

Abstract
Background
Microvascular endothelial cells (ECs) are central to an allograft’s immunogenicity. Cold ischemia and reperfusion injury associated with static cold storage and warm reperfusion activates ECs and increases the immunogenicity of the allograft. Following reperfusion, mitochondrial permeability transition pore (mPTP) opening contributes to mitochondrial dysfunction in the allograft, which correlates to alloimmune rejection. Current understanding of this relationship, however, centers on the whole allograft instead of ECs. This study aimed to elucidate the relationship between EC mPTP opening and their immuno-phenotype.
Methods
Mitochondrial metabolic fitness and glycolysis in ECs were assessed in parallel with metabolic gene microarray postreperfusion. NIM811 was used to inhibit mPTP opening to rescue mitochondrial fitness. The immunogenicity of NIM811-treated ECs was determined via levels of EC’s pro-inflammatory cytokines and allogeneic CD8+ T cell co-cultures. Finally, EC surface expression of adhesion, co-stimulatory, co-inhibitory, MHC-I molecules, and MHC-I machinery protein levels were characterized.
Results
Genes for glycolysis, tricarboxylic acid cycle, fatty acid synthesis, gluconeogenesis were upregulated at 6 hours postreperfusion but either normalized or downregulated at 24 hours postreperfusion. As mitochondrial fitness was reduced, glycolysis increased during the first 6 hours postreperfusion. EC treatment with NIM811 during the early postreperfusion period rescued mitochondrial fitness and reduced EC immunogenicity by decreasing CCL2, KC release, and VCAM-1, MHC-I, TAP1 expression.
Conclusions
Static cold storage and warm reperfusion leads to a reduction in mitochondrial fitness in microvascular ECs due to mPTP opening. Further, mPTP opening promotes increased EC immunogenicity that can be prevented by NIM811 treatment.
INTRODUCTION
Early insults to a transplanted allograft, including injury associated with the standard cold storage and warm reperfusion (CS-WR), contribute to poor long-term transplant outcomes.1–4 In addition, the effects of circulating memory T cell (Tmem) populations, which are spared from conventional induction immunosuppression, remain a barrier to achieving allograft tolerance.5–8 Microvascular endothelial cells (ECs) are central to the immunogenicity of allografts early posttransplantation and function as semi-professional antigen-presenting cells (APCs), interfacing between the donor allograft and the recipient immune system.9 After CS-WR, donor ECs are activated and recruit recipient immune cells, including circulating Tmem to the newly transplanted organ, creating an inflammatory microenvironment.1,2,4 Donor ECs adopt an immunogenic phenotype that directly activates recipient alloreactive Tmem,4,10 resulting in T cell infiltration into the allograft, within 24 hours, leading to eventual graft dysfunction.4,10–15 Given the importance of the donor microvascular endothelium, modulating the immunogenicity of ECs can potentially lead to graft tolerance.16,17
Mitochondrial physiology is essential to immune cell activation. Regulating the mitochondrial electron transport chain (ETC) alters the cellular response to immunologic stimuli.18 Ischemia-reperfusion injury (IRI) in transplantation impacts mitochondrial function of the allograft, by causing mitochondrial heterogeneity, ETC defects, reduction in mitochondrial membrane potential, and mitochondrial swelling.19–25 An important component regulating mitochondrial physiology is the mitochondrial permeability transition pore (mPTP) located on the inner mitochondrial membrane.26 In transplantation, its opening is implicated in the mitochondrial damage associated with preservation and reperfusion.23,27 Although numerous studies have investigated the detrimental effects of IRI on donor graft’s mitochondria, they do not fully elucidate the relationship between mitochondrial dysfunction and graft immunogenicity. Limited studies suggest an association between the allograft’s mitochondrial profile and the immunologic outcome.28–30 More importantly, the mPTP opening of allografts has been linked to CD8+ T cell infiltration and increased intra-graft perforin, granzyme, TNFα levels.28 However, these studies focus on the mitochondrial profile of the allograft’s parenchymal cells and not on microvascular ECs, the APCs that dictate early graft immunogenicity.28–30
We have previously shown that ischemia-reperfusion promotes ECs to release pro-inflammatory cytokines, and that sensitized “memory-like” allogeneic T cells co-cultured with these ECs release more interferon gamma (IFNγ).17 In the current study, we aimed to elucidate the impact of mPTP opening and mitochondrial dysfunction on the immunologic phenotype of microvascular ECs following static CS-WR.
MATERIALS AND METHODS
Cells and culture media
Immortalized FVB mouse cardiac microvascular ECs were purchased from Cedarlane (Ontario, Canada), and cultured in DMEM (Sigma-Aldrich, MO) supplemented with 5% fetal bovine serum (Sigma-Aldrich, MO) in T75 tissue-culture flasks in a 37°C incubator with humidified room air and 5% CO2.
In-vitro static cold storage model
A cell culture model that recapitulates CS-WR was used as previously described.17 Briefly, a confluent monolayer of ECs underwent a 6-hour period of cold ischemia in University of Wisconsin (UW) solution (Bridge to Life, SC) at 4°C in a sealed hypoxic chamber preflushed with pure nitrogen (Airgas, SC). After cold ischemia, UW was removed and replaced with warm culture media to stimulate reperfusion injury. For mPTP modulation, 1μM NIM811 was added to the reperfusion media.
Real-time quantitative gene expression microarrays
ECs underwent CS-WR as described, and at 6 hours or 24 hours postreperfusion, RNA was isolated from the cells using mirVana miRNA isolation (Life Technologies, CA), quantified, and cloned to cDNA using RT2 First Strand kit (Qiagen, Hilden, Germany). cDNA was added to 84-gene RT2 Profiler PCR Arrays for glucose metabolism (PAMM-006Z) or antigen-presenting cells (PAMM-406Z) (Qiagen, Hilden, Germany). Real-time PCR was detected with Bio-Rad CFX96 (Hercules, CA). Fold changes in gene expression were compared to normal cell controls.
Oxygen consumption and glycolytic flux assays
Commercial mitochondrial and glycolytic stress tests were performed to determine spare respiratory capacity and glycolysis using Seahorse Bioscience XF96 instruments (North Billerica, MA) as previously described.31 Briefly, ECs were subjected to CS-WR, and at the denoted timepoints postreperfusion, the cells were subjected to mitochondrial stress test with 3 injections: 1μM oligomycin, 1μM FCCP, 2μM rotenone/100nM antimycin A (Sigma-Aldrich, MO). Oxygen consumption rate measurements before oligomycin and after FCCP injections were used to calculate spare respiratory capacity. For glycolytic stress test, 3 injections were 10mM glucose, 1μM oligomycin, and 100mM 2-deoxyglucose (Sigma-Aldrich, MO). Extracellular acidification rate measurements before and after glucose injection were used to determine glycolysis in the cells.
mPTP modulation
mPTP inhibition by NIM811 (Norvatis, Basel, Switzerland) was performed at a 1μM dose, as described previously32 and based on our preliminary dosage determination (data not shown).
Memory CD8+ T cell generation and isolation
Male C57BL/6 mice (Jackson Laboratory, ME) were used for co-culture experiments. Animals were housed under conventional conditions at Medical University of South Carolina (MUSC, Charleston, SC). All procedures were approved by MUSC Committee for Animal Research in accordance with the NIH Guide for Care and Use of Laboratory Animals. C57BL/6 mice were injected i.p. with 3.5 × 106 ECs, and 3 weeks later spleens were removed. Immediately following spleen removal, splenocytes were isolated, and CD8+ T cells were purified using an untouched isolation kit (Miltenyi Biotec, CA) and an autoMACS Pro Separator (Miltenyi Biotec, CA).17 CD8+ T cell purity was confirmed to be >96% by flow cytometry using an anti-mouse CD8 APC/Cy7 antibody (BD Biosciences, NJ). The phenotype of purified T cells was confirmed by flow cytometry using fluorophore-conjugated antibodies against CD69 (Caltag, CA), CD44, CCR7, CD62L (BioLegend, CA), CD8 (BD Biosciences, NJ), and live cell gating was performed using 7AAD exclusion (Invitrogen, CA).
Endothelial-T-cell co-culture
ECs seeded in a 48-well plate at 30,000 cells/well were rested for 24 hours and subjected to static cold storage for 6 hours in UW. Subsequently, ECs were reperfused with warm media and co-incubated with 400,000 CD8+ T cells/well (at 1:14 target-to-T-cell ratio). 1μM NIM811 or vehicle control was added at the initiation of co-culture treating both ECs and T cells. In a second co-culture model, after cold storage, ECs were reperfused with warm media containing 1μM NIM811 or vehicle control. Six hours later ECs were washed and co-incubated with 400,000 CD8+ T cells/well (at 1:14 target-to-T-cell ratio). Supernatants were collected for ELISA after 7 days of co-culture in both models.
Cytokine analysis
MCP-1/CCL2, granzyme B, KC (R&D Systems, MN), and IFNγ, IL-6 (BD Biosciences, NJ) supernatant levels were determined by ELISA. MCP-1/CCL2 and KC levels were normalized to the total protein concentration of the corresponding group. Granzyme B and IFNγ levels were normalized to the number of ECs at the initiation of co-culture.
Flow cytometry
After cold storage, ECs were warm-reperfused with/without 1μM NIM811 for 6 or 24 hours. ECs were detached, washed, and resuspended in PBS supplemented with 1% BSA (Fisher Scientific, NH) and 10μg/mL DNAse I (Sigma-Aldrich, MO). ECs were then FcR-blocked at 10μL/mL (eBioscience, CA) and stained with fluorophore-conjugated antibodies against ICAM-1, VCAM-1, PD-L1, MHC-I (BioLegend, CA), E-selectin, CD80 (BD Biosciences, NJ), and CD86 (eBioscience, CA). Mean fluorescence intensity (MFI) was determined by Guava easyCyte 8HT flow cytometer (Merck Millipore, MA). Data were processed with FCS Express 4 (De Novo Software, CA).
Immunoblotting
After cold storage, ECs were warm-reperfused with/without 1μM NIM811 as described above for 6 or 24 hours. Cells were then lysed with M-PER buffer containing 1× phosphatase-protease cocktails (Thermo Scientific, MA). After BCA quantification (Thermo Scientific, MA), protein lysates were denatured and separated with 4-20% gradient SDS-PAGE, followed by a wet transfer to PVDF membrane (Bio-Rad Laboratories, CA). Because MHC-I assembly requires TAP1 to transport the antigen peptide into endoplasmic reticulum where calreticulin facilitates peptide loading, primary antibodies against TAP1, calreticulin (Cell Signaling, MA), and beta actin (Santa Cruz, TX) were used. Antibodies were detected using standard western blotting techniques and the signal was visualized by chemiluminescence.
Statistical analysis
All data are expressed as mean±SEM. Data analysis was performed using GraphPad Prism software version 7.03 for Windows (GraphPad, CA) unless otherwise specified. For analysis of glycolysis and mitochondrial fitness, multiple comparisons with two-way ANOVA were performed. For analysis of mitochondrial recovery with NIM811, multiple comparisons with one-way ANOVA were performed. For the remaining analyses, Student t-test was performed. All analyses were 2-sided, and p-values of less than 0.05 were considered statistically significant.
RESULTS
Cold Storage and Warm Reperfusion Induces Time-dependent Changes in Metabolic Gene Expression
Genetic screening for metabolic gene alterations was performed to elucidate the alterations of EC cellular metabolism in the setting of CS-WR. ECs were subjected to cold storage for 6 hours and reperfused with warm media for a period of 6 or 24 hours as previously published.17 At these time points, we performed gene microarray using Qiagen 84-gene chips for glucose metabolism. To define a biologically meaningful change in gene expression, we set the threshold to be either higher than 2-fold or lower than minus 2-fold compared to the normal cell control. Out of 84 genes that were screened (Table S1), at 6 hours postreperfusion 3 genes encoding enzymes for glycolysis (Eno1, Eno3, Aldoa), 2 genes of tricarboxylic acid (TCA) cycle (Aco1, Aco2), 1 gene of fatty acid synthesis (Acly), and 1 gene of gluconeogenesis (G6pc3) were upregulated. Interestingly, at 24 hours postreperfusion, the genes encoding enzymes for glycolysis, TCA cycle, fatty acid synthesis, and gluconeogenesis were either normalized (Eno1, Eno3, Aldoa, Aco2, Acly, G6pc3) or downregulated (Aldoc, Idh1), except for Aco1, which remained upregulated (Figures 1A–D). In addition, at 24 hours postreperfusion, gene microarray analysis showed a downregulation of Pdk2 encoding pyruvate dehydrogenase kinase, a regulatory enzyme that normally facilitates pyruvate conversion into lactate. Pdk2 downregulation correlated to an increase in pyruvate shunting into the TCA cycle (Figure 1E). Also at 24 hours postreperfusion, Pygm encoding an enzyme for glycogenolysis was upregulated (Figure 1F). Collectively, screening results using gene microarray suggest the highest degree of metabolic gene alterations occurred during the first 6 hours postreperfusion.
Figure 1. Metabolic Gene Expression Postreperfusion.
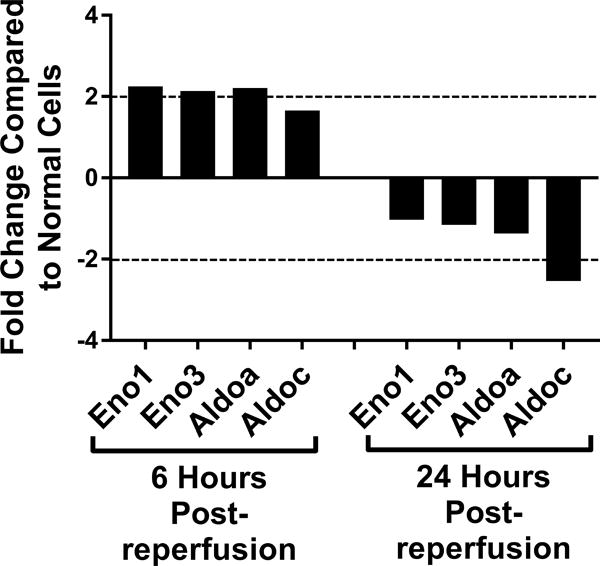
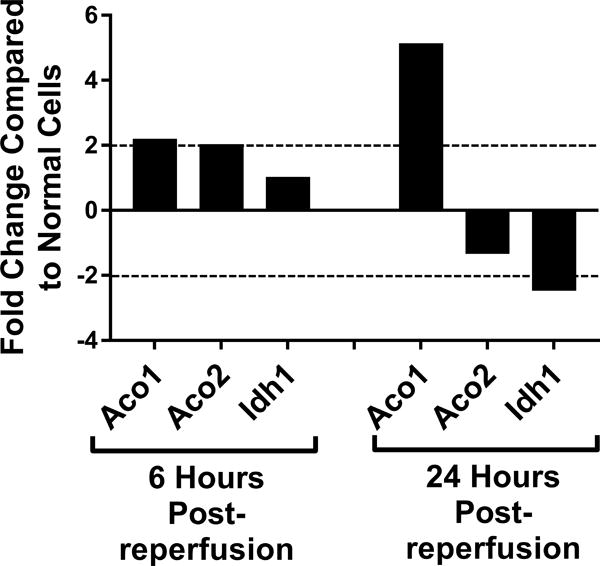
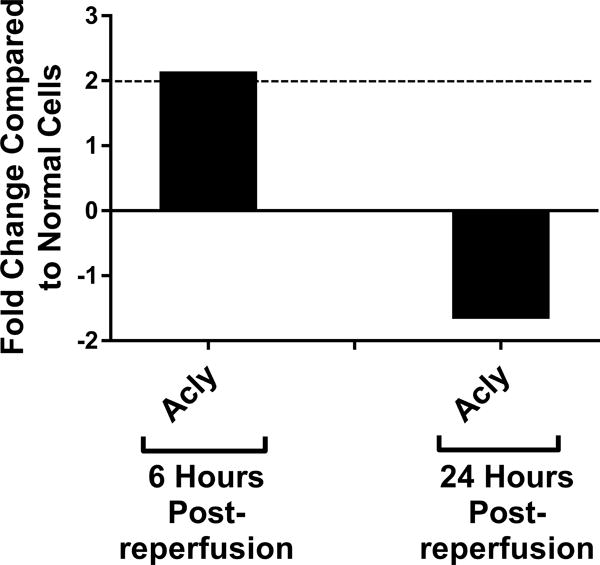
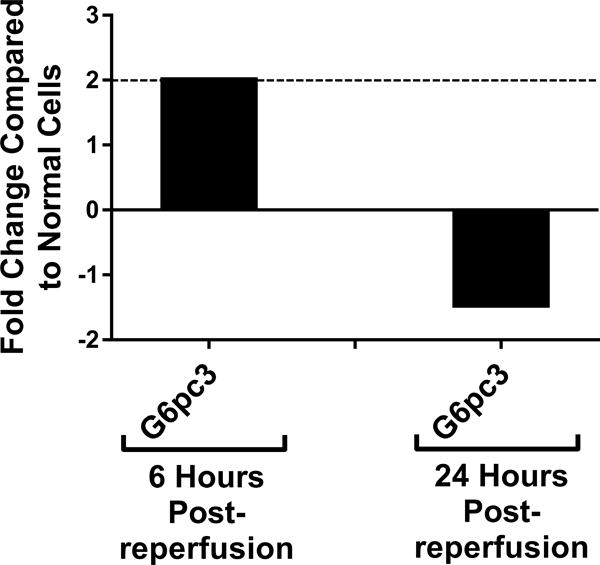
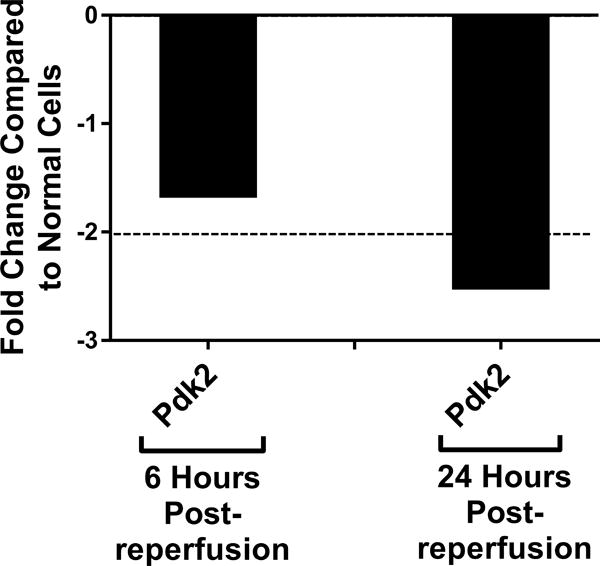
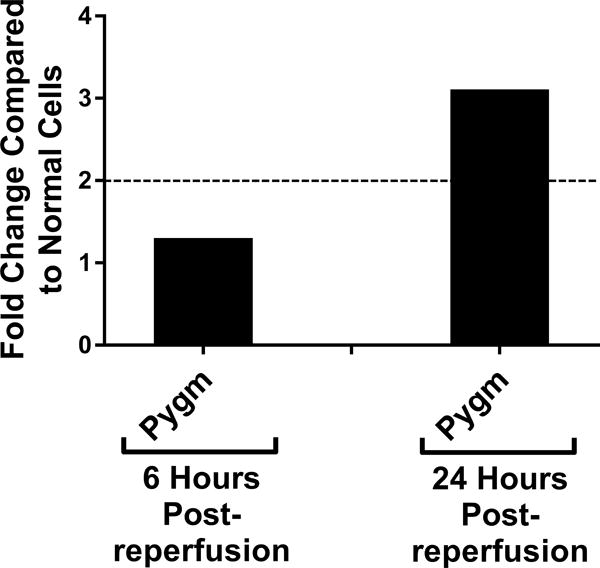
Cold storage and warm reperfusion induces genes encoding enzymes for (A) glycolysis (Eno1, Eno3, Aldoa), (B) tricarboxylic acid (TCA) cycle (Aco1, Aco2), (C) fatty acid synthesis (Acly), and (D) gluconeogenesis (G6pc3) to be upregulated at 6 hours postreperfusion. At 24 hours postreperfusion, the genes of glycolysis, TCA cycle, fatty acid synthesis, and gluconeogenesis are either normalized (Eno1, Eno3, Aldoa, Aco2, Acly, G6pc3) or downregulated (Aldoc, Idh1), except for Aco1, which remains upregulated. At 24 hours postreperfusion, (E) Pdk2 encoding pyruvate dehydrogenase kinase is downregulated, allowing more pyruvate to be shunted into the TCA cycle, and (F) Pygm encoding an enzyme for glycogenolysis is upregulated. (n=3)
Cold Storage and Warm Reperfusion Induces Time-dependent Changes in Glycolysis and Mitochondrial Fitness
To further characterize the metabolic function of ECs after CS-WR, we utilized Seahorse bioenergetic flux technology to assess glycolysis, expressed in extracellular acidification rate (ECAR), and mitochondrial metabolic fitness, expressed in oxygen consumption rate (OCR), at 6-hour intervals during the first 24 hours postreperfusion. These data show that glycolysis increased during the first 6 hours postreperfusion, then normalized, and slightly depressed around 20-24 hours postreperfusion (Figures 2A, 2B). Mitochondrial metabolic fitness, reflected by mitochondrial spare respiratory capacity (SRC), was decreased during the first 24 hours postreperfusion, with the most pronounced decrease observed during the first 12 hours postreperfusion (Figures 2C, 2D).
Figure 2. Glycolysis and Mitochondrial Metabolic Fitness Postreperfusion.
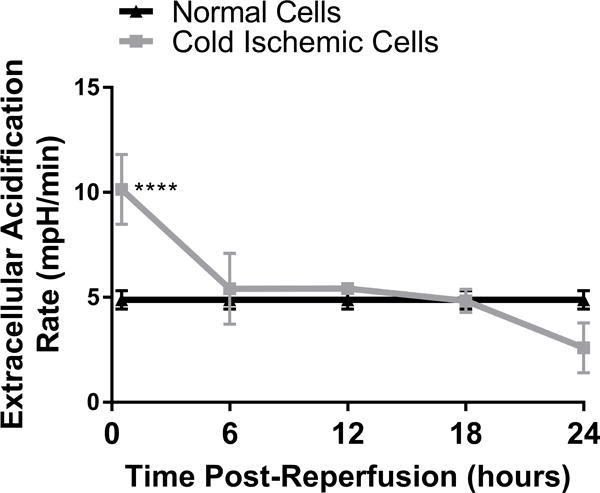
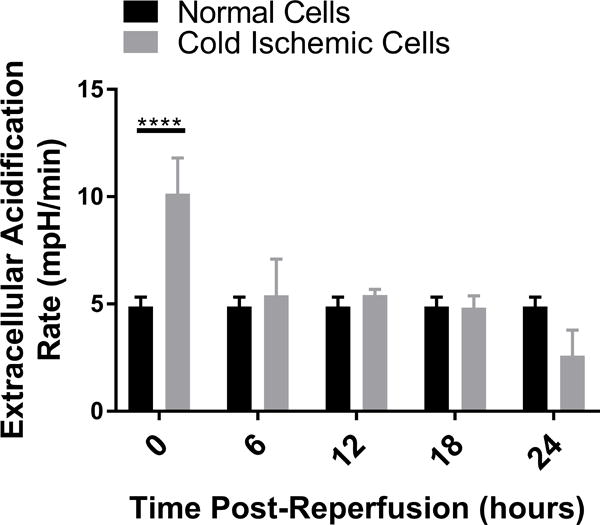
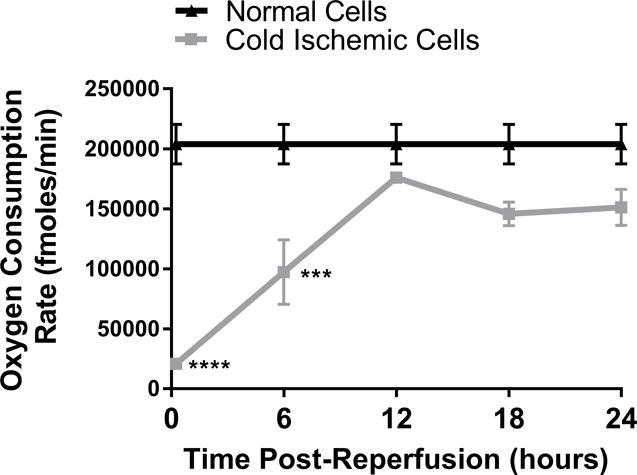
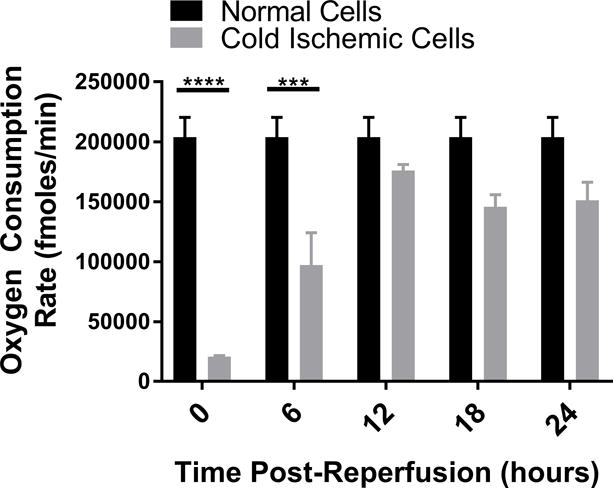
Cold storage and warm reperfusion increases glycolysis during the first 6 hours postreperfusion (A: line graph, B: bar graph) and decreases mitochondrial metabolic fitness during the first 24 hours postreperfusion, with the most pronounced effects observed during the first 12 hours postreperfusion (C: line graph, D: bar graph). (n=3, ***p<0.001, ****p<0.0001).
Inhibiting mPTP Opening Recovers Mitochondrial Metabolic Fitness
IRI is known to induce mitochondrial permeability transition pores (mPTP) to open upon reperfusion, thus compromising mitochondrial integrity.26,27 As the oxygen consumption flux assay suggested a reduction in mitochondrial metabolic fitness, we next examined the implication of mPTP opening upon reperfusion in our cold storage model. ECs were subjected to cold storage for 6 hours, followed by warm reperfusion with media containing NIM811, a specific mPTP opening inhibitor, or vehicle control for 6 hours, and mitochondrial SRC was evaluated. Our data reveal that inhibiting mPTP opening during the postreperfusion period led to full mitochondrial recovery (NIM811 Postreperfusion vs Normal: 196578±11417 vs 214675±5548 fmoles/min, p=0.3593, not significant; Cold Ischemic vs Normal: 157607±7562 vs 214675±5548 fmoles/min, p=0.0021) (Figure 3).
Figure 3. Mitochondrial Recovery when mPTP Opening Is Inhibited.
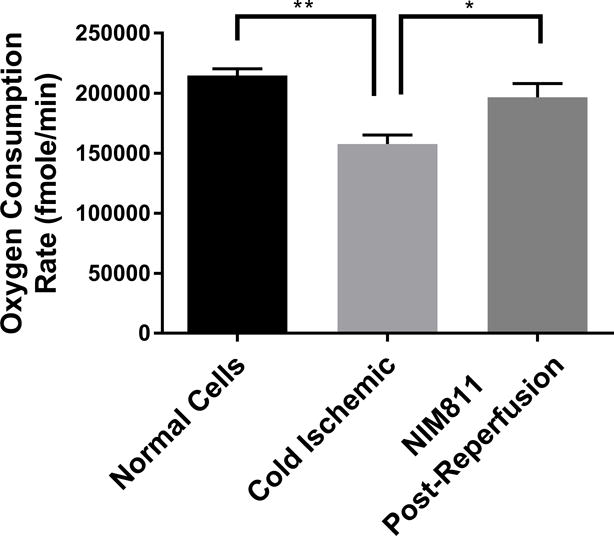
Treating ECs with 1μM NIM811 during the postreperfusion period facilitates mitochondrial recovery. Mitochondrial spare respiratory capacity was assessed at 6 hours postreperfusion. (n=6, *p<0.01, **p<0.005)
Cold Storage and Warm Reperfusion Induces a Pro-Inflammatory Phenotype in ECs, and Inhibiting mPTP Opening Reduces EC Inflammation
Given that our data suggested that metabolic changes occurred between 0 to 12 hours following reperfusion in our model, we focused on EC immunologic phenotype during the early postreperfusion period to establish whether there was any discernable association between metabolic alterations and immune function. Using Qiagen 84-gene chips for antigen-presenting cells, we assessed gene alterations in ECs exposed to 6 hours of cold storage followed by 6 hours of reperfusion. Analyzing the 84 genes (Table S2) that were screened using the same threshold criteria outlined above, we determined that 3 genes (Tlr7, Ccl2, Csf2) met our standard of a meaningful biological change. Ccl2 and Csf2, which encode monocyte attractant protein (MCP-1, also known as CCL2), and granulocyte-macrophage colony stimulating factor (GM-CSF), were upregulated, while Tlr7 was downregulated at 6 hours postreperfusion (Figure 4A). As a means of validation, we analyzed supernatants from EC cultures subjected to cold storage for CCL2 at 6 and 12 hours postreperfusion. In keeping with our array data, CCL2 was significantly increased, compared to the normal cell control at 6 hours postreperfusion (0.09041±0.003587 vs 0.04579±0.0007381 pg/mL per μg/mL of total protein concentration, p<0.0001) and at 12 hours postreperfusion (0.1084±0.0007052 vs 0.04583±0.001224 pg/mL per μg/mL of total protein concentration, p<0.0001) (Figure 4B). We next tested the hypothesis that inhibiting mPTP opening in ECs, and thus recovering mitochondrial fitness, would reduce EC inflammatory profile. After cold storage, ECs were treated with NIM811 or vehicle control for 6 or 12 hours immediately upon warm reperfusion. Using CCL2 levels as our inflammatory readout, we demonstrated that NIM811 treatment was associated with a significant reduction in CCL2 at 12 hours postreperfusion compared to the cold ischemic group (0.05021±0.0003124 vs 0.1084±0.0007052 pg/mL per μg/mL of total protein concentration, p<0.0001) (Figure 4C). In addition to cytokines identified by gene array, we analyzed KC, a mouse homologue of human IL-8 and IL-6, which are key cytokines associated with IRI.33 We could discern no detectable levels for IL-6 in our culture system (data not shown) but demonstrated similar trends for KC as noted with CCL2. IRI in cold ischemic cells at 6 and 12 hours postreperfusion resulted in a significant increase in KC levels compared to controls (0.1284±0.00155 vs 0.0553±0.001566 pg/mL per μg/mL of total protein concentration, p=0.0007) and (0.2161±0.002613 vs 0.08772±0.001751 pg/mL per μg/mL of total protein concentration, p<0.0001), respectively (Figure 4D). Treatment with NIM811 upon reperfusion resulted in a significant reduction of KC levels at 12 hours postreperfusion compared to untreated controls (0.07178±0.005298 vs 0.2161±0.002613, p<0.0001) (Figure 4E).
Figure 4. Immunologic Phenotype of ECs Postreperfusion.
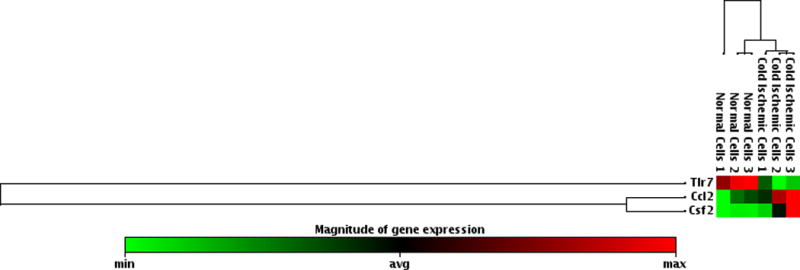
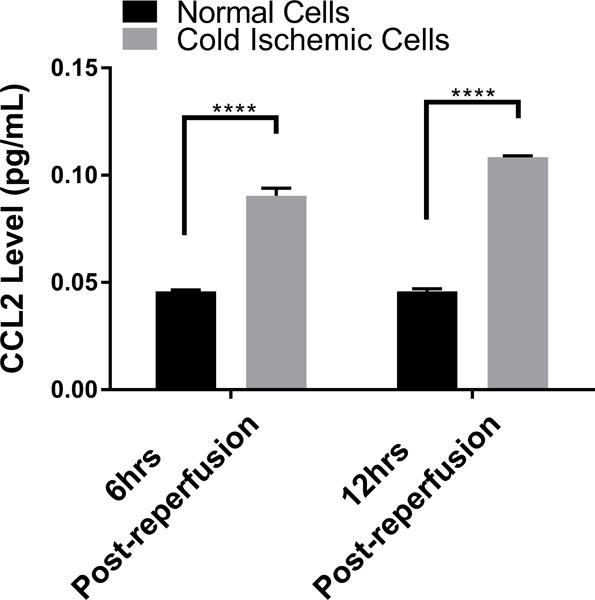
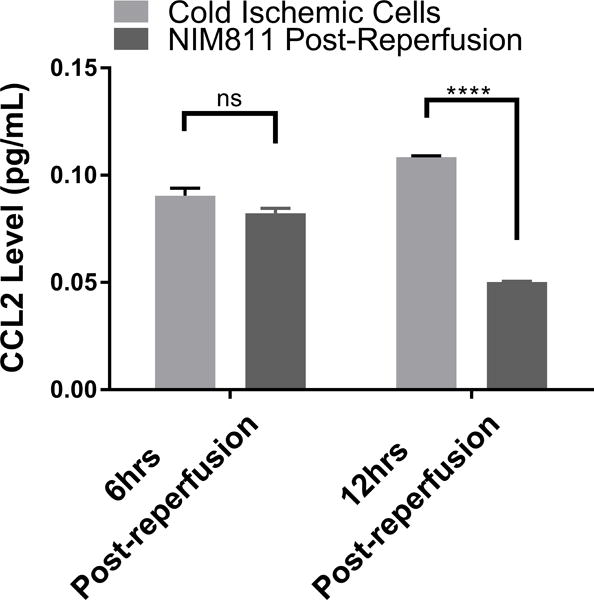
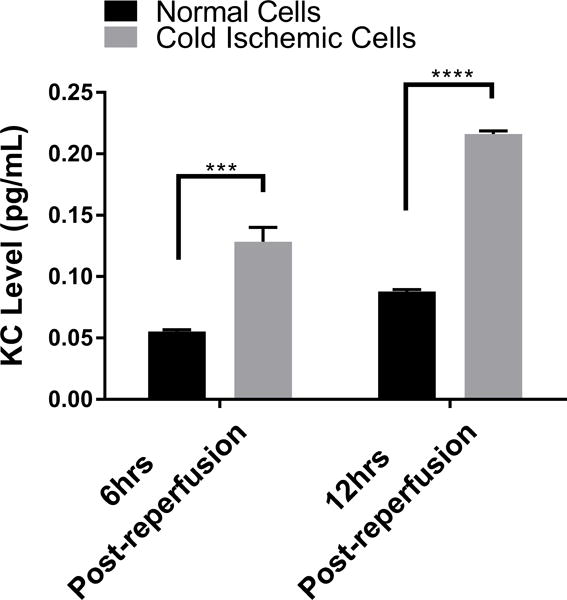
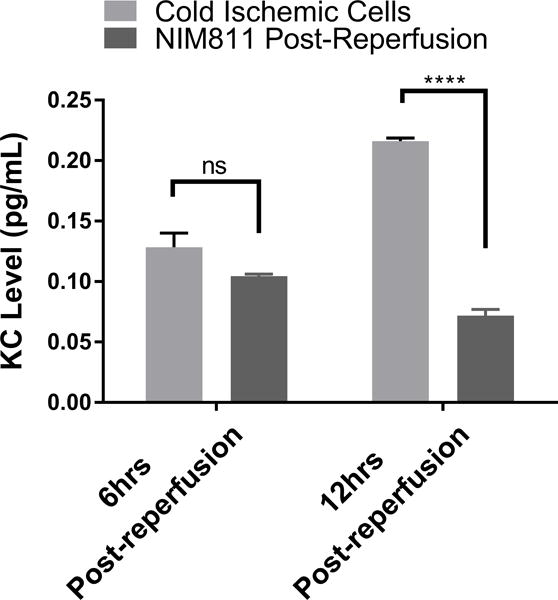
(A) Cold storage and warm reperfusion induces Ccl2 and Csf2 to be upregulated and Tlr7 to be downregulated at 6 hours postreperfusion. (B) (D) At the protein level, supernatant CCL2 and KC are increased during the first 12 hours postreperfusion. (C) (E) Treating ECs with NIM811 during the postreperfusion period reduces the CCL2 and KC levels. CCL2 and KC protein levels were normalized to the total protein concentration of the corresponding group. (n=3, ***p<0.001, ****p<0.0001).
Inhibiting mPTP Opening Reduces EC Immunogenicity to Allogeneic T cells
We next sought to determine whether inhibiting mPTP opening to facilitate mitochondrial recovery could alter EC contact-dependent immunogenicity. We performed a series of co-culture experiments with ECs and presensitized allogeneic CD8+ T cells. In these experiments, we presensitized C57BL/6 mice with ECs to develop a skewed memory/effector CD8 population for our co-culture studies. Phenotypic characterization of CD8+ T cells isolated from presensitized mouse spleens confirmed a higher percentage of T cells that were positive for CD69, CD44, and CD62L CCR7, a central memory phenotype, compared to unsensitized controls (Figure S1). To recapitulate posttransplant treatment with NIM811, as described previously,28 we subjected ECs to cold storage for 6 hours, followed by warm reperfusion in the presence of T cells. 1μM NIM811 or vehicle control was added at the beginning of the co-cultures, and supernatants were collected after 7 days (Figures 5A, 5B). In addition, to determine whether modulating mPTP opening specifically in ECs would yield any protective effects, ECs were first subjected to cold storage for 6 hours, followed by warm reperfusion to mimic organ storage and reperfusion injury. ECs were reperfused with media containing 1μM NIM811 or vehicle control for 6 hours. After the postreperfusion treatment with NIM811 to facilitate EC mitochondrial recovery, the treatment was withdrawn and ECs were co-cultured with presensitized CD8+ T cells for 7 days (Figures 5C, 5D). IFNγ and granzyme B were used as markers of allogeneic T cell cytotoxicity. Our results demonstrate that in both co-culture models, NIM811-treated groups were associated with a marked reduction in IFNγ and granzyme B, compared to controls (Figure 5A: undetectable vs 0.01228±0.001108 pg/mL per EC, p=0.0004; Figure 5B: undetectable vs 0.006595±0.0007836 pg/mL per EC, p=0.0011; Figure 5C: 0.009984±0.0005183 vs 0.02433±0.002209 pg/mL per EC, p=0.0032; Figure 5D: 0.007887±0.0007087 vs 0.0267±0.001871 pg/mL per EC, p=0.0007).
Figure 5. Co-cultures between NIM811-Treated ECs and Presensitized Allogeneic CD8+ T cells.
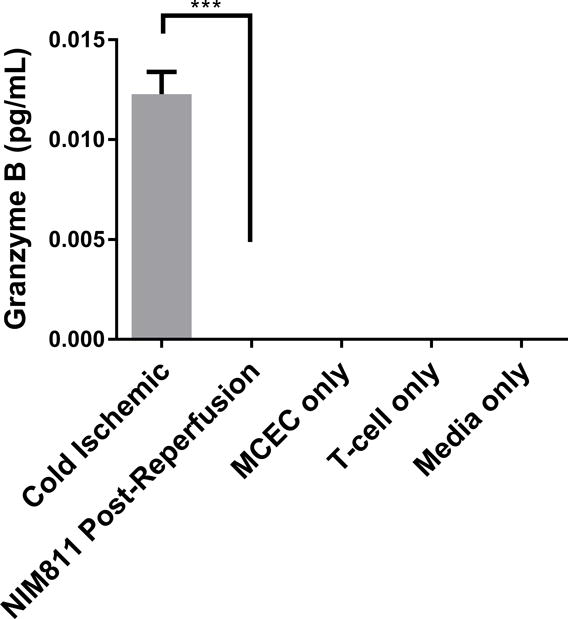
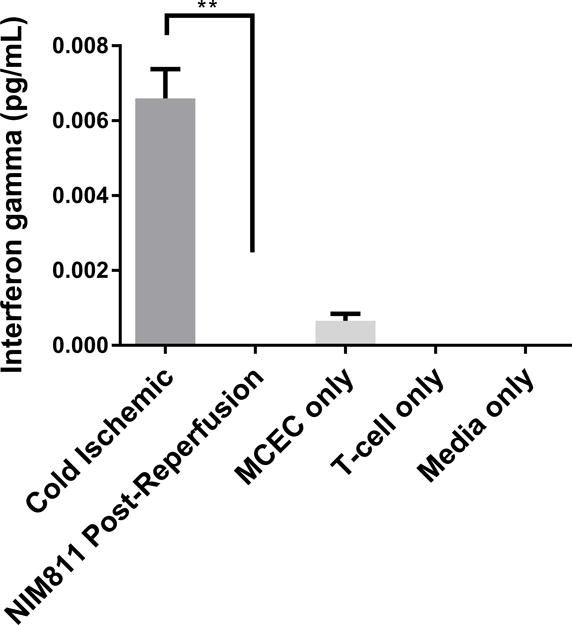
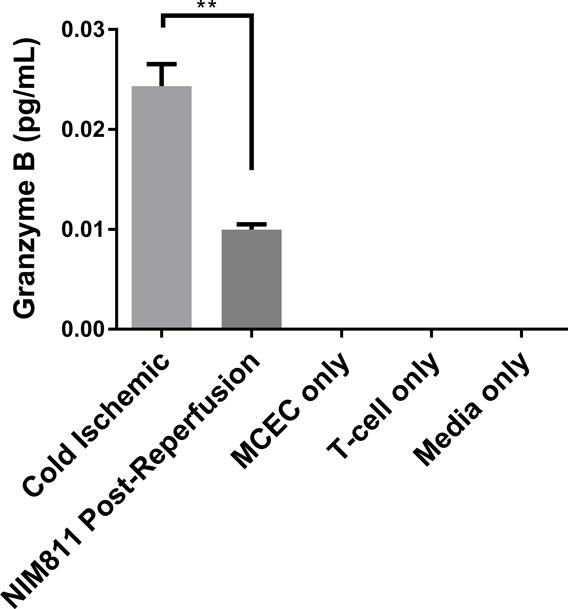
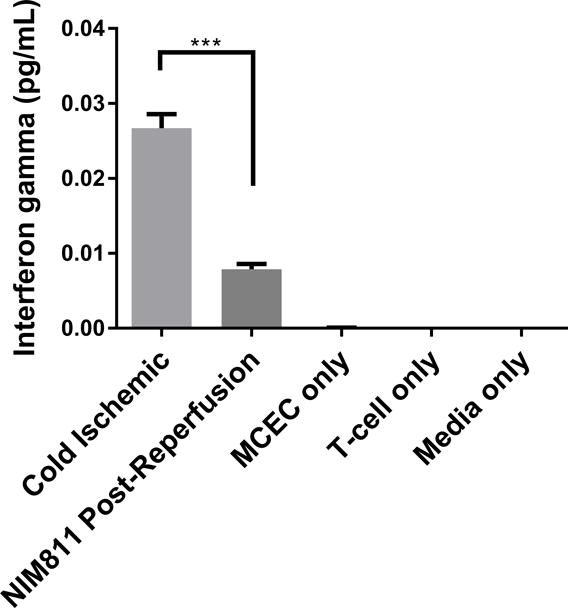
(A), (B) Treating ECs and T cells with NIM811 for 7 days during the co-culturing period reduces granzyme B and interferon gamma released by the co-cultured T cells. (C), (D) Inhibiting mPTP opening in ECs for 6 hours during the postreperfusion period to facilitate mitochondrial recovery before T cells are added reduces granzyme B and interferon gamma released by the co-cultured T cells. Granzyme B and interferon gamma levels were normalized to the number of ECs at the initiation of co-culture. (n=3, **p<0.005, ***p<0.001)
Inhibiting mPTP Opening Reduces Adhesion and MHC-I Surface Expression, and Decreases MHC-I Antigen-Presenting Machinery Expression
NIM811 treatment of ECs postreperfusion resulted in reduced T cell responses. To investigate further the mechanism, we analyzed the impact of NIM811 on endothelial adhesion, co-stimulatory, and co-inhibitory molecule expression at 6 and 24 hours postreperfusion. As determined by flow cytometry, NIM811 treatment had no impact on the expression of the co-stimulatory (CD80, CD86) or co-inhibitory (PD-L1) molecules (Tables 1, 2). Analysis of type I and II EC adhesion molecules showed at 24 hours postreperfusion, NIM811 significantly reduced VCAM-1 expression, a molecule that is important for T cell adhesion and immune-synapse formation (Figures 6A, 6B). No other adhesion molecules were impacted by NIM811 (Tables 1, 2). We also found that MHC-I surface expression decreased at 6 hours postreperfusion (Figures 6C, 6D), which led us to assess MHC-I antigen-presenting machinery expression levels, specifically of the 2 proteins TAP1 and calreticulin. NIM811 transiently reduced TAP1 expression at 6 hours postreperfusion, with levels returning to normal at 24 hours postreperfusion (Figure 6E). No changes were observed with calreticulin expression (Figure 6F).
Table 1.
Flow Cytometric Analysis of EC Surface Adhesion, Co-stimulatory, Co-inhibitory, and MHC-I Molecules at 6 Hours Postreperfusion.
| 6 HOURS POSTREPERFUSION | |||
|---|---|---|---|
| Molecule Name | Activated Cell Control (MFI) | Cold Ischemic Cells (MFI) | NIM811-Treated Cells (MFI) |
| VCAM-1 | 413.2 ±42.12 | 195.7 ±3.53 | 190.4 ±7.60 |
| ICAM-1 | 24.42 ±0.51 | 10.96 ±0.57 | 11.08 ±0.40 |
| E-selectin | 14.76 ±0.11 | 13.13 ±0.12 | 13.32 ±0.25 |
| PD-L1 | 34.65 ±0.42 | 14.01 ±0.28 | 14.02 ±0.14 |
| CD80 | 24.55 ±1.90 | 32.99 ±1.20 | 28.66 ±0.96 |
| CD86 | 9.607 ±0.25 | 10.51 ±0.27 | 11.62 ±0.51 |
| MHC I | 482.2 ±43.48 | 138.2 ±0.98 | 127.5 ±1.15 ** |
n=3, Cold Ischemic Cells vs NIM811-Treated Cells
p<0.005, Student t-test.
Table 2.
Flow Cytometric Analysis of EC Surface Adhesion, Co-stimulatory, Co-inhibitory, and MHC-I Molecules at 24 Hours Postreperfusion.
| 24 HOURS POSTREPERFUSION | |||
|---|---|---|---|
| Molecule Name | Activated Cell Control (MFI) | Cold Ischemic Cells (MFI) | NIM811-Treated Cells (MFI) |
| VCAM-1 | 833.9 ±71.89 | 180.7 ±3.80 | 127.1 ±5.14 ** |
| ICAM-1 | 20.45 ±0.55 | 10.35 ±0.30 | 9.86 ±0.30 |
| E-selectin | 14.1 ±0.03 | 11.76 ±0.66 | 10.55 ±0.13 |
| PD-L1 | 30.82 ±0.36 | 12.44 ±0.06 | 12.27 ±0.12 |
| CD80 | 12.83 ±0.32 | 14.88 ±1.29 | 13.12 ±0.53 |
| CD86 | 5.46 ±0.03 | 5.00 ±0.02 | 4.93 ±0.08 |
| MHC I | 540 ±31.09 | 117.3 ±0.79 | 109.5 ±5.09 |
n=3, Cold Ischemic Cells vs NIM811-Treated Cells
p<0.005, Student t-test
Figure 6. Expression of VCAM-1 and MHC-I Antigen-Presenting Machinery.
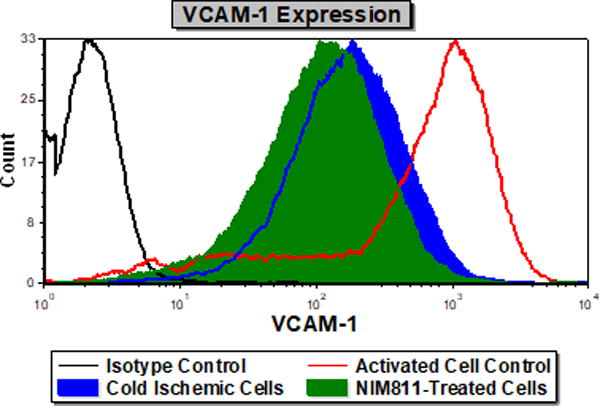
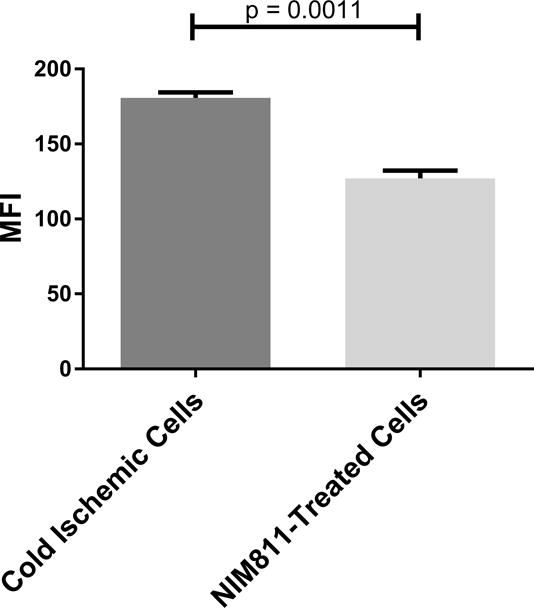
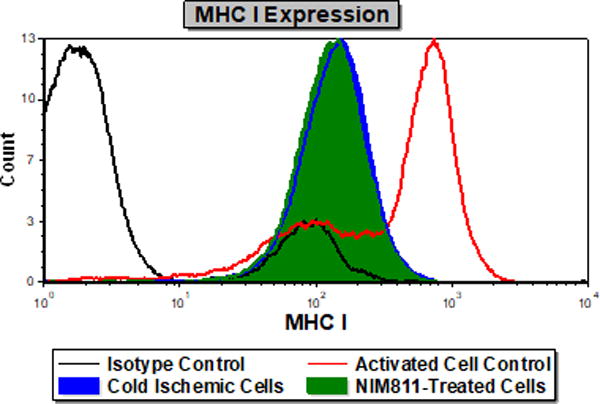
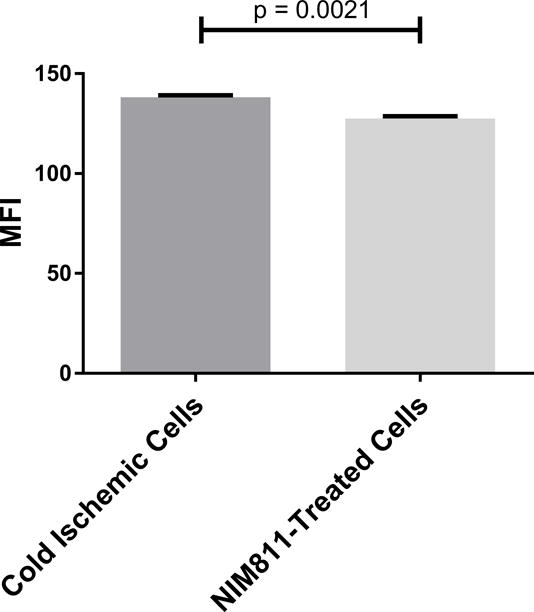
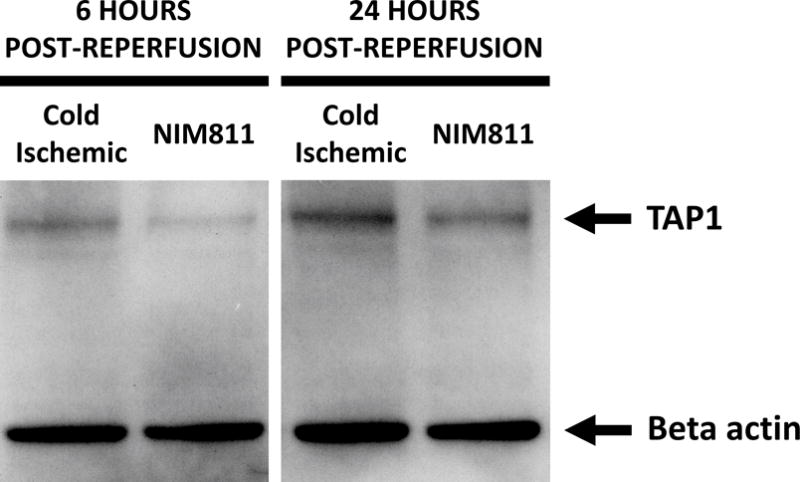
Treating ECs with NIM811 during the postreperfusion period reduces VCAM-1 expression at 24 hours postreperfusion, shown by representative histogram (A), and MFI quantification (B) (n=3, p=0.0011), and reduces MHC-I surface expression at 6 hours postreperfusion, shown by representative histogram (C), and MFI quantification (D) (n=3, p=0.0021), as well as decreases the expression of TAP1 at 6 hours postreperfusion (E), but not calreticulin (F) (representative immunoblot of n=3).
DISCUSSION
Allograft immunogenicity relies on microvascular ECs that interface between the donor organ and the recipient’s immune system.9 Early insults to these cells associated with CS-WR injure ECs, causing them to release damage-associated molecular patterns as well as pro-inflammatory cytokines and chemokines, creating an intra-graft inflammatory microenvironment.1,2,4 As APCs, microvascular ECs can activate recipient alloreactive Tmem, which mediate allograft rejection.4,10–15 Previously, using an in vitro model that recapitulates the early insults encountered in transplantation, we have shown that CS-WR induces ECs to adopt an immunogenic phenotype.17 Here, we utilized the same model to investigate the underlying mechanisms that promote increased immunogenicity in ECs.
Recent studies have shown an important link between the APCs’ cellular metabolism and their immunologic function. For example, immunogenic dendritic cells utilize less mitochondrial oxidative phosphorylation and more glycolysis for survival and function.34–36 On the other hand, tolerogenic dendritic cells display increased mitochondrial metabolism and decreased glycolysis.37,38 In transplantation, CS-WR has been reported to alter mitochondrial function of the allograft, by compromising mitochondrial respiratory machinery, causing mitochondrial swelling, and reducing mitochondrial membrane potential.19–25 We therefore hypothesized that mitochondrial dysfunction following CS-WR could alter the immunologic phenotype of microvascular ECs in a metabolic-dependent manner. Our data indicate that, in ECs, mitochondrial activity is markedly decreased while glycolysis is increased during the first 6 hours postreperfusion, likely to compensate for the lack of mitochondrial output. These results strikingly mirror the metabolic profile of immunogenic DCs. Furthermore, recent data suggest that recipient T cells infiltrate the allograft within the first 24 hours postreperfusion.4,10–15 Consistent with this temporal pattern, our data indicate that EC mitochondrial dysfunction occurs during the first 24 hours postreperfusion. More importantly, pronounced changes in mitochondrial activities and glycolysis during the first 6 hours align with the temporal expression patterns of pro-inflammatory chemokines already reported in literature.13 Glycolytic and mitochondrial gene upregulation in ECs at 6 hours postreperfusion is likely an adaptive response to compromised mitochondrial fitness, as these genes are later normalized or downregulated at 24 hours postreperfusion. The temporal pattern of gene expression appears to corroborate the notion that early metabolic changes are associated with a pro-inflammatory phenotype.
mPTP opening following reperfusion plays a key role in IRI, causing mitochondrial uncoupling and loss of membrane potential, thus compromising respiratory function.26 Published data in rat liver transplantation suggest that animals treated with inhibitors targeting mPTP opening protects the ETC and facilitates mitochondrial recovery.23 The role of mPTP opening in transplant immunity, however, is yet to be completely elucidated. Treating mice with an mPTP opening inhibitor after transplantation has been shown to reduce CD8+ T cell infiltration into the allografts; yet, these studies did not delineate how mitochondrial dysfunction and mPTP opening in an allograft’s microvascular ECs could impact the immunologic outcome.28 Our data indicate that inhibiting mPTP opening in ECs with NIM811 during the early period after reperfusion not only fully recovers EC mitochondrial fitness, but also significantly reduces the levels of pro-inflammatory cytokines released by the ECs. In addition, the immunogenicity to allogeneic T cells exhibited by ECs are dampened by inhibiting mPTP opening. To confirm that restoration of mitochondrial fitness by inhibiting mPTP opening would reduce EC contact-dependent immunogenicity, we utilized 2 experimental approaches. First, to recapitulate posttransplant treatment with NIM811, as described previously by Gomez et al,28 we co-cultured ECs with allogeneic CD8+ T cells immediately upon reperfusion in the presence of 1μM NIM811 treatment for 7 days. NIM811 was administered at the initiation of co-culture treating both ECs and T cells. Expectedly, the levels of cytotoxic proteins released by the co-cultured T cells were undetectable. These findings however did not reflect the significance of modulating mPTP opening in microvascular ECs. Thus, in the second co-culture model, following CS-WR, we treated ECs with 1μM NIM811 for 6 hours, as mitochondrial metabolic fitness is reduced the most during this postreperfusion period, and withdrew the treatment before T cells were added. After 7 days in co-culture, the levels of cytotoxic proteins released by the co-cultured T cells were significantly decreased. These findings suggest that the immuno-protective effect delivered by mPTP opening inhibition is EC-dependent and further highlight the importance of modulating EC immunogenicity.
NIM811 was chosen because of its specific affinity for cyclophilin D, a subunit of the mPTP,26 thus avoiding the immunosuppressive effects often observed with other mPTP opening inhibitors (such as cyclosporin A) that could confound our studies. Analysis of adhesion molecules and MHC-I machinery demonstrates that NIM811 treatment has effects on EC VCAM-1, MHC-I and TAP1 expression. VCAM-1 is associated with T cell adhesion and immune-synapse formation.39 Its expression is regulated by ERK1/2 pathway, hyperactivation of which can antagonize VCAM-1 expression on EC surface.40 On the other hand, targeting cyclophilin D leads to activation of ERK1/2 in mouse hearts.41 Based on these observations and our data, it is possible that NIM811 inhibiting cyclophilin D leads to ERK1/2 hyperactivation and consequently downregulation of VCAM-1, as observed in our model. In addition, TAP1 consumes energy to transport peptides into endoplasmic reticulum.42 We speculate that by facilitating mitochondrial recovery through inhibition of mPTP opening, more cellular energy is produced and thus TAP1 functions more efficiently, eliminating the need of expressing TAP1 at a high level in NIM811-treated group compared to the cold ischemic group. TAP1 reduced expression implies fewer MHC I molecules loaded with peptides, and consequently, MHC-I surface expression is also reduced.
Previous attempts in transplantation to inhibit mPTP opening focus on the whole allograft.23,28 Here, by understanding the implication of mPTP opening and mitochondrial dysfunction in EC immunogenicity, specific approaches to target microvascular endothelium, such as normothermic or hypothermic machine perfusion, can be devised to achieve tolerance. For future studies, in-vivo approaches to improve mitochondrial fitness during the first 6 hours postreperfusion, such as mitochondrial transplant and induced mitochondrial biogenesis, are areas for further investigation.
In summary, the current standard of care employing CS-WR leads to mPTP opening and mitochondrial dysfunction in the microvascular ECs of allografts. Altered cellular metabolism affects EC immune functions, and consequently activates recipient circulating T cells, promoting allograft rejection. Knowing these mechanisms, future therapeutic approaches will focus on improving mitochondrial fitness of microvascular ECs to achieve allograft tolerance.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. John J. Lemasters for his generous gift of NIM811 and insights into mPTP opening, Ms. Gyda Beeson for her technical input to troubleshoot oxygen consumption and glycolytic flux assays, Dr. Shilpak Chatterjee for his assistance with gene microarrays, Dr. Jiangting Hu for her insights into mPTP opening.
Funding:
NIH/NIBIB K08 EB019495-01A1
NIH/NHLBI Predoctoral Fellowship T32 HL007260
NIH/MSTP Predoctoral Fellowship T32 GM008716
NIH/NCATS KL2 TR001452 & UL1 TR001450
Patterson Barclay Memorial Foundation
Abbreviations
- APCs
antigen-presenting cells
- CS-WR
cold storage and warm reperfusion
- ECAR
extracellular acidification rate
- ECs
endothelial cells
- ETC
electron transport chain
- IFNγ
interferon gamma
- IRI
ischemia-reperfusion injury
- MFI
mean fluorescent intensity
- mPTP
mitochondrial permeability transition pore
- MUSC
Medical University of South Carolina
- OCR
oxygen consumption rate
- SRC
spare respiratory capacity
- TCA
tricarboxylic acid
- Tmem
memory T cells
- UW
University of Wisconsin
Footnotes
Authorship statement:
D.T. participated in research design, performance of the research, data analysis, and manuscript preparation.
C.A., S.N. participated in research design, data analysis, and manuscript preparation.
J.K. participated in research design and data analysis.
S.M. participated in research design.
S.E. participated in performance of the research
Disclosure: The authors declare no conflicts of interest.
References
- 1.van der Woude FJ, Schnuelle P, Yard BA. Preconditioning strategies to limit graft immunogenicity and cold ischemic organ injury. J Investig Med. 2004;52(5):323–329. doi: 10.1136/jim-52-05-32. [DOI] [PubMed] [Google Scholar]
- 2.Laskowski I, Pratschke J, Wilhelm MJ, Gasser M, Tilney NL. Molecular and cellular events associated with ischemia/reperfusion injury. Ann Transplant. 2000;5(4):29–35. [PubMed] [Google Scholar]
- 3.Jansen MA, Otten HG, de Weger RA, Huibers MM. Immunological and Fibrotic Mechanisms in Cardiac Allograft Vasculopathy. Transplantation. 2015;99(12):2467–2475. doi: 10.1097/TP.0000000000000848. [DOI] [PubMed] [Google Scholar]
- 4.Su CA, Iida S, Abe T, Fairchild RL. Endogenous memory CD8 T cells directly mediate cardiac allograft rejection. Am J Transplant. 2014;14(3):568–579. doi: 10.1111/ajt.12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pearl JP, Parris J, Hale DA, et al. Immunocompetent T-cells with a memory-like phenotype are the dominant cell type following antibody-mediated T-cell depletion. Am J Transplant. 2005;5(3):465–474. doi: 10.1111/j.1600-6143.2005.00759.x. [DOI] [PubMed] [Google Scholar]
- 6.Kirk AD, Mannon RB, Kleiner DE, et al. Results from a human renal allograft tolerance trial evaluating T-cell depletion with alemtuzumab combined with deoxyspergualin. Transplantation. 2005;80(8):1051–1059. doi: 10.1097/01.tp.0000174341.49741.8f. [DOI] [PubMed] [Google Scholar]
- 7.Trzonkowski P, Zilvetti M, Friend P, Wood KJ. Recipient memory-like lymphocytes remain unresponsive to graft antigens after CAMPATH-1H induction with reduced maintenance immunosuppression. Transplantation. 2006;82(10):1342–1351. doi: 10.1097/01.tp.0000239268.64408.84. [DOI] [PubMed] [Google Scholar]
- 8.Zeevi A, Husain S, Spichty KJ, et al. Recovery of functional memory T cells in lung transplant recipients following induction therapy with alemtuzumab. Am J Transplant. 2007;7(2):471–475. doi: 10.1111/j.1600-6143.2006.01641.x. [DOI] [PubMed] [Google Scholar]
- 9.Abrahimi P, Liu R, Pober JS. Blood Vessels in Allotransplantation. Am J Transplant. 2015;15(7):1748–1754. doi: 10.1111/ajt.13242. [DOI] [PubMed] [Google Scholar]
- 10.Epperson DE, Pober JS. Antigen-presenting function of human endothelial cells. Direct activation of resting CD8 T cells. J Immunol. 1994;153(12):5402–5412. [PubMed] [Google Scholar]
- 11.El-Sawy T, Miura M, Fairchild R. Early T cell response to allografts occurring prior to alloantigen priming up-regulates innate-mediated inflammation and graft necrosis. Am J Pathol. 2004;165(1):147–157. doi: 10.1016/s0002-9440(10)63283-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schenk AD, Nozaki T, Rabant M, Valujskikh A, Fairchild RL. Donor-reactive CD8 memory T cells infiltrate cardiac allografts within 24-h posttransplant in naive recipients. Am J Transplant. 2008;8(8):1652–1661. doi: 10.1111/j.1600-6143.2008.02302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morita K, Miura M, Paolone DR, et al. Early chemokine cascades in murine cardiac grafts regulate T cell recruitment and progression of acute allograft rejection. J Immunol. 2001;167(5):2979–2984. doi: 10.4049/jimmunol.167.5.2979. [DOI] [PubMed] [Google Scholar]
- 14.Shiao SL, McNiff JM, Pober JS. Memory T cells and their costimulators in human allograft injury. J Immunol. 2005;175(8):4886–4896. doi: 10.4049/jimmunol.175.8.4886. [DOI] [PubMed] [Google Scholar]
- 15.Manes TD, Pober JS. Antigen presentation by human microvascular endothelial cells triggers ICAM-1-dependent transendothelial protrusion by, and fractalkine-dependent transendothelial migration of, effector memory CD4+ T cells. J Immunol. 2008;180(12):8386–8392. doi: 10.4049/jimmunol.180.12.8386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang C, Yi T, Qin L, et al. Rapamycin-treated human endothelial cells preferentially activate allogeneic regulatory T cells. J Clin Invest. 2013;123(4):1677–1693. doi: 10.1172/JCI66204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nadig SN, Dixit SK, Levey N, et al. Immunosuppressive nano-therapeutic micelles downregulate endothelial cell inflammation and immunogenicity. RSC advances. 2015;5(54):43552–43562. doi: 10.1039/C5RA04057D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pearce EL, Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. 2013;38(4):633–643. doi: 10.1016/j.immuni.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuznetsov AV, Schneeberger S, Seiler R, et al. Mitochondrial defects and heterogeneous cytochrome c release after cardiac cold ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2004;286(5):H1633–1641. doi: 10.1152/ajpheart.00701.2003. [DOI] [PubMed] [Google Scholar]
- 20.Kuznetsov AV, Schneeberger S, Renz O, et al. Functional heterogeneity of mitochondria after cardiac cold ischemia and reperfusion revealed by confocal imaging. Transplantation. 2004;77(5):754–756. doi: 10.1097/01.tp.0000115346.85679.34. [DOI] [PubMed] [Google Scholar]
- 21.Thatte HS, Rousou L, Hussaini BE, Lu XG, Treanor PR, Khuri SF. Development and evaluation of a novel solution, Somah, for the procurement and preservation of beating and nonbeating donor hearts for transplantation. Circulation. 2009;120(17):1704–1713. doi: 10.1161/CIRCULATIONAHA.108.808907. [DOI] [PubMed] [Google Scholar]
- 22.Yang C, Xu H, Cai L, et al. Donor pretreatment with adenosine monophosphate-activated protein kinase activator protects cardiac grafts from cold ischaemia/reperfusion injury. Eur J Cardiothorac Surg. 2016;49(5):1354–1360. doi: 10.1093/ejcts/ezv413. [DOI] [PubMed] [Google Scholar]
- 23.Plin C, Haddad PS, Tillement JP, Elimadi A, Morin D. Protection by cyclosporin A of mitochondrial and cellular functions during a cold preservation-warm reperfusion of rat liver. Eur J Pharmacol. 2004;495(2–3):111–118. doi: 10.1016/j.ejphar.2004.05.042. [DOI] [PubMed] [Google Scholar]
- 24.Duval M, Plin C, Elimadi A, et al. Implication of mitochondrial dysfunction and cell death in cold preservation--warm reperfusion-induced hepatocyte injury. Can J Physiol Pharmacol. 2006;84(5):547–554. doi: 10.1139/y06-014. [DOI] [PubMed] [Google Scholar]
- 25.Shrum S, MacMillan-Crow LA, Parajuli N. Cold Storage Exacerbates Renal and Mitochondrial Dysfunction Following Transplantation. J Kidney. 2016;2(1) [PMC free article] [PubMed] [Google Scholar]
- 26.Halestrap AP, Richardson AP. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol. 2015;78:129–141. doi: 10.1016/j.yjmcc.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 27.Schipper DA, Marsh KM, Ferng AS, Duncker DJ, Laman JD, Khalpey Z. The Critical Role of Bioenergetics in Donor Cardiac Allograft Preservation. J Cardiovasc Transl Res. 2016;9(3):176–183. doi: 10.1007/s12265-016-9692-2. [DOI] [PubMed] [Google Scholar]
- 28.Gomez L, Raisky O, Chalabreysse L, Verschelde C, Bonnefoy-Berard N, Ovize M. Link between immune cell infiltration and mitochondria-induced cardiomyocyte death during acute cardiac graft rejection. Am J Transplant. 2006;6(3):487–495. doi: 10.1111/j.1600-6143.2005.01219.x. [DOI] [PubMed] [Google Scholar]
- 29.Schneeberger S, Amberger A, Mandl J, et al. Cold ischemia contributes to the development of chronic rejection and mitochondrial injury after cardiac transplantation. Transplant Int. 2010;23(12):1282–1292. doi: 10.1111/j.1432-2277.2010.01126.x. [DOI] [PubMed] [Google Scholar]
- 30.Zepeda-Orozco D, Kong M, Scheuermann RH. Molecular Profile of Mitochondrial Dysfunction in Kidney Transplant Biopsies Is Associated With Poor Allograft Outcome. Transplant Proc. 2015;47(6):1675–1682. doi: 10.1016/j.transproceed.2015.04.086. [DOI] [PubMed] [Google Scholar]
- 31.Ferrick DA, Neilson A, Beeson C. Advances in measuring cellular bioenergetics using extracellular flux. Drug Discov Today. 2008;13(5–6):268–274. doi: 10.1016/j.drudis.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 32.Chen J, Liu DG, Wang H, et al. NIM811 downregulates transforming growth factorbeta signal transduction in vivo and in vitro. Mol Med Rep. 2016;13(1):522–528. doi: 10.3892/mmr.2015.4572. [DOI] [PubMed] [Google Scholar]
- 33.Park SW, Chen SW, Kim M, D’Agati VD, Lee HT. Human heat shock protein 27-overexpressing mice are protected against acute kidney injury after hepatic ischemia and reperfusion. Am J Physiol Renal Physiol. 2009;297(4):F885–894. doi: 10.1152/ajprenal.00317.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krawczyk CM, Holowka T, Sun J, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115(23):4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Everts B, Amiel E, van der Windt GJ, et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood. 2012;120(7):1422–1431. doi: 10.1182/blood-2012-03-419747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Everts B, Amiel E, Huang SC, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat Immunol. 2014;15(4):323–332. doi: 10.1038/ni.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hackstein H, Taner T, Zahorchak AF, et al. Rapamycin inhibits IL-4--induced dendritic cell maturation in vitro and dendritic cell mobilization and function in vivo. Blood. 2003;101(11):4457–4463. doi: 10.1182/blood-2002-11-3370. [DOI] [PubMed] [Google Scholar]
- 38.Sim WJ, Ahl PJ, Connolly JE. Metabolism Is Central to Tolerogenic Dendritic Cell Function. Mediators Inflamm. 2016;2016:2636701. doi: 10.1155/2016/2636701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chakraborty S, Hu SY, Wu SH, Karmenyan A, Chiou A. The interaction affinity between vascular cell adhesion molecule-1 (VCAM-1) and very late antigen-4 (VLA-4) analyzed by quantitative FRET. PloS one. 2015;10(3):e0121399. doi: 10.1371/journal.pone.0121399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang C, Qin L, Manes TD, Kirkiles-Smith NC, Tellides G, Pober JS. Rapamycin antagonizes TNF induction of VCAM-1 on endothelial cells by inhibiting mTORC2. The J Exp Med. 2014;211(3):395–404. doi: 10.1084/jem.20131125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klawitter J, Seres T, Pennington A, Beatty JT, Klawitter J, Christians U. Ablation of Cyclophilin D Results in an Activation of FAK, Akt, and ERK Pathways in the Mouse Heart. J Cell Biochem. 2017;118(9):2933–2940. doi: 10.1002/jcb.25947. [DOI] [PubMed] [Google Scholar]
- 42.Abele R, Tampe R. The ABCs of immunology: structure and function of TAP, the transporter associated with antigen processing. Physiology (Bethesda) 2004;19:216–224. doi: 10.1152/physiol.00002.2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.