

Abstract
The identification of protein ligands from a DNA-encoded library is commonly conducted by an affinity selection assay. These assays are often not validated for robustness, raising questions about selections that fail to identify ligands and the utility of enrichment values for ranking ligand potencies. Here, we report a method for optimizing and utilizing affinity selection assays to identify potent and selective peptidic ligands to the highly related chromodomains of CBX proteins. To optimize affinity selection parameters, statistical analyses (Z′ factors) were used to define the ability of selection assay conditions to identify and differentiate ligands of varying affinity. A DNA-encoded positional scanning library of peptidomimetics was constructed around a trimethyllysine-containing parent peptide, and parallel selections against the chromodomains from CBX8 and CBX7 were conducted over three protein concentrations. Relative potencies of off-DNA hit molecules were determined through a fluorescence polarization assay and were consistent with enrichments observed by DNA sequencing of the affinity selection assays. In addition, novel peptide-based ligands were discovered with increased potency and selectivity to the chromodomain of CBX8. The results indicate low DNA tag bias and show that affinity-based in vitro selection assays are sufficiently robust for both ligand discovery and determination of quantitative structure–activity relationships.
Keywords: DNA-encoded libraries, affinity selection assay, chromobox (CBX) proteins, chromodomains, peptidomimetics
Introduction
In vitro selections allow for libraries of DNA–small molecule conjugates to be collectively assayed for a desired function, usually binding to a target protein, as a single pooled sample.1 With DNA-encoded libraries, this is generally accomplished using an affinity pull-down approach with an immobilized target protein.2 This approach has led to the identification of numerous hits for drug development efforts and allows for the screening of libraries at a much lower cost per compound than traditional high-throughput screening (HTS).3 In contrast to assays for HTS, robustness parameters for in vitro selection assays are not well established. In the case of HTS, the considerations are primarily well-to-well variability. The in vitro selection, however, is a one-well assay, which provides the advantage that assay variability is applied equally to each compound in the library. Still, a number of considerations remain that contribute to the robustness of in vitro selection assays and their potential for success. These factors can include DNA tag bias, differential enrichment of ligands of varying affinities, absolute recovery of ligands, and fold enrichment over nonligands (signal to background). In this report, we investigate the optimization of affinity selection assays for CBX chromodomains (ChDs) and validate these assays in the selection of ligands from a positional scanning library (PSL) of peptides and peptidomimetics.
An affinity binding selection using an immobilized, pure protein target is the predominant approach to ligand discovery from DELs. Altered equilibrium effects of solid supports and unknown specific activities of immobilized proteins make it difficult to predict an effective protein concentration. This makes empirical assay optimization the best approach, but it is not always possible. In order to optimize selection parameters prior to a DEL selection, control selections can be conducted using a known DNA-linked ligand to your target protein and monitoring absolute recovery and enrichment by quantitative PCR (qPCR) relative to a nonligand.4–6 Much like increased DNA recovery or phage titer can indicate library convergence in SELEX and phage display approaches, qPCR quantitation of total DNA recovery after a selection assay can be used to indicate the successful enrichment of binders.7,8 The differential enrichment of ligands based on their affinity constants to the target protein is highly desirable, yet difficult to achieve. Without incurring dramatic losses of ligands from the library (no less than 0.1% recovery), a given affinity selection assay has a limited dynamic range for differential enrichment based on affinity (~1–2 orders of magnitude).9 In addition, it is difficult to determine how significant small changes in enrichment are. Setting selection stringency appropriately is critical. Prior work from Neri and colleagues has demonstrated a correlation between the normalized sequence counts and affinities from a series of serum albumin binders identified from a 100,000-membered DEL.10 Selections from more complex libraries have often failed to show correlations, which is likely due to complexities introduced from high variation in synthetic yield among library members.11 Further evaluation of selection assays is needed as the use of DELs continues to increase in ligand discovery.
We are interested in using DELs to develop tool compounds to probe the chromatin binding activities of the polycomb repressive complex 1 (PRC1). This complex contains one of five ChD-containing CBX paralogs (CBX2, CBX4, CBX6, CBX7, or CBX8).12 The ChDs of the CBX proteins localize PRC1 on the genome by recognition and binding to trimethylated lysine 27 of histone H3.13 PRC1 binding initiates chromatin compaction, which leads to transcriptional silencing. Although the same basic epigenetic regulation mechanism has been proposed for all five CBX paralogs, they have nonredundant functions in development14 and cancer.15 In particular, the CBX8 protein has unique roles in breast cancer,16 lymphoma,17 glioblastoma multiforme,18 and leukemia with MLL translocations.19 The polycomb CBX paralogs have high sequence and structural similarity to the heterochromatin protein 1 (HP1) CBX proteins: CBX1, CBX3, and CBX5.12 The similarities of CBX ChDs (Suppl. Fig. 1) has made development of specific chemical probes and inhibitors challenging. In addition, the shallow, poorly defined binding pockets of ChDs are less amenable to targeting, with conventional small molecules hampering drug design efforts.20 Recent studies with modified peptides (5- to 6-mers) have identified potent (<1 μM) inhibitors for the CBX4, CBX6, and CBX7 ChDs, yet specific (>5-fold selectivity) ligands have only been identified for CBX6 ChD.21–24 Neither potent nor specific ligands have been reported for the CBX8 ChD. The lack of paralog-specific inhibitors has limited the understanding of the CBX proteins in cancer and developmental biology. Further development of more drug-like and selective ligands to the CBX family would provide valuable chemical probes for deciphering paralog-specific functions, as well as validation of these proteins as therapeutic targets.20–26 CBX7 has been the primary target of previous studies, and development of selective ligands against other CBX or ChD-containing proteins has been less explored. The most potent inhibitor of CBX7 to date is the peptide ligand UNC3866 with a reported Kd of 97 nM, which shows 6- to 18-fold selectivity to CBX7 over other CBX proteins and ChDs.21
Structure–activity relationships (SARs) for short peptide ligands binding to the ChD of CBX7 have been explored by the labs of Hof23–25 and Frye.21,22 Briefly, a hydrogen bond donor, such as serine, is favored at the C-terminal residue at the P(1) position (relative to trimethyllysine [Kme3]). The C-terminus is hypothesized to be solvent exposed when bound to the ChD and was the site of a fluorophore attachment for use in fluorescence anisotropy assays.22,23,25 Lipophilic amino acids at the (–1) position are favored,22,25 and the (–2) position is proposed to be the most important site for specificity toward specific CBX homologs. CBX7 tolerates only alanine at this position, but CBX6 and CBX8 additionally tolerated both 2-aminobutyric acid (ethyl side chain) and valine.23 The large lipophilic residues (e.g., Phe) are preferred at the (–3) amino acid, yet this side chain is proposed to be solvent exposed.22 N-terminal benzoyl caps, particularly those with lipophilic 4-substitutions, have been demonstrated to be crucial for increasing the affinity of peptide inhibitors for CBX7.21–25 While these examples provide extensive SAR around targeting CBX proteins, CBX7 was the primary target of these studies, and selectively against other CBX or ChD-containing proteins has been less explored.
The extensive SARs of these CBX7 ligands provide a valuable data set to validate the capabilities of an affinity selection assay. In this report, we used three previously reported ligands for CBX7 to optimize affinity selection assays for CBX7 and CBX8. We then performed parallel synthesis of a DNA-encoded PSL. Parallel selection assays to both CBX7 and CBX8 ChDs were conducted with the library under three conditions with the dual goals of assay validation and informing the development of selective ligands to CBX8.
Materials and Methods
Oligonucleotides were purchased from IDT (Coralville, IA) or Bioneer (Alameda, CA) and used as provided. Analytical high-performance liquid chromatography (HPLC) separations were completed using an Agilent 1100 system with detection at 260 nm using a water/MeCN gradient containing 100 mM triethylammonium acetate, pH 5.5. Preparative HPLC separations were completed using a Varian ProStar system with detection at 260 and 280 nm using a water/MeOH gradient containing 0.75% hexafluoroisopropanol, 0.0035% triethylamine, pH 7.0. Reagents and solvents were used as received from commercial sources.
Preparation of 96 Single 140-Mer dsDNA Constructs
The integrated polymerase chain assembly (PCA)–PCR experiments were used to generate 96 single-gene barcode DNA constructs using a modified procedure from TerMaat et al.27 For each reaction, six pairs of complementary 40-mer DNA oligonucleotides (Suppl. File 1) were used. Six 40-mer oligos were pooled and used as templates for PCA. Each 5.0 μL PCA reaction contained 0.2 μM of each template 40-mer, with the following: 1.0 mM dNTPs, 0.1 U/μL of Vent DNA polymerase in 1× DNA polymerase buffer (NEB). All thermocycle procedures were as follows: 3 min at 94 °C, then cycling for denaturation at 94 °C for 15 s, annealing at 58 °C for 15 s, extension at 72 °C for 30 s, and a final extension of 72 °C for 5 min after 20 cycles. Each 50 μL PCR contained the entire 5 μL PCA reaction, 0.2 mM of each dNTP, 0.4 μM of each end primer, and 0.025 U/μL Dream Taq DNA polymerase in 1× Dream Taq buffer (Thermo Fisher, Waltham, MA). The successive PCR went for 20 cycles using the same thermocycling conditions as PCA. Following PCR, each reaction was purified by a solid-phase reversible immobilization (SPRI) purification using Sera-Mag Carboxylate-Modified Magnetic SpeedBeads (GE Healthcare, Pittsburgh, PA) as previously reported28 and quantified by UV absorbance at 260 nm.
CBX ChD Protein Expression and Purification
Addgene plasmid 25241 (CBX7) and a CBX8 plasmid (provided by Cheryl Arrowsmith)13 were transformed into chemically competent BL21 CodonPlus RIL Escherichia coli cells (Stratagene, La Jolla, CA) as N-terminal His6-tagged proteins. Bacterial growth was completed at 37 °C in LB media to OD600 = 2.0, followed by reducing the temperature to 16 °C over 30–60 min, and induced with 1 mM IPTG for 16 h. Cells were collected by centrifugation and resuspended in ChD binding buffer (20 mM Tris, pH 8, 150 mM NaCl, 0.01% Tween-20, 20 mM imidazole) with 1.0 mM PMSF. Bacteria pellets were stored at –20 °C until needed. Pellets were thawed on ice for 10 min in ChD binding buffer with 1 mg/mL CHAPS and 1 mM PMSF, and lysed by sonication (15 W, 30 s on, followed by 30 s off, twice, followed by 20 W for 1 min). The solubilized fraction was collected by centrifugation at 5000g for 20 min at 4 °C. Meanwhile, Ni-NTA Agarose resin (QIAGEN, Venlo, Netherlands) was washed with H2O and equilibrated with ChD binding buffer. The soluble fraction was incubated with the prewashed Ni-NTA agarose resin at 4 °C for 2 h. The resin was then washed 3× with ChD purification buffer (20 mM Tris, pH 8, 150 mM NaCl, 0.01% Tween-20, 1 mM PMSF). Proteins were eluted by the addition of 0.5 M imidazole to ChD purification buffer. The elution was diluted with glycerol, flash frozen, and stored at –80 °C until needed. Protein purity was assessed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and concentration was determined by UV absorbance at 280 nm.
Selections of DNA-Linked Molecules
A frozen pellet from an induced 5 mL E. coli culture with CBX7-His6 ChD was suspended in 300 μL of ice-cold lysis buffer (20 mM Tris, pH 8, 150 mM NaCl, 1 mg/mL CHAPS, 0.02% Tween-20, 1 mM PMSF) and lysed by sonication for 2 min (3 s on, 3 s off) at 30% power while on ice. The lysate was collected after centrifugation at 4000g at 4 °C. Meanwhile, 21 μL of His Mag Sepharose Ni (Ni-NTA-Sepharose–magnetic beads [MBs]) were prewashed with 3× 21 μL of purification buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 20 mM imidazole, 1 mM PMSF, 0.02% Tween-20). The soluble lysate was then combined with 12 μL of prewashed Ni-NTA-Sepharose-MBs and incubated at 4 °C for 1 h. The MBs were separated and washed in 5× 11 μL of purification buffer. After the last wash, the MBs were suspended in 12 μL of purification buffer. The CBX7-bound MBs were split and diluted to yield 10 μL of 1× (~50 μM CBX7), 10 μL of 1/10× (~5 μM CBX7), and 10 μM 1/20× (~2.5 μM CBX7). The MBs were separated and 10 μL of the DNA premix (50 nM Bz-140-mers nos. 72–96 [nonligand], 0.5 nM 4-BrBA-F-A-I-Kme3-S-140-mers nos. 1–24 [high-affinity ligand], 0.5 nM Ac-F-A-I-Kme3-S-140-mers nos. 25–48 [medium-affinity ligand], 0.5 nM Bz-A-I-Kme3-S-140-mers nos. 49–96 [low-affinity ligand] in 20 mM Tris, pH 8, 150 mM NaCl, 10 mM MgCl2, 0.02% Tween-20, 1 mg/mL bovine serum albumin [BSA], 1 mg/mL sheared salmon sperm DNA) was added to all four samples (mock [no protein/MBs only], 50 μM CBX7, 5 μM CBX7, and 2.5 μM CBX7) and allowed to incubate at room temperature (RT) for 1 h. The MBs were then separated and washed 5× in 10 μL of the above buffer. DNA conjugates and protein were eluted by incubating the MBs for 5 min at RT in the above buffer with 0.5 M imidazole. Each elution was collected and prepared for PCR and next-generation sequencing (NGS). The above procedure was repeated identically with CBX8.
Preparation of Kme3-Ser-CPF
The first two residues of the CBX consensus sequence were synthesized in bulk as previously described.29 Briefly, 150 nmol of NH2-5′-CPF in DEAE bind buffer (10 mM HOAc and 0.005% Triton X-100) was split between six cartridges. Each contained 220 μL of 50% DEAE Sepharose slurry in 50% ethanol and was prewashed with DEAE bind buffer. The DNA-loaded cartridges were washed 3× in 3 mL of MeOH. Fmoc–amino acid coupling was achieved by incubating the cartridges in 1 mL of 50 mM Fmoc–amino acid, 50 mM EDC-HCl, and 5 mM HOAt in 40% DMF/60% MeOH for 30 min at RT, with double couplings. After couplings, the cartridges were washed 3× in 3 mL of MeOH, followed by 3× in 3 mL of DMF. Fmoc deprotection was achieved by incubating the cartridges in 1 mL of 20% piperidine in DMF for 30 min at RT, and then washed with 3× 3 mL of DMF, 3× 3 mL of MeOH, and 1 mL of DEAE bind buffer after the final coupling. The DNA was eluted and collected by passing 1 mL of DEAE elution buffer (1.5 M NaCl and 0.005% Triton X-100) through each cartridge. The crude conjugate was desalted and concentrated to dryness.
Scanning Positional Library Synthesis
The purified Kme3-Ser-CPF conjugate was suspended in 4.8 mL of DEAE bind buffer. To 96 wells in a 384-well filter plate, 20 μL of DEAE Sepharose was added and washed 3× in 90 μL of DEAE bind buffer. To each well, 50 μL of Kme3-Ser-CFF solution (approximately 1 nmol conjugate per well) was added and washed 3× in 90 μL of MeOH. Briefly, Fmoc–amino acids were coupled using 50 mM Fmoc–amino acid, 50 mM EDC-HCl, 5 mM HOAt in 40% DMF/60% DMF for 30 min at RT with double coupling and deprotected by 20% piperidine in DMF for 30 min at RT. Peptoid couplings were achieved with 100 mM sodium chloroacetate and 150 mM DMTMM-Cl in MeOH for 30 min at RT with double coupling, followed by displacement by incubation with 1 M primary amine in DMSO for 16 h at RT. Wells were washed 3× in 90 μL of MeOH and 3× in 90 μL of DMF between each step. Following the final chemistry step, wells were washed 3× in 90 μL of DMF, 3× in 90 μL of MeOH, and in 90 μL of DEAE bind buffer. DNA conjugates were eluted by incubating 2× in 40 μL of DEAE elution buffer in each well for 5 min at RT, and then collected by centrifugation. Each conjugate was then attached to a unique 140-mer dsDNA template sequence by PCR individually (1× DreamTaq Buffer, 0.5 μM CFF-conjugate [PSL member], 0.5 μM CPR, 0.2 mM dNTPs, 0.05 ng/μL template, and 0.025 U/μL). All PCRs were pooled and purified by SPRI and quantified by UV absorbance at 260 nm.
Scanning Positional Library Selection against CBX7 and CBX8
The selection was carried out as described above for test DNA-linked molecules with slight modifications. The pre-selection mixture contained 0.5 nM Bz-140-mer on DNA construct 97 (nonligand control), 0.5 nM 4-BrBA-F-A-I-Kme3-S-140-mer (high-affinity ligand) on DNA construct 98, and 50 nM CBX scanning positional library–DNA conjugates (approximately 0.5 nM of each library member) in 20 mM Tris, pH 8, 150 mM NaCl, 10 mM MgCl2, 0.02% Tween-20, 1 mg/mL BSA, 1 mg/mL sheared salmon sperm DNA. The mixture was allowed to incubate with MBs at RT for 1 hour. The MBs were then separated and washed 5× in 10 μL of the above buffer. DNA conjugates were eluted by incubating the MBs for 5 min at RT in the above buffer with 0.5 M imidazole. Each elution was collected and prepared for PCR and NGS as described above.
Fluorescence Polarization Assay of PSL Hits against CBX7 and CBX8
Fluorescence polarization (FP) was measured by titration of CBX ChDs to a fluorescein isothiocyanate (FITC)–labeled probe as previously reported.25 Binding and competition FP assays were performed in black 384-well plates with optical bottoms. The FITC-labeled probe was kept constant at 100 nM with 0.4 μM CBX7 or 4 μM CBX8 with varying amounts of peptide, and in triplicate. Raw data were analyzed using GraphPad Prism 7 (GraphPad, La Jolla, CA) following a one-site total binding competition model, with any outliers (95% confidence interval) being excluded.
Fluorescence Polarization Assay of PSL Hits against CBX ChD Panel
FP assays were conducted as above with slight modifications. The FITC-labeled probe was kept constant at 100 nM. The protein concentrations used were selected based on the relative affinity of the CBX protein for the FITC probes. The concentrations used were 10 μM (CBX1), 1 μM (CBX2), 0.4 μM (CBX4, CBX6, CBX7), and CBX8 (4 μM).
Results and Discussion
We first set out to validate our affinity-based selection strategy using purified CBX ChDs and known DNA-linked ligands. Using purified protein domains with various fusion tags on nonporous MBs proved challenging and gave low and highly variable enrichments and recoveries of ligands. We found significant improvements by switching to a porous affinity support (Sepharose) and following a “just-in-time” protein purification strategy.30 Frozen E. coli cell pellets expressing CBX-His6 ChDs were lysed, and protein was bound to Ni-NTA-Sepharose-linked MBs (GE Healthcare) immediately before a selection. The selection stringency was modulated by using fewer beads that were saturated with the ChDs for selections at 1×, 1/10×, and 1/20× amounts of beads. Based on the bead capacity and the assay volume, this would yield 50, 5.0, and 2.5 μM protein concentrations, respectively, if free in solution. To perform selections, the DNA–ligand pool (20 nM nonligand, 0.2 nM each ligand) was added to each bead sample and allowed to equilibrate before five washes to remove unbound and nonligands. The immobilized protein (and any bound ligand–DNA conjugates) was then eluted by the addition of imidazole. For assays by DNA sequencing, the elution was submitted to PCR for amplification and addition of sequencing adapters with selection-identifying index sequences. Comparison of the enrichment of each ligand relative to the nonligand over three target protein concentrations for CBX7 and CBX8 is summarized in Figure 1 and Supplemental Figure S2.
Figure 1.
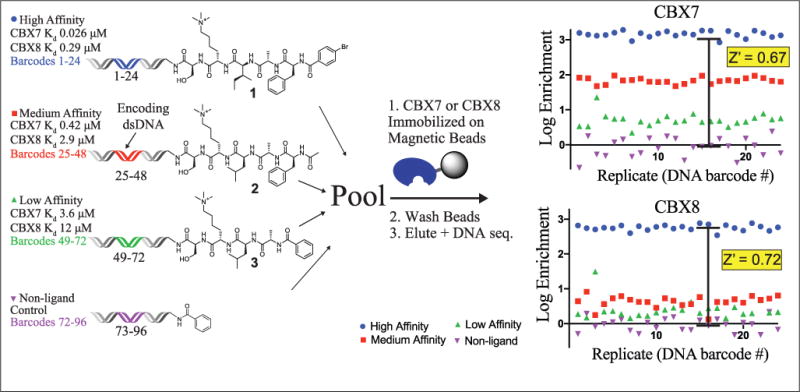
Optimized selection scheme with His6-tagged CBX7 and CBX8 using three on-DNA peptide ligands and nonligand.
Enrichment and recovery of ligands on single, unique DNA constructs at both 1× and 1/10× bead amounts were assessed by qPCR (Suppl. Table 1). This approach yielded acceptable enrichments and recoveries of ligands with varying affinities to both CBX7 and CBX8 ChDs. Overall, lower enrichments and recoveries were observed with lower ligand affinity and were only slightly lower with the lower protein level. In contrast to our previous work with affinity selections with protein immobilized on nonporous supports,29 these results do not model well to an equilibrium binding selection.9,31 This suggests that the effective concentration of protein within a bead may be a more relevant parameter than the overall amount within the solution. An additional contributing factor may be limited diffusion in and out of porous beads.
To evaluate additional bias from encoding DNA and detection by DNA sequencing, control ligands and the nonligand control were placed on 24 unique constructs each. In comparing selection results, we used the Z′ statistic commonly employed in the evaluation of HTS assay robustness as a convenient statistical measure of effect size.32 While in HTS the Z′ factor reflects both signal-to-background ratio (S/B) and well-to-well variability, in the DNA-encoded ligand selection employed here, it reflects S/B and variability associated with preparation, amplification, and detection of unique DNA barcodes. In all selections, the nonligand was depleted, and the high-affinity ligand was consistently the highest enriching molecule with Z′ factors of >0.5, indicating robust signal by HTS metrics (Fig. 1, Suppl. Figs. S2 and S3). In the CBX7 selections, the medium-affinity ligand was moderately enriched and the low-affinity ligand was poorly enriched. The differential enrichment of these two ligands varied between the high and low protein concentrations, illustrating the ability of the selection stringency to differentiate ligands of varying affinities. In the CBX8 selections, both the medium- and low-affinity ligands were well distinguished from the high-affinity ligand, having Z′ > 0.5 at both protein concentrations. Interestingly, both high-affinity ligands enriched similarly, despite the large difference in affinity to CBX7 and CBX8. At the low protein concentration, enrichments observed for compound 2 for CBX8 and compound 3 for CBX7 were quite similar, which is consistent with the similar affinities to their respective targets (Fig. 1). In contrast, enrichments of compound 1 for CBX8 and compound 2 for CBX7 differed by approximately 10-fold despite a less than 2-fold difference in affinity. This result may indicate the affinity range with high differential enrichment or may suggest that the effective concentration of the immobilized CBX7 ChD is slightly lower than the CBX8 ChD, and highlight the value of empirical assay validation. The poor enrichment of the medium- and low-affinity ligands in the CBX8 selections illustrates the difference in affinities for these ligands between CBX7 and CBX8.
As an additional demonstration of the robustness of CBX ChD selections with DNA-encoded ligands and to further expand the SAR around inhibitory Kme3-containing peptides, a PSL of 96 unique DNA-barcoded peptides and peptidomimetics was prepared. Four positions of a known peptide ligand25 were varied, as shown in Figure 2A. Building blocks used in the scanning positional library were chosen to confirm previously published SAR for CBX7 and CBX8,21–25 and also to expand on this known SAR using unnatural monomers. Non-α-amino acids, D-amino acids, N-alkyl glycine peptoids, and heterocyclic carboxylic acid caps were included to potentially identify new inhibitors. Briefly, the dipeptide Kme3-S was synthesized on a 5′-amine-modified 20-mer oligonucleotide and used as starting material for library synthesis. The PSL was prepared in parallel on solid phase with the DNA bound noncovalently to ion exchange resin (DEAE Sepharose), as previously described (Fig. 2B).5 Each of the four variable positions included 24 building blocks. After synthesis, peptide–oligonucleotide conjugates were eluted from DEAE with salt and used directly in a PCR amplification with a unique 140-mer DNA template. PCRs were pooled, and DNA purified in a PCR cleanup step. A series of three selections were then completed in parallel against both CBX7 and CBX8 to assess both relative enrichment differences and selectivities between the two proteins. Selections to both CBX7 and CBX8 were performed identically to the model selection experiments and included the nonligand control. The enrichment of each library member was calculated relative to the nonligand and displayed as a dot-blot plot for each position, shown in Figure 3 for the 2.5 μM selections, and for all protein concentrations in Supplemental Figures S6–S9.
Figure 2.
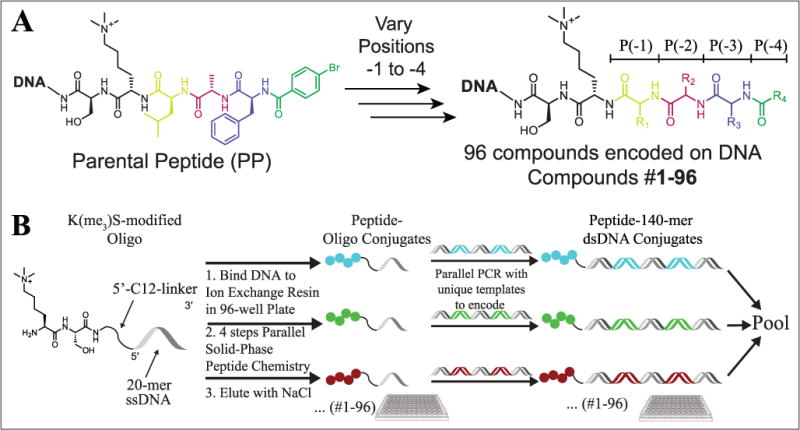
(A) On-DNA PSL parental peptide (PP) sequence. Colored amino acids indicate varied amino acids at the P(–1), P(–2), P(–3), and P(–4) positions. The invariant amino acids (Ser and Kme3) are shown in black. (B) Parallel synthesis of on-DNA PSL members.
Figure 3.

Dot blot of enrichments of DNA-encoded PSL with selection against either CBX8 or CBX7 at the lowest (~2.5 μM) protein concentration. The color of each dot represents the log enrichment relative to the nonligand control included in each selection.
For the selection of hits for subsequent study, both the enrichment (relative to the nonligand control) of the library member compared with the parental peptide and the selectivity for CBX8 over CBX7 (i.e., having greater enrichment for CBX8 than CBX7) were considered. The synthon corresponding to the parental peptide is the first library member at each position (compounds 1, 25, 49, and 72). Enriched synthons in position –1 (parental synthon: Leu), were generally lipophilic α-amino acids. Non-α-amino acids were poorly enriched for both CBX7 and CBX8. Compound 3 (P[–1] Ile) was highly enriched for both CBX7 and CBX8, having an average 1.5-fold higher enrichment for CBX8 over CBX7. Compound 6 (Aic) demonstrated the same preference for CBX8 over CBX7 and was also enriched at a similar level; however, the non-aryl derivative, compound 11, was poorly enriched for both CBX7 and CBX8, which is inconsistent with previously published work.25 Compound 19 (P[–1] homoproline) was moderately enriched and peptoids (compounds 21–23) were weakly enriched for CBX7 only, demonstrating increased selectivity toward CBX7 over CBX8. The selectivity of compound 2 (P[–1] Phe) for CBX7 over CBX8 (fourfold based on this work) corresponds well with previous work demonstrating that Tyr has increased selectivity for CBX7 over CBX8 at this position.23
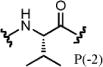
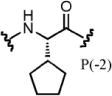
The second position has previously been demonstrated to give the highest degree of selectivity between CBX ChDs and is hypothesized to be driven by a single-residue difference in the protein (Val13 in CBX4 and CBX7; Ala13 in CBX2, CBX6, and CBX8). The additional space within this binding pocket of CBX8 allows for accommodation of slightly larger or branched side chains in place of the parental synthon Ala.23 Enrichments of compounds 28 (P[–2] Val) and 35 (P[–2] cyclopentylglycine) support this hypothesis, as they have the highest degree of selectivity for CBX8 over CBX7, more than any other synthons at any of the positions. Interestingly, Gly (compound 30) had the highest degree of selectivity for CBX7 at position –2.
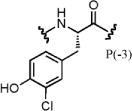
Synthons tested in position –3 (parental synthon: Phe) that were highly enriched were generally Phe derivatives. Almost all compounds with P(–3) variants were enriched to a greater degree with CBX7 than CBX8, with the exception of compound 61 (3-chlorotyrosine). Several non-α-amino acids had moderate to high enrichments with CBX7, including the four peptoid synthons (nos. 69–72), suggesting that this position may be amenable to such modifications to increase selectivity for CBX7 over CBX8 (and potentially other ChDs).
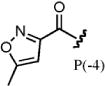
The importance of a 4-lipophilic substituted benzoyl cap at position –4 has been previously demonstrated.22–25 The parental synthon (4-BrBA) demonstrated greater enrichment over the unsubstituted benzoyl cap (no. 89) for both CBX7 and CBX8. Similarly, the 4-tertbutyl benzoyl cap explored by Frye and co-workers21,22 demonstrated improved enrichment. Compound 81 (P[–4] 5-methylisoxazole-3-carboxylic acid) was highly enriched for both CBX7 and CBX8 and demonstrated the highest selectivity for CBX8 over CBX7, though only marginally. Interestingly, compound 82 (P[–4] 1H-pyrazole-3-carboxylic acid) had a high selectivity for CBX7 over CBX8. These results suggest potential for differences in hydrogen-bonding patterns that are exploited by the isoxazole or pyrazole heterocycles that can give rise to this selectivity. A summary of the SAR observed by DNA sequencing is shown in Figure 4B.
Figure 4.
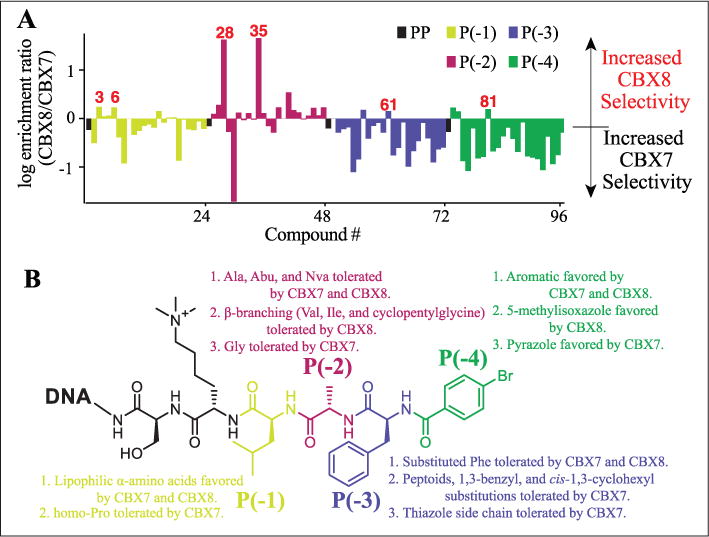
(A) Comparison of PSL members for selectivity toward CBX8 or CBX7. (B) Summary of SAR observed from on-DNA PSL selections against CBX7 and CBX8.
The majority of the substitutions explored in the library favored increased CBX7 selectivity over the parental peptide. Prior work has shown the utility of parallel selection of a DEL against homologous target proteins to identify ligands with desired selectivity properties.33 A comparison of the ratios of the enrichments of each compound for CBX8 over CBX7 at the most stringent selection conditions (Fig. 4A) quickly indicates the changes in selectivity from the parental compound. As previously demonstrated,23 modifications at position –2 accounted for the highest degree of selectivity between CBX7 and CBX8 between all the library members.
To validate the selectivity observed in the parallel selections of the DNA-encoded scanning positional library, six library members that showed increased selectivity and fold enrichment for CBX8 over CBX7, relative to the parental peptide, were synthesized off-DNA using standard solid-phase peptide synthesis (SPPS) methods. These peptides were compared using a competitive FP binding assay23,25 with CBX7 and CBX8, summarized in Table 1. Substitution of Ile for Leu in position –1 (PSL-3) demonstrated increased affinity and is consistent with previously published work,25 as well as a marginal increase in selectivity for CBX8 over CBX7. Interestingly, PSL-6, having Aic in place of Leu at position –1, did not compete with the probe for either CBX7 or CBX8. PSL-28 demonstrated the highest degree of selectivity for CBX8, having no apparent binding to CBX7. PSL-35 demonstrated binding to CBX8, yet appeared to aggregate the protein or probe at high concentration (Suppl. Fig. 5), complicating the determination of an IC50. Modification of position –3 to 3-chlorotyrosine (PSL-61) from Phe increased the potency from PSL-PP. Very little selectivity for CBX8 over CBX7 was observed with PSL-61 itself; however, it demonstrated an improvement in selectivity for CBX8 relative to the off-DNA parental peptide (PSL-PP). Acylation of the inhibitory peptide with 5-methylisoxazole-3-carboxylic acid (PSL-81) instead of 4-BrBA demonstrated improved potency for CBX8 over PSL-PP and an increase in selectivity for CBX8, as suggested by the relative enrichments.
Table 1.
Comparison of Enrichments from Selection at the Lowest Protein Concentration (~2.5 μM) and IC50 Values Determined by Fluorescence Polarization to CBX7 and CBX8 of Hits Prepared Off-DNA by SPPS.
| Compound | Log Enrichment CBX8 | Low/High Ratio | CBX8 IC50 (μM) | Log Enrichment CBX7 | Low/High Ratio | CBX7 IC50 (μM) | 8/7 Enrichment Ratio |
|---|---|---|---|---|---|---|---|
| PSL-PP | 2.9 | 1.0 | 11 | 3.1 | 1.0 | 0.90 | 0.60 |
| PSL-3 | 3.3 | 1.1 | 4.8 | 3.1 | 1.4 | 0.085 | 1.7 |
| |||||||
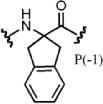
| PSL-6 | 3.3 | 1.1 | >500 | 3.0 | 1.0 | >500 | 1.1 |
| |||||||
| PSL-28 | 3.1 | 1.0 | 3.7 | 1.5 | 0.74 | >500 | 42 |
| |||||||
| PSL-35 | 2.4 | 1.0 | Agg. | 0.71 | 0.56 | Agg. | 46 |
| |||||||
| PSL-61 | 3.4 | 1.1 | 4.8 | 3.2 | 1.1 | 0.23 | 1.4 |
| |||||||
| PSL-81 | 3.3 | 1.0 | 4.5 | 3.1 | 1.0 | 1.7 | 1.6 |
| |||||||
Low/high ratio = ratio of enrichments at low protein concentration (~2.5 μM) to high protein concentration (~50 μM). The enrichment of the parental peptide was averaged from the four encoded peptides. IC50 values were determined from n = 3 samples at 0.4 μM CBX7 ChD or 4 μM CBX8 ChD. Compound PSL-28 caused probe aggregation (agg.) at high concentrations.
Overall, with the exception of PSL-6, the results obtained from the off-DNA validation correlated well with the enrichments obtained from the parallel selections against CBX7 and CBX8. To further determine the selectivity of these peptides to other CBX ChDs, competition FP assays were completed against CBX1, CBX2, CBX4, CBX6, CBX7, and CBX8 for PSL-28, PSL-61, and PSL-81, summarized in Table 2. The valine modification at position –2 (PSL-28) was tolerated by CBX1, CBX4, CBX6, and CBX8. Very little selectivity was observed from the 3-chlorotyrosine modification (PSL-61). The 5-methylisoxazole cap (PSL-81) was also well tolerated across the CBX proteins, with binding preferences for CBX2, CBX6, and CBX7.
Table 2.
Comparison IC50 Values (μM) for Fluorescein-Labeled Probe Displacement (100 nM) as Determined by Fluorescence Polarization to CBX1 (10 μM); CBX2 (1 μM); CBX4, CBX6, CBX7 (0.4 μM); and CBX8 (4 μM), n = 3.
| Compound | CBX Protein Chromodomain
|
|||||
|---|---|---|---|---|---|---|
| CBX1 | CBX2 | CBX4 | CBX6 | CBX7 | CBX8 | |
| PSL-28 | 9.5 | >100 | 9.6 | 7.5 | 49 | 14 |
| PSL-61 | 6.3 | 4.5 | 2.0 | 3.6 | 1.5 | 11 |
| PSL-81 | 58 | 7.3 | 32 | 8.1 | 4.3 | 29 |
For many DEL applications, it would be desirable for selection assay enrichments to correlate with affinities of molecules when synthesized off-DNA. Analysis of DNA-encoded library selections has shown how highly variable synthetic yields can lead to high false-negative rates and poor correlations of enrichments to target affinities.9,31 Given the similarly high-yielding reactions of Fmoc–amino acid chemistry on DNA,34 the contributions of variable yields to the enrichments of compounds in this peptide library should be minimized. An approach to address the complication of variable synthetic yield is to perform selections at multiple protein concentrations and to use the ratio of enrichments at high and low protein concentrations to triage compounds. We compared the ratio of enrichments at high versus low protein concentration with the enrichment at low concentration for ligands that enriched greater than 10-fold to CBX7 (Suppl. Fig. 10). A strong correlation was observed, suggesting that enrichment from the low protein concentration would correlate well to affinity. The most egregious outlier was compound 5 (P[–1] cyclopentylglycine), which maintained a high enrichment ratio of 0.99 but had poor enrichment (log E of 1.0). This would suggest that the compound is, in fact, a high-affinity ligand, despite the low enrichment values. These low values may have been due to an unusually low synthetic yield. This is additionally supported by similar compounds from Hof and coworkers with cyclopentylglycine at P(–1), which showed high affinity to CBX7.25 An additional outlier with a high enrichment ratio but poor enrichment values was compound 66 (P[–3] cycloleucine), which is an α-disubstituted amino acid. The steric hindrance of these amino acids is well known to provide low coupling yields.35
The robustness of affinity selection assays to the ChDs of CBX7 and CBX8 was evaluated using a set of three known ligands and a nonligand control on a number of DNA constructs. We found that DNA tag bias was sufficiently minimal to allow confident discrimination of compound affinity using enrichment values. Compound enrichment of lower-affinity ligands was more dependent on the protein concentration in the selection than the higher-affinity ligand. Using optimized selection conditions, parallel selections of a DNA-encoded peptidomimetic PSL against both CBX7 and CBX8 were completed. These selections allowed for a further validation of the selection assays by comparison of ligand enrichment SAR to the affinity SAR values in published literature. Specifically, a subset of peptides displayed high selectivity for CBX8 over CBX7 through the incorporation of β-branched amino acids at the (–2) position, which was suggested by previous work.22,23 Also consistent with our enrichment data was the previously illustrated importance of 4-substituted benzoic acid derivatives for affinity to CBX7.22,25 The use of parallel selections against homologous target proteins allowed for the assessment of ligand selectivity of all the library compounds using the first-pass assay. We found several heterocyclic carboxylic acids at the (–4) position that gave rise to selectivity between CBX7 and CBX8, with the 5-methylisoxazole-3-carboxylic acid cap improving both potency and selectivity toward CBX8. Relative potencies determined in follow-up assays of off-DNA hits were consistent with the enrichments observed in the DNA sequencing data of the in vitro selection assays for CBX8. These studies lay the groundwork for future studies exploring additional modifications to improve potency and selectivity toward CBX8 against other CBX ChD proteins. Broadly, our results show how carefully conducted in vitro selection assays can be useful not only for ligand discovery but also for finer-grained determination of quantitative SARs.
Supplementary Material
Acknowledgments
We would like to thank Aktan Alpsoy for help with protein expression and Dr. Dongwook Kim for helpful discussions and suggestions.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We gratefully acknowledge support from Purdue University. The Purdue University Mass Spectrometry and Genome Sequencing Shared Resources are supported by P30 CA023168 from the National Institutes of Health. E.C.D. is supported by the V Foundation for Cancer Research (V2014-004 and D2016-030) and the NIH (CA207532). F.H. is supported by Cancer Research Society (grant 19284).
Footnotes
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
ORCID
Kyle E. Denton https://orcid.org/orcid.org/0000-0003-1236-9174
Casey J. Krusemark https://orcid.org/0000-0003-2964-3520
References
- 1.Chan AI, McGregor LM, Liu DR. Novel Selection Methods for DNA-Encoded Chemical Libraries. Curr Opin Chem Biol. 2015;26:55–61. doi: 10.1016/j.cbpa.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hale P. A Handbook for DNA-Encoded Chemistry. John Wiley & Sons; Hoboken, NJ: 2014. S. Screening Large Compound Collections; pp. 281–317. [Google Scholar]
- 3.Goodnow RA, Dumelin CE, Keefe AD. DNA-Encoded Chemistry: Enabling the Deeper Sampling of Chemical Space. Nat Rev Drug Discov. 2016;16:131–147. doi: 10.1038/nrd.2016.213. [DOI] [PubMed] [Google Scholar]
- 4.Belyanskaya SL, Ding Y, Callahan JF, et al. Discovering Drugs with DNA-Encoded Library Technology: From Concept to Clinic with an Inhibitor of Soluble Epoxide Hydrolase. Chembiochem. 2017;18:837–842. doi: 10.1002/cbic.201700014. [DOI] [PubMed] [Google Scholar]
- 5.Krusemark CJ, Tilmans NP, Brown PO, et al. Directed Chemical Evolution with an Outsized Genetic Code. PLoS One. 2016;11:1–16. doi: 10.1371/journal.pone.0154765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denton KE, Krusemark CJ. Crosslinking of DNA-Linked Ligands to Target Proteins for Enrichment from DNA-Encoded Libraries. Medchemcomm. 2016;7:2020–2027. doi: 10.1039/C6MD00288A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wrenn SJ, Weisinger RM, Halpin DR, et al. Synthetic Ligands Discovered by In Vitro Selection. J Am Chem Soc. 2007;129:13137–13143. doi: 10.1021/ja073993a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y, Zimmermann G, Scheuermann J, et al. Quantitative PCR Is a Valuable Tool to Monitor Performance of DNA-Encoded Chemical Library Selections. Chembiochem. 2017;18:848–852. doi: 10.1002/cbic.201600626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Satz AL. DNA Encoded Library Selections and Insights Provided by Computational Simulations. ACS Chem Biol. 2015;10:2237–2245. doi: 10.1021/acschembio.5b00378. [DOI] [PubMed] [Google Scholar]
- 10.Franzini RM, Ekblad T, Zhong N, et al. Identification of Structure-Activity Relationships from Screening a Structurally Compact DNA-Encoded Chemical Library. Angew Chem Int Ed. 2015;54:3927–3931. doi: 10.1002/anie.201410736. [DOI] [PubMed] [Google Scholar]
- 11.Satz AL. Simulated Screens of DNA Encoded Libraries: The Potential Influence of Chemical Synthesis Fidelity on Interpretation of Structure-Activity Relationships. ACS Comb Sci. 2016;18:415–424. doi: 10.1021/acscombsci.6b00001. [DOI] [PubMed] [Google Scholar]
- 12.Connelly KE, Dykhuizen EC. Compositional and Functional Diversity of Canonical PRC1 Complexes in Mammals. Biochim Biophys Acta Gene Regul Mech. 2017;1860:233–245. doi: 10.1016/j.bbagrm.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Kaustov L, Ouyang H, Amaya M, et al. Recognition and Specificity Determinants of the Human Cbx Chromodomains. J Biol Chem. 2011;286:521–529. doi: 10.1074/jbc.M110.191411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klauke K, Radulovic V, Broekhuis M, et al. Polycomb Cbx Family Members Mediate the Balance between Haematopoietic Stem Cell Self-Renewal and Differentiation. Nat Cell Biol. 2013;15:353–362. doi: 10.1038/ncb2701. [DOI] [PubMed] [Google Scholar]
- 15.Koppens M, van Lohuizen M. Context-Dependent Actions of Polycomb Repressors in Cancer. Oncogene. 2015;35:1341–1352. doi: 10.1038/onc.2015.195. [DOI] [PubMed] [Google Scholar]
- 16.Chung CY, Sun Z, Mullokandov G, et al. Cbx8 Acts Non-Canonically with Wdr5 to Promote Mammary Tumorigenesis. Cell Rep. 2016;16:472–486. doi: 10.1016/j.celrep.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Béguelin W, Teater M, Gearhart MD, et al. EZH2 and BCL6 Cooperate to Assemble CBX8-BCOR Complex to Repress Bivalent Promoters, Mediate Germinal Center Formation and Lymphomagenesis. Cancer Cell. 2016;30:197–213. doi: 10.1016/j.ccell.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li G, Warden C, Zou Z, et al. Altered Expression of Polycomb Group Genes in Glioblastoma Multiforme. PLoS One. 2013;8:e80970. doi: 10.1371/journal.pone.0080970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan J, Jones M, Koseki H, et al. CBX8, a Polycomb Group Protein, Is Essential for MLL-AF9-Induced Leukemogenesis. Cancer Cell. 2011;20:563–575. doi: 10.1016/j.ccr.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santiago C, Nguyen K, Schapira M. Druggability of Methyl-Lysine Binding Sites. J Comput Aided Mol Des. 2011;25:1171–1178. doi: 10.1007/s10822-011-9505-2. [DOI] [PubMed] [Google Scholar]
- 21.Stuckey JI, Dickson BM, Cheng N, et al. A Cellular Chemical Probe Targeting the Chromodomains of Polycomb Repressive Complex 1. Nat Chem Biol. 2016;12:180–187. doi: 10.1038/nchembio.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stuckey JI, Simpson C, Norris-Drouin JL, et al. Structure-Activity Relationships and Kinetic Studies of Peptidic Antagonists of CBX Chromodomains. J Med Chem. 2016;59:8913–8923. doi: 10.1021/acs.jmedchem.6b00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milosevich N, Gignac MC, McFarlane J, et al. Selective Inhibition of CBX6: A Methyllysine Reader Protein in the Polycomb Family. ACS Med Chem Lett. 2016;7:139–144. doi: 10.1021/acsmedchemlett.5b00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simhadri C, Gignac MC, Anderson CJ, et al. Structure–Activity Relationships of Cbx7 Inhibitors, Including Selectivity Studies against Other Cbx Proteins. ACS Omega. 2016;1:541–551. doi: 10.1021/acsomega.6b00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Simhadri C, Daze KD, Douglas SF, et al. Chromodomain Antagonists That Target the Polycomb-Group Methyllysine Reader Protein Chromobox Homolog 7 (CBX7) J Med Chem. 2014;57:2874–2883. doi: 10.1021/jm401487x. [DOI] [PubMed] [Google Scholar]
- 26.Ma RG, Zhang Y, Sun TT, et al. Epigenetic Regulation by Polycomb Group Complexes: Focus on Roles of CBX Proteins. J Zhejiang Univ Sci B. 2014;15:412–428. doi: 10.1631/jzus.B1400077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.TerMaat JR, Pienaar E, Whitney SE, et al. Gene Synthesis by Integrated Polymerase Chain Assembly and PCR Amplification Using a High-Speed Thermocycler. J Microbiol Methods. 2009;79:295–300. doi: 10.1016/j.mimet.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jetson RR, Krusemark CJ. Sensing Enzymatic Activity by Exposure and Selection of DNA-Encoded Probes. Angew Chem Int Ed Engl. 2016;55:9562–9566. doi: 10.1002/anie.201603387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Denton KE, Krusemark CJ. Crosslinking of DNA-Linked Ligands to Target Proteins for Enrichment from DNA-Encoded Libraries. Med Chem Commun. 2016;7:2020–2027. doi: 10.1039/C6MD00288A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Machutta CA, Kollmann CS, Lind KE, et al. Prioritizing Multiple Therapeutic Targets in Parallel Using Automated DNA-Encoded Library Screening. Nat Commun. 2017;8:16081. doi: 10.1038/ncomms16081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Satz AL. Simulated Screens of DNA Encoded Libraries: The Potential Influence of Chemical Synthesis Fidelity on Interpretation of Structure-Activity Relationships. ACS Comb Sci. 2016 doi: 10.1021/acscombsci.6b00001. [DOI] [PubMed] [Google Scholar]
- 32.Zhang J-H. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 33.Franzini RM, Nauer A, Scheuermann J, et al. Interrogating Target-Specificity by Parallel Screening of a DNA-Encoded Chemical Library against Closely Related Proteins. Chem Commun. 2015;51:8014–8016. doi: 10.1039/c5cc01230a. [DOI] [PubMed] [Google Scholar]
- 34.Halpin DR, Lee JA, Wrenn SJ, et al. DNA Display III. Solid-Phase Organic Synthesis on Unprotected DNA. PLoS Biol. 2004;2:E175. doi: 10.1371/journal.pbio.0020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, Gabriele E, Samain F, et al. Optimized Reaction Conditions for Amide Bond Formation in DNA-Encoded Combinatorial Libraries. ACS Comb Sci. 2016;18:438–443. doi: 10.1021/acscombsci.6b00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.