in subsite C (Fig. 2a, Table 1). Fitting of paroxetine in the opposite orientation with the benzodioxol in subsite C, and the fluorophenyl in subsite B yields a poor fit to the electron density and produces clashes in subsite A and C (Supplementary Fig. 2). The sertraline and fluvoxamine structures were solved in complex with ts3 at 3.5 and 3.8 Å respectively. Omit maps and anomalous difference Fourier maps were used to position sertraline with the dichloro phenyl ring in subsite B and the amine and tetrahydronaphthalene groups in subsites A and C, respectively (Fig. 2b–c). The density for fluvoxamine was not continuous but we found density for the aminoethyloxime and trifluoro aromatic groups in subsites A and B (Fig. 2d); no density was observed for the methyoxybutyl moiety, likely due to its flexibility. A Polder ‘omit’ map34 was used to further recover fluvoxamine density and position it more accurately within subsites A and B. Placement of residues which are involved in drug binding at the central site was guided by electron density features and by chemical interactions with the drug (Supplementary Fig. 3). No density was observed in the allosteric site with the sertraline and fluvoxamine complexes, consistent with sertraline being a less potent allosteric inhibitor of the dissociation of citalopram from the central site compared to citalopram13. At present there is no information on whether fluvoxamine binds to the allosteric site and influences ligand dissociation from the central site.
Figure 2. Antidepressant binding in the central binding site.
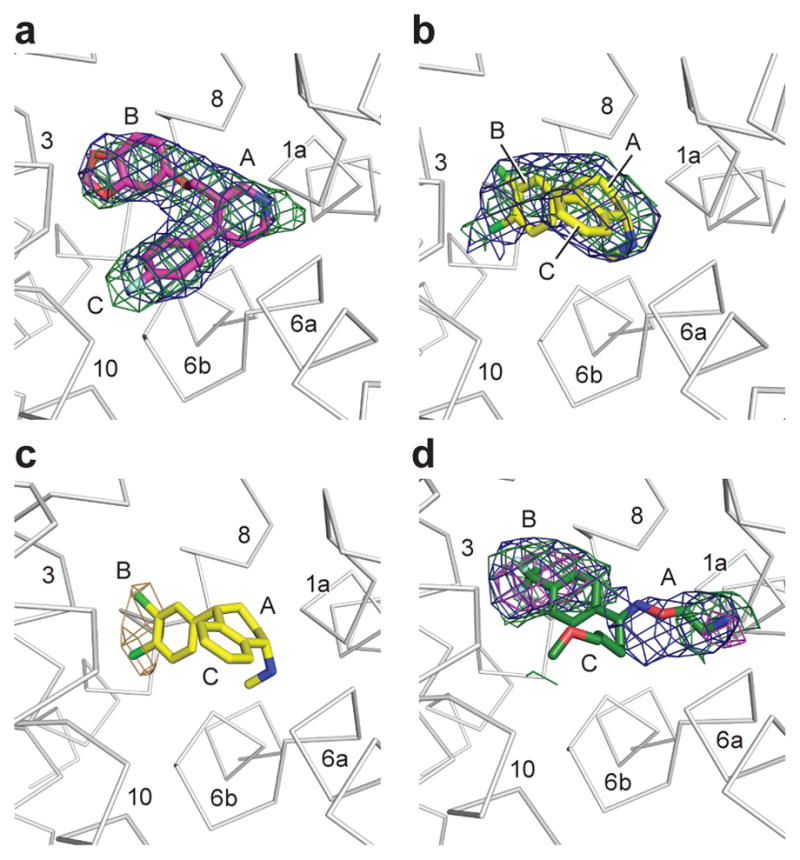
a, Shown is the ‘omit’ electron density (green mesh) associated with paroxetine, contoured at 4σ. Electron density derived from a Polder ‘omit’ electron density is shown in blue mesh and is contoured at 7σ. b, This shows electron density for sertraline derived from an ‘omit’ map, contoured at 2.5σ; the Polder ‘omit’ electron density is contoured at 5σ. c, Anomalous difference electron density (light brown), derived from sertraline, is contoured at 3σ. d, Density for fluvoxamine derived from an omit electron density map and contoured at 1σ (green) and 2σ (magenta), with the Polder omit electron density contoured at 3σ. The approximate positions of subsites A, B, and C are shown.
Table 1.
Data collection and refinement statistics.
| Paroxetine ts2 6AWN |
Fluvoxamine ts3 6AWP |
Sertraline ts3 6AWO |
Sertraline ts3 6AWQ |
|
|---|---|---|---|---|
| Data collection | APS24-IDE | APS24-IDE | APS24-IDE | ALS 5.0.2 |
| Space group | C2221 | C2221 | C2221 | C2221 |
| Cell dimensions | ||||
| a, b, c (Å) | 129.2, 162.8, 140.8 | 129.5, 163.9, 141.0 | 129.0, 162.0, 140.8 | 130.6, 165.9, 142.7 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Wavelength | 0.979 | 0.979 | 0.979 | 1.771 |
| Resolution (Å) | 53.17-3.62 (3.73-3.62) | 30.00-3.80 (3.94-3.80) | 40.0-3.53 (3.62-3.53) | 20.00-4.05 (4.14-4.05) |
| Rmeas (%) | 6.2 (41.6) | 6.2 (49.0) | 5.8 (58.9) | 10.4 (59.8) |
| I/σI | 9.0 (1.7) | 8.4 (1.9) | 13.1 (1.7) | 11.1 (2.5) |
| Completeness (%) | 92.6 (90.7) | 95.5 (95.7) | 98.7 (86.7) | 98.1 (98.1) |
| Redundancy | 2.6 (2.2) | 3.6 (3.0) | 5.5 (3.2) | 10.1 (6.9) |
| CC1/2 (%) | 99.9 (21.9) | 99.9 (28.3) | 99.9 (16.0) | 99.9 (20.3) |
| Refinement | ||||
| Resolution (Å) | 53.26-3.62 (3.75-3.62) | 28.96-3.80 (3.94-3.80) | 38.91-3.53 (3.66-3.53) | |
| No. reflections | 16974 (1529) | 14768 (1328) | 18289 (1615) | |
| Rwork/Rfree (%) | 25.6 (37.8)/26.3 (37.0) | 24.6 (40.3)/26.9 (43.7) | 27.3 (41.4)/28.4 (42.3) | |
| No. atoms | 7703 | 7680 | 7678 | |
| Protein | 7599 | 7598 | 7598 | |
| Ligand/ion | 104 | 81 | 79 | |
| Water | 0 | 1 | 1 | |
| B-factors | ||||
| Protein | 157.7 | 206.4 | 196.9 | |
| Ligand/ion | 151.9 | 206.6 | 187.3 | |
| Water | N/A | 225.0 | 182.0 | |
| R.m.s deviations | ||||
| Bond lengths (Å) | 0.009 | 0.009 | 0.008 | |
| Bond angles (°) | 1.08 | 1.01 | 0.96 | |
| Ramachandran plot | ||||
| Favored (%) | 95.5 | 95.2 | 95.0 | |
| Allowed (%) | 4.5 | 4.8 | 5.0 | |
| Disallowed (%) | 0 | 0 | 0 | |
The structure of the ts2 SERT variant in complex with paroxetine revealed only minor perturbations in the pose of the inhibitor and the conformations of residues involved in ligand binding in comparison to the ts3 – paroxetine structure (Fig. 3a,b). We believe this is because the hydroxyl group of threonine 439 faces subsite B, as does the serine at this position, and thus both are in a position to participate in interactions with ligands in the central site. We speculate that the ts2 variant has a higher affinity for paroxetine because, at least in part, the hydroxyl of Ser439 is closer to a ligand benzodioxol oxygen (3.9 Å) in comparison to the hydroxyl of Thr439 (5.2 Å). A recent study using photo-cross linking also favored a placement of paroxetine which is consistent with our structures35 however, further studies, at higher resolution, will be required to more definitively define transporter-ligand interactions.
Figure 3. Antidepressant recognition.

a, Overall view of the ts2 variant of the serotonin transporter in cartoon representation, Thr439 and paroxetine are depicted as sticks and spheres. b, Interactions of paroxetine (magenta) with residues in the central binding site of the ts2 variant. The position of paroxetine in the ts3 variant is overlaid in grey. c, Interactions of sertraline (yellow) with residues in the central binding site. d, Interactions of fluvoxamine (green) with residues in the central binding site.
The amine groups of sertraline and fluvoxamine also occupy subsite A, interacting with Asp98 and Try95 (Fig. 3c,d) residues crucial for drug binding36–38. In subsite B, Ser439 is within 4.5 and 3.9 Å of the halide atoms of sertraline and fluvoxamine while aromatic interactions are formed between Tyr176 and the drugs. The side chain of Ile172 adopts a similar conformation as it does in the paroxetine state, fitting snuggly between substituents in subsites B and C. In subsite C, Phe341 forms a face-to-face interaction with the naphthalene ring of sertraline while Phe335 forms a face-to-edge interaction with the naphthalene moiety. In the fluvoxamine complex, Phe34126 assumes a similar rotameric conformation as in the sertraline complex while the conformation of Phe335 is closer to that observed in the paroxetine structures.
DISCUSSION
Comparison of SSRI-complexes reveals diversity in drug binding
Comparisons of the structures of the sertraline and fluvoxamine complexes to the previously determined structures of SERT with paroxetine and S-citalopram reveal important differences in the position of residues in subsite C. In the sertraline complex, relative to the paroxetine and S-citalopram complexes, Phe341 has flipped ‘upward’ to fill a space that is unoccupied by ligand and to interact with the naphthalene ring of sertraline; Phe335 undergoes a rotation of ~90° about its Cβ-Cγ bond (chi2) in order to accommodate the conformational change of Phe341 (Fig. 4a). In the fluvoxamine complex, Phe341 is in a similar position as the sertraline complex, but Phe335 adopts a conformation more similar to that observed in the paroxetine and S-citalopram complexes (Fig. 4b,c). We also note that Thr497 is also shifted by ~1 Å (Cα-Cα distance) in the sertraline and fluvoxamine complexes in comparison to the paroxetine complexes, likely because paroxetine is the only drug with a fluorinated substituent in subsite C, a substituent which interacts with Thr497.
Figure 4. Comparisons of SSRI binding poses and central binding site structures.

a, Comparison of ts3 sertraline (yellow) and ts2 paroxetine (magenta) complexes. b, Comparison of ts3 fluvoxamine (green) and ts2 paroxetine binding. c, ts3 fluvoxamine vs. ts3 sertraline. d, Superposition of ts3 sertraline and ts3 S-citalopram (orange). e, ts3 fluvoxamine vs. ts3 S-citalopram. f, ts2 paroxetine vs. ts3 S-citalopram. 2D representation of selected drug binding residues, panels g–j. Distances of each residue to various positions of drug are shown for g, ts2 paroxetine h, ts3 sertraline i, ts3 fluvoxamine and j, ts3 S-citalopram.
Positioning of antidepressant in the central site
Superposition of all four SSRI complexes revealed different placement of the various drug substituents within the central binding site (Fig. 4d–j) resulting in differences in van der Waals (>4 Å), aromatic (4.5–7 Å)39, and ionic (~4 Å)40 interactions. We caution that due to the modest resolution, the accuracy of these measurement will contain some degree of error. We have estimated the standard uncertainty in the ligand position by calculating the coordinate error measured by the diffraction precision index41. For the paroxetine ligand, the uncertainty is 0.4 Å, sertraline 0.5 Å, fluvoxamine 0.6 Å, and S-citalopram 0.3 Å. In subsite A, the distance from the amine of sertraline and S-citalopram to Asp98 is longer while the distance to the amine of fluvoxamine and paroxetine is shorter. Conversely, the positioning of the amine to Tyr95 is farther for fluvoxamine and paroxetine whereas the amine of S-citalopram and sertraline is more equally posed between Asp98 and Tyr95. In subsite B, the meta chlorine of sertraline and the fluorine of S-citalopram engages with Ser439 while the fluorines of fluvoxamine and the oxygen of paroxetine form even closer interactions with Ser439/Thr439 though the distance is not close enough for a hydrogen-bond to the halides42. The shorter distance to Ser439/Thr439 in the fluvoxamine complex and in the ts2 versus the ts3 paroxetine complexes may also explain, at least in part, the differences in affinity observed for fluvoxamine and fluoxetine in comparison to other SSRIs which do not have a trifluoro group in subsite B. The aromatic substituents of the SSRIs are arranged ~90° relative to Try176 and Ile172 in subsite B, but the distances to Tyr176 and Ile172 are divergent between inhibitors. In subsite C, the naphthalene group of sertraline is localized ‘higher’ than the benzofuran and fluorophenyl groups of S-citalopram and paroxetine.
Outward-open state reveals effect of human mutations
A rare autism-associated mutation in DAT (Thr356Met) has recently been shown to stabilize the outward-open conformation in LeuT and induces hyperactive locomotion in flies43. Dopamine uptake of the DAT mutant is markedly reduced resulting from the catalysis of dopamine efflux. The equivalent residue in SERT is Thr371 which is located in TM7 a half-turn away from the Na1 site. Thr371 likely interacts with Tyr289 in TM5 and substitution of a larger methionine side-chain would disrupt this interaction which could prevent closure of the extracellular gate.
Conclusion
Taken together, the SERT-SSRI complexes provide new insights into how pharmacophores of different drugs participate in interactions within the central binding site – all acting to stabilize the outward-open conformation of the transporter. Using structure analysis we can determine more precisely how mutations alter the function of neurotransmitter transporters. We anticipate that these studies will provide a blueprint for the development of new therapeutic agents for the treatment of depression and anxiety disorders.
ONLINE METHODS
SERT constructs
The ts3 variant20,21 contains the thermostabilizing mutations Tyr110Ala, Ile291Ala, Thr439Ser, as well as mutation of surface-exposed cysteines Cys554, Cys580, and Cys622 to alanine. The ts3 SERT gene is then fused to sequences to express a C-terminal GFP fluorophore followed by a twin Strep and, lastly, a His10 purification tag. The mutation of Ser439 to threonine reverts the thermostabilizing mutation at position 439 of the ts3 construct to a threonine to then yield the ts2 construct.
Expression and purification
SERT constructs were expressed by using baculovirus-mediated transduction of mammalian HEK293S GnTI− cells44. Cells were solubilized in a buffer composed of 50 mM Tris (pH 8), 150 mM NaCl, 20 mM n-dodecyl-β-D-maltoside (DDM), and 2.5 mM cholesterol hemisuccinate (CHS). Detergent-solubilized SERT was purified via Strep-Tactin affinity chromatography in a buffer composed of 20 mM Tris pH 8, 100 mM NaCl (TBS) containing 1 mM DDM, 0.2 mM CHS, 5% glycerol, 25 μM lipid (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine, and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol at a molar ratio of 1:1:1). Following elution from the column, the resulting protein was digested by thrombin and EndoH. SERT was then combined with the recombinant antibody fragment (Fab) 8B6 in a molar ratio of 1:1.2 (SERT:8B6) and the complex was purified by size-exclusion chromatography in TBS supplemented with 40 mM n-octyl β-D-maltoside, 0.5 mM CHS, 5% glycerol and 25 μM of the lipid mixture described above. Prior to crystallization, the purified SERT-8B6 complex was concentrated to 2 mg/ml; inhibitors were added to the protein solution to reach a final concentration of 50 μM and an additional amount of 8B6 Fab was added so that the final concentration of the added Fab was 1 μM.
Crystallization
The SERT-Fab complex crystals were grown by hanging drop vapor diffusion at 4 °C using a ratio of 2 μl protein to 1 μl reservoir solution, the latter of which contained 50–100 mM Tris pH 8.5, 25–75 mM Li2SO4, 25–75 mM Na2SO4, 33.5 – 36 % PEG 400 and 0.5% 6-aminohexanoic acid.
Data collection and structure refinement
Crystals were directly flash cooled in liquid nitrogen. Data was collected at the Advanced Photon Source (Argonne National Laboratory, beamline 24-ID-E) and at the Advanced Light Source (Lawrence Berkeley National Laboratory, beamline 5.0.2). X-ray data sets were processed using XDS45. Molecular replacement was performed using coordinates (PDB 5I6X code) from the prior structure determination of SERT9 using the computer program PHASER46. Several iterations of refinement and manual model building were carried out using PHENIX47 and Coot48 until the models converged to acceptable R-factors and stereochemistry. Polder ‘omit’ maps were calculated by excluding bulk solvent as previously described34.
Radioligand binding assays
Saturation binding experiments employed the scintillation proximity assay (SPA)49 and a solution containing 10 nM SERT, 0.5 mg/ml Cu-Ysi beads, TBS, 1 mM DDM, 0.2 mM CHS, and [3H-paroxetine] from 0.15–20 nM. Competition binding experiments were also performed using SPA, in the same buffer as the saturation binding experiments and ligands at concentrations of 5 nM for 3H-paroxetine and at 0.01–100,000 nM for the cold competitors. Each data point was measured in triplicate and each experiment was performed three times. The error bars for each data point represent the standard error of the mean. Ki values were determined using the Cheng-Prusoff equation50 in Graphpad Prism.
Supplementary Material
Acknowledgments
We thank L. Vaskalis for assistance with figures, H. Owen for help with manuscript preparation and all Gouaux laboratory members for discussion. We acknowledge the staff of the Berkeley Center for Structural Biology at the Advanced Light Source and the Northeastern Collaborative Access Team at the Advanced Photon Source for assistance with data collection. J.A.C. has support from a Banting postdoctoral fellowship from the Canadian Institutes of Health Research. We are particularly grateful to Bernie and Jennifer LaCroute for their generous support, as well as for funding from the National Institutes of Health (NIH) (5R37MH070039). E.G. is an Investigator with the Howard Hughes Medical Institute.
Footnotes
AUTHOR CONTRIBUTIONS
J.A.C. and E.G. designed the project. J.A.C. performed protein purification, crystallography, and biochemical assays. J.A.C. and E.G. wrote the manuscript.
COMPETING FINANCIAL INTERESTS STATEMENT
The authors declare no competing financial interests.
DATA AVAILABILITY AND ACCESSION CODE AVAILABILITY STATEMENTS
The coordinates for the structure have been deposited in the Protein Data Bank under the accession codes 6AWN (ts2-paroxetine), 6AWP (ts3-fluvoxamine), 6AWO (ts3-sertraline), 6AWQ (ts3-sertraline-anomalous) (see Table 1 for details).
References
- 1.Kristensen AS, et al. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacological reviews. 2011;63:585–640. doi: 10.1124/pr.108.000869. [DOI] [PubMed] [Google Scholar]
- 2.Berger M, Gray JA, Roth BL. The expanded biology of serotonin. Annu Rev Med. 2009;60:355–366. doi: 10.1146/annurev.med.60.042307.110802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hahn MK, Blakely RD. The functional impact of SLC6 transporter genetic variation. Annu Rev Pharmacol Toxicol. 2007;47:401–441. doi: 10.1146/annurev.pharmtox.47.120505.105242. [DOI] [PubMed] [Google Scholar]
- 4.Watts SW, Morrison SF, Davis RP, Barman SM. Serotonin and blood pressure regulation. Pharmacological reviews. 2012;64:359–388. doi: 10.1124/pr.111.004697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brenner B, et al. Plasma serotonin levels and the platelet serotonin transporter. Journal of neurochemistry. 2007;102:206–215. doi: 10.1111/j.1471-4159.2007.04542.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Broer S, Gether U. The solute carrier 6 family of transporters. Br J Pharmacol. 2012;167:256–278. doi: 10.1111/j.1476-5381.2012.01975.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rudnick G. Ion-coupled neurotransmitter transport: thermodynamic vs. kinetic determinations of stoichiometry. Methods in enzymology. 1998;296:233–247. doi: 10.1016/s0076-6879(98)96018-9. [DOI] [PubMed] [Google Scholar]
- 8.Penmatsa A, Wang KH, Gouaux E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature. 2013;503:85–90. doi: 10.1038/nature12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coleman JA, Green EM, Gouaux E. X-ray structures and mechanism of the human serotonin transporter. Nature. 2016 doi: 10.1038/nature17629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- 11.Singh SK, Piscitelli CL, Yamashita A, Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science. 2008;322:1655–1661. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wennogle LP, Meyerson LR. Serotonin modulates the dissociation of [3H]imipramine from human platelet recognition sites. European journal of pharmacology. 1982;86:303–307. doi: 10.1016/0014-2999(82)90333-8. [DOI] [PubMed] [Google Scholar]
- 13.Chen F, et al. Characterization of an allosteric citalopram-binding site at the serotonin transporter. Journal of neurochemistry. 2005;92:21–28. doi: 10.1111/j.1471-4159.2004.02835.x. [DOI] [PubMed] [Google Scholar]
- 14.Plenge P, et al. Steric hindrance mutagenesis in the conserved extracellular vestibule impedes allosteric binding of antidepressants to the serotonin transporter. The Journal of biological chemistry. 2012;287:39316–39326. doi: 10.1074/jbc.M112.371765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kahlig KM, et al. Amphetamine induces dopamine efflux through a dopamine transporter channel. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:3495–3500. doi: 10.1073/pnas.0407737102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramamoorthy S, et al. Antidepressant- and cocaine-sensitive human serotonin transporter: molecular cloning, expression, and chromosomal localization. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:2542–2546. doi: 10.1073/pnas.90.6.2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersen J, Kristensen AS, Bang-Andersen B, Stromgaard K. Recent advances in the understanding of the interaction of antidepressant drugs with serotonin and norepinephrine transporters. Chem Commun (Camb) 2009:3677–3692. doi: 10.1039/b903035m. [DOI] [PubMed] [Google Scholar]
- 18.Mojtabai R, Olfson M. Proportion of antidepressants prescribed without a psychiatric diagnosis is growing. Health Aff (Millwood) 2011;30:1434–1442. doi: 10.1377/hlthaff.2010.1024. [DOI] [PubMed] [Google Scholar]
- 19.Vetulani J, Nalepa I. Antidepressants: past, present and future. European journal of pharmacology. 2000;405:351–363. doi: 10.1016/s0014-2999(00)00565-3. [DOI] [PubMed] [Google Scholar]
- 20.Green EM, Coleman JA, Gouaux E. Thermostabilization of the human serotonin transporter in an antidepressant-Bound Conformation. PloS one. 2015;10:e0145688. doi: 10.1371/journal.pone.0145688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coleman JA, Green EM, Gouaux E. Thermostabilization, Expression, Purification, and Crystallization of the Human Serotonin Transporter Bound to S-citalopram. Journal of visualized experiments: JoVE. 2016 doi: 10.3791/54792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis BA, Nagarajan A, Forrest LR, Singh SK. Mechanism of Paroxetine (Paxil) Inhibition of the Serotonin Transporter. Scientific reports. 2016;6:23789. doi: 10.1038/srep23789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andersen J, et al. Molecular determinants for selective recognition of antidepressants in the human serotonin and norepinephrine transporters. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:12137–12142. doi: 10.1073/pnas.1103060108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H, et al. Structural basis for action by diverse antidepressants on biogenic amine transporters. Nature. 2013;503:141–145. doi: 10.1038/nature12648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou Z, et al. Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nature structural & molecular biology. 2009;16:652–657. doi: 10.1038/nsmb.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andersen J, et al. Mutational mapping and modeling of the binding site for (S)-citalopram in the human serotonin transporter. The Journal of biological chemistry. 2010;285:2051–2063. doi: 10.1074/jbc.M109.072587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gabrielsen M, et al. Molecular mechanism of serotonin transporter inhibition elucidated by a new flexible docking protocol. European journal of medicinal chemistry. 2012;47:24–37. doi: 10.1016/j.ejmech.2011.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grouleff J, Ladefoged LK, Koldso H, Schiott B. Monoamine transporters: insights from molecular dynamics simulations. Frontiers in pharmacology. 2015;6:235. doi: 10.3389/fphar.2015.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koldso H, Grouleff J, Schiott B. Insights to ligand binding to the monoamine transporters-from homology modeling to LeuBAT and dDAT. Frontiers in pharmacology. 2015;6:208. doi: 10.3389/fphar.2015.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andersen J, et al. Molecular basis for selective serotonin reuptake inhibition by the antidepressant agent fluoxetine (Prozac) Molecular pharmacology. 2014;85:703–714. doi: 10.1124/mol.113.091249. [DOI] [PubMed] [Google Scholar]
- 31.Tavoulari S, Forrest LR, Rudnick G. Fluoxetine (Prozac) binding to serotonin transporter is modulated by chloride and conformational changes. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:9635–9643. doi: 10.1523/JNEUROSCI.0440-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koban F, et al. A salt bridge linking the first intracellular loop with the C terminus facilitates the folding of the serotonin transporter. The Journal of biological chemistry. 2015;290:13263–13278. doi: 10.1074/jbc.M115.641357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sucic S, et al. Switching the clientele: a lysine residing in the C terminus of the serotonin transporter specifies its preference for the coat protein complex II component SEC24C. The Journal of biological chemistry. 2013;288:5330–5341. doi: 10.1074/jbc.M112.408237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liebschner D, et al. Polder maps: improving OMIT maps by excluding bulk solvent. Acta crystallographica. Section D, Structural biology. 2017;73:148–157. doi: 10.1107/S2059798316018210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rannversson H, Andersen J, Bang-Andersen B, Stromgaard K. Mapping the Binding Site for Escitalopram and Paroxetine in the Human Serotonin Transporter Using Genetically Encoded Photo-Cross-Linkers. ACS chemical biology. 2017;12:2558–2562. doi: 10.1021/acschembio.7b00338. [DOI] [PubMed] [Google Scholar]
- 36.Henry LK, et al. Tyr-95 and Ile-172 in transmembrane segments 1 and 3 of human serotonin transporters interact to establish high affinity recognition of antidepressants. The Journal of biological chemistry. 2006;281:2012–2023. doi: 10.1074/jbc.M505055200. [DOI] [PubMed] [Google Scholar]
- 37.Barker EL, et al. High affinity recognition of serotonin transporter antagonists defined by species-scanning mutagenesis. An aromatic residue in transmembrane domain I dictates species-selective recognition of citalopram and mazindol. The Journal of biological chemistry. 1998;273:19459–19468. doi: 10.1074/jbc.273.31.19459. [DOI] [PubMed] [Google Scholar]
- 38.Sorensen L, et al. Interaction of antidepressants with the serotonin and norepinephrine transporters: mutational studies of the S1 substrate binding pocket. The Journal of biological chemistry. 2012;287:43694–43707. doi: 10.1074/jbc.M112.342212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anjana R, et al. Aromatic-aromatic interactions in structures of proteins and protein-DNA complexes: a study based on orientation and distance. Bioinformation. 2012;8:1220–1224. doi: 10.6026/97320630081220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kumar S, Nussinov R. Close-range electrostatic interactions in proteins. Chembiochem: a European journal of chemical biology. 2002;3:604–617. doi: 10.1002/1439-7633(20020703)3:7<604::AID-CBIC604>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 41.Kumar KSD, GM, Satheesh SN, Radha P, Pavithra S, Thulaa Tharshan KPS, Helliwell JR, Sekara K. Online_DPI: a web server to calculate the diffraction precision index for a protein structure. Journal of applied crystallography. 2015;48:939–942. [Google Scholar]
- 42.Steiner T. Hydrogen-Bond Distances to Halide Ions in Organic and Organometallic Crystal Structures: Up-to-date Database Study. Acta Cryst. 1998;B54:8. [Google Scholar]
- 43.Hamilton PJ, et al. De novo mutation in the dopamine transporter gene associates dopamine dysfunction with autism spectrum disorder. Molecular psychiatry. 2013;18:1315–1323. doi: 10.1038/mp.2013.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goehring A, et al. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nature protocols. 2014;9:2574–2585. doi: 10.1038/nprot.2014.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kabsch W. Xds. Acta crystallographica. Section D, Biological crystallography. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bunkoczi G, et al. Phaser.MRage: automated molecular replacement. Acta crystallographica. Section D, Biological crystallography. 2013;69:2276–2286. doi: 10.1107/S0907444913022750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta crystallographica. Section D, Biological crystallography. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta crystallographica. Section D, Biological crystallography. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quick M, Javitch JA. Monitoring the function of membrane transport proteins in detergent-solubilized form. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:3603–3608. doi: 10.1073/pnas.0609573104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochemical pharmacology. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.