

Abstract
Cardiovascular complications are the major cause of mortality in patients with diabetes. This is closely associated with both macrovascular and microvascular complications of diabetes, which lead to organ injuries in diabetic patients. Previous studies have consistently demonstrated the beneficial effects of relaxin treatment for protection of the vasculature, with evidence of antioxidant and anti-remodeling actions. Relaxin enhances nitric oxide, prostacyclin and endothelium-derived hyperpolarization (EDH)-type-mediated relaxation in various vascular beds. These effects of relaxin on the systemic vasculature, coupled with its cardiac actions, reduce pulmonary capillary wedge pressure and pulmonary artery pressure. This results in an overall decrease in systemic and pulmonary vascular resistance in heart failure patients. The anti-fibrotic actions of relaxin are well established, a desirable property in the context of diabetes. Further, relaxin ameliorates diabetic wound healing, with accelerated angiogenesis and vasculogenesis. Relaxin-mediated stimulation of vascular endothelial growth factor (VEGF) and stromal cell-derived factor 1-α, as well as regulation of metalloproteinase expression, ameliorates cardiovascular fibrosis in diabetic mice. In the heart, relaxin is a cardioprotective molecule in several experimental animal models, exerting anti-fibrotic, anti-hypertrophy and anti-apoptotic effects in diabetic pathologies. Collectively, these studies provide a foundation to propose the therapeutic potential for relaxin as an adjunctive agent in the prevention or treatment of diabetes-induced cardiovascular complications. This review provides a comprehensive overview of the beneficial effects of relaxin, and identifies its therapeutic possibilities for alleviating diabetes-related cardiovascular injury.
Keywords: relaxin, diabetes, vasculopathy, endothelial dysfunction, remodeling, cardiomyopathy
Introduction
Diabetes mellitus is a major risk factor for the development of cardiovascular diseases. This disease affects an estimated 6.4% of adults worldwide, but this number is likely to increase to 7.7% by 2030 (Shaw et al., 2010). Diabetes leads to microvascular complications such as kidney failure, lower limb amputation, and blindness (Forbes and Fotheringham, 2017). Macrovascular complications of diabetes include peripheral vascular disease, myocardial infarction, stroke, and congestive heart failure. In the context of diabetic cardiomyopathy, left ventricular diastolic dysfunction at an early stage which subsequently progress to systolic dysfunction is closely associated with cardiac hypertrophy and fibrosis (Boudina and Abel, 2007). We and others have reviewed the key mechanisms and strategies to target diabetic cardiomyopathy (Boudina and Abel, 2010; Huynh et al., 2014; Ritchie et al., 2017; Tate et al., 2017b). It is however worth noting that there are still no effective therapies to limit the progression of diabetes-induced cardiovascular complications. Here, we propose a small peptide hormone, relaxin, which shows a promising role in protecting the cardiovascular system (Leo et al., 2016a; Sarwar et al., 2017). The possibilities of relaxin as a therapy for diabetes-related complications are also discussed.
Cardiovascular Complications of Diabetes
Diabetic cardiomyopathy is characterized by damage to the myocardium, in particular diastolic dysfunction (Pappachan et al., 2013), where the heart is unable to relax and undergo filling during the diastolic part of the cardiac cycle. Pathological processes underlying this include cardiac hypertrophy, neuronal abnormalities and myocardium stiffening as a result of collagen cross-linking and extracellular matrix deposition (Iimoto et al., 1988). Activation of the sympathetic nervous system as a result of insulin resistance leads to impaired kidney and vascular function, an effect that is associated with the onset of hypertension (Reaven et al., 1996). For example, an FDA-approved glucose-lowering drug for the treatment of type 2 diabetes, the sodium-glucose co-transporter 2 (SGLT2) antagonist empagliflozin, exhibits promising actions in reducing cardiovascular burden by limiting the sympathetic outflow to the kidneys (Sano, 2018). Diabetes-induced collagen cross-linking that accumulates within the myocardium can ultimately lead to structural remodeling of the heart tissue, including cardiac fibrosis. Mechanisms that contribute to the phenotype of the diabetic myocardium include (but are not limited to) impairments in function of type 2 ryanodine receptors (RyR2) (Santulli et al., 2015), increased oxidative stress, activation of the renin-angiotensin-aldosterone system (RAAS), endothelin-1 upregulation, with mitochondrial dysfunction, inflammation and endoplasmic reticulum stress (Huynh et al., 2014; Prakoso et al., 2017; Tate et al., 2017a,b). Reactive oxygen species (ROS) directly damage proteins, DNA and lipids, produce reactive lipid peroxidases, and can generate reactive nitrogen species, all which contribute to diabetic cardiomyopathy (Baynes and Thorpe, 1999). In diabetes, the over-activation of the RAAS increases angiotensin II (Ang II) production in cardiac fibroblasts and cardiomyocytes, to impair cardiac structure and function (Lavrentyev et al., 2007; Singh et al., 2008). Similarly, these pathological pathways also play a major role in diabetes-associated vasculopathy. The most well-known functional characteristic of diabetes-induced vascular complications is endothelial dysfunction, where there is an imbalance in vascular homeostasis, causing a shift toward vasoconstrictor state and reduced vasodilation. Hyperglycemia leads to the imbalance of vasoprotective molecules such as nitric oxide (NO), prostacyclin (PGI2), and endothelium-derived hyperpolarization (EDH) pathways, and causes overproduction of ROS or vasoconstrictor, thus leading to endothelial dysfunction (Johnstone et al., 1993; Guzik et al., 2002; Angulo et al., 2003; Vanhoutte et al., 2017). Both the cardiac (Bugger and Abel, 2014; Huynh et al., 2014; Tate et al., 2017b) and vascular complications (Forbes and Fotheringham, 2017; Shi and Vanhoutte, 2017) of diabetes have been extensively reviewed in detail elsewhere. Insulin resistance can be initiated by altered glucose metabolism and disturbance in fatty acid metabolism (McGarry, 1992), whereby there is an imbalance between fatty acid uptake and beta-oxidation in the skeletal muscle or heart, leading to impaired insulin signaling. Inhibition of both glucose metabolism and fatty acid uptake (Randle et al., 1963), and muscle glucose uptake via PKCζ activation (Samuel and Shulman, 2016) all contribute to the pathophysiology of diabetes.
Is Glucose-Lowering Sufficient?
Insulin therapy is the first line treatment for type 1 diabetic patients. One school of thought favors a role for insulin therapy in heart failure outcomes, particularly in the elderly (Sardu et al., 2014). However, this is countered by evidence from several studies in which diabetic patients on insulin therapy exhibit increased risk of developing heart failure (Nichols et al., 2005; Smooke et al., 2005; Mangiavacchi et al., 2008). Over the years, many approaches to safely treat type 2 diabetes have been developed. First-line glucose-lowering therapy for type 2 diabetes remains to be metformin (Holman, 2007). Recently, several new classes of drugs have been developed for treating type 2 diabetes, which include dipeptidyl peptidase-4 (DPP4) inhibitors, glucagon-like peptide-1 (GLP-1) receptor agonists and SGLT2 antagonists. These therapies can be added to first-line therapy if adequate glycemic control is not yet achieved, aiming to reduce and control blood glucose levels for as long as possible after diagnosis, and thereby potentially preventing the development and progression of cardiovascular complications. However, the majority of these conventional glucose-lowering drugs fail to prevent the progression of cardiovascular complications, despite their favorable effects on glycemic control. There has recently emerged the suggestion that GLP-1 receptor agonists may reduce cardiovascular complications, although this may be secondary to the reduction in glycated hemoglobin levels (Bethel et al., 2017). Excitingly however, the SGLT2 inhibitors empagliflozin and canagliflozin reduce heart failure hospitalization and may be the first glucose-lowering therapy to relieve some of the cardiovascular burden of diabetes as demonstrated in the EMPA-REG OUTCOME and CANVAS trials (Lytvyn et al., 2017). Hence, alternative therapies are needed to address the ever-increasing incidence of diabetes to prevent and/or treat diabetes-induced cardiovascular complications.
Protective Role for Relaxin in the Vasculature
An emerging molecule implicated in the protection of the cardiovascular system is the hormone relaxin. In rodents, the relaxin/insulin-like family peptide receptor 1 (RXFP1) is expressed in the aorta (Ng et al., 2015), small renal and mesenteric arteries (Novak et al., 2006). RXFP1 is predominantly localized in the endothelial cells of rat mesenteric artery and vein, vena cava and the aorta, but the distribution of this receptor is more abundant in the vascular smooth muscle cells of rat small pulmonary artery, femoral artery and vein (Jelinic et al., 2014). The differential distribution of RXFP1 in several vascular beds could serve as an important target for relaxin activity independent of any potential source of local relaxin production. The other members of the relaxin family peptide receptors comprise RXFP2, RXFP3, and RXFP4. Relaxin can weakly bind to and activate RXFP2 at higher concentrations, however, in human and rodents, relaxin has higher binding affinity to RXFP1. RXFP3, and RXFP4 on the other hand are structurally and functionally different to RXFP1 and RXFP2, with their corresponding ligands relaxin-3 and INSL5 (Bathgate et al., 2006). Traditionally, relaxin is known as a reproductive hormone that plays an important role in maternal adaptations to pregnancy, by elongating the pubic ligament at the end of pregnancy in rodents, as well as causing cervical ripening and nipple development (Sherwood, 2004). However, many actions of relaxin have also been extensively reported in several non-reproductive organs (Leo et al., 2016a; Samuel et al., 2016; Sarwar et al., 2017). Much of this has relied on the potent anti-fibrotic actions of relaxin (Samuel et al., 2016). Exogenous relaxin administration also has significant therapeutic benefits in animal models of hypertension, atherosclerosis and tobacco-related disease, often beyond its anti-fibrotic mechanisms, as detailed below.
In the vasculature, relaxin reduces myogenic reactivity and sensitivity to the α1-adrenergic vasoconstrictor, phenylephrine in Wistar Kyoto rats (WKY) and spontaneously hypertensive rats (SHR), accompanied by improved flow-mediated vasodilation (van Drongelen et al., 2013). This action is NO-dependent in normotensive, but not hypertensive rats (van Drongelen et al., 2013). In Ang II-induced hypertensive rats, relaxin reduces mean arterial pressure, albumin excretion and impairments in NO availability, effects attributed to reduced oxidative stress (Sasser et al., 2011). Furthermore, relaxin attenuates aortic remodeling to reduce wall thickness and collagen content (Xu et al., 2010). Inward remodeling of parenchymal arterioles is also ameliorated in hypertensive rats (Chan et al., 2013), thus increasing arterial compliance. These studies in hypertensive models clearly show the vasculoprotective effects of relaxin against hypertension-induced dysfunction and its potential role in reversing hypertension-associated fibrosis. More recently, relaxin treatment is reported to reverse cigarette smoke-induced endothelial dysfunction, an effect attributed to enhanced endothelial NO synthase (eNOS) expression (Pini et al., 2016). Relaxin infusion over 4 weeks also reverses the atherosclerosis-associated endothelial dysfunction in ApoE-/- mice, an effect attributed to reduction in angiotensin AT1 receptors and oxidative stress (Tiyerili et al., 2016). In addition, relaxin-mediated vasculoprotective effects are also evident in vascular preparations ex vivo, primarily through its action on endothelin B receptors (ETB), NO and PGI2 (Dschietzig et al., 2003; McGuane et al., 2011a; Dschietzig et al., 2012; Ng et al., 2016). In isolated small human gluteal (Fisher et al., 2002) and subcutaneous (McGuane et al., 2011b) arteries, relaxin induces rapid dilator actions. However, the rapid vasodilator effects of relaxin are not observed in all blood vessels such as the rat mesenteric arteries, despite the presence of RXFP1 (Leo et al., 2014b, 2016a, 2017). All of these studies provide an important insight into the protective mechanisms of relaxin against vascular dysfunction, where altered vasodilator and vasoconstrictor pathways are implicated, as well as modifying the vessel wall structure.
Cardiac Role for Relaxin
In addition to the above vasoprotective roles of relaxin, a plethora of literature has identified relaxin as a cardioprotective molecule, particularly through its anti-fibrotic and anti-hypertrophic actions. For example, relaxin ameliorates post-infarction fibrotic healing by reducing the density of mature scar tissue in the infarcted area (Samuel et al., 2011), and regulates cardiac fibroblast proliferation, differentiation and collagen deposition, thereby reversing cardiac fibrosis in vivo (Samuel et al., 2004). In SHR, relaxin administration for 2 weeks is sufficient to lower collagen content in the left ventricle, ameliorate left ventricular hypertrophy and dysfunction, as well as reverse cardiac fibrosis (Lekgabe et al., 2005). Furthermore, relaxin also prevents atrial fibrillation secondary to the amelioration of cardiac fibrosis and hypertrophy in SHR. This action is associated with changes in Na+ current density (Parikh et al., 2013; Henry et al., 2016). One of the well-reported mechanisms of relaxin as an anti-fibrotic molecule is its ability to up-regulate the Notch signaling pathway, whereby the transforming growth factor (TGF)-β/Smad 3 signaling is inhibited to prevent cardiac fibroblast-myofibroblast transition (Sassoli et al., 2013). In addition to the potent anti-fibrotic actions of relaxin in various disease animal models, relaxin has the ability to reduce oxidative stress, at least in part via protein kinase B (Akt) and extracellular signal-regulated kinase (ERK) signaling pathways, suppressing cardiomyocytes apoptosis and hypertrophy (Moore et al., 2007). Collectively, the actions of relaxin in pressure-overloaded animals may represent an attractive target of unloading the heart, via vasodilation and reducing systemic vascular resistance.
Role for Relaxin in the Context of Diabetes-Induced Vascular Complications
Further to the widely reported actions of relaxin in hypertensive animals, several lines of evidence suggest that relaxin may possess similar effects in diabetic animals. Relaxin ameliorates diabetic wound healing by accelerating angiogenesis and vasculogenesis through the stimulation of vascular endothelial growth factor (VEGF) and stromal cell-derived factor (SDF)1-α, as well as regulating metalloproteinase (MMP) expression to impede fibrosis in db/db mice (Bitto et al., 2013; Squadrito et al., 2013). These wound healing properties of relaxin are primarily attributed to induction of VEGF expression and subsequent angiogenesis that selectively target the wound site (Unemori et al., 2000). Relaxin also upregulates NO production in the vasculature, which may play an additional role in wound healing. Notably, relaxin treatment attenuates fibrosis in the heart of streptozotocin (STZ) Ren-2 diabetic rats by decreasing interstitial and total left ventricle collagen deposition, thereby reducing myocardial stiffness and improving overall left ventricular diastolic function (Samuel et al., 2008). Indeed, relaxin treatment inhibits both proliferation of cardiac fibroblasts and formation of type I and III fibrillar collagen induced by high glucose in vitro (Wang et al., 2009). Intriguingly, relaxin also been reported to attenuate skeletal muscle insulin resistance, accompanied by enhanced aortic endothelium-dependent relaxation in high fat-fed pre-diabetic mice (Bonner et al., 2013). The underlying mechanisms of relaxin actions in this context however are not further interrogated.
Our own studies have recently demonstrated that relaxin protects the mouse aorta and mesenteric arteries by restoring endothelial function under acute (high glucose) (Ng et al., 2016) and chronic hyperglycemia (Ng et al., 2017). Relaxin prevents high glucose-induced endothelial dysfunction in mouse aorta by suppressing oxidative stress and stimulating vasodilator PGI2 production, as well as changing the relative gene expression of the PGI2 receptor (IP) to thromboxane (TP) (Ng et al., 2016). Under high glucose conditions, PGI2, which normally acts as a vasodilator, reverts to a vasoconstrictor (Zhu et al., 2014), likely acting through the TP receptor. This ex vivo study on relaxin co-treatment under hyperglycemic conditions provided ‘proof-of-concept’ evidence that motivated us to further investigate the in vivo effects of relaxin utilizing chronic type 1 diabetic mice. It is well-established that chronic hyperglycemia induces endothelial dysfunction in an experimental model of diabetes (Pieper et al., 1995; Leo et al., 2011a), with downregulation of endothelium-derived NO. Consistent with the ex vivo data, relaxin treatment for 2 weeks following 10 weeks of untreated diabetes is sufficient to restore endothelial function in diabetic mouse aorta (Ng et al., 2017). This is underpinned by enhanced NO-mediated relaxation. Interestingly, relaxin treatment in this context failed to increase basal NO availability in both ex vivo and in vivo settings of hyperglycemia. This suggests that exogenous relaxin treatment selectively augments agonist stimulated NO-mediated relaxation pathways to preserve endothelial vasodilator function, without impacting on basal NO synthase activity in endothelial cells. In contrast to the findings in diabetic aortae, relaxin increases basal NOS activity and eNOS phosphorylation in healthy rat aortae following two days of continuous intravenous infusion (Leo et al., 2016b), leading to enhanced NO-mediated relaxation. The disparity of findings may be attributed to the difference between non-diabetic and diabetic settings, or deficits in the NOS co-factor, tetrahydrobiopterin (BH4), in diabetic aortae, as a result of oxidation by peroxynitrite produced from the interaction between superoxide and NO in the endothelium (Cai et al., 2005).
Diabetes also attenuates endothelial function in smaller resistance arteries (mesenteric arteries), by increasing the contribution of vasoconstrictor prostanoids to endothelial dysfunction and reducing EDH-type relaxation (Ng et al., 2017). It is worth noting that the role of EDH increases as the artery diameter decreases, thus the relaxation seen in mesenteric artery will be largely dependent on EDH, in contrast to larger blood vessel such as the aorta. Relaxin treatment for 2 weeks in diabetic mice reverses endothelial dysfunction (Ng et al., 2017). This is accompanied by a profound increase in the relative contribution of NO to endothelium-dependent relaxation. Although diabetes selectively impairs EDH-type relaxation in the mesenteric artery, relaxin treatment failed to restore EDH-type relaxation at least in the STZ C57BL/6 mouse model of diabetes. The mechanisms underpinning the impairment of EDH-type relaxation in diabetic mesenteric arteries are multi-factorial (Ding et al., 2005; Ding and Triggle, 2010; Leo et al., 2011b). One possible explanation is that relaxin is not able to restore the reduction in low-resistance electrical coupling between endothelial and vascular smooth muscle cells in the mouse small mesenteric arteries in the context of diabetes (Ding et al., 2005). Indeed, the mechanism of relaxin action is quite distinct to that of NO in reversing diabetes-induced endothelial dysfunction, possibly by reducing superoxide production and increasing eNOS dimerization. Relaxin treatment for two to five days in healthy rats significantly enhances NO-mediated relaxation in the mesenteric artery (van Drongelen et al., 2011, 2012, 2013; Jelinic et al., 2014; Leo et al., 2016b). Despite compelling evidence to suggest that relaxin treatment augments EDH-type relaxation in mesenteric arteries (Leo et al., 2014b) and cerebral parenchymal arterioles (Chan and Cipolla, 2011), with activation of intermediate conductance Ca2+-activated K+ channels, it is worth mentioning that these experiments were done in healthy male and non-pregnant female rats. Therefore, the discrepancies in animal species (mice vs. rats) and physiological conditions (diseased vs. healthy) may account for the differences in the mesenteric artery response to exogenous relaxin.
A key striking effect of relaxin treatment in diabetic mice is the normalization of vasoconstrictor prostanoids in the mesenteric artery. Consistent with this, our group previously shown that Rln-/- mice have altered prostanoid pathways in both the mesenteric artery (Leo et al., 2014a) and aorta (Ng et al., 2015). We suggest that relaxin likely restores diabetes-induced endothelial dysfunction by affecting the NO and prostanoid pathways, but not the EDH pathway, in both the aorta and mesenteric artery. In addition to these actions of relaxin in regulating vasodilator pathways, the hormone also suppresses the action of vasoconstrictors. Specifically, relaxin treatment markedly attenuates Ang II-induced contraction in the mesenteric artery of diabetic mice (Ng et al., 2017). It is well-established that diabetes leads to an imbalance of vasoactive factors with opposing actions, including NO and Ang II (Tousoulis et al., 2012), favoring vasoconstrictor actions, resulting in impaired vascular homeostasis. Relaxin may have a potential physiological antagonistic effect to the actions of AT-1 or AT-2 angiotensin receptor activation in the mesenteric artery (Marshall et al., 2016, 2017), counteracting diabetes-induced increased in Ang II contraction. It is feasible to speculate that relaxin counter-regulated the AT-1 mediated actions of Ang II by a mechanism which involved increased NO production. Indeed, relaxin treatment has been previously demonstrated to relieve Ang II-induced hypertension by increasing NO availability (Sasser et al., 2011), with possible concomitant reduction of oxidative stress (Sasser et al., 2014).
Roles of Relaxin in the Treatment of Diabetes-Induced Cardiac Remodeling
Left ventricular hypertrophy and fibrosis are associated with the progression of diabetic cardiomyopathy (Boudina and Abel, 2010). Relaxin treatment for the final 2 weeks of type 1 diabetes results in beneficial cardiovascular effects in diabetic mice, alleviating cardiomyocyte, and pro-hypertrophic gene expression hypertrophy (Ng et al., 2017). Interestingly, the cardioprotective effect of relaxin appears to selectively suppress cardiomyocyte hypertrophy, but not fibrosis, at least in the setting of a relatively short-term 2 weeks of relaxin administration, in direct contrast to the well-documented anti-fibrotic effects of relaxin in other animal models (Samuel et al., 2008, 2011, 2014). Relaxin is thought to inhibit the progression of fibrosis through suppression of TGF-β and downregulation of Smad2 (Samuel et al., 2014) and Smad3 phosphorylation (Sassoli et al., 2013). Given that left ventricular TGF-β gene expression was not elevated in placebo-treated type 1 diabetic mice in the above study (Ng et al., 2017), it is rational to speculate that the ability of relaxin to limit cardiac fibrosis may relate to its distinct action to restore physiological levels of TGF-β (Sassoli et al., 2013). Another possibility is that the anti-fibrotic properties of relaxin may be mediated by its ability to restore physiological levels of the inflammatory cytokine interleukin (IL)-6, as distinct to CTGF/TGF-β pathways, hence resulting in a moderate decrease in diabetes-induced interstitial cardiac collagen content as recently shown (Ng et al., 2017). Such speculation is consistent with that previously shown by Dschietzig et al. (2004), where relaxin suppressed endotoxin-induced increase in IL-6 in THP-1 cells in vitro (Dschietzig et al., 2004). Although relaxin fails to reverse diabetes-induced fibrosis in the left ventricle, it elicits potent anti-apoptotic actions in diabetic mouse myocardium (Ng et al., 2017). Consistent with this finding, Moore et al. (2007) also reported that relaxin treatment attenuated hydrogen peroxide-induced apoptosis in neonatal rat cardiomyocytes, an effect attributed to the actions of pro-survival proteins such as Akt and Bcl-2 (Moore et al., 2007). Taken together, relaxin treatment not only shows promising effects in the vasculature, but also in the heart in experimental settings of type 1 diabetes, by alleviating left ventricular hypertrophy and apoptosis.
Relaxin as an Adjunctive Treatment in Diabetes?
We and others have showed that relaxin has promising therapeutic effects to limit diabetes-induced cardiovascular complications with experimental evidence in in vitro and in vivo studies, as summarized in Table 1. Although the majority of rodent studies have utilized animal models that mimic type 1 diabetes, it remains necessary to determine whether or not relaxin exhibits similar actions in type 2 diabetes, utilizing spontaneously diabetic db/db mice or high fat-fed mice. This is important because type 2 diabetes accounts for 85% of patients affected by diabetes. The optimal relaxin treatment period in the context of type 2 diabetes also remains to be determined. In type 1 diabetes which tends to exhibit a juvenile onset, the majority of patients affected are still at a young age at time of diagnosis. Relaxin treatment in humans requires continuous intravenous infusion, hence it is not practical to start relaxin infusion in young children, where prolonged hospitalization might be required. In type 2 diabetes, the proportion of patients affected remain undiagnosed, or are considered pre-diabetic because blood glucose levels are not high enough to be classified as diabetic. Because these patients do not exhibit symptoms of severe diabetes when medical attention would be needed, it would not be feasible to treat with relaxin.
Table 1.
Cardiovascular effects of relaxin treatment under hyperglycemic conditions.
| Duration; dose; route | Vascular effects | References |
|---|---|---|
| 6.5 h; 15 μg/h; jugular vein | Mouse aorta: Increased carbachol-evoked relaxation in lean but not high fat-fed mice. | Bonner et al., 2013 |
| 3 weeks; 1 mg/kg/d; subcutaneous | Increased carbachol-evoked relaxation in high fat-fed mice. | |
| 72 h; 10 nM; in vitro | Mouse aorta: Prevented high glucose (30 mM)-induced endothelial dysfunction by increasing vasodilator prostacyclin and counteracting superoxide production. | Ng et al., 2016 |
| 2 weeks; 0.5mg/kg/d; subcutaneous | Mouse aorta: Increased nitric oxide-mediated ACh-evoked relaxation in STZ-induced diabetic mice. Mouse mesenteric artery: Reversed diabetes-induced endothelial dysfunction by increasing nitric oxide-mediated relaxation, normalizing the contribution of vasoconstrictor prostanoids, and reducing vasoconstrictor response to AngII. | Ng et al., 2017 |
| 30 min; 100 ng/mL; in vitro | Rat ventricular myocyte: Prevented high glucose (33 mM)-induced apoptosis and endoplasmic reticulum stress by reducing CHOP, cleaved caspase-8, -9, and -12 protein expression. | Zhang et al., 2015 |
| 48 h; 100 ng/mL; in vitro | Rat fibroblast: Inhibited high glucose (33 mM)-induced oxidative stress and collagen synthesis by decreasing collagen I and III, α-SMA, P2X7R-mediated NLRP3 inflammasome activation, IL-18, IL-1β, and cleaved caspase-1 expression. | Zhang et al., 2018 |
| 48 h; 0.1 mM; in vitro | Rat H9c2 cell line: Reduced high glucose (33 mM)-induced cardiomyocyte hypertrophy, oxidative stress and apoptosis by decreasing ANP, BNP, caspase-3, cytochrome C protein expression, and increasing Notch1, Hes1, and MnSOD expression. | Wei et al., 2018 |
| 72 h; 100 ng/mL; in vitro | Rat fibroblast: Reduced high glucose (25 mM)-induced fibroblast proliferation, procollagen I and III, and MMP2 and MMP9 production. | Wang et al., 2009 |
| 2 weeks; 0.5 mg/kg/d; subcutaneous | Rat left ventricle: Improved diastolic function and decreased myocardial stiffness by reducing α-SMA, TIMP-1 and increasing MMP-13 expression in STZ-induced transgenic mRen-2 rats. | Samuel et al., 2008 |
| 2 weeks; 2 μg/kg/d; subcutaneous | Rat left ventricle: Improved function by mitigating diabetes-induced apoptosis, fibrosis and inflammasome activation in STZ-induced diabetic rats. | Zhang et al., 2017 |
| 2 weeks; 0.5 mg/kg/d; subcutaneous | Mouse left ventricle: Suppressed hypertrophy and apoptosis through a reduction in BNP and Bax:Bcl2 expression in STZ-induced diabetic mice. | Ng et al., 2017 |
ACh, acetylcholine; AngII, angiotensin II; STZ, streptozotocin; CHOP, C/EBP homologous protein; α-SMA, alpha smooth muscle actin; P2X7R, ionotropic purinergic receptor; NLRP3, nucleotide-binding domain and leucine-rich repeat-containing family, pyrin domain containing 3; IL, interleukin; ANP, atrial natriuretic peptide; BNP, B-type natriuretic peptide; Hes1, hairy and enhancer of split-1; MnSOD, manganese superoxide dismutase; MMP, matrix metalloproteinase; TIMP, tissue inhibitor of metalloproteinase; Bcl2, B-cell lymphoma 2; Bax, Bcl2-associated X protein.
Although relaxin treatment exhibits fairly profound effects on the vasculature and heart in diabetic mice, its impact on metabolism remains controversial. Relaxin does not alter blood glucose levels, in either healthy rodents (Samuel et al., 2008; Bitto et al., 2013; Bonner et al., 2013; Wong et al., 2013) or in type 1 diabetic models (Samuel et al., 2008; Wong et al., 2013; Ng et al., 2017). However, in type 2 diabetic animal models, relaxin lowers blood glucose levels (Bitto et al., 2013; Bonner et al., 2013). Type 2 diabetes is closely associated with loss of insulin sensitivity and changes in peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α) function, particularly at the level of both biogenesis of mitochondria and glucose metabolism (Liang and Ward, 2006). Relaxin modulates PPARγ and PCG-1α signaling pathways, downstream of RXFP1 (Singh et al., 2015). Hence, it is logical to speculate that relaxin may exert metabolic actions in type 2 diabetic mice, modulating their glucose metabolism and/or mitochondria biogenesis. Although there is promising evidence of relaxin blood glucose-lowering actions in mice with type 2 diabetes, it is however important to note that relaxin is ineffective in type 1 diabetes, where insulin administration is required for glycemic control. Hence, relaxin may be an adjunctive agent with glucose-lowering therapy for preventing or treating diabetes. Indeed, relaxin exhibits synergistic effects with the angiotensin converting enzyme inhibitor, enalapril, to abrogate cardiac fibrosis in isoprenaline-induced mouse model (Samuel et al., 2014). Thus, it is possible that relaxin might have similar actions in diabetic mice and could be used in combination with hypoglycemic drugs such as metformin, liraglutide or insulin, to then reduce cardiovascular complications associated with diabetes. However, this hypothesis remains to be tested. It is also yet to be determined whether or not relaxin exhibits adverse effects when used in combination with these conventional anti-diabetic therapies.
Conclusion
Figure 1 shows an overview of the therapeutic potential of relaxin in the pathogenesis of diabetes. Although the conventional glucose-lowering therapies such as metformin and insulin are widely prescribed for the treatment of diabetes, they may not be sufficient to limit the associated cardiovascular complications caused by hyperglycemia. Therefore, we now present the hypothesis that alternative adjunctive agents such as relaxin may offer a promising role in the prevention or treatment of diabetes-related cardiovascular complications, opening up new avenues in addressing the ever-increasing incidence of diabetes and its consequences. Although pre-clinical studies suggest a potential promising role for relaxin to protect against diabetes-associated cardiovascular complications, there remain many unresolved questions relating to the efficacy, dosage and duration of relaxin administration for clinical use in diabetic patients. Clinical trials are needed to specifically determine the translational effects of relaxin in the setting of diabetes, as there are no data available addressing this patient population. As chronic administration of relaxin is not feasible due to its pharmacokinetics and short half-life in vivo (Chen et al., 1993a,b), recent development of small molecule relaxin mimetics such as B7-33 (Hossain et al., 2016) and ML290 (Xiao et al., 2013) now warrants such enhanced translation of the vasoprotective effects of relaxin in this setting.
FIGURE 1.
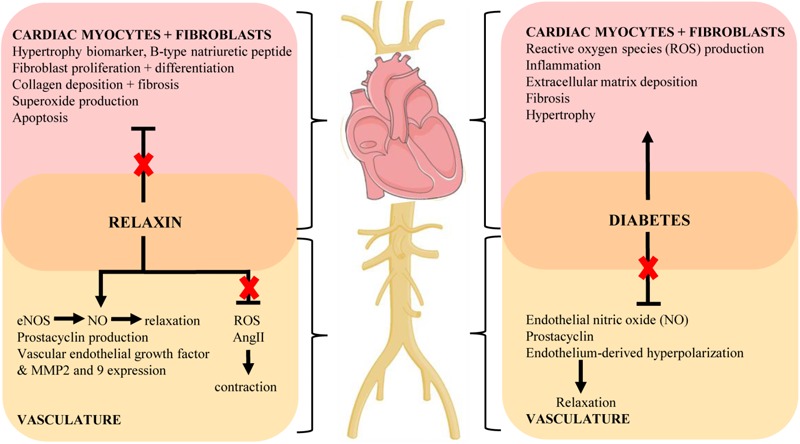
Key mechanisms of the cardiovascular complications of diabetes and proposed protective actions of relaxin to target these pathways (indicated by red cross). Diagram created using artwork provided by Servier Medical Art by Servier, licensed under a Creative Common License.
Author Contributions
HN and RR drafted the manuscript. All authors edited, revised, and approved the final version of this review.
Conflict of Interest Statement
The authors disclose that this research was partially funded by Novartis Pharma AG, who also provided the serelaxin as a condition of an Australian Research Council Linkage Grant (LP110200543). LP was also a paid consultant for Novartis Pharma AG.
Acknowledgments
This review forms part of HN’s Ph.D. thesis, and it is the only medium that first appeared online. The thesis can be accessed at http://hdl.handle.net/11343/123225 (Ng, 2016).
Footnotes
Funding. HN received a Melbourne International Research Scholarship, a Melbourne International Fee Remission Scholarship and a grant from the Albert Shimmins Fund. RR is a National Health and Medical Research Council of Australia Senior Research Fellow (ID1059960).
References
- Angulo J., Cuevas P., Fernandez A., Gabancho S., Allona A., Martin-Morales A., et al. (2003). Diabetes impairs endothelium-dependent relaxation of human penile vascular tissues mediated by NO and EDHF. 312 1202–1208. 10.1016/j.bbrc.2003.11.034 [DOI] [PubMed] [Google Scholar]
- Bathgate R. A., Ivell R., Sanborn B. M., Sherwood O. D., Summers R. J. (2006). International union of pharmacology LVII: recommendations for the nomenclature of receptors for relaxin family peptides. 58 7–31. 10.1124/pr.58.1.9 [DOI] [PubMed] [Google Scholar]
- Baynes J. W., Thorpe S. R. (1999). Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. 48 1–9. 10.2337/diabetes.48.1.1 [DOI] [PubMed] [Google Scholar]
- Bethel M. A., Patel R. A., Merrill P., Lokhnygina Y., Buse J. B., Mentz R. J., et al. (2017). Cardiovascular outcomes with glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes: a meta-analysis. 6 105–113. 10.1016/S2213-8587(17)30412-6 [DOI] [PubMed] [Google Scholar]
- Bitto A., Irrera N., Minutoli L., Calo M., Lo Cascio P., Caccia P., et al. (2013). Relaxin improves multiple markers of wound healing and ameliorates the disturbed healing pattern of genetically diabetic mice. 125 575–585. 10.1042/cs20130105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner J. S., Lantier L., Hocking K. M., Kang L., Owolabi M., James F. D., et al. (2013). Relaxin treatment reverses insulin resistance in mice fed a high-fat diet. 62 3251–3260. 10.2337/db13-0033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudina S., Abel E. D. (2007). Diabetic cardiomyopathy revisited. 115 3213–3223. 10.1161/circulationaha.106.679597 [DOI] [PubMed] [Google Scholar]
- Boudina S., Abel E. D. (2010). Diabetic cardiomyopathy, causes and effects. 11 31–39. 10.1007/s11154-010-9131-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugger H., Abel E. D. (2014). Molecular mechanisms of diabetic cardiomyopathy. 57 660–671. 10.1007/s00125-014-3171-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai S., Khoo J., Mussa S., Alp N. J., Channon K. M. (2005). Endothelial nitric oxide synthase dysfunction in diabetic mice: importance of tetrahydrobiopterin in eNOS dimerisation. 48 1933–1940. 10.1007/s00125-005-1857-5 [DOI] [PubMed] [Google Scholar]
- Chan S. L., Cipolla M. J. (2011). Relaxin causes selective outward remodeling of brain parenchymal arterioles via activation of peroxisome proliferator-activated receptor-gamma. 25 3229–3239. 10.1096/fj.10-175471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S. L., Sweet J. G., Cipolla M. J. (2013). Treatment for cerebral small vessel disease: effect of relaxin on the function and structure of cerebral parenchymal arterioles during hypertension. 27 3917–3927. 10.1096/fj.13-230797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S. A., Perlman A. J., Spanski N., Peterson C. M., Sanders S. W., Jaffe R., et al. (1993a). The pharmacokinetics of recombinant human relaxin in nonpregnant women after intravenous, intravaginal, and intracervical administration. 10 834–838. [DOI] [PubMed] [Google Scholar]
- Chen S. A., Reed B., Nguyen T., Gaylord N., Fuller G. B., Mordenti J. (1993b). The pharmacokinetics and absorption of recombinant human relaxin in nonpregnant rabbits and rhesus monkeys after intravenous and intravaginal administration. 10 223–227. [DOI] [PubMed] [Google Scholar]
- Ding H., Hashem M., Wiehler W. B., Lau W., Martin J., Reid J., et al. (2005). Endothelial dysfunction in the streptozotocin-induced diabetic apoE-deficient mouse. 146 1110–1118. 10.1038/sj.bjp.0706417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H., Triggle C. R. (2010). Endothelial dysfunction in diabetes: multiple targets for treatment. 459 977–994. 10.1007/s00424-010-0807-3 [DOI] [PubMed] [Google Scholar]
- Dschietzig T., Bartsch C., Richter C., Laule M., Baumann G., Stangl K. (2003). Relaxin, a pregnancy hormone, is a functional endothelin-1 antagonist: attenuation of endothelin-1-mediated vasoconstriction by stimulation of endothelin type-B receptor expression via ERK-1/2 and nuclear factor-kappaB. 92 32–40. 10.1161/01.RES.0000051884.27117.7E [DOI] [PubMed] [Google Scholar]
- Dschietzig T., Bartsch C., Stangl V., Baumann G., Stangl K. (2004). Identification of the pregnancy hormone relaxin as glucocorticoid receptor agonist. 18 1536–1538. 10.1096/fj.03-1120fje [DOI] [PubMed] [Google Scholar]
- Dschietzig T., Brecht A., Bartsch C., Baumann G., Stangl K., Alexiou K. (2012). Relaxin improves TNF-alpha-induced endothelial dysfunction: the role of glucocorticoid receptor and phosphatidylinositol 3-kinase signalling. 95 97–107. 10.1093/cvr/cvs149 [DOI] [PubMed] [Google Scholar]
- Fisher C., MacLean M., Morecroft I., Seed A., Johnston F., Hillier C., et al. (2002). Is the pregnancy hormone relaxin also a vasodilator peptide secreted by the heart? 106 292–295. [DOI] [PubMed] [Google Scholar]
- Forbes J. M., Fotheringham A. K. (2017). Vascular complications in diabetes: old messages, new thoughts. 60 2129–2138. 10.1007/s00125-017-4360-x [DOI] [PubMed] [Google Scholar]
- Guzik T. J., Mussa S., Gastaldi D., Sadowski J., Ratnatunga C., Pillai R., et al. (2002). Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. 105 1656–1662. 10.1161/01.CIR.0000012748.58444.08 [DOI] [Google Scholar]
- Henry B. L., Gabris B., Li Q., Martin B., Giannini M., Parikh A., et al. (2016). Relaxin suppresses atrial fibrillation in aged rats by reversing fibrosis and upregulating Na(+) channels. 13 983–991. 10.1016/j.hrthm.2015.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holman R. (2007). Metformin as first choice in oral diabetes treatment: the UKPDS experience. 13–20. [PubMed] [Google Scholar]
- Hossain M. A., Kocan M., Yao S. T., Royce S. G., Nair V. B., Siwek C., et al. (2016). A single-chain derivative of the relaxin hormone is a functionally selective agonist of the G protein-coupled receptor, RXFP1. 7 3805–3819. 10.1039/C5SC04754D [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh K., Bernardo B. C., McMullen J. R., Ritchie R. H. (2014). Diabetic cardiomyopathy: mechanisms and new treatment strategies targeting antioxidant signaling pathways. 142 375–415. 10.1016/j.pharmthera.2014.01.003 [DOI] [PubMed] [Google Scholar]
- Iimoto D. S., Covell J. W., Harper E. (1988). Increase in cross-linking of type I and type III collagens associated with volume-overload hypertrophy. 63 399–408. 10.1161/01.RES.63.2.399 [DOI] [PubMed] [Google Scholar]
- Jelinic M., Leo C. H., Post Uiterweer E. D., Sandow S. L., Gooi J. H., Wlodek M. E., et al. (2014). Localization of relaxin receptors in arteries and veins, and region-specific increases in compliance and bradykinin-mediated relaxation after in vivo serelaxin treatment. 28 275–287. 10.1096/fj.13-233429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone M. T., Creager S. J., Scales K. M., Cusco J. A., Lee B. K., Creager M. A. (1993). Impaired endothelium-dependent vasodilation in patients with insulin-dependent diabetes mellitus. 88 2510–2516. 10.1161/01.CIR.88.6.2510 [DOI] [PubMed] [Google Scholar]
- Lavrentyev E. N., Estes A. M., Malik K. U. (2007). Mechanism of high glucose induced angiotensin II production in rat vascular smooth muscle cells. 101 455–464. 10.1161/circresaha.107.151852 [DOI] [PubMed] [Google Scholar]
- Lekgabe E. D., Kiriazis H., Zhao C., Xu Q., Moore X. L., Su Y., et al. (2005). Relaxin reverses cardiac and renal fibrosis in spontaneously hypertensive rats. 46 412–418. 10.1161/01.HYP.0000171930.00697.2f [DOI] [PubMed] [Google Scholar]
- Leo C. H., Hart J. L., Woodman O. L. (2011a). 3’,4’-Dihydroxyflavonol reduces superoxide and improves nitric oxide function in diabetic rat mesenteric arteries. 6:e20813. 10.1371/journal.pone.0020813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo C. H., Hart J. L., Woodman O. L. (2011b). Impairment of both nitric oxide-mediated and EDHF-type relaxation in small mesenteric arteries from rats with streptozotocin-induced diabetes. 162 365–377. 10.1111/j.1476-5381.2010.01023.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo C. H., Jelinic M., Gooi J. H., Tare M., Parry L. J. (2014a). A vasoactive role for endogenous relaxin in mesenteric arteries of male mice. 9:e107382. 10.1371/journal.pone.0107382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo C. H., Jelinic M., Parkington H. C., Tare M., Parry L. J. (2014b). Acute intravenous injection of serelaxin (recombinant human relaxin-2) causes rapid and sustained bradykinin-mediated vasorelaxation. 3:e000493. 10.1161/jaha.113.000493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo C. H., Jelinic M., Ng H. H., Marshall S. A., Novak J., Tare M., et al. (2017). Vascular actions of relaxin: nitric oxide and beyond. 174 1002–1014. 10.1111/bph.13614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leo C. H., Jelinic M., Ng H. H., Tare M., Parry L. J. (2016a). Serelaxin: a novel therapeutic for vascular diseases. 37 498–507. 10.1016/j.tips.2016.04.001 [DOI] [PubMed] [Google Scholar]
- Leo C. H., Jelinic M., Ng H. H., Tare M., Parry L. J. (2016b). Time-dependent activation of prostacyclin and nitric oxide pathways during continuous i.v. infusion of serelaxin (recombinant human H2 relaxin). 173 1005–1017. 10.1111/bph.13404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H., Ward W. F. (2006). PGC-1alpha: a key regulator of energy metabolism. 30 145–151. 10.1152/advan.00052.2006 [DOI] [PubMed] [Google Scholar]
- Lytvyn Y., Bjornstad P., Udell J. A., Lovshin J. A., Cherney D. Z. I. (2017). Sodium glucose cotransporter-2 inhibition in heart failure: potential mechanisms, clinical applications, and summary of clinical trials. 136 1643–1658. 10.1161/circulationaha.117.030012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiavacchi M., Gasparini M., Genovese S., Pini D., Klersy C., Bragato R., et al. (2008). Insulin-treated type 2 diabetes is associated with a decreased survival in heart failure patients after cardiac resynchronization therapy. 31 1425–1432. 10.1111/j.1540-8159.2008.01206.x [DOI] [PubMed] [Google Scholar]
- Marshall S. A., Leo C. H., Girling J. E., Tare M., Beard S., Hannan N. J., et al. (2017). Relaxin treatment reduces angiotensin II-induced vasoconstriction in pregnancy and protects against endothelial dysfunctiondagger. 96 895–906. 10.1093/biolre/iox023 [DOI] [PubMed] [Google Scholar]
- Marshall S. A., Leo C. H., Senadheera S. N., Girling J. E., Tare M., Parry L. J. (2016). Relaxin deficiency attenuates pregnancy-induced adaptation of the mesenteric artery to angiotensin II in mice. 310 R847–R857. 10.1152/ajpregu.00506.2015 [DOI] [PubMed] [Google Scholar]
- McGarry J. D. (1992). What if Minkowski had been ageusic? An alternative angle on diabetes. 258 766–770. 10.1126/science.1439783 [DOI] [PubMed] [Google Scholar]
- McGuane J. T., Danielson L. A., Debrah J. E., Rubin J. P., Novak J., Conrad K. P. (2011a). Angiogenic growth factors are new and essential players in the sustained relaxin vasodilatory pathway in rodents and humans. 57 1151–1160. 10.1161/hypertensionaha.110.165027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuane J. T., Debrah J. E., Sautina L., Jarajapu Y. P., Novak J., Rubin J. P., et al. (2011b). Relaxin induces rapid dilation of rodent small renal and human subcutaneous arteries via PI3 kinase and nitric oxide. 152 2786–2796. 10.1210/en.2010-1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore X. L., Tan S. L., Lo C. Y., Fang L., Su Y. D., Gao X. M., et al. (2007). Relaxin antagonizes hypertrophy and apoptosis in neonatal rat cardiomyocytes. 148 1582–1589. 10.1210/en.2006-1324 [DOI] [PubMed] [Google Scholar]
- Ng H. H. (2016). Ph.D. thesis, University of Melbourne; Parkville VIC. [Google Scholar]
- Ng H. H., Jelinic M., Parry L. J., Leo C. H. (2015). Increased superoxide production and altered nitric oxide-mediated relaxation in the aorta of young but not old male relaxin-deficient mice. 309 H285–H296. 10.1152/ajpheart.00786.2014 [DOI] [PubMed] [Google Scholar]
- Ng H. H., Leo C. H., Parry L. J. (2016). Serelaxin (recombinant human relaxin-2) prevents high glucose-induced endothelial dysfunction by ameliorating prostacyclin production in the mouse aorta. 107 220–228. 10.1016/j.phrs.2016.03.011 [DOI] [PubMed] [Google Scholar]
- Ng H. H., Leo C. H., Prakoso D., Qin C., Ritchie R. H., Parry L. J. (2017). Serelaxin treatment reverses vascular dysfunction and left ventricular hypertrophy in a mouse model of Type 1 diabetes. 7:39604. 10.1038/srep39604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols G. A., Koro C. E., Gullion C. M., Ephross S. A., Brown J. B. (2005). The incidence of congestive heart failure associated with antidiabetic therapies. 21 51–57. 10.1002/dmrr.480 [DOI] [PubMed] [Google Scholar]
- Novak J., Parry L. J., Matthews J. E., Kerchner L. J., Indovina K., Hanley-Yanez K., et al. (2006). Evidence for local relaxin ligand-receptor expression and function in arteries. 20 2352–2362. 10.1096/fj.06-6263com [DOI] [PubMed] [Google Scholar]
- Pappachan J. M., Varughese G. I., Sriraman R., Arunagirinathan G. (2013). Diabetic cardiomyopathy: pathophysiology, diagnostic evaluation and management. 4 177–189. 10.4239/wjd.v4.i5.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh A., Patel D., McTiernan C. F., Xiang W., Haney J., Yang L., et al. (2013). Relaxin suppresses atrial fibrillation by reversing fibrosis and myocyte hypertrophy and increasing conduction velocity and sodium current in spontaneously hypertensive rat hearts. 113 313–321. 10.1161/circresaha.113.301646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieper G. M., Meier D. A., Hager S. R. (1995). Endothelial dysfunction in a model of hyperglycemia and hyperinsulinemia. 269(3 Pt 2) H845–H850. 10.1152/ajpheart.1995.269.3.H845 [DOI] [PubMed] [Google Scholar]
- Pini A., Boccalini G., Baccari M. C., Becatti M., Garella R., Fiorillo C., et al. (2016). Protection from cigarette smoke-induced vascular injury by recombinant human relaxin-2 (serelaxin). 20 891–902. 10.1111/jcmm.12802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakoso D., De Blasio M. J., Qin C., Rosli S., Kiriazis H., Qian H., et al. (2017). Phosphoinositide 3-kinase (p110alpha) gene delivery limits diabetes-induced cardiac NADPH oxidase and cardiomyopathy in a mouse model with established diastolic dysfunction. 131 1345–1360. 10.1042/cs20170063 [DOI] [PubMed] [Google Scholar]
- Randle P. J., Garland P. B., Hales C. N., Newsholme E. A. (1963). The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. 1 785–789. 10.1016/S0140-6736(63)91500-9 [DOI] [PubMed] [Google Scholar]
- Reaven G. M., Lithell H., Landsberg L. (1996). Hypertension and associated metabolic abnormalities–the role of insulin resistance and the sympathoadrenal system. 334 374–381. 10.1056/nejm199602083340607 [DOI] [PubMed] [Google Scholar]
- Ritchie R. H., Zerenturk E. J., Prakoso D., Calkin A. C. (2017). Lipid metabolism and its implications for type 1 diabetes-associated cardiomyopathy. 58 R225–R240. 10.1530/JME-16-0249 [DOI] [PubMed] [Google Scholar]
- Samuel C. S., Bodaragama H., Chew J. Y., Widdop R. E., Royce S. G., Hewitson T. D. (2014). Serelaxin is a more efficacious antifibrotic than enalapril in an experimental model of heart disease. 64 315–322. 10.1161/hypertensionaha.114.03594 [DOI] [PubMed] [Google Scholar]
- Samuel C. S., Cendrawan S., Gao X. M., Ming Z., Zhao C., Kiriazis H., et al. (2011). Relaxin remodels fibrotic healing following myocardial infarction. 91 675–690. 10.1038/labinvest.2010.198 [DOI] [PubMed] [Google Scholar]
- Samuel C. S., Hewitson T. D., Zhang Y., Kelly D. J. (2008). Relaxin ameliorates fibrosis in experimental diabetic cardiomyopathy. 149 3286–3293. 10.1210/en.2008-0250 [DOI] [PubMed] [Google Scholar]
- Samuel C. S., Summers R. J., Hewitson T. D. (2016). Antifibrotic actions of serelaxin - new roles for an old player. 37 485–497. 10.1016/j.tips.2016.02.007 [DOI] [PubMed] [Google Scholar]
- Samuel C. S., Unemori E. N., Mookerjee I., Bathgate R. A., Layfield S. L., Mak J., et al. (2004). Relaxin modulates cardiac fibroblast proliferation, differentiation, and collagen production and reverses cardiac fibrosis in vivo. 145 4125–4133. 10.1210/en.2004-0209 [DOI] [PubMed] [Google Scholar]
- Samuel V. T., Shulman G. I. (2016). The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. 126 12–22. 10.1172/jci77812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M. (2018). A new class of drugs for heart failure: SGLT2 inhibitors reduce sympathetic overactivity. 71 471–476. 10.1016/j.jjcc.2017.12.004 [DOI] [PubMed] [Google Scholar]
- Santulli G., Pagano G., Sardu C., Xie W., Reiken S., D’Ascia S. L., et al. (2015). Calcium release channel RyR2 regulates insulin release and glucose homeostasis. 125 1968–1978. 10.1172/jci79273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardu C., Marfella R., Santulli G. (2014). Impact of diabetes mellitus on the clinical response to cardiac resynchronization therapy in elderly people. 7 362–368. 10.1007/s12265-014-9545-9 [DOI] [PubMed] [Google Scholar]
- Sarwar M., Du X. J., Dschietzig T. B., Summers R. J. (2017). The actions of relaxin on the human cardiovascular system. 174 933–949. 10.1111/bph.13523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasser J. M., Cunningham M. W., Jr., Baylis C. (2014). Serelaxin reduces oxidative stress and asymmetric dimethylarginine in angiotensin II-induced hypertension. 307 F1355–F1362. 10.1152/ajprenal.00407.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasser J. M., Molnar M., Baylis C. (2011). Relaxin ameliorates hypertension and increases nitric oxide metabolite excretion in angiotensin II but not N(omega)-nitro-L-arginine methyl ester hypertensive rats. 58 197–204. 10.1161/hypertensionaha.110.164392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassoli C., Chellini F., Pini A., Tani A., Nistri S., Nosi D., et al. (2013). Relaxin prevents cardiac fibroblast-myofibroblast transition via notch-1-mediated inhibition of TGF-beta/Smad3 signaling. 8:e63896. 10.1371/journal.pone.0063896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw J. E., Sicree R. A., Zimmet P. Z. (2010). Global estimates of the prevalence of diabetes for 2010 and 2030. 87 4–14. 10.1016/j.diabres.2009.10.007 [DOI] [PubMed] [Google Scholar]
- Sherwood O. D. (2004). Relaxin’s physiological roles and other diverse actions. 25 205–234. 10.1210/er.2003-0013 [DOI] [PubMed] [Google Scholar]
- Shi Y., Vanhoutte P. M. (2017). Macro- and microvascular endothelial dysfunction in diabetes. 9 434–449. 10.1111/1753-0407.12521 [DOI] [PubMed] [Google Scholar]
- Singh S., Simpson R. L., Bennett R. G. (2015). Relaxin activates peroxisome proliferator-activated receptor gamma (PPARgamma) through a pathway involving PPARgamma coactivator 1alpha (PGC1alpha). 290 950–959. 10.1074/jbc.M114.589325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh V. P., Le B., Khode R., Baker K. M., Kumar R. (2008). Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. 57 3297–3306. 10.2337/db08-0805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smooke S., Horwich T. B., Fonarow G. C. (2005). Insulin-treated diabetes is associated with a marked increase in mortality in patients with advanced heart failure. 149 168–174. 10.1016/j.ahj.2004.07.005 [DOI] [PubMed] [Google Scholar]
- Squadrito F., Bitto A., Irrera N., Minutoli L., Caccia P., Altavilla D. (2013). Porcine derived relaxin stimulates new vessel formation and improves the disturbed wound healing of the genetically diabetic mice. 118(1 Suppl.) 66–70. [PubMed] [Google Scholar]
- Tate M., Deo M., Cao A. H., Hood S. G., Huynh K., Kiriazis H., et al. (2017a). Insulin replacement limits progression of diabetic cardiomyopathy in the low-dose streptozotocin-induced diabetic rat. 14 423–433. 10.1177/1479164117710390 [DOI] [PubMed] [Google Scholar]
- Tate M., Grieve D. J., Ritchie R. H. (2017b). Are targeted therapies for diabetic cardiomyopathy on the horizon? 131 897–915. 10.1042/cs20160491 [DOI] [PubMed] [Google Scholar]
- Tiyerili V., Beiert T., Schatten H., Camara B., Jehle J., Schrickel J. W., et al. (2016). Anti-atherosclerotic effects of serelaxin in apolipoprotein E-deficient mice. 251 430–437. 10.1016/j.atherosclerosis.2016.06.008 [DOI] [PubMed] [Google Scholar]
- Tousoulis D., Kampoli A. M., Stefanadis C. (2012). Diabetes mellitus and vascular endothelial dysfunction: current perspectives. 10 19–32. 10.2174/157016112798829797 [DOI] [PubMed] [Google Scholar]
- Unemori E. N., Lewis M., Constant J., Arnold G., Grove B. H., Normand J., et al. (2000). Relaxin induces vascular endothelial growth factor expression and angiogenesis selectively at wound sites. 8 361–370. 10.1111/j.1524-475X.2000.00361.x [DOI] [PubMed] [Google Scholar]
- van Drongelen J., Ploemen I. H., Pertijs J., Gooi J. H., Sweep F. C., Lotgering F. K., et al. (2011). Aging attenuates the vasodilator response to relaxin. 300 H1609–H1615. 10.1152/ajpheart.00360.2010 [DOI] [PubMed] [Google Scholar]
- van Drongelen J., van Koppen A., Pertijs J., Gooi J. H., Parry L. J., Sweep F. C., et al. (2012). Impaired vascular responses to relaxin in diet-induced overweight female rats. 112 962–969. 10.1152/japplphysiol.00470.2011 [DOI] [PubMed] [Google Scholar]
- van Drongelen J., van Koppen A., Pertijs J., Gooi J. H., Sweep F. C., Lotgering F. K., et al. (2013). Impaired effect of relaxin on vasoconstrictor reactivity in spontaneous hypertensive rats. 49 41–48. 10.1016/j.peptides.2013.08.020 [DOI] [PubMed] [Google Scholar]
- Vanhoutte P. M., Shimokawa H., Feletou M., Tang E. H. (2017). Endothelial dysfunction and vascular disease - a 30th anniversary update. 219 22–96. 10.1111/apha.12646 [DOI] [PubMed] [Google Scholar]
- Wang P., Li H. W., Wang Y. P., Chen H., Zhang P. (2009). Effects of recombinant human relaxin upon proliferation of cardiac fibroblast and synthesis of collagen under high glucose condition. 32 242–247. 10.1007/bf03346460 [DOI] [PubMed] [Google Scholar]
- Wei X., Yang Y., Jiang Y. J., Lei J. M., Guo J. W., Xiao H. (2018). Relaxin ameliorates high glucose-induced cardiomyocyte hypertrophy and apoptosis via the Notch1 pathway. 15 691–698. 10.3892/etm.2017.5448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S. E., Samuel C. S., Kelly D. J., Zhang Y., Becker G. J., Hewitson T. D. (2013). The anti-fibrotic hormone relaxin is not reno-protective, despite being active, in an experimental model of type 1 diabetes. 20 1029–1038. 10.2174/0929866511320090009 [DOI] [PubMed] [Google Scholar]
- Xiao J., Huang Z., Chen C. Z., Agoulnik I. U., Southall N., Hu X., et al. (2013). Identification and optimization of small-molecule agonists of the human relaxin hormone receptor RXFP1. 4:1953. 10.1038/ncomms2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Chakravorty A., Bathgate R. A., Dart A. M., Du X. J. (2010). Relaxin therapy reverses large artery remodeling and improves arterial compliance in senescent spontaneously hypertensive rats. 55 1260–1266. 10.1161/hypertensionaha.109.149369 [DOI] [PubMed] [Google Scholar]
- Zhang X., Fu Y., Li H., Shen L., Chang Q., Pan L., et al. (2018). H3 relaxin inhibits the collagen synthesis via ROS- and P2X7R-mediated NLRP3 inflammasome activation in cardiac fibroblasts under high glucose. 22 1816–1825. 10.1111/jcmm.13464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Ma X., Zhao M., Zhang B., Chi J., Liu W., et al. (2015). H2 and H3 relaxin inhibit high glucose-induced apoptosis in neonatal rat ventricular myocytes. 108 59–67. 10.1016/j.biochi.2014.11.004 [DOI] [PubMed] [Google Scholar]
- Zhang X., Pan L., Yang K., Fu Y., Liu Y., Chi J., et al. (2017). H3 relaxin protects against myocardial injury in experimental diabetic cardiomyopathy by inhibiting myocardial apoptosis, fibrosis and inflammation. 43 1311–1324. 10.1159/000481843 [DOI] [PubMed] [Google Scholar]
- Zhu N., Liu B., Luo W., Zhang Y., Li H., Li S., et al. (2014). Vasoconstrictor role of cyclooxygenase-1-mediated prostacyclin synthesis in non-insulin-dependent diabetic mice induced by high-fat diet and streptozotocin. 307 H319–H327. 10.1152/ajpheart.00022.2014 [DOI] [PubMed] [Google Scholar]