

Abstract
The mechanism of miR-30a in myocardial fibrosis in rats with myocardial infarction (MI) was investigated. rAAV9-miR-30a was constructed and transfected to heart via injecting into the left ventricular cavity of MI rats. The sham operation group, control group, miR-30a group and miR-30a-NC group were established. Besides, the 3′-UTR of CTGF was inserted into luciferase expression plasmid (pMir-report), then transfected into COS1 cells. miR-30a and control miRNA were, respectively, cotransfected into COS1 cells. The expression of luciferase was detected before and after knockdown of the binding site of miR-30a and the 3′-UTR of CTGF. Four weeks after MI surgery, cardiac function was measured by color Doppler echocardiography, including short axis shortening (FS) and left ventricular ejection fraction (LVEF); the myocardial collagen volume fraction (CVF) was observed by Masson's staining; deposition of collagen I and collagen III were evaluated by immunohistochemical stain; using real-time PCR to detect expression levels of miR-30a and CTGF; the expression of CTGF was observed by western blotting. In MI group, cardiac function was significantly improved, while the expression levels of CVF, collagen I and III, the ratio of type I/III collagen, CTGF were significantly reduced. After knockdown the binding site of miR-30a and the 3′-UTR of CTGF, luciferase expression in COS1 cells decreased significantly. miR-30a might inhibit the expression of CTGF by directly combining with the 3′-UTR site of CTGF after MI, thereby reduce the production of collagen in myocardia, inhibit myocardial fibrosis, then improve cardiac function.
Keywords: miR-30a, MI, myocardial fibrosis, CTGF, 3′-UTR
Introduction
Myocardial infarction (MI) leading to heart failure (HF) is a major cause of death in the world. The main center of HF is cardiac remodeling including cardiomyocyte hypertrophy, apoptosis and cardiac fibrosis (1). Cardiac fibrosis plays a great role in development of mid- and late-stage of HF, and it is a result of fibroblast proliferation and excessive extracellular collagen matrix deposition which leads to cardiac hypertrophy (2,3). Thus, inhibition of cardiac fibrosis is a critical therapeutic approach to treat HF after MI clinically.
MicroRNAs (miRNAs) are a relatively new class of 22-nt nonprotein-coding single-strand RNAs that control several cellular processes, such as cell growth, proliferation and differentiation. miRNAs have been shown to play a central role in the pathogenesis of cardiac fibrosis (4). Related research has been done previously. Li et al (5) reported that miR-29a-3p inhibited ET-1-induced cardiomyocyte hypertrophy via directly targeting the 3′-UTR of NFATc4 mRNA and silencing NFATc4 expression. Besides, Zhang et al (6) found that miR-151-5p could bind to the 3′-UTR of FXYD1 and inhibit its expression, then influence cardiovascular diseases. However, molecularly targeted therapy for MI in clinic has not been sufficiently studied.
Recently, several studies showed that miR-30a could inhibit myocardial fibrosis by regulating CTGF (7,8). The five miR-30 family members (miR-30a through miR-30e) are evolutionarily conserved and abundantly expressed in the mature heart, but they are significantly dysregulated in human HF, in experimental I/R, and in vitro after oxidative stress (9,10). Further, it has been reported that miR-30a levels are reduced in mouse models of hypertrophy, and in vitro downregulated miR-30a aggravates phenylephrine-induced cardiomyocyte hypertrophy by activating autophagy (11). However, the molecular mechanism of miR-30 attenuating cardiac fibrosis has not been reported previously.
In the present study, the aim was to test whether the putative miR-30a target sequence directly regulates protein levels of CTGF. We first constructed the Adeno Associated Virus (AAV) plasmid vector which carried the miR-30a gene of rat, then, we transfected the miR-30a into the left ventricular cavity of heart in MI rats. The 3′-UTR of CTGF was transfected into COS1 cells, miR-30a and control miRNA were cotransfected into COS1 cells, respectively. The expression of luciferase was detected before and after knockdown of the binding site of the miR-30a and the 3′-UTR of CTGF. The cardioprotective effect of miR-30a and the expression of luciferase in these hypertrophy models were observed. Last, we tried to explore possible miR-30a-regulated mechanism.
Materials and methods
Animals
Rats were purchased from Jiancheng Bioengineering Institute (Nanjing, China). Treatment of the rats used in this investigation conformed to the recommendations in the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH Publication no. 85-23, revised 1996) and the protocol was approved by the Animal Care Committee of Nanjing First Hospital (Nanjing, China).
rAAV9-miR-30a construction
RAAV9-miR-30a construction procedure was carried out as previously described in detail (12). Briefly, the expression vector was linearized and digested by restriction enzyme HindIII. After handled with calf intestinal alkaline phosphatase (CIP) for 1 h, the 5451 bp vector fragment was recycled to amplify the target gene. Primer design was as follows: Primer 1 was used to isolate miRNA-30a by PCR, its forward strand was 5′-GATCTCGAGCTCAAGTAGGGGCATATCTGAACGAGGCT-3′, and its reverse strand was 5′-GATTATCGATAAGCTTCAATGCTATAACACATTTCTTTGC-3′. Primer 2 was used to identify transformation by colony PCR, its forward strand was 5′-CATGGTCCTGCTGGAGTTCGTG-3′, and its reverse strand was 5′-CATAGCGTAAAAGGAGCAACA-3′. Then, miRNA30a DNA fragment was recombined with line carrier. After the sequence identified by colony PCR was correct, plasmid was extracted from inoculated colony.
In vivo miR-30a heart transfection and MI induction
At the beginning, we randomly divided 100 rats into four groups: Sham operation group, control group (injected with PBS), miR-30a-NC group (injected with blank AAV plasmid vector) and miR-30a group (injected with rAAV9-miR-30a). Before surgery, rats were anesthetized by intraperitoneal injection, intubated and supported by small animal ventilator. After that we opened the chest and exposed the heart. Then, PBS buffer, blank AAV plasmid vector and rAAV9-miR-30a were injected into left ventricular cavities of rats through the heart apex according to the grouping, respectively. Then the ascending aorta was clamped for 5–8 sec by non-traumatic hemostatic forceps gently for hemostasis, and immediately released after the heart rate was down (13). If the rats still had no signs of death after 10 min, MI model was established by left coronary artery ligation as reported. Briefly, the left anterior descending coronary artery was occluded with a nylon (6.0) suture at approximately 1 mm from its origin below the tip of the left atrium. Except that MI was not induced in rats of sham group, other rats underwent the same procedures. The chest was closed with a silk suture (14).
Echocardiographic evaluation
Four weeks after surgery, echocardiographic evaluation was performed by a blinded observer. Rats were firstly anaesthetized with 50 mg/kg sodium pentobarbital (Dolethal, Vétoquinol, France) via intraperitoneal injection, and then images were obtained using a Vevo 770 cardiac system (VisualSonics, Inc., Toronto, ON, Canada). The following parameters were evaluated: the left ventricular end-systolic dimension (LVESD), the left ventrical end-diastolic dimension (LVEDD), the fractional shortening (FS) and ejection fraction (EF) (15).
Masson's staining
The heart tissue sample was fixed in 10% buffered formalin and embedded in paraffin. Sections at 4 µm were stained with Masson's staining. The deposition of total collagen (green distributional area) was semi-quantitatively analyzed by ImageJ system software. The collagen volume fraction was calculated as the ratio of the total area of interstitial fibrosis to the entire visual field of the section. A minimum of 5 randomly selected areas per sample were observed at ×200 magnification, and the average value was calculated for statistical analysis (16).
Immunohistochemical analysis
The heart sections were deparaffinized in xylene (Thermo Fisher Scientific, Darmstadt, Germany) and rehydrated in graded ethanol, and then rinsed in PBS. After blocking endogenous peroxidase activity with 3% H2O2 for 10 min, tissue sections were boiled for the antigen epitope retrieval in 0.1 mol/l citrate buffer (pH 6.0) for 10 min. Then the sections were interacted with the 1:100 diluted primary antibodies (rabbit anti-rat collagen I, III polyclonal antibodies; ab34710, ab7778; Abcam, Cambridge, MA, USA) overnight at 4°C. After washing with PBS, the slides were then incubated with secondary antibody (goat anti-rabbit IgG polyclonal antibodies; 1:800; cat. no. DE0601; Biodee Biotechnology Co., Beijing, China)) for 1 h, following by a 15-min-incubation with Streptavidin Peroxidase (Lab Vision Corp., Fremont, CA, USA) and 1-min hematoxylin-stain (C.I. 75290, B.S.C. certification no. AcH-10; Aldrich, Milwaukee, WI, USA). Then, the slides were dehydrated by graded ethanol, dehydrated by xylene, and mounted with rhamsan gum.
The depositions of collagen I and collagen III (a yellow to brown color) were semi-quantitatively analyzed by ImageJ system software. The method of calculating collagen I and III volume fraction were the same as the total collagen (17).
Real-time PCR
Total RNA was extracted with the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions, and it was reverse transcribed into cDNA by using PrimeScript RT reagent kit (Takara Bio, Kyoto, Japan). Real-time PCR was performed using a 7500 Real-time PCR System (Applied Biosystems) with SYBR Premix Ex Taq kit (Takara Bio). PCR primer sequences were as followed: β-actin: (forward: 5′-GGTGGGAATGGGTCAGAAGG-3′, reverse: 5′-GGGGTACTTCAGGGTCAGGA-3′); CTGF (forward: 5′-GTGTGATGAGCCCAAGGAC-3′, reverse: 5′-TGACAATACCTTCTGCAGGC-3′). The miRNA-30 primer was introduced in the above-mentioned content. The following amplification conditions were used: predenaturation at 95°C for 15 sec, denaturation at 94°C for 30 sec, annealing at 60°C for 20 sec and extension at 72°C for 40 sec for 40 cycles. The relative changes of mRNA levels were normalized to β-actin and the miRNA-30 relative change was normalized to U6 (18).
Luciferase reporter gene assay
The CTGF 3′-UTR was inserted into a luciferase expression plasmid (pMir-report), then transfected into COS1 cells. miR-30a and control miRNA were transfected into COS1 cells (ICLC ATL01002, ATCC, USA) respectively. Then maintained in DMEM supplemented with 10% FBS, and 2 mM L-Glutamine at 37°C in 5% CO2 for 24 h. The expression levels of luciferase were tested by 1X CCLR (cell lysis reagent) luciferase assay reagent. Besides, after knockdown of the binding site of the miR-30a and the 3′-UTR of CTGF, miR-30a and control miRNA were transfected into COS1 cells, respectively, then the expression levels of luciferase were tested.
Western blot analysis
Protein was extracted from heart tissue and COS1 cells, respectively. The concentration of protein was measured using the Bradford method. The expression of CTGF and β-actin in heart tissue was detected, while the expression of CTGF and GAPDH in COS1 cells were detected by western blotting. The extracted protein (50 g) was electrophoresed on 10% SDS polyacrylamide gels and then transferred to nitrocellulose membranes. After blocking for 2 h at 4°C in buffer (10% fat-free milk in TBS-T), the membrane was incubated overnight with anti-CTGF (1:1,000; Abcam, ab6992), anti-β-actin (1:4,000; KeyGen Biotech, Nanjing, China), and anti-GAPDH (1:1,000; Abbkine, Redlands, CA, USA, A01025), respectively. The membranes were washed and incubated for 2 h at 4°C and conjugated with respective horseradish peroxidase (HRP)-conjugated secondary antibodies (goat anti-mouse IgG 1:5,000 and goat anti-rat IgG 1:5,000; Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 2 h at room temperature. Detection was performed using enhanced chemiluminescence (Thermo Fisher Scientific, Frederick, MD, USA) and photography (19).
Statistical analysis
Data are expressed as mean values ± standard deviation. Differences among groups were analyzed by one-way ANOVA using SPSS 13.0 (SPSS Inc., Chicago, IL, USA). The Student-Newman-Keul's test was used for pairwise comparisons of data. P<0.05 was considered to indicate a statistically significant difference.
Results
Heart miR-30a level changes in experimental groups
Four weeks after MI surgery, we used RT-PCR to measure the levels of miR-30a in different groups. miR-30a levels were both obviously reduced in PBS group and miR-30a-NC group (P<0.01 versus sham group). After injected with rAAV9-miR-30a in rat cavities, miR-30a level was notably increased compared with other three groups (P<0.01), suggesting that miR-30a was successfully transfected into rat hearts (Fig. 1).
Figure 1.
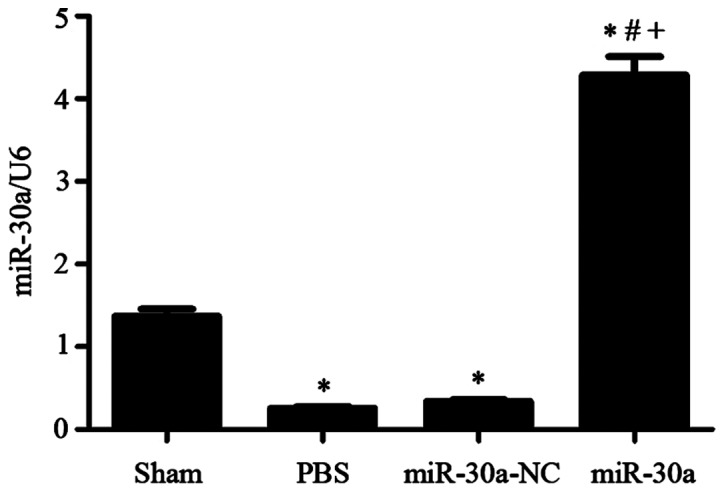
Heart miR-30a level changes in experimental groups. Sham, sham operation group n=15; PBS, MI group n=25; miR-30a-NC, blank AAV plasmid vector injected group n=22; miR-30a, rAAV9-miR-30a injected group n=26. *P<0.01 versus sham group. #P<0.01 versus PBS group. +P<0.01 versus miR-30a-NC group.
Effect of miR-30a on heart function in MI rats
Before these rats were sacrificed, we used echocardiography to determine heart diastolic and systolic function. LVESD and LVEDD, as indexes of diastolic function, were markedly increased in PBS group, miR-30a-NC group and miR-30a group (P<0.01 versus sham group). After injected with rAAV9-miR-30a, heart diastolic function was significantly improved (P<0.01 versus PBS group and miR-30a-NC group). Heart systolic function reflected by EF and FS was evidently decreased in PBS group, miR-30a-NC group and miR-30a group (P<0.01 versus sham group), and these indexes of miR-30a group were notably increased (P<0.01 versus PBS group and miR-30a-NC group). In conclusion, these results showed that miR-30a transfected into heart could significantly inhibit the changes of heart diastolic and systolic function in MI rats (Fig. 2).
Figure 2.

Effect of miR-30a on heart function in MI rats. (A) Sham, sham operation group n=15; (B) PBS, MI group n=25; (C) miR-30a-NC, blank AAV plasmid vector injected group n=22; (D) miR-30a, rAAV9-miR-30a injected group n=26. LVESD, left ventricular end-systolic dimension; LVEDD, left ventricular end-diastolic dimension; FS, fractional shortening; EF, ejection fraction.*P<0.01 versus sham group. #P<0.01 versus PBS group, +P<0.01 versus miR-30a-NC group.
Effect of miR-30a on collagen deposition in hearts of MI rats
The Masson's staining and immunohistochemical staining were performed to analyze the distribution of total collagen, collagen I and III. As a result, total collagen, collagen I and III were all obviously increased in PBS group and miR-30a-NC group and miR-30a group (P<0.01 versus sham group). However, they were all significantly ameliorated in miR-30a treatment group (P<0.01 versus PBS group and miR-30a-NC group). Altogether, these results indicated that miR-30a could attenuate cardiac fibrosis in MI rats (Fig. 3).
Figure 3.

Effect of miR-30a on collagen deposition in hearts of MI rats. (A and B) Masson's staining. (C and D) Collagen I, immunohistochemical stain. (E and F) Collagen III immunohistochemical stain. (A) a, sham, sham operation group n=15; b, PBS, MI group n=25; c, miR-30a-NC, blank AAV plasmid vector injected group n=22; d, miR-30a, rAAV9-miR-30a injected group n=26. *P<0.01 versus sham group. #P<0.01 versus PBS group. +P<0.01 versus miR-30a-NC group.
Effect of miR-30a on CTGF expression levels in hearts of MI rats
RT-PCR and western blotting were used to measure CTGF expression. In PBS group, miR-30a-NC group and miR-30a group, CTGF expression was markedly upregulated both at the transcriptional and translational level (P<0.01 versus sham group), but it was downregulated after treated with miR-30a, suggesting that miR-30a could ameliorate myocardial fibrosis through inhibition of CTGF expression in MI rats (Fig. 4).
Figure 4.

Effect of miR-30a on (A) CTGF mRNA and (B and C) protein expression in hearts of MI rats. Sham, sham operation group n=15; PBS, MI group n=25; miR-30a-NC, blank AAV plasmid vector injected group n=22; miR-30a, rAAV9-miR-30a injected group n=26. *P<0.01 versus sham group, #P<0.01 versus PBS group, +P<0.01 versus miR-30a-NC group.
CTGF is a direct target of miR-30a
The above suggests that CTGF is regulated by miR-30a (8). Deletion of the miRNA binding site abrogated the repressive effect of miR-30a on luciferase activity, which indicated that the expression of CTGF was extremely sensitive to the miR-binding sequence within the 3′-UTR. miR-30a also dose-dependently inhibited the synthesis of Flag-tagged MEF2C expression cassette which linked to the CTGF 3′-UTR binding sequence. However, there was no significant difference in the mutant CTGF 3′-UTR group with the miR-30a target site deleted (Fig. 5).
Figure 5.

CTGF is a direct target of miR-30a. (A) The 3′-UTR of CTGF: Human, Mouse, Rat; miR-30; (B) CTGF-3′-UTR: miR-30a n=13, control miRNA n=12; mutant 3′-UTR: miR-30a n=11, control miRNA n=9. (C) Western blotting with Flag-CTGF-3′-UTR.
Discussion
In this study, we found that miR-30a could improve cardiac dysfunction and myocardial fibrosis in rats with MI by inhibiting CTGF expression, which was previously unreported. AAV9 is proven to be the strong cardiotropic serotype in mouse and rat as compared to AAV1, 6, 7 and 8. AAV9. The transcoronary gene delivery is a more high-efficiency method than gene delivery by tail vein injection. Thus, we used AAV9 to transfer miR-30a to the heart by this method in the investigation. Four weeks after the operation, we found that the level of heart miR-30a after injected with rAAV9-miR-30a was obviously increased, indicating that heart miR-30a transfection was successful.
Yin et al (11) reported that miR-30a levels are reduced in transverse aortic constriction mice, and downregulated miR-30a aggravates phenylephrine-induced cardiomyocyte hypertrophy by activating autophagy in H9c2 cells. Liu and his colleges revealed that miR-30a could attenuate the Angiotensin II-induced upregulation of hypertrophy-related genes and decrease the cardiomyocyte surface area via targeting beclin 1 (20). Cardioprotective effect of triiodothyronine was associated with restored levels of miR-30a expression that correspond to p53 mRNA downregulation in rats with cardiac ischemia/reperfusion (9). miR-30a levels in this study were significantly reduced 4 weeks after MI surgery, and heart function and cardiac fibrosis were severely deteriorated, which was reversed by treatment with miR-30a. Besides, the expression level of serum miR-30a was closely correlated with the cardiac function, therefore it could be as a biomarker for indicating heart failure (21). Therefore, maintenance of miR-30a level may be associated with cardioprotection.
CTGF, a mitogen secreted by vascular endothelial cells, acts as integrins, proteoglycans, or tyrosine kinases (22). In normal physiological environment, CTGF is mainly involved in angiogenesis and cell differentiation. It is worth noting that CTGF could mediate the process of tissue repair and fibrosis under pathological conditions (23). Many experiments in vitro showed that CTGF could stimulate the proliferation of cardiac fibroblasts and increase the production of extracellular matrix (24). In the process of myocardial injury, repair and fibrosis, CTGF is an obvious activation sign for fibroblasts. Though the pro-fibrogenic effect of CTGF was weak, it provided a beneficial environment for other fibrotic stimuli (25,26). In our study, we measured heart CTGF expression in experimental groups. CTGF expression was markedly increased both at the transcriptional and translational level, which was decreased by miR-30 injection, revealing that miR-30 may also improve cardiac fibrosis. Related studies found that miR-30 could directly influence CTGF levels (27,28). In our study, luciferase expression in COS1 cells decreased significantly, after knockdown of miR-30a and CTGF binding sites. This indicated that the miR-30a regulated protein levels of CTGF combined with 3′-UTR directly, revealing that miR-30 may also improve cardiac fibrosis via binding 3-UTR of CTGF. Similarly to a previous study, miR-30 through specific binding to its 3-UTR influences CTGF levels (29).
In summary, miR-30a may inhibit the expression of CTGF in myocardium after MI by directly combining with the 3′-UTR of CTGF in myocardium after MI, thereby reducing the production of collagen in myocardia, inhibiting myocardial fibrosis, then improving cardiac functions.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
LC and NT contributed to the conception and design of the study. LC drafted the manuscript. QJ performed the data analyses. HZ and YR performed Masson's staining and immunohistochemical analysis. ZF performed and analyzed real-time PCR. NT reviewed the manuscript and gave his approval to the submitted and final versions. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The study was approved by the Ethics Committee of Nanjing First Hospital (Nanjing, China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123:37–45. doi: 10.1172/JCI62839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen C, Li R, Ross RS, Manso AM. Integrins and integrin-related proteins in cardiac fibrosis. J Mol Cell Cardiol. 2016;93:162–174. doi: 10.1016/j.yjmcc.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koshman YE, Patel N, Chu M, Iyengar R, Kim T, Ersahin C, Lewis W, Heroux A, Samarel AM. Regulation of connective tissue growth factor gene expression and fibrosis in human heart failure. J Card Fail. 2013;19:283–294. doi: 10.1016/j.cardfail.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Creemers EE, van Rooij E. Function and therapeutic potential of non-coding RNAs in cardiac fibrosis. Circ Res. 2016;118:108–118. doi: 10.1161/CIRCRESAHA.115.305242. [DOI] [PubMed] [Google Scholar]
- 5.Li M, Wang N, Zhang J, He HP, Gong HQ, Zhang R, Song TF, Zhang LN, Guo ZX, Cao DS, et al. MicroRNA-29a-3p attenuates ET-1-induced hypertrophic responses in H9c2 cardiomyocytes. Gene. 2016;585:44–50. doi: 10.1016/j.gene.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Wang R, Du W, Wang S, Yang L, Pan Z, Li X, Xiong X, He H, Shi Y, et al. Downregulation of miR-151-5p contributes to increased susceptibility to arrhythmogenesis during myocardial infarction with estrogen deprivation. PLoS One. 2013;8:e72985. doi: 10.1371/journal.pone.0072985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen LW, Zhu LL, Ji Q, Zhu H, Ren YZ, Fan ZG, Li XB, Gao XF, Zhang YJ, Tian NL. Relationship between myocardial microRNA-30a expression and myocardial fibrosis in rats post myocardial infarction. Zhonghua Xin Xue Guan Bing Za Zhi. 2016;44:443–449. doi: 10.3760/cma.j.issn.0253-3758.2016.05.015. (In Chinese) [DOI] [PubMed] [Google Scholar]
- 8.Yuan CT, Li XX, Cheng QJ, Wang YH, Wang JH, Liu CL. MiR-30a regulates the atrial fibrillation-induced myocardial fibrosis by targeting snail 1. Int J Clin Exp Pathol. 2015;8:15527–15536. [PMC free article] [PubMed] [Google Scholar]
- 9.Forini F, Kusmic C, Nicolini G, Mariani L, Zucchi R, Matteucci M, Iervasi G, Pitto L. Triiodothyronine prevents cardiac ischemia/reperfusion mitochondrial impairment and cell loss by regulating miR30a/p53 axis. Endocrinology. 2014;155:4581–4590. doi: 10.1210/en.2014-1106. [DOI] [PubMed] [Google Scholar]
- 10.Gutiérrez-Escolano A, Santacruz-Vázquez E, Gómez-Pérez F. Dysregulated microRNAs involved in contrast-induced acute kidney injury in rat and human. Ren Fail. 2015;37:1498–1506. doi: 10.3109/0886022X.2015.1077322. [DOI] [PubMed] [Google Scholar]
- 11.Yin X, Peng C, Ning W, Li C, Ren Z, Zhang J, Gao H, Zhao K. miR-30a downregulation aggravates pressure overload-induced cardiomyocyte hypertrophy. Mol Cell Biochem. 2013;379:1–6. doi: 10.1007/s11010-012-1552-z. [DOI] [PubMed] [Google Scholar]
- 12.Gao X, Ma YT, Yang YN, Xiang Y, Chen BD, Liu F, Du L. Recombinant adeno-associated virus serotype 9 transfection of rats H9C2 cells in vitro. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2010;26:18–20. (In Chinese) [PubMed] [Google Scholar]
- 13.Bish LT, Morine K, Sleeper MM, Sanmiguel J, Wu D, Gao G, Wilson JM, Sweeney HL. Adeno-associated virus (AAV) serotype 9 provides global cardiac gene transfer superior to AAV1, AAV6, AAV7, and AAV8 in the mouse and rat. Hum Gene Ther. 2008;19:1359–1368. doi: 10.1089/hum.2008.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalisz M, Baranowska B, Wolinska-Witort E, Maczewski M, Mackiewicz U, Tulacz D, Gora M, Martynska L, Bik W. Total and high molecular weight adiponectin levels in the rat model of post-myocardial infarction heart failure. J Physiol Pharmacol. 2015;66:673–680. [PubMed] [Google Scholar]
- 15.Bagno LL, Carvalho D, Mesquita F, Louzada RA, Andrade B, Kasai-Brunswick TH, Lago VM, Suhet G, Cipitelli D, Werneck-de-Castro JP, et al. Sustained IGF-1 secretion by adipose-derived stem cells improves infarcted heart function. Cell Transplant. 2016;25:1609–1622. doi: 10.3727/096368915X690215. [DOI] [PubMed] [Google Scholar]
- 16.Chen J, Huang C, Zhang B, Huang Q, Chen J, Xu L. The effects of carvedilol on cardiac structural remodeling: The role of endogenous nitric oxide in the activity of carvedilol. Mol Med Rep. 2013;7:1155–1158. doi: 10.3892/mmr.2013.1329. [DOI] [PubMed] [Google Scholar]
- 17.Abdel-Hamid AAM, Firgany A-D. Atorvastatin alleviates experimental diabetic cardiomyopathy by suppressing apoptosis and oxidative stress. J Mol Histol. 2015;46:337–345. doi: 10.1007/s10735-015-9625-4. [DOI] [PubMed] [Google Scholar]
- 18.Parimala M, Debjani M, Vasanthi HR, Shoba FG. Nymphaea nouchali Burm. f. hydroalcoholic seed extract increases glucose consumption in 3T3-L1 adipocytes through activation of peroxisome proliferator-activated receptor gamma and insulin sensitization. J Adv Pharm Technol Res. 2015;6:183–189. doi: 10.4103/2231-4040.165013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wojcik B, Harasim E, Zabielski P, Chabowski A, Gorski J. Effect of tachycardia on lipid metabolism and expression of fatty acid transporters in heart ventricles of the rat. J Physiol Pharmacol. 2015;66:691–699. [PubMed] [Google Scholar]
- 20.Pan W, Zhong Y, Cheng C, Liu B, Wang L, Li A, Xiong L, Liu S. MiR-30-regulated autophagy mediates angiotensin II-induced myocardial hypertrophy. PLoS One. 2013;8:e53950. doi: 10.1371/journal.pone.0053950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xia X. The relationship of serum miR-30a and heart failure in children with congenital heart disease. http://www.jcp-sh.org.cn/EN/abstract/abstract8665.shtml J Clin Pediatr. 2014;32:607–609. [Google Scholar]
- 22.Leask A, Abraham DJ. All in the CCN family: Essential matricellular signaling modulators emerge from the bunker. J Cell Sci. 2006;119:4803–4810. doi: 10.1242/jcs.03270. [DOI] [PubMed] [Google Scholar]
- 23.Shimo T, Nakanishi T, Nishida T, Asano M, Kanyama M, Kuboki T, Tamatani T, Tezuka K, Takemura M, Matsumura T, et al. Connective tissue growth factor induces the proliferation, migration, and tube formation of vascular endothelial cells in vitro, and angiogenesis in vivo. J Biochem. 1999;126:137–145. doi: 10.1093/oxfordjournals.jbchem.a022414. [DOI] [PubMed] [Google Scholar]
- 24.Wang B, Haldar SM, Lu Y, Ibrahim OA, Fisch S, Gray S, Leask A, Jain MK. The Kruppel-like factor KLF15 inhibits connective tissue growth factor (CTGF) expression in cardiac fibroblasts. J Mol Cell Cardiol. 2008;45:193–197. doi: 10.1016/j.yjmcc.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hayata N, Fujio Y, Yamamoto Y, Iwakura T, Obana M, Takai M, Mohri T, Nonen S, Maeda M, Azuma J. Connective tissue growth factor induces cardiac hypertrophy through Akt signaling. Biochem Biophys Res Commun. 2008;370:274–278. doi: 10.1016/j.bbrc.2008.03.100. [DOI] [PubMed] [Google Scholar]
- 26.Bonniaud P, Martin G, Margetts PJ, Ask K, Robertson J, Gauldie J, Kolb M. Connective tissue growth factor is crucial to inducing a profibrotic environment in ‘fibrosis-resistant’ BALB/c mouse lungs. Am J Respir Cell Mol Biol. 2004;31:510–516. doi: 10.1165/rcmb.2004-0158OC. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Duan L, Guo T, Gao Y, Tian L, Liu J, Wang S, Yang J. Downregulation of miR-30c promotes renal fibrosis by target CTGF in diabetic nephropathy. J Diabetes Complications. 2016;30:406–414. doi: 10.1016/j.jdiacomp.2015.12.011. [DOI] [PubMed] [Google Scholar]
- 28.Su Z, Wei G, Wei L, Liu J, Li X. Effects of rhBNP on myocardial fibrosis after myocardial infarction in rats. Int J Clin Exp Pathol. 2015;8:6407–6415. [PMC free article] [PubMed] [Google Scholar]
- 29.Duisters RF, Tijsen AJ, Schroen B, Leenders JJ, Lentink V, van der Made I, Herias V, van Leeuwen RE, Schellings MW, Barenbrug P, et al. miR-133 and miR-30 regulate connective tissue growth factor: Implications for a role of microRNAs in myocardial matrix remodeling. Circ Res. 2009;104:170–178. doi: 10.1161/CIRCRESAHA.108.182535. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.