

Abstract
The delivery of proteins into cells is a potential game changer for a wide array of therapeutic purposes, including cancer therapy, immunomodulation and treatment of inherited diseases. In this review, we present recently developed nanoassemblies for protein delivery that utilize strategies that range from direct assembly, encapsulation and composite formation. We will discuss factors that affect the efficacy of nanoassemblies for delivery from the perspective of both nanoparticles and proteins. Challenges in the field, particularly achieving effective cytosolar protein delivery through endosomal escape or evasion are discussed.
Graphical abstract
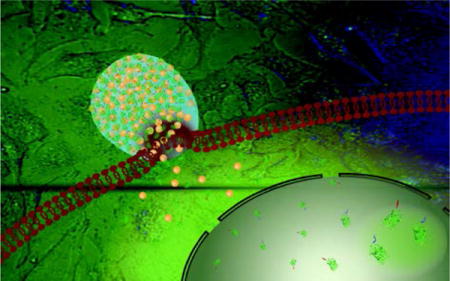
1. Introduction
Protein delivery holds enormous promise for therapeutic applications.1 Proteins can be used to introduce deficient proteins due to genetic defects or therapeutic proteins to ameliorate other disease states.2 To date, protein delivery strategies have focused primarily on extracellular targets, and are effective for delivery of proteins that have external receptors or endogenous mechanisms for cellular internalization. There are a wide range of potential applications, however, that require access to the cell cytosol. Accessing these targets necessitates the development of strategies to achieve intracellular delivery.2,3 However, intracellular protein delivery is challenging due to the intrinsic properties of proteins, such as high molecular weight and polarity4, which prevent proteins from crossing the cell membrane. Additionally, the poor stability and high clearance rate of most proteins reduce their therapeutic effect in vivo.
Different approaches have been used to deliver proteins into mammalian cells, including mechanical/physical methods, covalent protein modification, and nanocarrier systems.5 Mechanical/physical methods include microinjection and electroporation that effect protein delivery using special equipment to mechanically/physically puncture the cell membrane.6 Although these methods provide direct access of the protein to the cell cytosol, their use is limited because they are low-throughput, invasive, and not suitable for in vivo use. Covalent protein modification with cell-penetrating peptides (CPPs) is a common approach to facilitate access of proteins into the cell.7 The uptake of the protein-conjugated CPP often occurs through endocytosis. Therefore, delivered proteins are entrapped into endosomes and can eventually be released in the cytosol with the use of membrane-destabilizing agents, such as proteins/peptides (e.g. saporin, penetratin, polyhistidine) and chemicals (e.g. chloroquine, methylamine, polyethylenimine).8 However, these methods are often cytotoxic and unable to prevent protein degradation.9
Over the past decade, several types of nanocarriers for intracellular delivery of therapeutic proteins have been developed, including virus-like particles, polymeric nanoparticles, and inorganic nanoparticles.5,10 Among these carriers, inorganic nanoparticle-protein vehicles have generated considerable interest as a strategy to direct intracellular protein delivery.2,5 Nanoparticle-based delivery approaches offer several desirable qualities, including versatility, control over size and surface functionalization, minimal toxicity, long circulation time, efficient cellular uptake and targeting ability.11 Specifically, the choice of the inorganic core, size and surface functionality of the nanoparticle are critical to control the protein loading and modulate the uptake efficiency and mechanism (i.e. endocytosis followed by endosomal escape or direct cytosolic delivery by membrane fusion).
For delivery applications, proteins can be conjugated to nanoparticles using two different main approaches: covalent attachment12 or non-covalent interaction (e.g. electrostatic, hydrogen bonding, hydrophobic interactions).13 The first approach provides stable conjugates that can be used for a variety of applications.14 On the other hand, non-covalent conjugation is generally preferred for applications in which the activity of the protein is critical, and is the topic of this Tutorial Review. Non-covalent interactions between nanoparticles and proteins can be used to build supramolecular assemblies, in which the protein structure and activity is maintained through the delivery process.15 Additionally, these assemblies can be easily tuned by engineering both the protein and nanoparticle components to deliver different types of proteins or to modulate the mechanism by which these systems are uptaken by cells.
To date, several strategies to build nanoparticle-protein supramolecular architectures have been utilized, including proteins non-covalently bound to nanoparticles,13,16 proteins loaded into nanocapsules,17, 18 nanoparticle-stabilized capsules,19 and nanoparticle-protein nanocomposites.20,21 This review will focus on the relevant and successful non-covalent approaches for intracellular delivery of proteins, discussing the key factors involved in the formation of the different supramolecular assemblies and the efficiency of the delivery, such as size, surface functionality, and supramolecular interactions. Methods for generating stimuli responsive vehicles and how this capability aids protein delivery are also discussed.
2. Mechanisms of cellular uptake: endocytosis vs membrane fusion
Nanomaterials are typically taken up by cells via endocytosis. Endocytosis is an energy- dependent mechanism by which cells capture molecules, such as proteins, entrapping them into vesicular compartments (i.e. endosomes).22 Endosomal entrapment typically prevents access to the cytosol and nucleus.7,23 As most proteins are utilized in the nucleus and cytosol, endosomal escape is typically required for delivered proteins to perform their therapeutic function. Proteins can eventually escape to the cytosolic compartment by using carriers designed to disrupt the endosomal membrane, allowing proteins to enter the cytosol after being entrapped in endosomes (Fig. 1a).13,16 Membrane fusion mechanisms are an emerging strategy that provides direct access of the proteins to the cytosol, avoiding entrapment into endosomes. In this route, the supramolecular assembly fuses with the cell membrane, allowing direct transfer of the protein cargo to the cytosol, and potentially to the nucleus of the cell (Fig. 1b).19,20 Both endocytosis and membrane fusion have applications for which they are best suited. Endocytotic delivery is a directly useful strategy for applications such as targeted vaccine delivery into endosomes24 and for treating lysosomal diseases.25 In contrast, protein vectors that promote either endosomal escape or direct cytosolic delivery are particularly attractive for applications where proteins need to access the cytosol, nucleus, or other intracellular sites to exert therapeutic activity. These applications include gene editing, protein replacement therapy, and imaging.
Fig. 1.
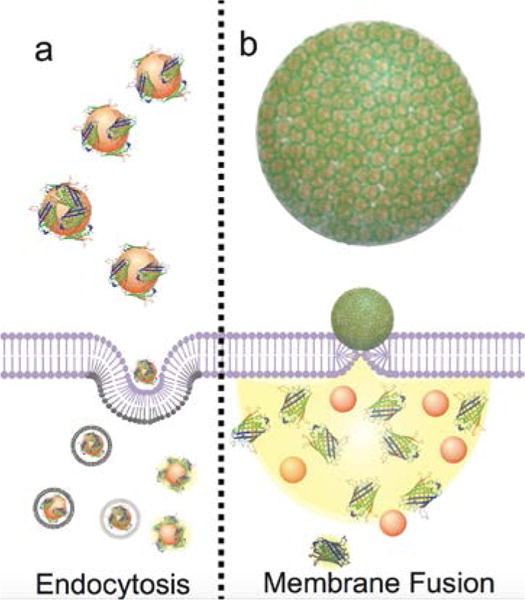
Schematic representation of two representative nanoparticle-protein supramolecular architectures and their delivery mechanisms. Proteins non-covalently bound to nanoparticles are taken up by the cell via endocytosis, following endosomal release (a), while nanoparticle-protein nanocomposites are taken up by the cell via membrane fusion mechanism with direct release of the protein in the cytosol (b).
A wide variety of nanoparticle-protein supramolecular assemblies have been developed to promote endosomal escape and potentially endosomal evasion, resulting in higher cytosolic delivery efficacy.26 The different strategies for creating supramolecular assemblies for protein delivery into the cytosol/nucleus will be discussed in the following sections.
3. Nanoparticle-protein supramolecular assembly strategies
Non-covalent conjugation methods can employ a range loading strategies including electrostatic attraction, host-guest encapsulation, and hydrophobic adsorption.13 Of these methods, electrostatic attraction is the most widely employed, as there are a multitude of ways to adjust the surface charge of both particles and proteins to create a compatible system.2,11 Fig. 1 summarizes two representative nanoparticle-protein supramolecular architectures obtained by self-assembly of proteins and inorganic nanoparticles and their mechanism of uptake. Inorganic nanoparticles can simply be conjugated with proteins (Fig. 1a) or employed to form more complex structures such as nanocapsules or nanocomposites (Fig. 1b).21,27 Generally, both the surface functionalization and the size of these systems determine their cellular uptake mechanism.2,5 Overall, nanoparticle-protein supramolecular assemblies must be soluble and stable under physiological conditions to allow delivery of the protein into cells.10 Therefore, it is necessary to design assemblies with hydrophilic exteriors. Moreover, an efficient delivery system is required to maintain the structure, avoiding release of the protein before cellular internalization. Different delivery strategies employing stable and soluble assemblies to promote endosomal escape or membrane fusion will be discussed in the following subsections.
3.1 Protein delivery to the cytosol by endosomal escape
Most nanoparticle-protein supramolecular assemblies are uptaken by cells via endocytic mechanism and are subsequently released into the cytosol through endosomal escape. Without effective endosomal escape, the delivered proteins will eventually be trafficked to the lysosome and degraded by cathepsins or transferred to an exosome and transported out of the cell entirely. A wide variety of strategies for endosomal escape have been developed using different inorganic nanoparticle core materials.
Silica is a versatile nanomaterial for delivery applications due to low toxicity and the ability to generate a wide range of particle structures. The use of hydrophobic smooth silica nanoparticles to immobilize and deliver functional proteins into cells was reported by Bale et al.,28 where 15 nm silica nanoparticles were functionalized with hydrophobic n- octadecyltrimethoxysilane (n-ODMS) to facilitate adsorption of proteins via hydrophobic interactions. Green fluoresecent protein (GFP) and fluorescently tagged bovine serum albumin (BSA) were used as model proteins to elucidate the mechanism of uptake, while RNase A and a monoclonal pAkt antibody were used to prove the retention of the protein’s biological function in the nanoparticle-protein conjugate. Fluorescence-activated cell sorting (FACS) analysis was used to characterize the uptake of the conjugates in MCF-7 human breast cancer cells. Rat neural stem cells, treated with various endocytosis pathway inhibitors, were used to investigate the nanoparticle uptake mechanism. However, confocal microscopy images of cells incubated with the conjugates showed punctate fluorescence, suggesting endosomal entrapment of the protein-nanoparticle conjugate.
More recently, Niu et al. have enhanced the protein loading in silica nanoparticles by using nanoparticles with tunable surface roughness.29 The control over the nanoparticle roughness, and the protein loading, was achieved by attaching small shell particles of different sizes to larger core particles. Specifically, uniform nonporous silica core nanoparticles, ~200 nm size, were first functionalized with aminosilane to generate positive surface charge and promote the attachment of the negatively charged shell particles by electrostatic interactions. Thereafter, the shell particles were fused to the core particles through migration and deposition of silica into the spaces between core and shell nanoparticles. The resulting rough nanoparticles were negatively charged and able to adsorb positively charged proteins of various molecular weights (Fig. 2). These proteins included cytochrome c (cyt C), IgG-F, a nonspecific rabbit IgG-A and the anti-pAkt antibody, revealing a correlation between the interspace size and the adsorption trend. Further functionalization of these rough silica nanoparticles with hydrophobic n-ODMS resulted in a slightly reduced net charge of the particles but a highly improved protein adsorption capability. Building upon that work, the same authors demonstrated that both surface roughness and hydrophobic modification could enhance cellular uptake via an endocytic mechanism.30 However, only the surface chemistry (i.e. n-ODMS) plays a role in the endosomal escape, independent of the surface roughness. The efficiency of endosomal escape was investigated using only TEM, so the actual mechanism of escape is unclear. Given that increasing hydrobicity of the particle surface increases escape, it is likely that there is an hydrophobic interaction between the surface and the endosome, leading to rupture of the membrane. Protein activity retention following conjugation was proved by measuring the binding activity of IgG-F with its complementary antigen, while endosomal escape was demonstrated by delivering anti-pAkt antibody, which inactivated pAkt and induced apoptosis in MCF-7 cells.
Fig. 2.

Comparison of strategies for the formation of “rough” surface silica NPs, using the “neck-enhancing approach” that utilizes smaller shell particles with high negative charge fused with the larger core particles (a) and traditional approaches based on electrostatic interaction between the core and shell particles that result in less stable nanocarriers. (b). The “neck-enhanced” particles were then used to deliver pAKT antibody to induce cell death in a cancer model (c).Reproduced from Ref. 29 with permission from The Royal Society of Chemistry.
In addition to solid silica nanoparticles, mesoporous silica nanoparticles (MSNs) or hollow mesoporous silica capsules (HMSCs) have garnered significant attention as protein delivery vehicles.31,32 MSNs can retain proteins within their nanoscale pores via electrostatic or hydrophobic interactions, protecting the guest protein from degradation. Moreover, by tuning the pore size and the surface functionality, a variety of proteins of different sizes and charges can be loaded and subsequently released into the cell. Typically, MSNs are uptaken by cells through endocytosis, which can be followed by partial endosomal release of the protein cargo. An example of this strategy was reported in a work by Lim et al., in which HMSCs were obtained by arrangement of cetyltrimethylammonium bromide (CTAB)-silicate nanosheets at the interface of an oil (cholesterol)-in-water emulsion.18 FITC-labeled proteins, such as glutathione S-transferase, BSA, yeast alcohol dehydrogenase and IgG, were loaded into the nanocapsules. While the delivery process was efficient, proteins were observed to be mostly predominately localized in endosomes.
A promising approach for creating silica capsules was recently reported by Yu and coworkers using a supra-assembly approach for synthesizing a silica vehicle of ~250 nm with large pores and an inner hollow cavity of ~170 nm.33 The formation of the vehicle occurred via self-assembly of small silica vesicles (~20 nm) at the interface of oil/water microemulsion droplets containing 3-aminopropyltriethoxysilane (APTES). Further packing between adjacent vesicles led to transformation into cone-type structures with large pores and a hollow cavity, named hollow dendritic mesoporous nanospheres (HDMSNs). Loading with IgG and β-Gal was then driven by electrostatic interactions between the positively charged amine groups on the HDMSN and the negatively charged proteins. While the functional activity of β- Gal was demonstrated, showing enzymatic activity inside the cell, the delivery of β-Gal labeled with rhodamine B isothiocyanate (β-Gal -RITC) occurred most likely via endocytosis with minimal escape, as demonstrated by the punctate fluorescence in confocal images.
Gold nanoparticles are promising platforms for protein delivery due to their biocompatibility and ease of functionalization with a wide range of ligands. This flexibility allows for specific tuning of the particle-protein interaction for more effective delivery. Ghosh et al., reported the use of peptide-coated gold nanoparticles with a core diameter of 2.5 nm as a carrier for β-Gactosidase (β-Gal), a membrane-impermeable negatively charged protein with high molecular weight (473 kDa).13 The gold nanoparticles were functionalized with a ligand comprised of three domains: an alkyl chain for stabilization of the hydrophobic gold core, a tetraethylene glycol (TEG) for preventing nonspecific interactions with biomolecules, and an external peptide-tag (Hys-Lys-Arg-Lys) for protein surface recognition and plasma membrane association through favorable electrostatic interaction (Fig. 3). Electrostatic interactions, as well as hydrogen-bonding interactions of the protein with the arginine units of the nanoparticle ligand are the driving forces in the formation of the nanoparticle-β-Gal complex. After internalization, the release of β-Gal adsorbed on the cationic particles was promoted by the presence of intracellular glutathione.34 While the protein maintained its activity after internalization, the uptake mechanism apparently occurred via endocytic pathway, as revealed by the resulting punctate fluorescence. Due to the incorporation of histidines on the nanoparticle surface, the particles were able to take advantage of the “proton sponge effect” to release some of the cargo from the endosomes.35 This endosomal escape mechanism utilizes an agent capable of accepting multiple protons (such as surface functionalization with histidine) from the acidic endosomal environment. This buffering effect of the agent causes an influx of the ions in the endosome, leading to swelling and eventually rupture. However, the efficiency of this mechanism is generally low, so the subsequent cytosolic release was limited.
Fig. 3.
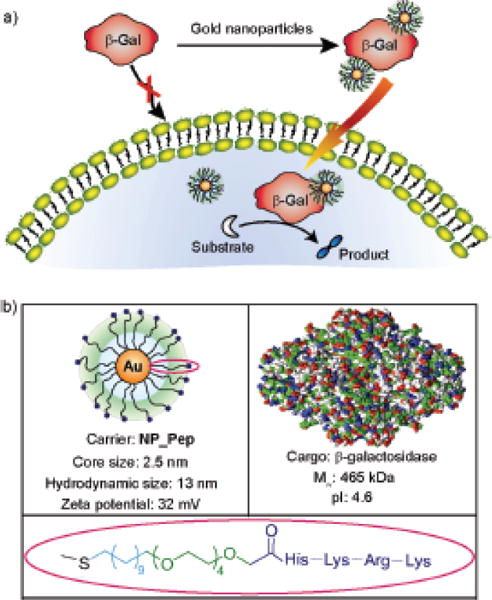
Ultrasmall gold nanoparticles functionalized with a modified small peptide effectively deliver negatively charged β-Gal with retention of enzyme activity. Reprinted with permission from Ref. 13 Copyright (2010) American Chemical Society.
A particularly promising approach for protein delivery with endosomal escape was reported by Das et al.16 In this work, polymer-coated calcium phosphate nanorods were used for delivery of negatively charged proteins such as BSA and RNase A. Calcium phosphate nanorods of ~35 nm length and 4–5 nm diameter were coated with polyacrylate monomers includinge 3-sulfopropyl methacrylate (anionic surface charge) and N-(3-aminopropyl) methacrylamide (cationic surface). In this work, FITC-labeled protein conjugated to polyacrylate-coated nanorods was efficiently delivered into the cytosol of HeLa cells with low cytotoxicity (Fig. 4). To prove nanorod-delivered proteins would retain functionality following uptake, RNase A was loaded onto the nanorods. Following delivery, the nanorod- delivered RNase A induced effective cellular apoptosis while free RNase A was ineffective due to poor cellular uptake. The proposed mechanism of endosomal escape involves dissolution of the calcium phosphate nanorods in the more acidic endosomes, which allows release of the protein from the endosome into the cytosol, as demonstrated by the diffuse fluorescence shown in Fig. 4.
Fig. 4.
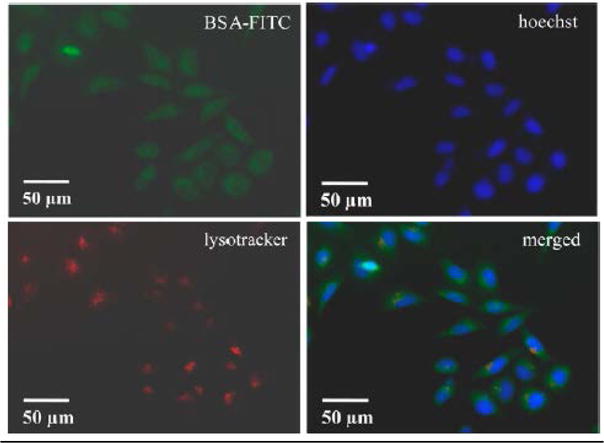
Confocal imagery of the delivery of FITC labeled BSA with calcium phosphate nanorods into HeLa cells. Calcium phosphate nanorods act as a pH-responsive endosomal escape system allowing for cytosolic delivery of BSA-FITC. Reprinted with permission from Ref. 16. Copyright (2016) American Chemical Society.
3.2 Direct delivery of proteins into the cytosol and nucleus
Fusion of nanoparticle-protein supramolecular assemblies with the cell membrane (membrane fusion) provides a promising strategy for intracellular protein delivery. Through facile modulation of both size and electrostatics, this approach allows direct access of proteins into the cytosol and, thence to intracellular targets, e.g. the nucleus.
As mentioned in the previous section, MSNs are typically taken up by cells through endocytosis, following endosomal escape. Yang et al. have reported a 230 nm nanovehicle in which MSNs are coated with a lipid bilayer (LB) containing lipopetides for targeted membrane fusion between liposomes and cells.27 Cuboidal MSNs were generated with 10 nm disk-shaped cavities able to accommodate the positively charged cytochrome C (cyt C) at pH 7.4 via electrostatic interactions with the negatively charged silanol groups on the surface of nanoparticles. Subsequent release of the protein was promoted by the high ionic strength inside the cell. Additional fusogenic LBs were incorporated into the MSNs to enhance colloidal stability, as well as prevent premature release of cyt C. Finally, a cholesterol-poly(ethylene glycol) lipopeptide was inserted in the LB, while cells were treated with the complementary lipopetide, the pair of which induced cellular uptake via a membrane fusion mechanism. The mechanism of uptake was demonstrated through studies of endocytosis inhibition, confirming that uptake occurred mainly via membrane fusion.
Nanoparticle-stabilized capsules (NPSC) provide a promising approach to achieving cytosolic delivery. Recent studies by Tang et al. have demonstrated the use of NPSCs as efficient, versatile delivery vehicles for direct cytosolic delivery of proteins such as GFP.19 These NPSCs utilize nano-emulsion structural motifs, in which gold nanoparticles functionalized with a cationic tetrapeptide bearing a guanidinium moiety (HKRK-AuNPs) act as stabilizers at the oil-water interface of a Pickering-type emulsion, to confer stability as well as functionality to the assembly.36 Generation of nanoscale emulsions to form NPSCs relies upon hydrogen bonding and electrostatic interaction between guanidinium moieties of the HKRK-AuNPs and the carboxylate groups on the oil core, allowing the formation of exceptionally small assemblies. Additional lateral stability in this system is given by the interaction between the anionic proteins and cationic HKRK-AuNPs, allowing for construction of stable NPSCs as small as 130 nm across (Fig. 5a-c). Through the combined effect of electrostatic interaction between the positively-charged ligand and the cell membrane, and the lipophilic nature of the oil core, these NPSCs are capable of cytosolic delivery of their protein cargo through membrane fusion. Confocal microscopy (and video) of NPSC-based delivery of GFP revealed homogeneous distribution of the protein throughout the cytosol. This work represents a generalizable approach for the direct cytosolic delivery of proteins in their native form. Furthermore, the authors have demonstrated the versatile nature of the NPSC-based protein delivery system by intracellular targeting of proteins to different subcellular compartments such as peroxisomes19 and the nucleus.37 In the latter work, the authors have exhibited a method to localize proteins into the nucleus using five naturally- derived nuclear localization sequences (NLS).37 These NLSs were fused to enhanced GFP and their nuclear trafficking was monitored in mammalian cells to compare the targeting efficiencies of the NLSs. One of these five sequences tagged to GFP delivered most efficiently into the nucleus, demonstrating nuclear accumulation and reaching import equilibrium within 6 minutes. NLS-tagged delivery systems lacked nuclear import under ATP-depleted conditions, implicating active transport of NLS-tagged proteins into the nucleus. Interestingly, Tang et al. developed the first reported strategy for nuclear targeting of a protein using a synthetic tag.38 Utilizing an aromatic benzyl boronate (BB) moiety, large proteins, which would otherwise be unable to passively diffuse across the nuclear membrane (≥60 kDa) were demonstrated to accumulate in the nucleus. A BB tag was conjugated to RNase A and red fluorescent protein from Discosoma sp. (dsRed, 112 kDa) to facilitate their delivery selectively into the nucleus (Fig. 5e). Quantification of delivery based on fluorescence revealed higher delivery in the nucleus as compared to the cytosol by ~300%, providing a method for facile, selective nuclear targeting of proteins. Mechanistic studies on this system revealed that the boronic acid group is responsible for the active nuclear import of the proteins, presenting the first example of active transport to the nucleus using a non-peptidic tag.
Fig. 5.
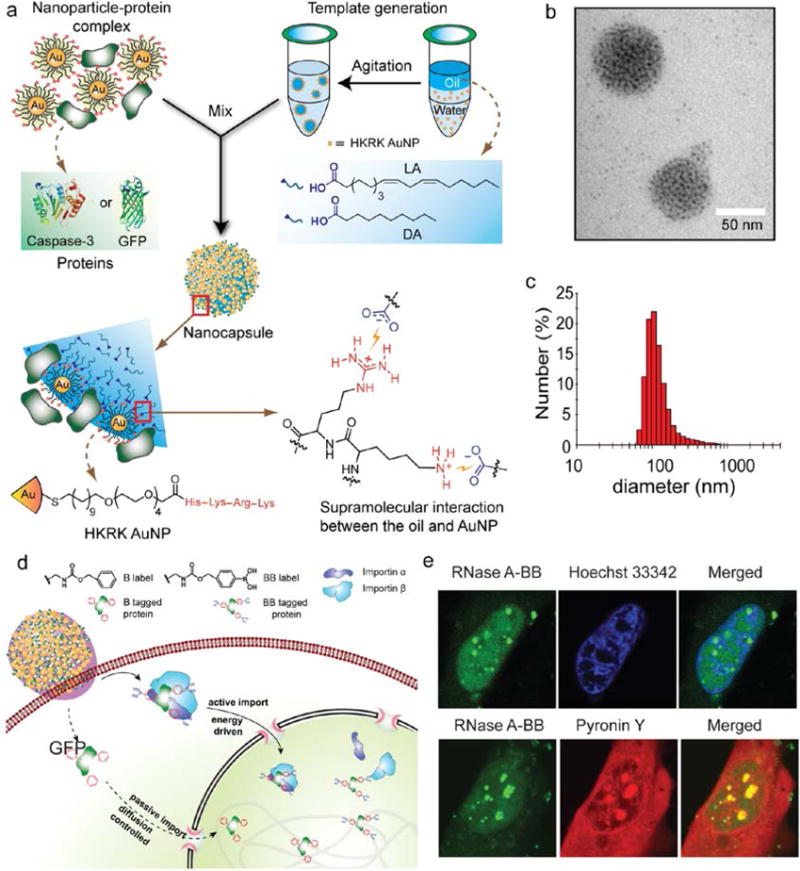
Design, preparation and delivery mechanism of NPSCs. a) Schematic showing the preparation of the protein-NPSC complex containing caspase-3 or GFP. b) TEM image of dried GFP-NPSC. c) DLS histogram of GFP-NPSCs indicating an average diameter of ∼130nm (Adapted with permission from Ref. 19. Copyright (2013) American Chemical Society). d) Schematic diagram showing the delivery of proteins tagged with benzyl boronate (BB) to the cytosol followed by translocation to the nucleus using active transport with boronate ligands and through passive diffusion using nonboronate analogues. e) Colocalization of RNase A-BB with Hoechst 33342, a DNA staining dye (top) and with Pyronin Y, a dsRNA staining dye (bottom) (Adapted with permission from Ref. 39 Copyright (2017) American Chemical Society).
Proteins and small molecules generally exhibit quite different physio-chemical properties, making complexation and co-delivery a real challenge for many delivery vehicles. Cytosolic delivery of therapeutic proteins, using NPSCs was demonstrated by Kim et al. through effective cytosolic delivery of caspase-3 (CASP3), an enzyme of therapeutic importance for its central role in regulating apoptosis.19,39 NPSC systems were further applied to the co-delivery of CASP3 and small molecule drugs (paclitaxel, PTX) using a single carrier. By loading PTX into the hydrophobic core, while simultaneously self-assembling CASP3 onto the shell of the capsule, the NP-Arg-based NPSCs were able to demonstrate synergistic behavior, with improved cytotoxicity over either therapeutic individually. This strategy for delivery presents a powerful therapeutic method, offering a ‘one-two’ punch for cellular therapy by integrating the activities of proteins and small molecule therapeutics.
Tuning the supramolecular chemistry of these NPSCs can also provide a simple method for optimization of fusion-based delivery conditions, the conditions of which will vary depending on the cargo. To improve delivery efficiency for large, highly charged proteins, the strength of the electrostatic interaction between the NPs and oil core was decreased. Re-formulation of the NPs ligands using 1-guanidino-2-(4-imidazole) propionic acid (GIPA) moieties was shown to facilitate more effective cytosolic payload release of β-Gal (464 kDa), while maintaining its capable delivery efficiency of dsRed, demonstrating the wide range of applicability for this system.40
Nanocomposite systems containing only nanoparticles and proteins have been developed for protein delivery applications as well. Nanocomposites containing only particle and protein are quite desirable as protein delivery agents due to their simplicity. Similar nanocomposite delivery systems involving polymers have also been reported in literature.21 One of the first examples of purely protein/particle mixed nanocomposites was reported by Khandelia et al., who developed a self-assembled nanocarrier based on gold nanoparticle-protein agglomerates.41 The formation of the nanocomposite occurred by spontaneous assembly of negatively charged citrate-capped gold nanoparticles and positively charged lysozyme. These agglomerates were capped with BSA to provide stability to the nanocomposite in biological fluids and potentially facilitate cellular uptake. Later, BSA was substituted with poly(lactic-co- glycolic acid (PLGA) to fabricate a biocompatible and protease degradation-free nanocarrier.42 Using electrostatic attraction and hydrophobic interactions, the authors demonstrated the ability of this system to incorporate more than one protein and host both hydrophobic and hydrophilic drugs. This multi-cargo loading utility of the vehicle was exploited to induce apoptosis of cancer cells, upon internalization of the entire complex by the cell.
The first protein-nanoparticle composite to be used successfully for direct cytosolic protein delivery was reported recently by Mout et al.20 The authors fabricated self-assembled superstructures through co-engineering of nanoparticles and recombinant proteins. These assemblies featured multiple layers of structural hierarchy between proteins and nanoparticles.43 Here, the authors engineered GFP by genetically incorporating a peptide chain comprised of 10 units of glutamic acid (E10-tag) at the C-terminus of the protein. These engineered GFPs (GFP-E10) were self-assembled with arginine-terminated gold nanoparticles (ArgNPs) through carboxylate-guanidinium interactions to generate hierarchical superstructures (Fig. 6b). Transmission electron microscopy (TEM) studies revealed three distinct layers of hierarchical organization of proteins and nanoparticles within the superstructures. The first layer is a “corona like” structure 10 nm in diameter where the ArgNPs are surrounded by multiple GFP-E10 proteins. The second layer of the hierarchical organization is the “granule like” structure (~20 nm diameter) evolved from the “corona like” structure. These granular structures were then assembled together to produce the final ~250- 350 nm diameter superstructure. Loading E10-tagged proteins featuring varying size and pi values compared to native protein (such as Histone 2A, Cre recombinase, single chain antibody fragment, scFv) generated identical hierarchical structures. Next, the authors screened these hierarchical spherical nanoassemblies for delivery efficiency in mammalian cells. Similarly to NPSCs, these supramolecular assemblies fused with cell membranes through interaction of Arg-NPs ligands with the membrane, releasing the E-tagged protein directly into the cytosol, bypassing endosomal entrapment (Fig. 6c). The generality of the method was demonstrated by effectively delivering five different E-10 tagged proteins (GFP, Histone 2A, Granzyme A, Cre recombinase and Prothymosin-α) with diverse charge, size, and function. Time-lapse confocal microscopy demonstrated that after contacting the cell membrane, encapsulated protein was quickly released into the cytosol reaching the opposite end of the cell in less than 30 s and visible in the nucleus after 90 s. Significantly, the authors have demonstrated a highly efficient gene editing strategy based on the co-delivery of the Cas9 protein and sgRNA into mammalian cells using this nanoparticle-protein nanocomposite delivery approach.44 In this work, the authors have inserted E-tags at the N-terminus of Cas9, and also appended a nuclear localization signal (NLS) tag at the C-terminus to enhance nuclear accumulation. When the E-tagged Cas9 was mixed with ArgNP, they spontaneously formed self-assembled superstructures via carboxylate-guanidinium binding. The authors reported that both the length of the E-tag and the molar ratio between Arg-NPs and Cas9-En play a crucial role both in the size of the self-assembly and the efficiency of cytosolic delivery. Specifically, through parametric variation of the E-tag length, the authors found that E-20 provided the most efficient delivery (90%) of Cas9 into the cytosol and nucleus of mammalian cells, providing optimization of the electrostatic interaction between the protein and the NPs and demonstrating effective cytosolic delivery. Significantly, the engineered Cas9 protein retained its activity after cytosolic delivery since this construct provided effective gene editing efficiency.
Fig. 6.

Cationic ArgNPs complex with E-tag functionalized proteins (a) self-assemble into nanocomposite with increasing complexity as more conjugate units are introduced into the structure (b) (Adapted with permission from Ref. 43. Copyright (2017) American Chemical Society). Mechanism of uptake via membrane fusion followed by translocation of the protein in the nucleus (c) (Adapted with permission from Ref. 44. Copyright (2017) American Chemical Society). Unmodified GFP (GFP-E0) is not delivered into cell (d) while cytosolic delivery is demonstrated with GFP-E10 in HeLa cells (e and f). Flow cytometry data showing GFP-En delivery efficiency (as mean fluorescence intensity, MFI) increased with the E-tag length, reaching maximum at GFP-E10 (g). ArgNP nanocomposites demonstrated delivery of GFP-E10 in a variety of cell lines representing multiple types of tissues (h) (Adapted with permission from Ref. 20. Copyright (2017) American Chemical Society).
4. Stimuli-responsive nanoparticle supramolecular assemblies for protein delivery
Stimuli-responsive nanoparticle-protein supramolecular assemblies have been developed to provide enhanced control of protein delivery and release. These systems include endogenous pH- or redox-responsive as well as external stimuli-responsive nanoparticle assemblies. Typically, redox-responsive assemblies are designed to promote release of the protein cargo from the nanocarrier after internalization into cells, based on the higher intracellular levels of reducing thiols pH-responsive carriers are used to promote release of the protein after endosomal entrapment, taking advantage of the lower pH inside endosomes/lysosomes. Finally, External stimuli-responsive systems can be designed to direct the carrier and the protein to a specific target and/or promote release of the protein in the cytosolic compartment after internalization of the carrier via endocytosis.
An example of redox-responsive supramolecular assembly was recently reported by Yao et al., in which biodegradable silica nanocapsules were used for hypoxia-triggered intracellular protein delivery.45 These silica nanocapsules were doped with a bis(orthosilicate)-containing azo monomer that under hypoxic conditions could be reduced by flavoproteins, leading to degradation of the silica shell and subsequent release of the encapsulated protein. The silica capsules were doped with a fluorescent dye and a “black hole” quencher as a turn-on imaging module for sensing cell hypoxia and protein release. The surface of this protein-encapsulated biodegradable silica nanoquencher was then coated with cell-penetrating poly(disulfide)s to promote delivery via an endocytosis-independent pathway. This system was employed for delivery of BSA, as model protein, RNase A, as active enzyme, and the native therapeutic antibody Cetuximab. These protein-loaded silica nanoquenchers were taken up by cells via thiol-mediated pathways, allowing direct access into the cytosol, following depolymerization of the poly(disulfide) coating by intracellular GSH. Finally, when cells became hypoxic, the imaging and the release modules were activated, allowing protein release and imaging. Interestingly, while uptake was not affected by the presence of endocytosis inhibitors and the fluorescence of the released FITC-labeled protein did not co-localize with endosomal tracking agents, the fluorescence showed in confocal images was mainly punctate indicating vesicular entrapment.
External stimuli, such as light irradiation and electromagnetic fields have also been employed to promote protein release or direct the nanoparticle to the target. Tang et al. have demonstrated that plasmonic nanoparticles (e.g. gold nanorods, GNRs) can be used for transdermal protein delivery induced by near infrared (NIR) laser irradiation,46 while Heinemann et al. showed that GNPs may cause a transient permeabilization of the cellular membrane after laser irradiation and promote uptake of proteins subsequently added.47 However, laser induced intracellular delivery of proteins conjugated to plasmonic nanoparticles is still largely unexplored.
Photochemical internalization (PCI) is a common release stimulus employing light. In this method, a photosensitizer is introduced in the nanocarrier that is taken up by cells via endocytosis. After exposure to the light, the photosensitizer produces singlet oxygen to destroy the endosomal membrane, promoting release of the entrapped protein. Several examples employing liposomes or polymeric nanocarriers have been reported for protein delivery.48 Febvay et al. have reported the use of mesoporous silica nanoparticles (size<200nm) for targeted cytosolic delivery by light-triggered endosome disruption.49 Authors have shown effective release of FITC-dextran loaded in nanoparticles, providing a promising approach for cytosolic release of cell-impermeable molecules. However, the effectiveness of PCI for protein delivery using inorganic nanoparticles has yet to be investigated.
Externally generated magnetic fields have been used for targeting therapeutic protein delivery as well as for promoting intracellular release by conjugation of the protein with magnetically-responsive nanoparticles. Chertok et al. reported the use of an external magnetic field as a targeting methodology for selective delivery of protein drug-loaded magnetic particles to brain tumors.50 In this work, β-Gal was modified with low molecular weight cationic polyethyleneimine (PEI) domains (β-Gal-PEI) to enable translocation across biological membranes and electrostatic interaction with the negatively charged polyelectrolyte heparin on the nanoparticle surface. Following tumor localization by magnetic targeting, β-Gal PEI can either desorb from the nanoparticle before translocating across the microvascular barrier or be delivered with the entire complex via PEI- mediated transcytosis. In vivo experiments in a rat glioma model demonstrated that β-Gal was selectively delivered into the brain tumor, but not into the normal brain parenchyma.
Beyond guiding the nanovehicle to the target, the induction of an external magnetic field has been used for activating the release of therapeutic proteins inside the cell. An example was reported by Omar et al.51 The authors designed a spherical 100 nm silica-iron oxide hybrid nanovector with large pores for high loading of proteins. These mesoporous nanoparticles where then surface-functionalized with aminopropyltriethoxysilane to allow electrostatic interaction with negatively charged labeled proteins, such as Alexa-Ferritin and mTFP-Ferritin (Fig. 7). The release of the proteins was then promoted by applying an external magnetic field. Specifically, the electromagnetic energy from the magnetic field was converted into thermal energy by the nanovectors, promoting the disruption of the electrostatic interactions between the negatively charged proteins and the positively charged superparamagnetic nanovectors. Full release of the proteins occurred by applying a magnetic field after only 3h at pH 5, mimicking the lysosome environment. Negligible release of the proteins was shown in physiological conditions, however, without the magnetic field. An example of a magnetically guided nanocomposite system for intracellular protein delivery has been recently reported by Kawasaki et al. by loading iron oxide nanoparticles and proteins into a nanogel.52 This magnetic nanogel chaperone (MC) was obtained by selfassembling cholesterol-bearing pullulan, a hydrophobic polysaccharide, into a nanogel and subsequently loading hydrophobized iron oxide nanoparticles. The proteins were then encapsulated within the matrix of MC through hydrophobic interactions. It was shown that the protein loading could be tuned by changing the concentration of the incorporated iron oxide nanoparticles. The obtained MCs loaded with Alexa488-BSA or FITC-insulin, as model proteins, were delivered into HeLa cells by applying a magnetic field and the protein cargo was released in the cell by replacement with cytosolic proteins. The delivery of active proteins such as caspase-3, as a therapeutic protein, and β-Gal, as hydrolytic enzyme for the activation of a model prodrug, was also shown, demonstrating the feasibility of the system to deliver different proteins. However, while co-localization studies of MC and BSA exhibited a decrease in the co-localization during the time, confocal images showed punctate fluorescence, suggesting vesicular entrapment and, therefore an uptake mechanism involving endocytosis. Another similar nanocomposite system, employing iron oxide nanoparticles embedded with a polymer, has been developed by Lee et al., for both protein delivery and multimodal imaging.21 The nanocomposite was obtained by self-assembly of redox-responsive polymer ligands and superparamagnetic iron oxide nanoparticles (SPIONs) 4 nm size, following complexation via electrostatic interactions with human serum albumin labeled with Cy5.5 (HAS-Cy5.5). The multifunctional polymer ligand was comprised of dopamine groups, as an anchor to facilitate self-assembly via high affinity binding of SPIONs, diethylenetriamine, to provide high protein loading capacity, and disulfide linkages that could be reduced by intracellular GSH to promote protein release. The resulting nanocomposite displayed a magnetic core, composed of SPIONs, and a polymeric hydrophilic shell (Fig. 8). While the cytosolic distribution of HSA-Cy5.5 inside the cells was limited, in vivo studies showed the ability of this system to act as a dual-modal imaging probe for tumor diagnosis.
Fig. 7.
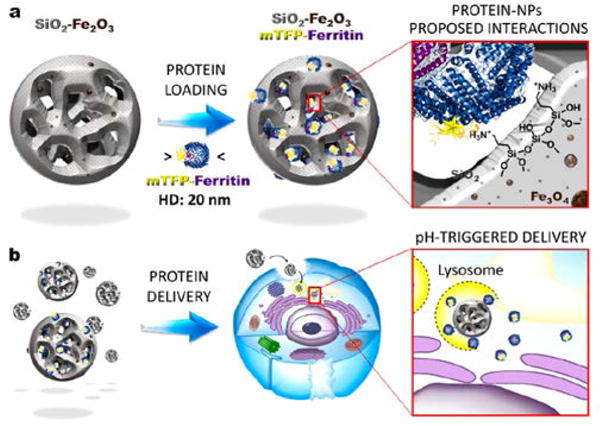
A hybrid silica-iron oxide nanoparticle utilizing an external magnetic field for tissue targeting and controlled cargo release: protein-nanoparticle interactions (a) and pH-triggered delivery into cells (b). (Reprinted with permission from Ref. 51 Copyright (2017) Elsevier).
Fig. 8.
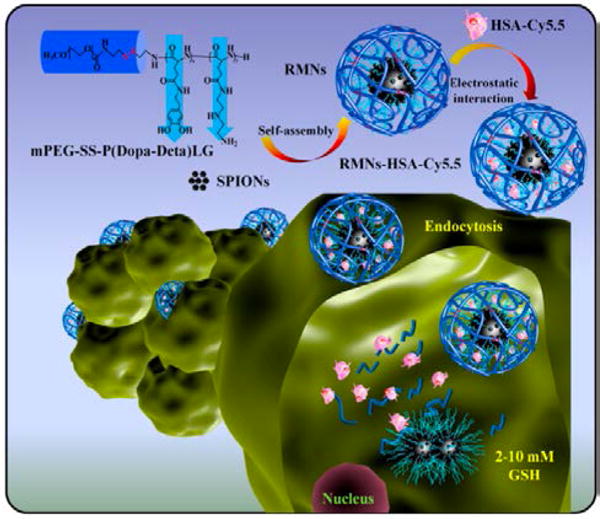
Schematic representation of the intracellular protein delivery with the nanocomposite made of magnetic core of self-assembled SPIONs and a polymeric hydrophilic shell. (Reprinted with permission from Ref.21. Copyright (2017) Elsevier).
Finally, NPSC delivery methods have been diversified from gold nanoparticles (AuNPs) to incorporate CdSe/ZnS core-shell quantum dot (QD) technology.53 QDs were laterally stabilized by electrostatic interaction with functionalized AuNPs, which are anchored to the fatty acid core through guanidinium-carboxylate interactions. This lipophilic core functions as a reservoir for endosome-disrupting agents. Polyhistidine-tagged protein systems were utilized for their metal-histidine coordination affinity that can be disrupted by competing molecules for triggered cargo release.
5. Summary
Nanoparticle-protein assemblies provide promising vectors for protein delivery. Size, surface functionality and the overall structure of the assembly play crucial roles in intracellular protein delivery. Specifically, nanoparticle-protein assemblies with sizes below 300 nm have been shown to be most effective for efficient delivery in terms of pharmacodynamics and cellular internalization efficiency. Surface functionality is involved in both the loading of the protein cargo through non-covalent interactions (e.g. electrostatic, hydrogen bonding, hydrophobic interactions) as well as in the interaction with the cell membrane. The overall structure of these vectors can be modulated to allow insertion of imaging or targeting modules, or to incorporate a hydrophobic core for lipophilic molecule loading or facilitating uptake.
Common challenges for protein delivery include: targeting a specific tissue to reduce offtarget effects and achieve effective cytosolic delivery. Endosomal entrapment of delivered proteins is a key issue that significantly decreases delivery efficacy for many vehicles. Nanocapsule and nanocomposite formulations that provide membrane fusion avoid the issue of endosomal entrapment and have increased delivery efficiency as a result. Stimuli- responsive nanoassemblies have also provided promising alternatives to achieve cytosolic delivery and targeting through induced release of the protein cargo from the carrier or endosomes by an external controllable force.
Overall, the diversity of nanoparticle structures provides numerous opportunities for addressing the engineering issues described above. Further advances can be provided through combination of co-engineered nanoparticles and proteins and stimuli-responsive modules, providing access to new structures featuring unique dynamic properties that will facilitate the development of new protein delivery strategies.
Supplementary Material
Key learning points.
Protein delivery holds significant therapeutic potential for a wide range of diseases.
Nanoparticles can be formulated with proteins through direct conjugation or through supramolecular assembly of nanocapsules or nanocomposites.
Cytosolic protein delivery is challenging due to endosomal entrapment.
Delivery of proteins through supramolecular assemblies can be passive or stimuli- induced.
Acknowledgments
This research was supported by the NIH (GM077173 and EB022641) and the NSF (CHE-1506725).
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Walsh G. Nat Biotech. 2010;28:917–924. doi: 10.1038/nbt0910-917. [DOI] [PubMed] [Google Scholar]
- 2.Gu Z, Biswas A, Zhao M, Tang Y. Chem Soc Rev. 2011;40:3638–3655. doi: 10.1039/c0cs00227e. [DOI] [PubMed] [Google Scholar]
- 3.Amer MH. Mol Cell Ther. 2014;2:27. doi: 10.1186/2052-8426-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leader B, Baca QJ, Golan DE. Nat Rev Drug Discovery. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 5.Fu A, Tang R, Hardie J, Farkas ME, Rotello VM. Bioconjugate Chem. 2014;25:1602–1608. doi: 10.1021/bc500320j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi SO, Kim YC, Lee JW, Park JH, Prausnitz MR, Allen MG. Small. 2012;8:1081–1091. doi: 10.1002/smll.201101747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brock R. Bioconjugate Chem. 2014;25:863–868. doi: 10.1021/bc500017t. [DOI] [PubMed] [Google Scholar]
- 8.Varkouhi AK, Scholte M, Storm G, Haisma HJ. J Control Release. 2011;151:220–228. doi: 10.1016/j.jconrel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 9.Qian Z, Dougherty PG, Pei D. Chem Comm. 2015;51:2162–2165. doi: 10.1039/c4cc09441g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Du J, Jin J, Yan M, Lu Y. Curr Drug Metab. 2012;13:82–92. doi: 10.2174/138920012798356862. [DOI] [PubMed] [Google Scholar]
- 11.Ray M, Lee YW, Scaletti F, Yu R, Rotello VM. Nanomedicine (Lond) 2017;12:941–952. doi: 10.2217/nnm-2016-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meńdez J, MoralesCruz M, Delgado Y, Figueroa CM, Orellano EA, Morales M, Monteagudo A, Griebenow K. Mol Pharmacol. 2014;11:102–111. doi: 10.1021/mp400400j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh P, Yang X, Arvizo R, Zhu ZJ, Agasti SS, Mo Z, Rotello VM. J Am Chem Soc. 2010;132:2642–2645. doi: 10.1021/ja907887z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar S, Jana AK, Dhamija I, Maiti M. J Drug Target. 2014;22:123–137. doi: 10.3109/1061186X.2013.844157. [DOI] [PubMed] [Google Scholar]
- 15.Aubin-Tam ME, Hwang W, Hamad-Schifferli K. Proc Natl Acad Sci USA. 2009;106:4095–4100. doi: 10.1073/pnas.0807299106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Das P, Jana NR. ACS Appl Mater Interfaces. 2016;8:8710–8720. doi: 10.1021/acsami.6b01667. [DOI] [PubMed] [Google Scholar]
- 17.Prasetyanto EA, Bertucci A, Septiadi D, Corradini R, Castro-Hartmann P, De Cola L. Angew Chem, Int Ed. 2016;55:3323–3327. doi: 10.1002/anie.201508288. [DOI] [PubMed] [Google Scholar]
- 18.Lim J-S, Lee K, Choi J-N, Hwang Y-K, Yun M-Y, Kim H-Jin, Won YS, Kim S-J, Kwon H, Huh S. Nanotechnology. 2012;23:085101. doi: 10.1088/0957-4484/23/8/085101. [DOI] [PubMed] [Google Scholar]
- 19.Tang R, Kim CS, Solfiell DJ, Rana S, Mout R, Velázquez-Delgado EM, Chompoosor A, Jeong Y, Yan B, Zhu ZJ, Kim C, Hardy JA, Rotello VM. ACS Nano. 2013;7:6667–6673. doi: 10.1021/nn402753y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mout R, Ray M, Tay T, Sasaki K, Yesilbag Tonga G, Rotello VM. ACS Nano. 2017;11:6416–6421. doi: 10.1021/acsnano.7b02884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang HY, Jang MS, Li Y, Lee JH, Lee DS. ACS Appl Mater Interfaces. 2017;9:19184–19192. doi: 10.1021/acsami.7b03747. [DOI] [PubMed] [Google Scholar]
- 22.Le Roy C, Wrana JL. Nat Rev Mol Cell Bio. 2005;6:112–126. doi: 10.1038/nrm1571. [DOI] [PubMed] [Google Scholar]
- 23.Johnson JR, Kocher B, Barnett EM, Marasa J, Piwnica-Worms D. Bioconjugate Chem. 2012;23:1783–1793. doi: 10.1021/bc300036z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wagner H, Heit A, Schmitz F, Bauer S. Curr Opin Biotechnol. 2004;15:538–542. doi: 10.1016/j.copbio.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 25.Parenti G. EMBO Mol Med. 2009;1:268–279. doi: 10.1002/emmm.200900036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.García-Fernández L, Garcia-Pardo J, Tort O, Prior I, Brust M, Casals E, Lorenzo J, Puntes Victor F. Nanoscale. 2017;9:6111–6121. doi: 10.1039/c7nr00947j. [DOI] [PubMed] [Google Scholar]
- 27.Yang J, Tu J, Lamers GEM, Olsthoorn RCL, Kros A. Adv Healthc Mater. 2017;6:1700759. doi: 10.1002/adhm.201700759. [DOI] [PubMed] [Google Scholar]
- 28.Bale SS, Kwon SJ, Shah DA, Banerjee A, Dordick JS, Kane RS. ACS Nano. 2010;4:1493–1500. doi: 10.1021/nn901586e. [DOI] [PubMed] [Google Scholar]
- 29.Niu Y, Yu M, Zhang J, Yang Y, Xu C, Yeh M, Taran E, Cheng Hou JJ, Graya PP, Yu C. J Mater Chem B. 2015;3:8477–8485. doi: 10.1039/c5tb01405k. [DOI] [PubMed] [Google Scholar]
- 30.Niu Y, Yu M, Meka A, Liu Y, Zhang J, Yang Y, Yu C, Mater J. Chem B. 2016;4:212–219. doi: 10.1039/c5tb01911g. [DOI] [PubMed] [Google Scholar]
- 31.Deodhar GV, Adams ML, Trewyn BG. Biotechnol J. 2017;12:1600408. doi: 10.1002/biot.201600408. [DOI] [PubMed] [Google Scholar]
- 32.Yu M, Gu Z, Ottewella T, Yu C, Mater J. Chem B. 2017;5:3241–3252. doi: 10.1039/c7tb00244k. [DOI] [PubMed] [Google Scholar]
- 33.Meka AK, Abbaraju PL, Song H, Xu C, Zhang J, Zhang H, Yu M, Yu C. Small. 2016;12:5169–5177. doi: 10.1002/smll.201602052. [DOI] [PubMed] [Google Scholar]
- 34.You CC, Verma A, Rotello VM. Soft Matter. 2006;2:190–204. doi: 10.1039/b517354j. [DOI] [PubMed] [Google Scholar]
- 35.Varkouhi AK, Scholte M, Storm G, Haisma HJ. J Controlled Release. 2010;151:220–228. doi: 10.1016/j.jconrel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 36.Yang XC, Samanta B, Agasti SS, Jeong Y, Zhu ZJ, Rana S, Miranda OR, Rotello VM. Angew Chem Int Ed. 2011;50:477–481. doi: 10.1002/anie.201005662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ray M, Tang R, Jiang Z, Rotello VM. Bioconjugate Chem. 2015;26:1004–1007. doi: 10.1021/acs.bioconjchem.5b00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tang R, Wang M, Ray M, Jiang Y, Jiang Z, Xu Q, Rotello VM. J Am Chem Soc. 2017;139:8547–8551. doi: 10.1021/jacs.7b02801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim CS, Mout R, Zhao Y, Yeh YC, Tang R, Jeong Y, Duncan B, Hardy JA, Rotello VM. Bioconjugate Chem. 2015;26:950–954. doi: 10.1021/acs.bioconjchem.5b00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang R, Jiang Z, Ray M, Hou S, Rotello VM. Nanoscale. 2016;8:18038–18041. doi: 10.1039/c6nr07162g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khandelia R, Jaiswal A, Ghosh SS, Chattopadhyay A. Small. 2013;9:3494–3505. doi: 10.1002/smll.201203095. [DOI] [PubMed] [Google Scholar]
- 42.Khandelia R, Jaiswal A, Ghosh SS, Chattopadhyay A. J Mater Chem B. 2014;2:6472–6477. doi: 10.1039/c4tb00800f. [DOI] [PubMed] [Google Scholar]
- 43.Mout R, Yesilbag Tonga G, Wang L-S, Ray M, Roy T, Rotello VM. ACS Nano. 2017;11:3456–3462. doi: 10.1021/acsnano.6b07258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mout R, Ray M, Yesilbag Tonga G, Lee Y-W, Tay T, Sasaki K, Rotello VM. ACS Nano. 2017;11:2452–2458. doi: 10.1021/acsnano.6b07600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yuan P, Zhang H, Qian L, Mao X, Du S, Yu C, Peng B, Yao SQ. Angew Chem Int Ed Engl. 2017;56:12481–12485. doi: 10.1002/anie.201705578. [DOI] [PubMed] [Google Scholar]
- 46.Tang H, Kobayashi H, Niidome Y, Mori T, Katayama Y, Niidome T. J Control Release. 2013;171:178–183. doi: 10.1016/j.jconrel.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 47.Heinemann D, Kalies S, Schomaker M, Ertmer W, Murua Escobar H, Meyer H, Ripken T. Nanotechnology. 2014;25:245101. doi: 10.1088/0957-4484/25/24/245101. [DOI] [PubMed] [Google Scholar]
- 48.Selbo PK, Weyergang A, Høgset A, Norum OJ, Berstad MB, Vikdal M, Berg K. J Control Release. 2010;148:2–12. doi: 10.1016/j.jconrel.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 49.Febvay S, Marini DM, Belcher AM, Clapham DE. Nano Lett. 2010;10:2211–2219. doi: 10.1021/nl101157z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chertok B, David AE, Yang VC. Biomaterials. 2011;32:6245e6253. doi: 10.1016/j.biomaterials.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Omar H, Croissant JG, Alamoudi K, Alsaiari S, Alradwan I, Majras MA, Martins P, Laamarti R, Eppinger J, Moosa B, Almalik A, Khashab NM. J Control Release. 2017;259:187–194. doi: 10.1016/j.jconrel.2016.11.032. [DOI] [PubMed] [Google Scholar]
- 52.Kawasaki R, Sasaki Y, Katagiri K, Mukai S, Sawada S, Akiyoshi K. Angew Chem Int Ed. 2016;55:11377–11381. doi: 10.1002/anie.201602577. [DOI] [PubMed] [Google Scholar]
- 53.Yeh YC, Tang R, Mout R, Jeong Y, Rotello VM. Angew Chem, Int Ed. 2014;53:5137–5141. doi: 10.1002/anie.201400559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.