

Abstract
Human pluripotent stem cells (hPSCs) can be directed to differentiate into skeletal muscle progenitor cells (SMPCs). However, the myogenicity of hPSC-SMPCs relative to human fetal or adult satellite cells remains unclear. HPSC-SMPCs derived by directed differentiation are less functional in vitro and in vivo compared to human satellite cells. Utilizing RNA-SEQ, we identified cell surface receptors ERBB3 and NGFR that demarcate myogenic populations, including PAX7 progenitors in human fetal development and hPSC-SMPCs. We demonstrated that hPSC skeletal muscle is immature, but inhibition of TGF-β signaling during differentiation improved fusion efficiency, ultrastructural organization, and expression of adult myosins. This enrichment and maturation strategy restored dystrophin in hundreds of dystrophin-deficient myofibers after engraftment of CRISPR/Cas9-corrected Duchenne muscular dystrophy hiPSC-SMPCs. The work provides an in-depth characterization of human myogenesis, and identifies candidates that improve the in vivo myogenic potential of hPSC-SMPCs to levels equal to directly-isolated human fetal muscle cells.
Introduction
Directed differentiation of human pluripotent stem cells (hPSCs), including human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs), seeks to recapitulate development to form cell lineages similar to human in vivo counterparts. Directed differentiation of hPSCs to specific lineages for cell therapies is showing promise in clinical settings and in preclinical animal models for diseases ranging from cardiac myopathy to macular degeneration, diabetes, and Parkinson's disease1-3. However, for many cell lineages, directed differentiation results in progeny that are heterogeneous and functionally immature compared to cells derived during normal human development4-6. Immature cells derived from hPSCs may function inappropriately, diminishing their utility for modeling human disease and/or cell replacement therapies7.
While several directed differentiation protocols generate skeletal muscle cells from hPSCs8-13, their developmental stage or functional similarity to fetal or adult human skeletal muscle is unknown. Longstanding protocols used to differentiate skeletal muscle from hPSCs require viral-mediated overexpression of transcription factors such as MYOD14,15, PAX716, or PAX317, limiting generation of truly representative myogenic progenitors. Skeletal myogenesis in vivo relies on tightly controlled spatial and temporal cues to ensure timely embryonic transitions through the presomitic mesoderm, somites, and dermomyotome to form the myotome18. Although recent studies have followed human developmental cues to differentiate hPSCs to somites in vitro19,20,10, none have generated sufficient quantity or quality of skeletal muscle progenitor cells (SMPCs) or satellite cells (SCs)from hPSCs.
SCs are endogenous skeletal muscle stem cells responsible for the formation of new muscle and are indispensable for maintenance and repair21, and are thought to arise during the secondary wave of fetal myogenesis22. Fetal muscle cells are unique from adult SCs in their ability to retain Pax7 in a niche-independent, non-quiescent state23. While a single transplanted SC can give rise to several hundred myofibers24, it remains unclear whether fetal or adult SCs are more regenerative in vivo, or whether directed differentiation of hPSC-SMPCs should seek to attain fetal-like or adult-like SCs. A better understanding of human SC biology and regenerative potential from multiple stages of development could enable derivation of more myogenic hPSC-SMPCs. Cell replacement is a promising therapy for many muscle diseases, including Duchenne muscular dystrophy (DMD), a degenerative muscle disease caused by lack of dystrophin. Autologous transplantation of CRISPR/Cas9-corrected hiPSC-SMPCs25 for DMD patients could repopulate diseased or damaged myofibers with donor SMPCs/SCs to restore muscle function over the patient's lifetime.
To improve our understanding of the developmental and functional status of hPSC-SMPCs, we compared established directed differentiation protocols8-11 to muscle cells across multiple stages of human development, including fetal weeks 8-18 and adult SCs. Fetal muscle cells were also profiled to other musculoskeletal tissues by RNA-SEQ, enabling identification of muscle-specific receptors, ERBB3 and NGFR. Our strategy enabled isolation of PAX7+ hPSC-SMPCs with robust engraftment in vivo, including the ability to restore dystrophin in mdx-NSG mice, and could potentially improve delivery of gene-editing therapies for skeletal muscle diseases. Importantly, this work improves our understanding of human fetal muscle progenitors and provides a developmental identity of hPSC-SMPCs relative to equivalent cells in vivo.
Results
HPSC-SMPCs have reduced fusion efficiency compared to their human muscle-derived counterparts
To evaluate myogenic potential of hPSC-SMPCs relative to fetal and adult SCs, multiple directed differentiation protocols were evaluated8-11, and two selected for further analysis10,11. Both methods consistently generated muscle cells expressing myogenic transcription factors (TFs) PAX7, MYF5, MYOD, and MYOG and spontaneously contracting myotubes (Figure S1). HPSC-SMPCs were dissociated/replated and compared to equivalent numbers of muscle cells isolated from human fetal weeks 9, 14, and 17, and adult skeletal muscle tissues (Figure 1A). Cells isolated from adult muscle formed myotubes most efficiently, and contained the most nuclei (24.5 nuclei/myotube, p<0.05). The fusion indices and nuclei per myotube from fetal weeks 14-17 were significantly greater than fetal week 9 and hPSC-SMPCs (p<0.05), while week 9 fetal muscle cells were not statistically different from hPSC-myotubes (p=0.3).
Figure 1. hPSC-SMPCs have reduced in vitro and in vivo myogenic potential relative to fetal or adult SCs.

A. Human muscle obtained from fetal week 9-17, adult 25yrs or hPSCs differ in ability to form MYHC+ myotubes in vitro (red). PAX7 (white), MYOD (green) and DAPI (blue) are also shown. Scale bar equals 200μm. Fusion index (% of nuclei within MYHC+ cells ≥3 nuclei/total nuclei) and nuclei per myotube are greatest in adult and fetal, while hPSCs primarily had ≤2 MYHC nuclei/mm2; (mean ± SEM; N=3 adultor N=5 fetal tissues, N=3 hPSC independent directed differentiations;One-way ANOVA posthoc Tukey; *p<0.05). B. Co-localization of PAX7 (white) and MYOD (green) differ across developmental stages (shown by arrows). Pie charts show the proportion of PAX7 and MYOD expression in each cell type (N=3 adult and N=5 fetal tissues, N=3 hPSC independent directed differentiations). Scale bar equals 50μm. C. To quantify engraftment, we assessed the total number of human (h) cells (Lamin A/C+, red) as well as the number of fused human myofibers (H-LaminA/C+Spectrin+, red) and H-dystrophin (green) thirty days post-engraftment. Graphs quantify the mean ± SEM number of fused human myofibers from multiple cross sections along the length of the mdx-NSG TA muscle (N=3 uncultured fetal, N=4 cultured fetal, N=4 hPSC-SMPC; where N=number of mice engrafted per group; Kruskal-Wallis ranks tests; *p<0.05), and the maximum number of engrafted myofibers in a single cross section (One-way ANOVA posthoc Tukey; *p<0.05).
In myogenic development, cells undergoing proliferation and differentiation co-exist26. To better understand the timing and heterogeneity of myogenic TF expression during myotube differentiation, PAX7 and MYOD were evaluated (Figure 1B). Indeed, all fetal muscle cultures contained PAX7+MYOD+ SMPCs. Myotubes derived from adult SCs expressed PAX7+ or MYOD+ single nuclei, but few PAX7+MYOD+ nuclei were present in adult cultures. HPSC-SMPCs from either directed differentiation method contained the largest fraction of PAX7+MYOD+ nuclei, and many MYOD+ nuclei were not contained within MYHC+ myotubes. Together, these data suggest that hPSC-SMPCs inefficiently differentiate to form myotubes and/or represent a distinct developmental stage.
HPSC-SMPCs have limited engraftment potential in vivo compared to human fetal myogenic progenitors
Directed differentiation of hPSCs has yet to generate SMPCs with robust engraftment potential in vivo. Previous reports show that cultured human fetal muscle cells also engraft inefficiently27. Notably, directly-isolated adult SCs engraft more efficiently than cultured SCs28, but engraftment of directly-isolated human fetal muscle cells has never been tested. We therefore utilized cultured and uncultured human fetal muscle cells (all directly-isolated mononuclear cells) to benchmark hPSC-SMPC engraftment potential, and assess whether the developmental state or culturing of fetal cells correlated with changes in engraftment. In parallel to cultured and uncultured fetal muscle cells, hPSC-SMPCs were transplanted into the tibialis anterior (TA) of cardiotoxin-pretreated mdx-NSG mice, which are immunocompromised and lack dystrophin expression (Figure 1C). At 30 days, several hundred hPSC-SMPCs, marked by human (h)-LaminA/C, were detected in the muscle. However, most had not fused with the host muscle (as marked by h-LaminA/C+Spectrin+ and h-Dystrophin+) nor did they reside in the SC position. Instead, most h-LaminA/C+ hPSC-SMPCs were found in the perimysium and epimysium of the host TA muscle (Figure 1C). In contrast, both cultured and uncultured fetal cells resulted in increased number of hdystrophin+ cells compared to hPSC-SMPCs (p<0.05, Table S1). While restoration of hdystrophin+ myofibers by cultured fetal muscle was less than 1% of host TA, uncultured fetal cell engraftment was significantly more efficient, restoring 10-15% h-dystrophin+ myofibers in host muscle, a level of dystrophin expression that has been reported to result in functional improvement in mouse30,31,32.
To evaluate the in vivo myogenic potential of hPSC-SMPCs compared to cultured and directly-isolated fetal muscle cells, we counted engrafted myofibers at eleven points (0-10 millimeters) throughout the TA of mdx-NSG mice, and ranked all counts using Kruskal-Wallis analysis of variance tests29 (Figure 1C). Directly-isolated fetal muscle cells had significantly higher engraftment efficiency than cultured fetal cells or hPSC-SMPCs (p<0.0001). These data suggest that generating SMPCs resembling directly-isolated fetal muscle could improve the inferior engraftment potential of hPSC-SMPCs.
Enriching for HNK1-NCAM+ SMPCs increases myogenic cell numbers but does not increase myogenicity in vivo
To potentially enrich for cells with increased myogenecity, we applied a previously described sorting strategy that removes HNK1+ cells (a neuroectodermal marker) and selects for neural cell adhesion molecule (NCAM)8,33. NCAM is also expressed by human fetal muscle27 and adult SCs34. We isolated HNK1-NCAM+ cells and evaluated their myogenic potential using two directed differentiation methods10,11. HNK1-NCAM+ enrichment increased PAX7 and MYF5 expression by approximately 1.7-fold compared to unsorted hPSC-SMPCs (p<0.05, Figure 2A). In both protocols, HNK1-NCAM+ SMPCs could be grown in SkBM2+FGF2, and when induced to differentiate, the number of MYHC+ cells increased compared to dissociated/replated SMPCs (p<0.05, Figure 2B).
Figure 2. HNK1-NCAM+ increases myogenic cell numbers but does not increase myogenicity in vivo.
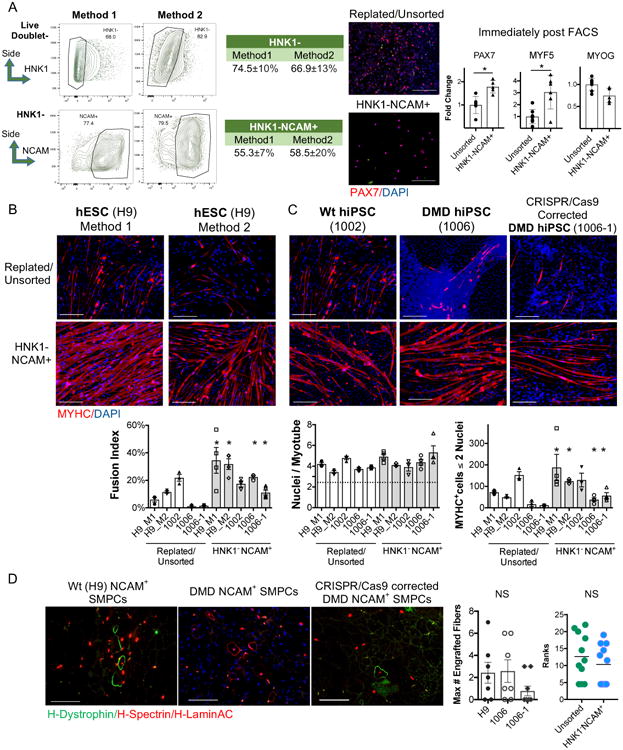
A. HNK1-NCAM+ FACS sorted hPSC-SMPCs have increased PAX7 (red) and MYF5 expression compared to replated/unsorted day 50 SMPCs by immunofluorescence (IF) and QPCR (Method 1; N=6 unsorted or N=5 NCAM hPSC-SMPCs from independent directed differentiations; mean ± SEM; two-tailed t-test; *p<0.05). Scale bar equals 100μm. FACS plots show mean ± SD of HNK1- and HNK1-NCAM+ as percentage of live non-doublet cells. B. HNK1-NCAM+ cells show increased myotube differentiation when hPSC-SMCPs are derived from two methods. C. HPSC myotubes differentiate independent of dystrophin expression. IF shows MYHC (red) and DAPI. Scale bar equals 200μm. Graph shows quantification of myotube fusion from all hPSC lines, (N=3 independent hPSC-myotube experiments, mean ± SEM, two-tailed t-test of NCAM vs. unsorted for each hPSC line; *p<0.05). D. HNK1-NCAM+ wild type, DMD and CRISPR corrected hPSC-SMPCs all engraft inefficiently in vivo. IF and quantification of H-LaminA/C+H-Spectrin+ (red) and H-dystrophin (green) are shown. Graphs quantify the maximum number of engrafted myofibers in a single cross section (left; mean ± SEM, two-tailed t-test; NS p>0.05), and the mean ± SEM of engrafted myofibers (N=7 mice per group) from multiple cross sections along the length of the muscle (right; Mann-Whitney U-test; NS p>0.05).
HPSC line variability is known to greatly affect the propensity of in vitro differentiation35,36 and some reports suggest DMD hiPSCs inefficiently differentiate to skeletal muscle in vitro33. After NCAM sorting, hiPSC-SMPCs from wild type, DMD and a previously generated CRISPR/Cas9-corrected DMD line25, could be differentiated to produce equivalent numbers of myotubes (Figure 2C). All hiPSC lines differentiated less well than the H9 hESC line. Thus, the absence or presence of dystrophin did not affect myogenic differentiation in vitro.
We next evaluated whether sorting on HNK1-NCAM+could improve the in vivo engraftment potential of hPSC-SMPCs. However, unlike in vitro, NCAM+ sorting did not improve engraftment compared to unsorted SMPCs (Figure 2D). Irradiation was unable to improve hPSC-SMPC engraftment potential (Table S1). In summary, enriching hPSC-SMPCs for HNK1-NCAM+ does not increase engraftment potential despite modestly increasing myogenic potential in vitro.
RNA Sequencing reveals HNK1-NCAM+ hPSC-SMPCs are heterogeneous and more differentiated than fetal muscle progenitor cells
To better understand functional differences between fetal myogenic cells, hPSC-SMPCs, and their differentiated progeny, we performed RNA-SEQ on unsorted, directly-isolated fetal muscle cells (all mononuclear), NCAM+ cultured fetal muscle cells and hPSC-SMPCs, and myotubes generated from both fetal muscle and hPSCs (Figure 3A). Genes related to myogenic progenitor identity, including PAX7 and MYF5, were enriched in cultured fetal muscle cells compared to hPSC-SMPCs (Table S2). In contrast, hPSC-SMPCs demonstrated substantial enrichment of TFs and structural proteins related to myogenic differentiation, such as MYOG. Principle component analysis confirmed hPSC-SMPCs clustered more closely with myotubes (Figure 3B). Moreover, Gene Ontology (GO) analysis identified multiple categories associated with development that were enriched in NCAM+ hPSC-SMPCs, suggesting that this population contains embryonic cell types. NCAM+ cultured fetal cells were also enriched for gene categories related to cell migration (Table S2). Comparison of directly-isolated fetal muscle cells to hPSC-SMPCs generated similar results, and GO analysis revealed potential factors, including HGF and LIF, associated with supporting fetal muscle (Table S3). While evaluation of all mononuclear cells from directly-isolated fetal muscle compared to cultured cells provided a comprehensive analysis of cells contained within the highest in vivo myogenic potential (Table S4), identification of potential surface markers was not feasible because of contaminating cell types (endothelium, hematopoietic) present in the unsorted fetal muscle cell samples.
Figure 3. RNA-SEQ identifies unique gene signatures in fetal and hPSC-derived muscle.
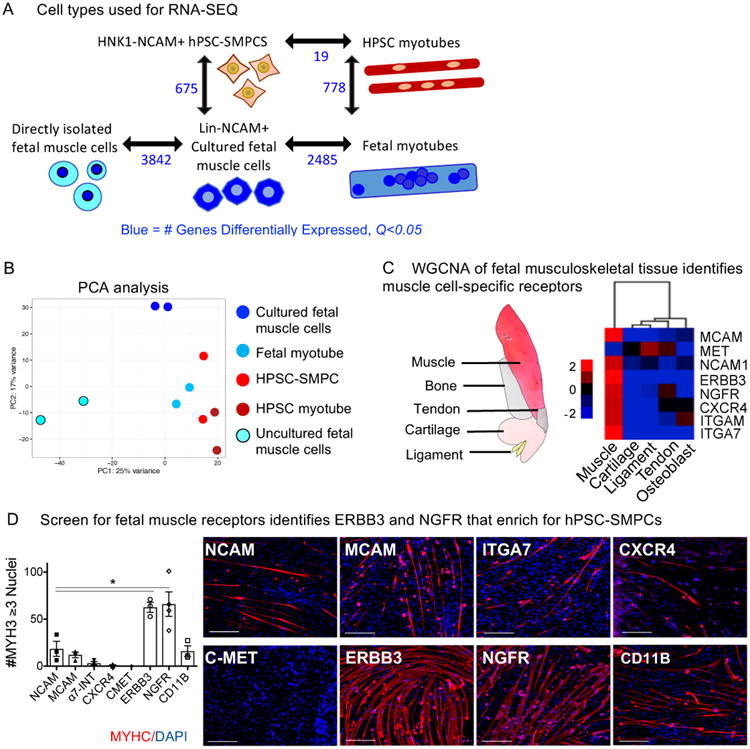
A. Differential gene expression using CuffDiff of all profiled cell types are shown (N=2, q<0.05, blue). B. Principal Component Analysis (PCA) analysis of the 5 cell types. Gene lists and key biological processes upregulated in hPSC-SMPCs, cultured fetal, or directly-isolated fetal muscle cells are shown in Tables S2-S4 (N=2; q<0.05). C. Illustration of profiled fetal musculoskeletal tissue is shown. WGCNA identification of candidate cell surface receptors enriched in fetal muscle cells versus other musculoskeletal tissues is shown. D. CRISPR/Cas9-corrected DMD hiPSC-SMPCs (1006-1) were sorted on eight candidate subpopulations and were fused in vitro and stained for MYHC (red) and DAPI (blue). Graph shows quantification of myotube differentiation (N=3 independent hPSC-myotube experiments; mean ± SEM; One-way ANOVA posthoc Dunnett; *p<0.05).
Muscle tissue-specific surface markers enable enrichment of myogenic cells from hPSC-SMPCs
To identify putative surface markers enriched on myogenic cells versus other skeletogenic lineages, we utilized the results of a Weighted Gene Co-Expression Network Analysis (WGCNA) performed across five fetal musculoskeletal tissues: muscle, bone, cartilage, ligament and tendon (Figure 3C and Ferguson et al., Submitted). In WGCNA, as modules of genes are defined based on their expression levels in each cell type, we chose candidate cell surface markers associated with myogenesis and/or enriched in the muscle module for further evaluation as possible markers of hPSC-SMPCs (Figure 3C). We next screened candidate receptors for ability to increase myogenic potential using the 1006-1 CRISPR/Cas9-corrected DMD-hiPSC line. Of the populations tested, cells positive for NGFR (CD271) and ERBB3 were the most myogenic and able to form myotubes more efficiently than NCAM+ cells in vitro (p<0.05, Figure 3D). Therefore, we chose these as two candidate surface markers to improve isolation of hPSC-SMPCs.
ERBB3 and NGFR surface marker levels demarcate a switch between primary and secondary fetal myogenesis and enrich for PAX7+ fetal muscle progenitor cells
RNA-SEQ identified two markers specific to week 17 fetal muscle capable of enriching hPSC-SMPCs. Based on our data, hPSC-SMPCs most closely resemble week 8-9 (late primary) muscle progenitors in their functional capacity. We hypothesized that ERBB3+ and NGFR+ cells would be present throughout human developmental myogenesis. As early as fetal week 8, a myogenic population of ERBB3+NGFR+ cells were identified that persisted throughout the time points examined (Figures 4A and S2). At weeks 8-9, the ERBB3+NGFR+ population enriched for several myogenic TFs (p<0.05, N=3). This is important because other known fetal markers, NCAM+MCAM+ and CD82, were either non-specific to muscle or not expressed at these time points, respectively. NCAM+MCAM+ and CD82 began to co-express with ERBB3+NGFR+ cells at fetal week 11.5 (Figure 4 and S2).
Figure 4. Increased myogenic ability resides in the ERBB3+NGFR+ fraction of human fetal muscle during primary and secondary myogenesis.
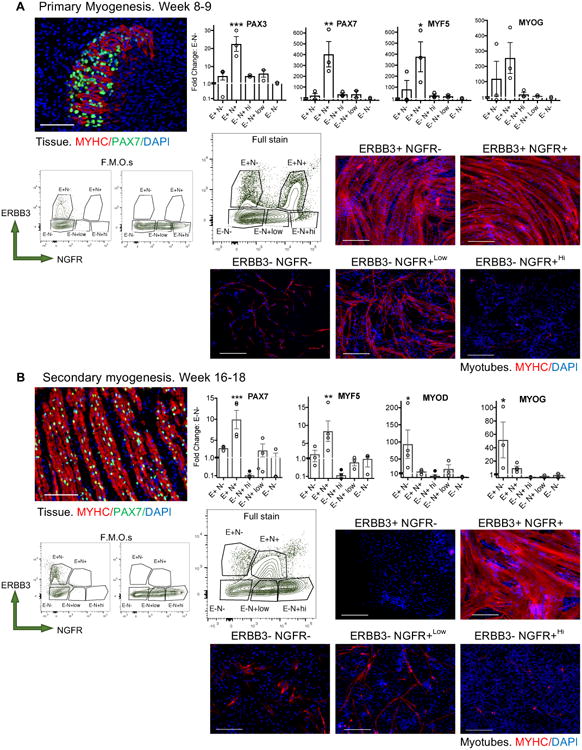
Five subpopulations of human myogenic cells were identified based ERBB3 and NGFR expression during A. primary myogenesis (weeks 8-10) and B. secondary myogenesis (fetal weeks 16-18).IF of fetal week 9 skeletal muscle tissue demonstrated establishment of the limb myofibers, and week 17 tissue demonstrated maturation to multinucleated myofibers. IF shows MYHC (red), PAX7 (green), and DAPI (blue). Scale bar equals 100μm.FACS plots of isotype controls and full stain (live, non-doublet CD45-CD31-CD235a- cells) show ERBB3+NGFR+ populations lose NGFR+Hi positivity during the transition from primary and secondary myogenesis. Statistical analyses of all fetal populations identified by FACS are shown in Figure S2. Graphs show fold change of myogenic gene expression of immediately sorted subpopulations compared to ERBB3-NGFR- cells (N=3 week 8-10 tissues and N=5 week 16-18 tissues; mean ± SEM; One-way ANOVA posthoc Dunnett; increase: *p<0.05, **p<0.01, ***p<0.001). Sorted populations were expanded in SkBM-2 for 24 hours and induced to differentiate to myotubes in N2 media for 5 days, highlight differences in in vitro myogenic potential between progenitors and subpopulations at different stages of human myogenesis. Myotubes were stained with MYHC (red) and DAPI (blue) (N=3 independent hPSC-myotube experiments). Scale bar equals 200μm.
By fetal week 17, two distinct ERBB3 subpopulations were identified containing myogenic activity (Figure 4B). ERBB3+NGFR+ cells enriched for PAX7 and MYF5 by 8-10-fold compared to ERBB3-NGFR- cells, (p<0.01, N=5), while ERBB3+NGFR- cells enriched for MYOD and MYOG by 40-fold (p<0.05); NGFR+LoERBB3- cells weakly expressed myogenic TFs, but did contain a population with some myogenic potential. Primary and secondary fetal muscle progenitors could also be distinguished by their positivity for ERBB3 and NGFR, as expression of NGFR was much higher during weeks 8-9 (Figure S2). Thus, the combination of these markers distinguished fetal muscle progenitors at different stages of development and differentiation. Upon sorting and differentiation to form myotubes, the ERBB3+NGFR+ fraction reached close to 100% fusion efficiency within 4 days, where as NGFR+LoERBB3- generated few myotubes (Figure 4B). ERBB3+NGFR- cells did not proliferate or form myotubes. We concluded that fetal ERBB3+NGFR+ cells are specifically enriched for PAX7 expressing myogenic progenitors during fetal myogenesis.
ERBB3 and NGFR enrich for PAX7 and myogenic capacity in hPSC-SMPCs
Based on ERBB3 and NGFR expression on fetal myogenic progenitors, we evaluated the ability of these markers to enrich hPSC-SMPCs. In all directed differentiation protocols tested, an ERBB3+NGFR+ SMPC population was detected as early as day 27 (Figure S3). By 50 days of directed differentiation, both protocols produced two distinct ERBB3 subpopulations, distinguished by NGFR expression, that resembled secondary myogenesis (Figure 5). Sorting these subpopulations revealed that ERBB3+NGFR+ hPSC-SMPCs enriched for PAX7 and MYF5 by 20-fold compared to ERBB3-NGFR- cells (p<0.001, N=4), while ERBB3+NGFR- cells were enriched for MYOD and MYOG (p<0.001). As expected, ERBB3+NGFR+SMPCs could be induced to form homogeneous myotubes. These data demonstrate a striking similarity between the surface marker phenotypes of fetal myogenic progenitors and hPSC-SMPCs undergoing directed differentiation.
Figure 5. Increased myogenic ability resides in the ERBB3+NGFR+ fraction of SMPCs from multiple hPSC lines and directed differentiation protocols.

Up to four myogenic populations were identified based on ERBB3 and NGFR expression after hPSC-SMPC directed differentiation. FACS gating of fluorescent minus one (F.M.O.) controls and full staining demonstrate variability and need to optimize ERBB3+NGFR+ sorting strategies across multiple hPSC lines and directed differentiation protocols. Shown are live non-doublet HNK1- cells from hESC, wild type, DMD and CRIPSR/Cas9 corrected hiPSC lines after 50 days of directed differentiation from method 1, or 27 days of directed differentiation from method 2 (method 1:N=5 H9, N=3 CDMD 1002, N=2 CDMD 1003, N=3 CDMD 1003-49, method 2: N=2 H9; where N=number of independent directed differentiations). Surface marker FACS analyses of hPSC-SMPCs from methods 1 and 2 are shown in Figure S3. Graphs show fold change of myogenic gene expression of immediately sorted subpopulations compared to ERBB3-NGFR- cells (N=3; mean ± SEM; One-way ANOVA posthoc Dunnett; increase: *p<0.05, ***p<0.001). Sorted subpopulations were expanded in SkBM-2 for 72 hours and induced to differentiate in N2 media for 5 days. IF evaluating the myogenic potential of each sorted and differentiated population is shown using MYH3 (red) and DAPI (blue) (N=3 independent hPSC-myotubes experiments). Scale bar equals 200μms. ERBB3+NGFR+ mark the majority of myogenic cells. FACS plots demonstrate in some protocols all ERBB3+ cells are positive for NGFR+.
To test the versatility of these markers, four hPSC lines, including DMD hiPSCs and isogenic CRISPR/Cas9-corrected DMD hiPSCs, were evaluated. Across all lines, myogenic potential was highly enriched in the ERBB3+NGFR+ subpopulation, as defined by expression of myogenic TFs and differentiation potential in vitro (Figure 5). However, double positivity based on NGFR levels varied across cell lines. Of note, upon passaging, NGFR levels increase and are associated with decreased myogenic potential in fetal and hPSC-SMPCs. Thus this previously unrecognized combination of surface markers informs on the SMPC differentiation state, in which the ERBB3+fraction was consistently the most myogenic.
TGF-β inhibition improves hPSC-SMPC differentiation and myotube fusion
Despite enabling myogenic enrichment, differentiation of ERBB3+NGFR+ populations isolated from hPSCs yielded myotubes that were often thinner and contained fewer nuclei than later stage fetal or adult counterparts. To understand the basis for these differences, we further analyzed the RNA-SEQ data comparing hPSC-myotubes and fetal myotubes to their progenitors. These data revealed that activators of TGF-β signaling decreased during the course of fetal myotube differentiation (Figure 6A). In contrast, hPSC-SMPCs had higher expression of TGF-β signaling genes (e.g. TGFβ1, ACVR1B and MSTN) and hPSC myotubes failed to downregulate TGF-β signaling (MSTN, INHBA and TGFβ2) relative to fetal myotubes.
Figure 6. Inhibition of TGFβ signaling induces hPSC skeletal muscle maturation.
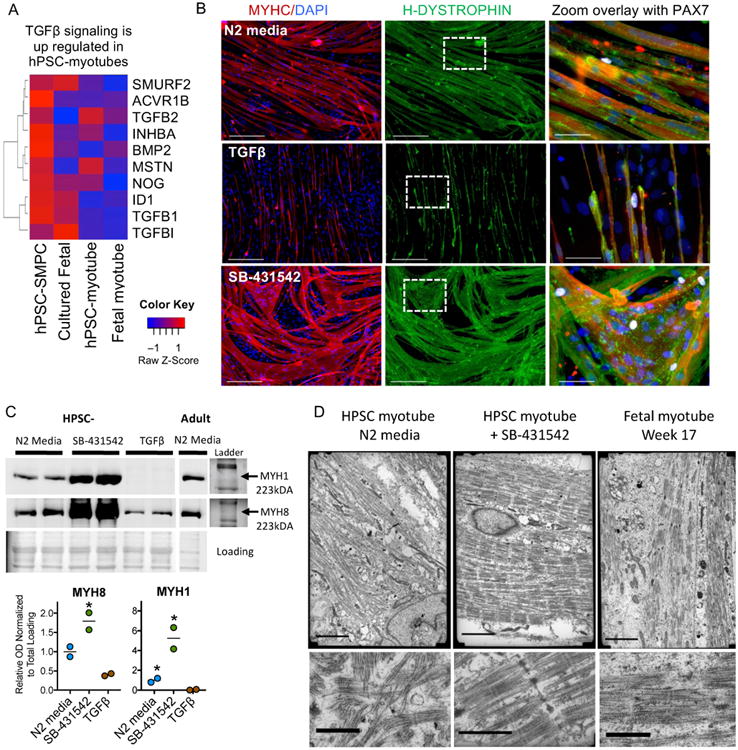
A. Heat map of TGF-β signaling genes reveals high TGF-β in hPSC myotubes as determined by RNA-SEQ FPKM values. B. ERBB3+ SMPCs from 1006-1 CRISPR/Cas9-corrected DMD hiPSCs were differentiated in N2 media alone, with recombinant TGF-β1, or in the presence of TGFβ inhibitor SB-431542 (N=6). HPSC-myotubes were stained with MYHC (red), H-Dystrophin (green), PAX7 (white), and DAPI (blue). Scale bar equals 200μms. Zoom images show no change in PAX7 expression with either treatment but an increase in myotube formation was seen with TGFβ inhibition. C. Western blot analysis showing TGFβ inhibitor SB-431542 treated hPSC-SMPCs have increased expression of MYH1 and MYH8 compared to N2 media alone or after treatment with recombinant TGFβ1 (N=2 independent myotube experiments; student t-test; *p<0.05). Ponceau S stained gel is shown for loading control. Ladder and unprocessed blots are included in Supplementary Figure 7. D. Transmission electron microscopy of fetal week 17 or NGFR+ hPSC-myotubes ± TGFβi demonstrate increased sarcomere organization and z-disk patterning after SB-431542 treatment (N=2 independent myotube experiments). Scale bar equals 3μms (top).
To test the function of TGF-β signaling in hPSC-derived myotube differentiation, we manipulated TGF-β during hPSC-SMPC to myotube formation. Compared to N2 media alone, addition of TGF-β1 completely inhibited myotube formation (p<0.05). In contrast, TGF-β inhibition (TGF-βi) using SB-431542 or A83-01 significantly increased hPSC-myotube fusion and produced morphology similar to late stage fetal myotubes (Figures 6 and S4). Single PAX7+ cells remained in hPSC-myotube cultures in the presence of either TGF-β1 or TGF-βi (Figure 6B), suggesting TGF-βi does not deplete PAX7+ SMPCs.
TGF-β regulation of hPSC myotube differentiation is independent of dystrophin expression. In two independent DMD and isogenic CRISPR/Cas9-corrected DMD hiPSC lines (1006 and 1003), the fusion indices of hPSC-SMPCs increased by 2-5 fold with TGF-βi compared to no treatment (Figure S4; p<0.05). These data demonstrate that inhibition of improper TGF-β levels during hPSC-SMPC differentiation can improve myogenesis in vitro.
TGF-β inhibition promotes hPSC myotube maturation in vitro
In mouse, TGF-β signaling is a potent negative regulator of fetal muscle maturity22,39; however, TGF-β has never been evaluated in human development. To test whether ectopic TGF-β signaling inhibited hPSC-SMPCs from forming more mature myotubes, we measured embryonic (MYH3), fetal (MYH8), and adult (MYH1) myosins at both the RNA and protein levels40. We found without TGF-βi, hPSC-myotubes predominately expressed embryonic and fetal myosins. Although ERBB3+NGFR+ enrichment increased expression of embryonic and fetal myosins relative to NCAM sorting (p<0.01), enrichment did not increase adult myosin expression (Figure S5).
Upon TGF-βi, expression of all myosins increased in hPSC-myotubes, and expression of MYH8 and MYH1 in ERBB3+NGFR+ hPSC myotubes equaled or surpassed that of fetal myotubes, respectively (Figure S5). Western blot analysis confirmed that TGF-βi also increased MYH8 and MYH1 protein levels in hPSC myotubes relative to adult myotubes (p<0.05, Figures 6C and S7). Interestingly, TGF-βi was more effective at inducing myosin expression in hPSC-SMPCs than in fetal or adult myogenic progenitors, which is in agreement with mouse embryonic and fetal myogenic progenitors22.
As another measure of maturation, we evaluated ultrastructural changes in hPSC-myotube morphology relative to fetal myotubes by transmission electron microscopy. In the absence of TGF-βi, NGFR+ hPSC-myotubes displayed disorganized sarcomere patterning (Figure 6D), while TGF-βi promoted formation of better-organized sarcomeres and z-disk patterning. In contrast, untreated fetal myotubes evidenced limited organization. These data demonstrate that TGF-βi facilitates hPSC-myotube maturity, and additively increases myogenicity of enriched hPSC-SMPC subpopulations.
In vivo engraftment of ERBB3+ hiPSC-SMPCs restores dystrophin to levels approaching uncultured fetal muscle
We hypothesized lack of engraftment by NCAM+ hPSC-SMPCs was due to the immature nature and heterogeneity of transplanted cells. We therefore tested whether ERBB3 or NGFR enrichment could improve engraftment in mdx-NSG mice. Single marker enrichment strategiesusingCRISPR/Cas9-corrected DMD hiPSC lines25 were tested as these may have more therapeutic relevance. Compared to NCAM+ sorting, both NGFR+ and ERBB3+ SMPCs significantly increased the number of engrafted myofibers as shown by h-LaminA/C+Spectrin+Dystrophin+ positivity (Table S1, p<0.05). These data demonstrated that ERBB3+ SMPCs are strongly enriched for cells capable of generating muscle in vivo.
As shown in Figure 6, myotubes generated from enriched SMPCs are immature due to precocious TGF-β signaling, and TGF-βi additively increased SMPC myogenicity. Co-delivery of TGF-βi during injection and for two weeks following transplantation increased numbers of h-LaminA/C+Spectrin+Dystrophin+ myofibers in all SMPCs subpopulations tested (Table S1). Mice injected with ERBB3+ hiPSC-SMPCs and treated with TGF-βi, improved the maximum number of h-dystrophin+ fibers to 137±22 per cross section (Figure 7A). Across all points measured, this represented a 50-fold greater engraftment than NCAM+ cellsalso treated with TGF-βi (p<0.001). A percentage of h-LaminA/C+ cells remained positive for myogenic or non-myogenic markers after engraftment in mdx-NSG mice at 30 days (Figure S6).
Figure 7. In vivo engraftment of CRISPR/Cas9-corrected DMD hiPSC-SMPCs restores dystrophin to levels approaching uncultured fetal muscle.
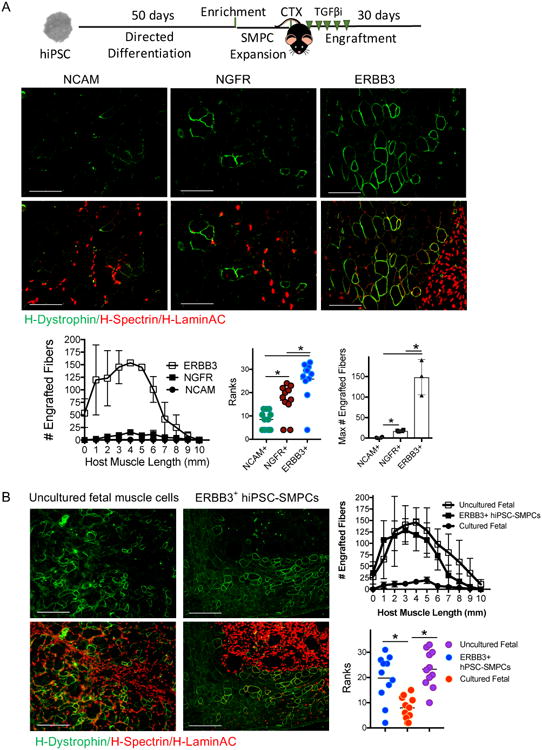
A. CRISPR/Cas9-corrected DMD hiPSC-SMPCs were enriched for surface markers and upon engraftment were co-delivered with the TGFβi (SB-431542). IF of h-Lamin A/C (red) denotes human cells, and j-Spectrin (red) and h-dystrophin (green) denote areas fused with mdx-NSG muscle fibers thirty days post engraftment. Graphs show the average ± SEM number of engrafted human+ myofibers from multiple cross sections along the length of the muscle (N=3 NCAM, N=4 NGFR, N=4 ERBB3 engrafted mice per group; Kruskal-Wallis ranks tests; *p<0.05, **p<0.01), and maximum number of engrafted myofibers in a single cross section (One-way ANOVA posthoc Tukey; *p<0.05). B. CRISPR/Cas9-corrected ERBB3+ SMPCs plus TGFβi engraft equivalent to uncultured (directly-isolated) fetal muscle cells. Graphs show the mean ± SEM number of engrafted myofibers from multiple cross sections along the length of the muscle (N=3 fetal SMPCs, N=4 ERBB3+ SMPCs engrafted mice per group; Kruskal-Wallis ranks tests; *p<0.05).
To evaluate how our hPSC-SMPC enrichment and maturation strategy compared with in vivo myogenic potential of cultured or directly-isolated fetal muscle cells, we quantified the number of engrafted myofibers at intervals throughout the TA of mdx-NSG mice, using Kruskal-Wallis rankstests (Figure 7B). ERBB3+ hPSC-SMPCs enabled engraftment surpassing cultured fetal muscle (p<0.003) and approached levels equivalent to directly-isolated fetal cells (p=0.43).Together, these results demonstrate a major advance in the ability to obtain engraftable SMPCs from hPSCs.
Discussion
Formany cell lineages, human development serves as a pivotal guide to generate equivalent progenitor cells from hPSCs41,42. Profiling hPSC-SMPCs to human fetal muscle established that directed differentiation produces embryonic myocytes that are less functional than human fetal or adult SCs. The best described marker for isolating hPSC-SMPCs, NCAM, is expressed on many cell types during development including neural cells, muscle progenitors, myotubes and other mesodermal progenitors37 and is not sufficient for progenitor enrichment in directed differentiation cultures. Enrichment of muscle progenitors using the surface markers identified in this study and targeting dysregulated signal pathways identified in RNA-SEQ data, enabled maturation of hPSC-SMPCs with significantly greater myogenic potential in vitro and in vivo.
Since we found that hPSC-SMPCs are embryonic, we evaluated progenitors from primary and secondary human myogenesis to identify more relevant enrichment markers. We demonstrated ERBB3+NGFR+ expression enriches for PAX7+ and MYF5+ cells during the first and second trimesters of human development, including at time points when other surface markers cannot distinguish fetal muscle progenitors43. Shifts in ERBB3+NGFR+ expression levels reliably demarcated transitions between early and late waves of human fetal myogenesis, and correlated to the establishment of primary limb myofibers or maturation of secondary fetal myofibers. ERBB3+NGFR+ expression was also able to demarcate progenitors from more differentiated MYOD+ cells; thus, expression levels of this marker combination may serve as an important tool for studying human SMPC biology. Upon searching a recent fetal dataset, we found ERBB3 is also expressed by human fetal MCAM+ cells43. Likewise, we found these markers enrich for myogenic SMPCs from heterogeneous hPSC cultures across multiple directed differentiation protocols10,11,13,19.
While ERBB3 has been identified in the initial phases of adult mouse SC activation44,45, the mechanism by which ERBB3 regulates muscle progenitors during human fetal myogenesis prior to adult SC generation or quiescence, and in hPSC-SMPCs, is unknown. In mouse development, deficiencies in Neuregulin-1 signaling, the primary ligand of ERBB3, results in loss of self-renewal of fetal muscle progenitors46 and suggests ERBB3 signaling may regulate the balance of fetal myogenic cell fate by preventing precocious SMPC differentiation. We hypothesize that NRG1 and ERBB3 signaling may also have important roles in specification and support of PAX7 cells including in hPSC-SMPC cultures. In mouse studies, muscle progenitor cells expressing ERBB3 or NGFR are responsive to paracrine signals secreted by neural crest or neuronal cells46,47. During hPSC directed differentiation, SMPCs may be supported by a local micro-environment that includes three dimensional cellular structures and neuronal cells48.As several biological processes associated with neuronal cells and transmission of neural impulses were identified by RNA-SEQ in hPSC-SMPC cultures, we expect that neuronal cells are important for hPSC myogenesis throughout directed differentiation.
A critical bottleneck to all hPSC directed differentiation protocols is the need to mature progenitors towards functionally relevant cell types. In mouse development, activation of TGF-β signaling prevents embryonic muscle progenitor cells from undergoing secondary myogenesis39,49. In a similar manner, we found several members of the TGF-β superfamily were dysregulated in hPSC-myotubes compared to human fetal myotubes. Our data provide multiple lines of evidence that TGF-βi is a major driver of hPSC maturation towards secondary or tertiary myogenesis. Additional strategies or factors that promote SMPC transition to SCs will be required as TGF-βi is only important for enhancing maturation of differentiated myotubes in vitro or myofibers in vivo.
DMD is a devastating muscle disease caused by out-of-frame mutations in the gene encoding dystrophin. We have shown that CRISPR/Cas9 mediated genetic deletion of DMD-hiPSCs can restore the dystrophin reading frame25. However, targeting muscle stem cells with CRISPR/Cas9 in vivo is inefficient50. Correction of patient-derived hiPSCs ex vivo and subsequent differentiation to a human SC equivalent offers one potential route for delivery of SMPCs resulting in dystrophin restoration after transplantation. We have shown directly-isolated human fetal muscle cells can restore several hundred dystrophin+ myofibers to levels speculated to be required for functional clinical gain30,31,32.We also demonstrated proof of concept that CRISPR/Cas9-corrected DMD hiPSC-SMPCs can be directed to differentiate and restore dystrophin in mdx-NSG mice to levels approaching those of directly-isolated fetal cells. It will be important to understand differences in PAX7+ cell states during development, as fetal cells maintain PAX7 expression independent of the adult SC niche23. Deriving methods to expand and support PAX7+ cells from fetal, adult, or hPSC-SMPCs without activating MYOD, and understanding regulation of self-renewal potential, will be required to develop a cell replacement therapy for diseases including DMD51.
In summary, directed differentiation protocols which more closely model human development may better recapitulate cell-specific lineages and thus lead to better in vitro models or improved clinical utility. We utilized human fetal myogenesis to identify previously unreported candidates for enriching and maturing hPSCs myogenic activity in vitro and in vivo. Furthering our understanding of human developmental myogenesis will provide inroads to regenerative approaches for muscle diseases including in combination with gene-edited patient derived hiPSCs.
Supplementary Material
Figure S1. Directed differentiation of human pluripotent stem cells to skeletal muscle progenitor cells.
Figure S2. Characterization of ERBB3 and NGFR expression at multiple stages of human developmental myogenesis.
Figure S3. Characterization of ERBB3 and NGFR across hPSC-SMPC directed differentiation.
Figure S4. TGFβ inhibition induces myotube fusion in hPSC-SMPCs and acts independently from dystrophin expression.
Figure S5. Evaluation of myosin expression by muscle tissue and myotubes derived from adult, fetal, and hPSCs ± TGFβi.
Figure S6. Expression of myogenic and non-myogenic markers by engrafted hPSC-SMPCs. Related to Figure 7.
Figure S7. Unprocessed scan of western blot gels and loading controls.
Acknowledgments
The authors would like to thank Shahab Younesi, Jamie Marshall, Michael Emami, Elena Korsakova, Kholoud Saleh, Valerie Rezek, Jane Wen, and Chino Kumagai-Cresse for helping with stem cell culture and mouse experiments. Thanks to Ekatrina Mokhonova for performing the western blots. We would also like to thank Jenny Morgan's lab for training on cell engraftments. The following cores were utilized: Center for Duchenne Muscular Dystrophy (CDMD) Muscle Phenotyping and Imaging Core, High Throughput and Cell Repository Core, and Bioinformatics and Genomics Core; the Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research at UCLA (BSCRC) Flow Cytometry Core; the UCLA Technology Center for Genomics and Bioinformatics, Jonsson Comprehensive Cancer Center (JCCC) Electron Microscopy Core, and Center for AIDS Research (CFAR) Flow Cytometry Core (NIH P30CA016042, 5P30AI028697), the UCLA Humanized Mouse Core (CFAR, NIAID AI028697). M.R.H. is the recipient of a BSCRC and Schaffer Fellowship, a CDMD-Cure Duchenne Fellowship, and a CDMD-NIH Paul Wellstone Center Training Fellowship (U54 AR052646). D.E. was funded by an NIH grant K01AR061415, DOD grant W81XWH-13-1-0465 and CIRM grant RB5-07230.Funding was provided by NIAMS R01AR064327 to A.D.P., UCLA Muscular Dystrophy Core Center grant (NIAMS 5P30AR05723), the CDMD at UCLA, the NIH/NCATS, UCLA CTSI (UL1TR000124), BSCRC Research Award (A.D.P), Rose Hills Foundation Research Award to A.D. P., and a CIRM Inception and CIRM Quest (DISC1-08823 and DISC2-08824 to A.D.P.).
Footnotes
Author Contributions: Conceptualization, M.R.H., B.VH., A.D.P.; Methodology, M.R.H., B.VH., M.J.S.; Formal Analysis, A.E., B.VH., S.F.N., M.R.H.; Investigation, M.R.H., B.VH., J.H., K.P., W.F., M.J., H.X., C.S.Y.; Writing – Original Draft, M.R.H., A.D.P.; Writing – Review & Editing, M.R.H., A.D.P.; B.VH., J.H., K.P., C.S.Y., M.J.S., S.F.N.; Funding Acquisition, M.R.H., A.D.P., D.E.; Resources, D.E.; Supervision, A.D.P.
References
- 1.Chong JJH, et al. Human embryonic-stem-cell-derived cardiomyocytes regenerate non-human primate hearts. Nature. 2014;510:273–277. doi: 10.1038/nature13233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schwartz SD, et al. Human embryonic stem cell-derived retinal pigment epithelium in patients with age-related macular degeneration and Stargardt's macular dystrophy: follow-up of two open-label phase 1/2 studies. The Lancet. 2014;385:509–516. doi: 10.1016/S0140-6736(14)61376-3. [DOI] [PubMed] [Google Scholar]
- 3.Steinbeck JA, et al. Optogenetics enables functional analysis of human embryonic stem cell-derived grafts in a Parkinson's disease model. Nat Biotech. 2015;33:204–209. doi: 10.1038/nbt.3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hrvatin S, et al. Differentiated human stem cells resemble fetal, not adult, β cells. Proc Natl Acad Sci U S A. 2014;111:3038–3043. doi: 10.1073/pnas.1400709111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Witty AD, et al. Generation of the epicardial lineage from human pluripotent stem cells. Nat Biotech. 2014;32:1026–1035. doi: 10.1038/nbt.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang X, Pabon L, Murry CE. Engineering adolescence: maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Res. 2014;114:511–523. doi: 10.1161/CIRCRESAHA.114.300558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jonsson MK, et al. Application of human stem cell-derived cardiomyocytes in safety pharmacology requires caution beyond hERG. J Mol Cell Cardiol. 2012;52:998–1008. doi: 10.1016/j.yjmcc.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 8.Borchin B, Chen J, Barberi T. Derivation and FACS-mediated purification of PAX3+/PAX7+ skeletal muscle precursors from human pluripotent stem cells. Cell Reports. 2013;21:620–631. doi: 10.1016/j.stemcr.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu C, et al. A zebrafish embryo culture system defines factors that promote vertebrate myogenesis across species. Cell. 2013;155:909–921. doi: 10.1016/j.cell.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chal J, et al. Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat Biotech. 2015;33:962–969. doi: 10.1038/nbt.3297. [DOI] [PubMed] [Google Scholar]
- 11.Shelton M, et al. Derivation and Expansion of PAX7-Positive Muscle Progenitors from Human and Mouse Embryonic Stem Cells. Stem Cell Reports. 2014;3:516–529. doi: 10.1016/j.stemcr.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chal J, et al. Generation of human muscle fibers and satellite-like cells from human pluripotent stem cells in vitro. Nat Protoc. 2016;11:1833–1850. doi: 10.1038/nprot.2016.110. [DOI] [PubMed] [Google Scholar]
- 13.Swartz EW, et al. A Novel Protocol for Directed Differentiation of C9orf72-Associated Human Induced Pluripotent Stem Cells Into Contractile Skeletal Myotubes. Stem Cells Transl Med. 2016;5:1461–1472. doi: 10.5966/sctm.2015-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abujarour R, et al. Myogenic differentiation of muscular dystrophy-specific induced pluripotent stem cells for use in drug discovery. Stem Cells Transl Med. 2014;3:149–160. doi: 10.5966/sctm.2013-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kimura E, et al. Cell-lineage regulated myogenesis for dystrophin replacement: a novel therapeutic approach for treatment of muscular dystrophy. Hum Mol Genet. 2008;17:2507–2517. doi: 10.1093/hmg/ddn151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darabi R, et al. Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell. 2012;10:610–619. doi: 10.1016/j.stem.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Darabi R, et al. Functional skeletal muscle regeneration from differentiating embryonic stem cells. Nat Med. 2008;14:134–143. doi: 10.1038/nm1705. [DOI] [PubMed] [Google Scholar]
- 18.Sambasivan R, Tajbakhsh S. Skeletal muscle stem cell birth and properties. Seminars in Cell & Developmental Biology. 2007;18:870–882. doi: 10.1016/j.semcdb.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 19.Xi H, et al. In Vivo Human Somitogenesis Guides Somite Development from hPSCs. Cell Rep. 2017;18:1573–1585. doi: 10.1016/j.celrep.2017.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loh KM, et al. Mapping the Pairwise Choices Leading from Pluripotency to Human Bone, Heart, and Other Mesoderm Cell Types. Cell. 2016;166:451–467. doi: 10.1016/j.cell.2016.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin H, Price F, Rudnicki MA. Satellite Cells and the Muscle Stem Cell Niche. Physiological Reviews. 2013;93:23–67. doi: 10.1152/physrev.00043.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biressi S, et al. Myf5 expression during fetal myogenesis defines the developmental progenitors of adult satellite cells. Dev Biol. 2013;379:195–207. doi: 10.1016/j.ydbio.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tierney Matthew T, et al. Autonomous Extracellular Matrix Remodeling Controls a Progressive Adaptation in Muscle Stem Cell Regenerative Capacity during Development. Cell Reports. 2016;14:1940–1952. doi: 10.1016/j.celrep.2016.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sacco A, Doyonnas R, Kraft P, Vitorovic S, Blau HM. Self-renewal and expansion of single transplanted muscle stem cells. Nature. 2008;456:502–506. doi: 10.1038/nature07384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Young Courtney S, et al. A Single CRISPR-Cas9 Deletion Strategy that Targets the Majority of DMD Patients Restores Dystrophin Function in hiPSC-Derived Muscle Cells. Cell Stem Cell. 2016;18:533–540. doi: 10.1016/j.stem.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tierney MT, Sacco A. Satellite Cell Heterogeneity in Skeletal Muscle Homeostasis. Trends Cell Biol. 2016;26:434–444. doi: 10.1016/j.tcb.2016.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Castiglioni A, et al. Isolation of progenitors that exhibit myogenic/osteogenic bipotency in vitro by fluorescence-activated cell sorting from human fetal muscle. Stem Cell Reports. 2014;2:92–106. doi: 10.1016/j.stemcr.2013.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Montarras D, et al. Direct Isolation of Satellite Cells for Skeletal Muscle Regeneration. Science. 2005;309:2064–2067. doi: 10.1126/science.1114758. [DOI] [PubMed] [Google Scholar]
- 29.Chan Y, Walmsley RP. Learning and understanding the Kruskal-Wallis one-way analysis-of-variance-by-ranks test for differences among three or more independent groups. Phys Ther. 1997;77:1755–1762. doi: 10.1093/ptj/77.12.1755. [DOI] [PubMed] [Google Scholar]
- 30.Cerletti M, et al. Highly Efficient, Functional Engraftment of Skeletal Muscle Stem Cells in Dystrophic Muscles. Cell. 2008;134:37–47. doi: 10.1016/j.cell.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Godfrey C, et al. How much dystrophin is enough: the physiological consequences of different levels of dystrophin in the mdx mouse. Hum Mol Genet. 2015;24:4225–4237. doi: 10.1093/hmg/ddv155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharp PS, Bye-a-Jee H, Wells DJ. Physiological characterization of muscle strength with variable levels of dystrophin restoration in mdx mice following local antisense therapy. Mol Ther. 2011;19:165–171. doi: 10.1038/mt.2010.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi IY, et al. Concordant but Varied Phenotypes among Duchenne Muscular Dystrophy Patient-Specific Myoblasts Derived using a Human iPSC-Based Model. Cell Rep. 2016;15:2301–2312. doi: 10.1016/j.celrep.2016.05.016. [DOI] [PubMed] [Google Scholar]
- 34.Xu X, et al. Human Satellite Cell Transplantation and Regeneration from Diverse Skeletal Muscles. Stem Cell Reports. 2015;5:419–434. doi: 10.1016/j.stemcr.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cahan P, Daley GQ. Origins and implications of pluripotent stem cell variability and heterogeneity. Nat Rev Mol Cell Biol. 2013;14:357–368. doi: 10.1038/nrm3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Osafune K, et al. Marked differences in differentiation propensity among human embryonic stem cell lines. Nat Biotechnol. 2008;26:313–315. doi: 10.1038/nbt1383. [DOI] [PubMed] [Google Scholar]
- 37.Evseenko D, et al. Mapping the first stages of mesoderm commitment during differentiation of human embryonic stem cells. Proc Natl Acad Sci U S A. 2010;107:13742–13747. doi: 10.1073/pnas.1002077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prashad SL, et al. GPI-80 defines self-renewal ability in hematopoietic stem cells during human development. Cell Stem Cell. 2015;16:80–87. doi: 10.1016/j.stem.2014.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cusella-De Angelis MG, et al. Differential response of embryonic and fetal myoblasts to TGF beta: a possible regulatory mechanism of skeletal muscle histogenesis. Development. 1994;120:925–933. doi: 10.1242/dev.120.4.925. [DOI] [PubMed] [Google Scholar]
- 40.Schiaffino S, Rossi AC, Smerdu V, Leinwand LA, Reggiani C. Developmental myosins: expression patterns and functional significance. Skelet Muscle. 2015;5:22. doi: 10.1186/s13395-015-0046-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pagliuca Felicia W, et al. Generation of Functional Human Pancreatic β Cells In Vitro. Cell. 2104;159:428–439. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maroof AM, et al. Directed differentiation and functional maturation of cortical interneurons from human embryonic stem cells. Cell Stem Cell. 2013;12:559–572. doi: 10.1016/j.stem.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alexander MS, et al. CD82 Is a Marker for Prospective Isolation of Human Muscle Satellite Cells and Is Linked to Muscular Dystrophies. Cell Stem Cell. 2016;19:800–807. doi: 10.1016/j.stem.2016.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Figeac N, Serralbo O, Marcelle C, Zammit PS. ErbB3 binding protein-1 (Ebp1) controls proliferation and myogenic differentiation of muscle stem cells. Dev Biol. 2014;386:135–151. doi: 10.1016/j.ydbio.2013.11.017. [DOI] [PubMed] [Google Scholar]
- 45.Golding JP, Calderbank E, Partridge TA, Beauchamp JR. Skeletal muscle stem cells express anti-apoptotic ErbB receptors during activation from quiescence. Exp Cell Res. 2007;313:341–356. doi: 10.1016/j.yexcr.2006.10.019. [DOI] [PubMed] [Google Scholar]
- 46.Ho Andrew, Tri V, et al. Neural Crest Cell Lineage Restricts Skeletal Muscle Progenitor Cell Differentiation through Neuregulin1-ErbB3 Signaling. Developmental Cell. 2011;21:273–287. doi: 10.1016/j.devcel.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 47.Deponti D, et al. The Low-Affinity Receptor for Neurotrophins p75(NTR) Plays a Key Role for Satellite Cell Function in Muscle Repair Acting via RhoA. Molecular Biology of the Cell. 2009;20:3620–3627. doi: 10.1091/mbc.E09-01-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Esteves de Lima J, Bonnin MA, Birchmeier C, Duprez D. Muscle contraction is required to maintain the pool of muscle progenitors via YAP and NOTCH during fetal myogenesis. Elife. 2016;5 doi: 10.7554/eLife.15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manceau M, et al. Myostatin promotes the terminal differentiation of embryonic muscle progenitors. Genes Dev. 2008;22:668–681. doi: 10.1101/gad.454408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arnett AL, et al. Adeno-associated viral (AAV) vectors do not efficiently target muscle satellite cells. Mol Ther Methods Clin Dev. 2014;1:1–10. doi: 10.1038/mtm.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quarta M, et al. An artificial niche preserves the quiescence of muscle stem cells and enhances their therapeutic efficacy. Nat Biotechnol. 2016;34:752–759. doi: 10.1038/nbt.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Directed differentiation of human pluripotent stem cells to skeletal muscle progenitor cells.
Figure S2. Characterization of ERBB3 and NGFR expression at multiple stages of human developmental myogenesis.
Figure S3. Characterization of ERBB3 and NGFR across hPSC-SMPC directed differentiation.
Figure S4. TGFβ inhibition induces myotube fusion in hPSC-SMPCs and acts independently from dystrophin expression.
Figure S5. Evaluation of myosin expression by muscle tissue and myotubes derived from adult, fetal, and hPSCs ± TGFβi.
Figure S6. Expression of myogenic and non-myogenic markers by engrafted hPSC-SMPCs. Related to Figure 7.
Figure S7. Unprocessed scan of western blot gels and loading controls.