

Abstract
Over the last decade, an increasing proportion of circulating human influenza A(H3N2) viruses exhibited haemagglutination activity that was sensitive to neuraminidase inhibitors. This change in haemagglutination as compared to older circulating A(H3N2) viruses prompted an investigation of the underlying molecular basis. Recent human influenza A(H3N2) viruses were found to agglutinate turkey erythrocytes in a manner that could be blocked with either oseltamivir or neuraminidase-specific antisera, indicating that agglutination was driven by neuraminidase, with a low or negligible contribution of haemagglutinin. Using representative virus recombinants it was shown that the haemagglutinin of a recent A(H3N2) virus indeed had decreased activity to agglutinate turkey erythrocytes, while its neuraminidase displayed increased haemagglutinating activity. Viruses with chimeric and mutant neuraminidases were used to identify the amino acid substitution histidine to arginine at position 150 flanking the neuraminidase catalytic site as the determinant of this neuraminidase-mediated haemagglutination. An analysis of publicly available neuraminidase gene sequences showed that viruses with histidine at position 150 were rapidly replaced by viruses with arginine at this position between 2005 and 2008, in agreement with the phenotypic data. As a consequence of neuraminidase-mediated haemagglutination of recent A(H3N2) viruses and poor haemagglutination via haemagglutinin, haemagglutination inhibition assays with A(H3N2) antisera are no longer useful to characterize the antigenic properties of the haemagglutinin of these viruses for vaccine strain selection purposes. Continuous monitoring of the evolution of these viruses and potential consequences for vaccine strain selection remains important.
Keywords: Influenza A(H3N2) virus, neuraminidase, haemagglutination, oseltamivir, balance, inhibition
Introduction
Influenza A viruses are responsible for the majority of seasonal influenza virus infections in humans [1], with A(H3N2) and A(H1N1pdm09) being the current predominant virus subtypes. During influenza epidemics, severe illness occurs in up to five million people worldwide each year, with an estimated 500 000 deaths [2–4]. The almost continuous accumulation of amino acid changes in the surface glycoprotein haemagglutinin (HA) and neuraminidase (NA) of seasonal influenza viruses facilitates viral escape from antibody-mediated immunity of the human host induced by prior infections or vaccinations. This antigenic evolution facilitates multiple reinfections of the same individual during a lifetime and makes a biannual review process necessary to keep the seasonal influenza vaccine components up to date [5].
Influenza virus particles contain two major transmembrane glycoproteins on the surface: HA and NA. Both HA and NA interact with host cells during infection, but with opposing activities. The globular head of the HA molecule contains a receptor-binding site that interacts with N-acetylneuraminic acid (sialic acid, SA), the virus receptor on host cells, to initiate infection [6, 7]. Binding of HA to SAs on erythrocytes can result in haemagglutination, i.e. clumping of red blood cells, the process from which HA obtained its name. In contrast, the viral NA protein has enzymatic activity to cleave SAs from host cells and from otherwise self-aggregating viral progeny, ensuring efficient release of virus particles during the budding process [7, 8]. A functional balance between the activities of HA and NA is important because of their opposing roles during influenza virus replication. Viruses with reduced or no NA activity can accumulate compensatory substitutions in the HA gene upon serial passage that result in a lower affinity of HA to cellular receptors [7–10]. The HA–NA balance does not necessarily only relate to the efficiency of binding and cleavage, it can also relate to substrate specificity [11]. HA and NA derived from avian influenza viruses bind SAs attached via α-2,3 linkage to the terminal galactose of glycans (α-2,3SA), while the HA of human viruses has a preference for α-2,6-linked sialic acids (α-2,6SA) [12]. The NA activity of human influenza viruses retains a preference for α-2,3SA, potentially to allow cleavage of α-2,3SA present in the mucus of the human respiratory tract [13].
Changes in HA affinity to erythrocytes from different species have been observed occasionally since influenza A(H3N2) viruses have been circulating in the human population [14–17]. Since the early 2000s, several groups have observed changes in the haemagglutination behaviour of influenza A(H3N2) viruses, i.e. reduced haemagglutination of commonly used erythrocytes (turkey, chicken and guinea pig erythrocytes) and poor inhibition of haemagglutination by reference post-infection ferret antisera [11, 18–22]. In addition to the changes in the haemagglutination behaviour of HA, the NA activity of influenza A(H3N2) viruses isolated between 2005 and 2009 was also affected [18]. The amino acid substitution aspartic acid to glycine at position 151 (151DG) in NA was shown to cause binding of NA to erythrocytes, which could be reversed by addition of the NA- inhibitor oseltamivir in the haemagglutination assay [18]. However, the vast majority of recent A(H3N2) viruses with similar changes in haemagglutination behaviour did not have 151DG in NA. Here, we describe another amino acid substitution, histidine to arginine at position 150 (150HR), flanking the catalytic site of NA, that confers haemagglutination activity upon recent A(H3N2) viruses via NA.
Results
Changes in the haemagglutination activity of A(H3N2) virus isolates from 2000 to 2016
Driven by our own observations and previous reports [18–20], the haemagglutination activity of 83 influenza A(H3N2) viruses isolated in MDCK cells from 2000 until 2016 was investigated (Table S1, available in the online Supplementary Material), from which a representative subset is shown in Table 1. Agglutination titres were measured with turkey and human type O erythrocytes in the presence or absence of the NA-inhibitor oseltamivir. All viruses isolated from 2000 to 2003 had normal haemagglutination activity and none displayed a substantial difference in titres in the presence or absence of oseltamivir. In 2004 and 2005, some virus isolates displayed low agglutination titres with turkey erythrocytes. From 2006 onwards, viruses were isolated that lost their ability to agglutinate turkey erythrocytes in the presence of oseltamivir, suggesting that haemagglutination was (partially) dependent on NA. In subsequent years, the proportion of virus isolates that did not agglutinate turkey erythrocytes in the presence of oseltamivir increased, approaching 100 % by 2009. Moreover, around this time an increasing proportion of virus isolates also lost the ability to agglutinate human type O erythrocytes in the presence of oseltamivir. From 2012 onwards, the vast majority of virus isolates showed a loss of agglutination of turkey erythrocytes in the presence of oseltamivir or did not agglutinate turkey erythrocytes at all.
Table 1. Haemagglutination titres for representative virus isolates from 2000 to 2016 using two erythrocyte sources, in the absence or presence of oseltamivir.
| Virus isolate* | Haemagglutination titre (HAU per 25 µl) | |||
|---|---|---|---|---|
| Turkey erythrocytes | Human type O erythrocytes | |||
| Oseltamivir | Oseltamivir | |||
| − | + | − | + | |
| A/NL/056/00 | 32 | 16 | 32 | 32 |
| A/NL/118/01 | 24 | 24 | 32 | 32 |
| A/NL/001/02 | 64 | 64 | 48 | 48 |
| A/NL/109/03 | 64 | 64 | >64 | >64 |
| A/NL/023/04 | 32 | 24 | 64 | 64 |
| A/NL/093/05 | 48 | 48 | 64 | 64 |
| A/NL/020/06 | 64 | <2 | 64 | 24 |
| A/NL/198/06 | 64 | 64 | 64 | 64 |
| A/NL/004/07 | 48 | <2 | >64 | 32 |
| A/NL/038/07 | 64 | 64 | 64 | 48 |
| A/NL/379/08 | >64 | <2 | 64 | 4 |
| A/NL/761/09 | 64 | <2 | 64 | 8 |
| A/NL/034/10 | 48 | <2 | 64 | 16 |
| A/NL/713/11 | 64 | <2 | 64 | 2 |
| A/NL/003/12 | 4 | <2 | 8 | 2 |
| A/NL/075/12 | 32 | <2 | 48 | <2 |
| A/NL/2249/13 | 24 | <2 | nt | nt |
| A/NL/252/14 | 32 | <2 | nt | nt |
| A/NL/584/15 | 32 | <2 | nt | nt |
| A/NL/354/16 | 8 | 2 | nt | nt |
nt, Not tested.
*Representative strains for each year.
Effect of neuraminidase in haemagglutination-inhibition assays
The oseltamivir-mediated inhibition of the haemagglutination of influenza viruses indicated a potential role for NA in the haemagglutination process. If true, this would pose problems for haemagglutination inhibition (HI) assays and complicate the antigenic characterization of the HA of circulating viruses for the purpose of influenza vaccine strain selection. A pair of virus isolates was used to investigate the role of NA in HI assays, one with normal haemagglutination activity (A/NL/109/03) and one that lost haemagglutination activity in the presence of oseltamivir (A/NL/761/09). We used post-infection ferret antisera raised against A(H3N2) virus isolates from the years 2003 and 2009, and ferret antisera raised against A(H7N2) recombinant viruses that contained 2003 and 2009 NA genes. The A(H7N2) post-infection antisera were raised to test the influence of NA-specific antibodies independently of HA-specific antibodies [23]. Haemagglutination by influenza virus A/NL/109/03 was inhibited by both A(H3N2) post-infection ferret antisera, but not by the A(H7N2) sera, which represents a normal HA-dependent HI pattern (Table 2). Haemagglutination by A/NL/761/09 was inhibited poorly by the A(H3N2)-specific antisera and displayed similar HI antibody titres upon incubation with A(H7N2)-specific antisera. Thus, for A/NL/761/09 this HI assay measured the inhibition of NA-mediated rather than HA-mediated haemagglutination, consistent with a dominant role for NA in the agglutination of turkey erythrocytes for A/NL/761/09. The findings that haemagglutination was blocked by oseltamivir and by NA-specific ferret antisera indicate that haemagglutination was mediated by NA and not by HA.
Table 2. Haemagglutination inhibition titres with ferret antisera in the absence of oseltamivir.
| Virus | Post-infection ferret sera raised against | ||||
|---|---|---|---|---|---|
| A/NL/109/03 (H3N2) |
A/NL/282/09 (H3N2) |
rH7NA03* (H7N2) |
rH7NA09* (H7N2) |
||
| A/NL/109/03 (H3N2) | H3N2 | 2560 | 960 | <10 | <10 |
| A/NL/761/09 (H3N2) | H3N2 | 20 | 160 | 30 | 160 |
*Recombinant H7 virus with NA from 2003 or 2009 viruses [23].
Decreased haemagglutination activity via HA and increased haemagglutination activity via NA of a 2009 A(H3N2) virus
Recombinant viruses with combinations of HA and NA of A/NL/109/03 (2003 HA and 2003 NA) and A/NL/761/09 (2009 HA and 2009 NA) were generated to assess the contribution of HA and NA in the agglutination of turkey erythrocytes. Recombinant viruses with the wild-type HA–NA combination of A/NL/109/03 or A/NL/761/09 on a A/Puerto Rico/8/34 genetic background showed the same haemagglutination patterns as the natural virus isolates, i.e. a recombinant virus carrying the 2003 HA and NA did not display a loss of haemagglutination upon addition of oseltamivir, while the haemagglutination titre of a recombinant virus carrying the 2009 HA and NA dropped from 32 to below 2 (Fig. 1). When the HA of the 2003 virus was combined with the NA of the 2009 virus, the haemagglutination titre was not affected by oseltamivir. In contrast, a recombinant virus carrying the HA of the 2009 virus and the NA of the 2003 virus did not show any haemagglutination in the absence or presence of oseltamivir. These results indicated that two separable phenotypic changes occurred in the 2009 A(H3N2) virus as compared to the 2003 virus; HA lost its ability to agglutinate turkey erythrocytes efficiently and NA gained the ability to bind to turkey erythrocytes. To assess the specificity of said NA binding to red blood cells, we used turkey erythrocytes that were reconstituted to contain either α-2,3SA or α-2,6SA alone, and found that haemagglutination by viruses with the 2009 NA occurred primarily via α-2,6SA (Table S2).
Fig. 1.
Haemagglutination titres of recombinant A(H3N2) viruses with various combinations of 2003 and 2009 HA and NA genes, in the presence (+) or absence (−) of oseltamivir.
NA binding to turkey erythrocytes is due to amino acid substitution 150HR next to the NA catalytic site
Two chimeric NAs were designed to identify which molecular changes in NA were responsible for the ability to agglutinate turkey erythrocytes, thereby splitting the seven amino acid differences observed between the 2003 and 2009 NA (150HR, 194VI, 215IV, 310HY, 370SL, 372LS and 387KN) over two chimeras (Fig. 2). The chimeric NAs were used to generate recombinant viruses on an A/Puerto Rico/8/1934 genetic background with either the 2003 or the 2009 HA gene. Regardless of the chimeric NA used, the recombinant virus with the 2003 HA displayed normal haemagglutination, which was not affected by the addition of oseltamivir (Table 3). Unfortunately, we were not able to rescue a recombinant virus with one of the chimeric NAs (NA-1) and the HA of the 2009 virus isolate. However, the other chimeric NA (NA-2), along with the 2009 HA, yielded a viable recombinant virus with haemagglutination activity that was sensitive to the addition of oseltamivir, suggesting that the NA-dependent agglutination of turkey erythrocytes was determined by one to three amino acid substitutions in the N-terminal half of NA (150HR, 194VI and 215IV). These three amino acid substitutions were then introduced individually in the 2003 NA that naturally lacked NA-mediated agglutination activity and recombinant viruses with the HA of the 2009 virus were generated. Recombinant viruses with the wild-type 2003 NA or the 2003 NA with substitutions 194VI or 215IV failed to agglutinate turkey erythrocytes. In contrast, the recombinant virus with substitution 150HR in 2003 NA was able to agglutinate turkey erythrocytes, and this haemagglutination was sensitive to the addition of oseltamivir (Table 3). Thus, we conclude that the 150HR substitution was the major determinant of NA-mediated haemagglutination of the 2009 A(H3N2) virus. The 2003 NA with the 150HR substitution was binding via α-2,6SA and also via α-2,3SA (Table S2).
Fig. 2.

Schematic representation of the cDNA of chimeric NAs based on the NA of A/NL/109/03 and A/NL/761/09. The seven amino acid differences between the two NA proteins are shown, as well as the EcoRV restriction site that was used to construct the chimeras. NCR, non-coding region.
Table 3. Agglutination of turkey erythrocytes by recombinant viruses with chimeric and mutant NA genes in the presence or absence of oseltamivir.
| Recombinant virus | Haemagglutination titre (HAU per 25 µl) | ||
|---|---|---|---|
| HA | NA | − Oseltamivir | + Oseltamivir |
| A/NL/109/03 | NA-1 | 64 | 64 |
| A/NL/109/03 | NA-2 | 32 | 32 |
| A/NL/761/09 | NA-1 | Not rescued | |
| A/NL/761/09 | NA-2 | 32 | <2 |
| A/NL/761/09 | A/NL/109/03 | <2 | <2 |
| A/NL/761/09 | A/NL/109/03–150HR | 16 | <2 |
| A/NL/761/09 | A/NL/109/03-194VI | <2 | <2 |
| A/NL/761/09 | A/NL/109/03-215IV | <2 | <2 |
Amino acid substitution 150HR in NA became dominant in recent A(H3N2) viruses
The occurrence of the amino acids histidine and arginine at position 150 of NA was investigated using all of the available full-length NA gene segment sequences of human A(H3N2) full-virus genomes from 2000 to 2016 (Fig. 3). From 2000 to 2005, the dominant amino acid at position 150 in NA was histidine. In 2006 and 2007, either histidine or arginine was found at position 150 in NA. From 2008 onwards, arginine became the dominant amino acid at position 150. This occurrence of amino acid substitution 150HR in NA coincided temporally with the functional changes in haemagglutination behaviour observed in A(H3N2) viruses (Tables 1 and S1). Of the 5942 NA sequences investigated, 1687 sequences were generated by the direct sequencing of clinical samples and 900 sequences were generated upon virus isolation in MDCK cells, but this history had no effect on the proportion of histidine and arginine at position 150 in the dataset (data not shown), indicating that substitution 150HR was not the result of passaging viruses in cell cultures.
Fig. 3.
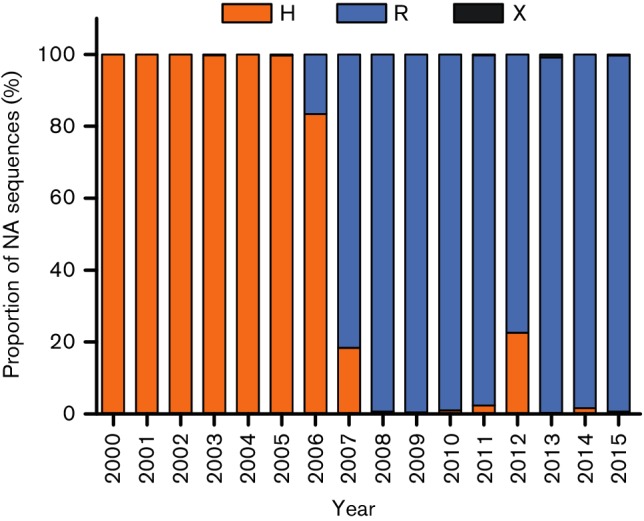
Proportion of human A(H3N2) viruses with substitutions at amino acid position 150 in NA, from 2000 until 2015. A total of 5942 NA sequences available from full genomes, including 900 from MDCK-passaged viruses and 1687 derived from clinical specimens, were analysed. No full-genome sequences were available for human A(H3N2) viruses from 2016. NA genes with histidine (H), arginine (R) or any other amino acid (X) at position 150 are shown in orange, blue and grey respectively.
Discussion
Here it is shown that an increasing proportion of recent influenza A(H3N2) viruses agglutinated commonly used erythrocytes in a manner that can be blocked by the NA-inhibitor oseltamivir or NA-specific antibodies. This unusual haemagglutination behaviour was the result of two phenomena: a decrease in haemagglutination activity by HA and the acquisition of haemagglutination activity by NA. Amino acid substitution 150HR in NA was found to be responsible for the agglutination of turkey and human type O erythrocytes and this substitution has recently become dominant in A(H3N2) viruses. Fluctuations of the haemagglutination activity of HA have been observed in the past, due to amino acid changes in the HA of influenza A(H3N2) and A(H1N1) viruses that led to the inability to agglutinate chicken erythrocytes [14–16]. Because erythrocytes of different animal species contain different oligosaccharide structures on their surface, changes in the receptor specificity of HA of influenza viruses have been shown to correlate with differences in haemagglutination behaviour [17]. However, changes in haemagglutination behaviour do not necessarily affect the ability of influenza viruses to bind to host cells during natural infection, as the nature and density of the oligosaccharides that are present on the surface of erythrocytes do not necessarily reflect those of the glycans present on host cells and tissues. In addition, if changes in HA were to diminish the role of HA in virus attachment, influenza viruses would still have to rely on HA during virus infection because of its role in the fusion of the viral and endosomal membranes [6].
Amino acid substitution 150HR in NA, which was identified to allow the NA-mediated agglutination of erythrocytes that can be blocked by oseltamivir and NA-specific antibodies, is adjacent to amino acid 151, a position at which another substitution (151DG) has been previously described to have a similar effect in influenza A(H3N2) viruses [18]. Amino acid positions 150 and 151 are located in the 150-loop (residues 147–152) at the edge of the catalytic site [24, 25]. However, residue 151, unlike residue 150, is categorized as a catalytic site residue and is believed to stabilize the transition state intermediate [26]. The position of these residues in or close to the catalytic site and the inhibition of NA-mediated binding by oseltamivir suggest that the binding of NA to turkey erythrocytes is mediated via the catalytic site and not via haemadsorption sites, which are secondary sialic acid-binding sites that have been described for some avian NAs [27–31]. At the moment it is not clear what mechanism is involved in the 150HR-mediated binding of erythrocytes and what functional changes are caused in NA by this amino acid substitution. Amino acid substitution 151DG in NA has been shown to decrease NA activity, while at the same time increasing NA affinity to α-2,3SA [18, 20]. Interestingly, another amino acid substitution located in the 150-loop of NA, 147GR, has been described to mediate subtype N1 NA binding to erythrocytes [32]. Amino acid substitution 147GR did not dramatically influence NA enzymatic kinetics. However, in contrast to 151DG, the effect of 147GR on NA properties was only assessed with the surrogate substrate MUNANA. Nevertheless, it is important to note that the canonical function of NA was not affected by substitutions 151DG and 147GR, showing that these NAs still function as sialidases.
The in vivo biological significance of the amino acid substitutions in NA that have led to functional changes in the in vitro haemagglutination behaviour of influenza viruses remains unclear. However, it is important to note that influenza A(H3N2) viruses continue to cause epidemics in humans, with apparently no or little impact from these in vitro properties. While amino acid substitution 147GR in NA emerged in a recombinant laboratory strain and was only found in a small number of natural virus isolates [32], various amino acids are found at position 151 of NA in different influenza A(H3N2) viruses [33]. However, it was found that most substitutions from 151D to different amino acids were acquired upon propagation of viruses in the laboratory [18, 33–36] and that the majority of clinical samples that were not passaged in culture possessed 151D [33, 36]. Here, we investigated the occurrence of the amino acids histidine and arginine at NA position 150 in all publicly available full-genome sequences of human A(H3N2) influenza viruses since the year 2000 and found an almost complete shift from histidine to arginine over the years, regardless of whether the sequences were derived from clinical specimens or passaged viruses.
As a consequence of the loss of haemagglutination activity via HA and the increase in haemagglutination activity via NA, the antigenic characterization of HA proteins of recent influenza A(H3N2) viruses using HI assays for the biannual selection of influenza vaccine strains has become problematic [5]. In fact, for recent A(H3N2) viruses, the HI assay appears to predominantly measure the effect of antibodies against NA (Table 2). It is therefore important to assess whether the viruses to be used in HI assays display sensitivity to oseltamivir in haemagglutination assays. If haemagglutination is found to be sensitive to inhibition by oseltamivir, virus (micro)neutralization assays could potentially be an alternative method for the antigenic characterization of influenza viruses [37, 38]. However, virus (micro)neutralization assays would only be an alternative if changes in the NA of recent H3N2 viruses did not mediate binding to cells and/or if NA neutralizing antibodies did not prevent this binding. Therefore, further investigations are needed to monitor the evolution of currently circulating H3N2 viruses and to improve our understanding of the potential consequences for the vaccine strain selection process.
Methods
Viruses
Influenza A(H3N2) viruses from 2000 until 2016 were isolated from clinical specimens in Madin–Darby canine kidney (MDCK) cells in the context of the Dutch national influenza virus surveillance programme. Virus stocks were harvested once cytopathic effects due to virus infection were observed in nearly 100 % of the cells. Virus stocks were frozen at −80 °C. The full-length HA and NA genes of two of the influenza A viruses, A/Netherlands/109/2003 (A/NL/109/2003, H3N2) and A/Netherlands/761/2009 (A/NL/761/2009, H3N2), were cloned in a modified pHW2000 expression plasmid [39]. Single or multiple amino acid substitutions were introduced using a QuickChange site-directed mutagenesis kit (Stratagene), according to the manufacturer's instructions. Recombinant viruses with various HA–NA combinations, as well as chimeric and mutant NA genes, were generated in the context of all other gene segments of influenza virus A/Puerto Rico/8/1934 using 293T cell transfection as previously described [40]. Virus stocks were passaged in MDCK cells up to three times. The HA and NA gene segments of all recombinant viruses were sequenced to verify the genetic modifications.
Cells
MDCK cells were grown at 37 °C and 5 % CO2 in Eagle's minimal essential medium (EMEM; Lonza) supplemented with 10 % foetal calf serum (FCS; Sigma), 100 IU ml−1 penicillin (Lonza), 100 µg ml−1 streptomycin (Lonza), 2 mM glutamine (Lonza), 10 mM HEPES buffer (Lonza), 1.5 mg ml−1 sodium bicarbonate (Lonza) and 0.1 mM non-essential amino acids (Lonza). Human embryonic kidney 293T cells were grown at 37 °C and 5 % CO2 in Dulbecco's modified Eagle medium (DMEM; Lonza) supplemented with 10 % FCS, 100 IU ml−1 penicillin, 100 mg ml−1 streptomycin, 2 mM glutamine, 1 mM sodium pyruvate (Gibco) and 0.1 mM non-essential amino acids.
Haemagglutination assay
To test haemagglutination, 50 µl virus stock was diluted using serial two fold dilutions in PBS. PBS (25 µl), with or without 20 nM oseltamivir (Roche), was added and incubated for 5 min at room temperature. Next, 25 µl of 1 % turkey or human type O erythrocytes in PBS was added and the cell and virus mixture was incubated for 1 h at 4 °C before the haemagglutination titres were read.
Sequencing of HA and NA gene segments
RNA of 200 µl virus-containing sample was isolated using a High Pure RNA isolation kit (Roche) according to the manufacturer's instructions. Complementary DNA (cDNA) synthesis was carried out with 22 µl viral RNA, non-coding region (NCR) primers and SuperScriptIII reverse transcriptase (Invitrogen). Subsequently, the HA or NA segment of influenza viruses was amplified with segment-specific primers. Sanger sequencing was performed on a 3130xl genetic analyser (Applied Biosystems, Hitachi).
Haemagglutination inhibition assay
Haemagglutination inhibition (HI) assays were carried out as described previously [41, 42]. A(H3N2) ferret antisera were obtained upon intranasal inoculation with A/Netherlands/109/2003 or A/Netherlands/282/2009, as described previously [41]. A(H7N2) ferret antisera were obtained upon intranasal inoculation with recombinant viruses based on A/Netherlands/219/2003 with the HA of A/chicken/Netherlands/33/2003 and the NA of either A/Netherlands/213/2003 or A/Netherlands/69/2009, as described previously [23]. Pre-treatment of antisera with receptor-destroying enzyme (Vibrio cholerae NA) was carried out at 37 °C overnight, followed by inactivation at 56 °C for 1 h. Antisera were diluted in two fold steps with a starting dilution of 1 : 20, mixed with 25 µl virus stock of four haemagglutinating units (HAU) and incubated at 37 °C for 30 min. Turkey erythrocytes (1 % in PBS, 25 μl) were added and incubated for 1 h at 4 °C before the haemagglutination inhibition titres were read. The reciprocal value of the highest dilution of antiserum that still completely inhibited agglutination was used to express HI titres.
Sequence analysis of NA amino acid position 150
All full-length NA gene segment sequences of human A(H3N2) full-virus genomes in the time period 2000–2016 were obtained from the EpiFlu database http://www.gisaid.org (n=5942). No full-virus genomes were available for 2016. The selection criteria were type A subtype H3 and N2 viruses from humans, with collection dates between 1 January 2000 and 1 January 2017. Sequences were aligned using the multiple-sequence alignment software mafft v7. Amino acids at position 150 were identified as histidine (H), arginine (R), or any other amino acid (X), and the relative proportion per year was calculated. For the discrimination of sequences from original material (n=1687) or from MDCK-passaged material (n=900), sequences with any of the following descriptions were included in the respective analysis: for original material [P0, clinical specimen, direct, isolated_directly_ from_host,_no_passage, original, original_specimen, original_specimen_uncultured_in_VTM, direct_clinical_sample); for MDCK-passaged material MDCK, MDCK_1, MDCK_2, MDCK_3, MDCK_4, MDCK_5, MDCK_6, MDCK/1, MDCK1, MDCK1_(H3N2), MDCK2, MDCK3, MDCK4, MDCK5, MDCKp1, MDCKp2, MDCKp3, MDCKX,_MDCK1, MDCKX, MDCK1, mdckx, mdck2, passage_details:_MDCK1, passage_details:_MDCK2, passage_details:_P1_MDCK, passage_details:_P3_MDCK, Plaque_from_MDCK_(H3N2)].
Haemagglutination assay with modified turkey erythrocytes
Sialic acids were removed from turkey erythrocytes by incubation with Vibrio cholerae NA (VCNA; Roche) as described previously [43]. In brief, a suspension of 1 % turkey erythrocytes in PBS was incubated at 37 °C for 1 h with 50 mU VCNA (1 mU µl−1) and 10 µl of 0.1 M CaCl2. Successful treatment to remove sialic acids was verified by the absence of the agglutination of erythrocytes for each virus. Resialylation of α-2,3- and α-2,6-linked sialic acids was performed using 0.5 mU of α2,3-(N)-sialyltransferase (Sigma-Aldrich) or 25 mU of α2,6-(N)-sialyltransferase (Sigma-Aldrich) and 1.5 mM CMP-sialic acid (Merck-Millipore) at 37 °C in 75 µl for 2 h. After a washing step, the erythrocytes were resuspended in PBS containing 1 % bovine serum albumin to a final concentration of 0.5 %. Resialylation was confirmed by haemagglutination assays with viruses of known receptor specificity [A/Puerto Rico/8/1934 with HA of A/Indonesia/5/2005 (H5N1), specific for α-2,3-linked sialic acids, or A/Netherlands/213/2003 (H3N2), specific for α-2,6-linked sialic acids]. To assess the receptor specificity of the viruses of interest, standard haemagglutination assays were carried out using modified erythrocytes. Viruses that did not agglutinate normal erythrocytes with a titre of 64 were used undiluted in this assay.
Funding information
This work was supported by NIAID/NIH contract HHSN272201400008C.
Acknowledgements
We thank Theo Bestebroer for technical assistance. We gratefully acknowledge the originating laboratories, where specimens were first obtained, and the submitting laboratories, where sequence data were generated and submitted to the EpiFlu Database of the Global Initiative on Sharing All Influenza Data (GISAID), who provided data for Fig. 3. These contributors are listed in Table S3 and the data are accessible from the GISAID website (http://platform.gisaid.org).
Conflicts of interest
The authors declare that there are no conflicts of interest.
Ethical statement
Animals were housed and experiments were conducted in strict compliance with European guidelines (EU Directive on Animal Testing 86/609/EEC) and Dutch legislation (Experiments on Animals Act, 1997). All animal experiments were approved by the independent animal experimentation ethical review committee ‘Stichting DEC Consult’ (Erasmus MC permit number EUR3385). The DEC considers the application and pays careful attention to the effects of the intervention on the animal and its discomfort and weighs this against the social and scientific benefit to humans or animals. The researcher is required to keep the effects of the intervention to a minimum, based on the three Rs (refinement, replacement, reduction).
Supplementary Data
Footnotes
Abbreviations: HA, haemagglutinin; HAU, haemagglutination unit; HI, haemagglutination inhibition; NA, neuraminidase; NCR, non-coding region; SA, sialic acid.
Three supplementary tables are available with the online Supplementary Material.
References
- 1.Nelson MI, Holmes EC. The evolution of epidemic influenza. Nat Rev Genet. 2007;8:196–205. doi: 10.1038/nrg2053. [DOI] [PubMed] [Google Scholar]
- 2.Monto AS. Influenza: quantifying morbidity and mortality. Am J Med. 1987;82:20–25. doi: 10.1016/0002-9343(87)90556-0. [DOI] [PubMed] [Google Scholar]
- 3.Stöhr K. Influenza—WHO cares. Lancet Infect Dis. 2002;2:517. doi: 10.1016/S1473-3099(02)00366-3. [DOI] [PubMed] [Google Scholar]
- 4.Thompson WW, Comanor L, Shay DK. Epidemiology of seasonal influenza: use of surveillance data and statistical models to estimate the burden of disease. J Infect Dis. 2006;194:S82–S91. doi: 10.1086/507558. [DOI] [PubMed] [Google Scholar]
- 5.WHO Influenza vaccine viruses and reagents. WHO. 2017. www.who.int/influenza/vaccines/virus/en/
- 6.Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- 7.Wagner R, Matrosovich M, Klenk HD. Functional balance between haemagglutinin and neuraminidase in influenza virus infections. Rev Med Virol. 2002;12:159–166. doi: 10.1002/rmv.352. [DOI] [PubMed] [Google Scholar]
- 8.Liu C, Eichelberger MC, Compans RW, Air GM. Influenza type A virus neuraminidase does not play a role in viral entry, replication, assembly, or budding. J Virol. 1995;69:1099–1106. doi: 10.1128/jvi.69.2.1099-1106.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mitnaul LJ, Matrosovich MN, Castrucci MR, Tuzikov AB, Bovin NV, et al. Balanced hemagglutinin and neuraminidase activities are critical for efficient replication of influenza A virus. J Virol. 2000;74:6015–6020. doi: 10.1128/JVI.74.13.6015-6020.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richard M, Erny A, Caré B, Traversier A, Barthélémy M, et al. Rescue of a H3N2 influenza virus containing a deficient neuraminidase protein by a hemagglutinin with a low receptor-binding affinity. PLoS One. 2012;7:e33880. doi: 10.1371/journal.pone.0033880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gulati U, Wu W, Gulati S, Kumari K, Waner JL, et al. Mismatched hemagglutinin and neuraminidase specificities in recent human H3N2 influenza viruses. Virology. 2005;339:12–20. doi: 10.1016/j.virol.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Wright P, Neumann G, Kawaoka Y. Orthomyxoviruses. In: Howley PM, Knipe DM, editors. Fields Virology. 1st ed. Wolters Kluwer: Lippincott Williams & Wilkins; 2013. pp. 1785–1837. (editors) [Google Scholar]
- 13.Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk HD. Neuraminidase is important for the initiation of influenza virus infection in human airway epithelium. J Virol. 2004;78:12665–12667. doi: 10.1128/JVI.78.22.12665-12667.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morishita T, Kobayashi S, Miyake T, Ishihara Y, Nakajima S, et al. Host-specific hemagglutination of influenza A (H1N1) virus. Microbiol Immunol. 1993;37:661–665. doi: 10.1111/j.1348-0421.1993.tb01689.x. [DOI] [PubMed] [Google Scholar]
- 15.Nobusawa E, Ishihara H, Morishita T, Sato K, Nakajima K. Change in receptor-binding specificity of recent human influenza A viruses (H3N2): a single amino acid change in hemagglutinin altered its recognition of sialyloligosaccharides. Virology. 2000;278:587–596. doi: 10.1006/viro.2000.0679. [DOI] [PubMed] [Google Scholar]
- 16.Medeiros R, Escriou N, Naffakh N, Manuguerra JC, van der Werf S. Hemagglutinin residues of recent human A(H3N2) influenza viruses that contribute to the inability to agglutinate chicken erythrocytes. Virology. 2001;289:74–85. doi: 10.1006/viro.2001.1121. [DOI] [PubMed] [Google Scholar]
- 17.Ito T, Suzuki Y, Mitnaul L, Vines A, Kida H, et al. Receptor specificity of influenza A viruses correlates with the agglutination of erythrocytes from different animal species. Virology. 1997;227:493–499. doi: 10.1006/viro.1996.8323. [DOI] [PubMed] [Google Scholar]
- 18.Lin YP, Gregory V, Collins P, Kloess J, Wharton S, et al. Neuraminidase receptor binding variants of human influenza A(H3N2) viruses resulting from substitution of aspartic acid 151 in the catalytic site: a role in virus attachment? J Virol. 2010;84:6769–6781. doi: 10.1128/JVI.00458-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin YP, Xiong X, Wharton SA, Martin SR, Coombs PJ, et al. Evolution of the receptor binding properties of the influenza A(H3N2) hemagglutinin. Proc Natl Acad Sci USA. 2012;109:21474–21479. doi: 10.1073/pnas.1218841110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu X, Mcbride R, Nycholat CM, Yu W, Paulson JC, et al. Influenza virus neuraminidases with reduced enzymatic activity that avidly bind sialic acid receptors. J Virol. 2012;86:13371–13383. doi: 10.1128/JVI.01426-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Baalen CA, Els C, Sprong L, van Beek R, van der Vries E, et al. Detection of nonhemagglutinating influenza A (H3) viruses by enzyme-linked immunosorbent assay in quantitative influenza virus culture. J Clin Microbiol. 2014;52:1672–1677. doi: 10.1128/JCM.03575-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumari K, Gulati S, Smith DF, Gulati U, Cummings RD, et al. Receptor binding specificity of recent human H3N2 influenza viruses. Virol J. 2007;4:42. doi: 10.1186/1743-422X-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Westgeest KB, Bestebroer TM, Spronken MI, Gao J, Couzens L, et al. Optimization of an enzyme-linked lectin assay suitable for rapid antigenic characterization of the neuraminidase of human influenza A(H3N2) viruses. J Virol Methods. 2015;217:55–63. doi: 10.1016/j.jviromet.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russell RJ, Haire LF, Stevens DJ, Collins PJ, Lin YP, et al. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443:45–49. doi: 10.1038/nature05114. [DOI] [PubMed] [Google Scholar]
- 25.Wu Y, Qin G, Gao F, Liu Y, Vavricka CJ, et al. Induced opening of influenza virus neuraminidase N2 150-loop suggests an important role in inhibitor binding. Sci Rep. 2013;3:1551. doi: 10.1038/srep01551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taylor NR, von Itzstein M. Molecular modeling studies on ligand binding to sialidase from influenza virus and the mechanism of catalysis. J Med Chem. 1994;37:616–624. doi: 10.1021/jm00031a011. [DOI] [PubMed] [Google Scholar]
- 27.Uhlendorff J, Matrosovich T, Klenk HD, Matrosovich M. Functional significance of the hemadsorption activity of influenza virus neuraminidase and its alteration in pandemic viruses. Arch Virol. 2009;154:945–957. doi: 10.1007/s00705-009-0393-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laver WG, Colman PM, Webster RG, Hinshaw VS, Air GM. Influenza virus neuraminidase with hemagglutinin activity. Virology. 1984;137:314–323. doi: 10.1016/0042-6822(84)90223-X. [DOI] [PubMed] [Google Scholar]
- 29.Kobasa D, Rodgers ME, Wells K, Kawaoka Y. Neuraminidase hemadsorption activity, conserved in avian influenza A viruses, does not influence viral replication in ducks. J Virol. 1997;71:6706–6713. doi: 10.1128/jvi.71.9.6706-6713.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hausmann J, Kretzschmar E, Garten W, Klenk HD. N1 neuraminidase of influenza virus A/FPV/Rostock/34 has haemadsorbing activity. J Gen Virol. 1995;76:1719–1728. doi: 10.1099/0022-1317-76-7-1719. [DOI] [PubMed] [Google Scholar]
- 31.Sung JC, van Wynsberghe AW, Amaro RE, Li WW, McCammon JA. Role of secondary sialic acid binding sites in influenza N1 neuraminidase. J Am Chem Soc. 2010;132:2883–2885. doi: 10.1021/ja9073672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hooper KA, Bloom JD. A mutant influenza virus that uses an N1 neuraminidase as the receptor-binding protein. J Virol. 2013;87:12531–12540. doi: 10.1128/JVI.01889-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee HK, Tang JW, Kong DH, Loh TP, Chiang DK, et al. Comparison of mutation patterns in full-genome A/H3N2 influenza sequences obtained directly from clinical samples and the same samples after a single MDCK passage. PLoS One. 2013;8:e79252. doi: 10.1371/journal.pone.0079252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deyde VM, Sheu TG, Trujillo AA, Okomo-Adhiambo M, Garten R, et al. Detection of molecular markers of drug resistance in 2009 pandemic influenza A (H1N1) viruses by pyrosequencing. Antimicrob Agents Chemother. 2010;54:1102–1110. doi: 10.1128/AAC.01417-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okomo-Adhiambo M, Nguyen HT, Sleeman K, Sheu TG, Deyde VM, et al. Host cell selection of influenza neuraminidase variants: implications for drug resistance monitoring in A(H1N1) viruses. Antiviral Res. 2010;85:381–388. doi: 10.1016/j.antiviral.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 36.Chambers BS, Li Y, Hodinka RL, Hensley SE. Recent H3N2 influenza virus clinical isolates rapidly acquire hemagglutinin or neuraminidase mutations when propagated for antigenic analyses. J Virol. 2014;88:10986–10989. doi: 10.1128/JVI.01077-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Baalen CA, Jeeninga RE, Penders GH, van Gent B, van Beek R, et al. ViroSpot microneutralization assay for antigenic characterization of human influenza viruses. Vaccine. 2017;35:46–52. doi: 10.1016/j.vaccine.2016.11.060. [DOI] [PubMed] [Google Scholar]
- 38.Lin Y, Gu Y, Wharton SA, Whittaker L, Gregory V, et al. Optimization of a micro-neutralisation assay and its application in antigenic characterisation of influenza viruses. Influenza Other Respir Viruses. 2015:331–340. doi: 10.1111/irv.12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci USA. 2000;97:6108–6113. doi: 10.1073/pnas.100133697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Wit E, Spronken MI, Bestebroer TM, Rimmelzwaan GF, Osterhaus AD, et al. Efficient generation and growth of influenza virus A/PR/8/34 from eight cDNA fragments. Virus Res. 2004;103:155–161. doi: 10.1016/j.virusres.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 41.Koel BF, Burke DF, Bestebroer TM, van der Vliet S, Zondag GC, et al. Substitutions near the receptor binding site determine major antigenic change during influenza virus evolution. Science. 2013;342:976–979. doi: 10.1126/science.1244730. [DOI] [PubMed] [Google Scholar]
- 42.Hirst GK. Studies of antigenic differences among strains of influenza A by means of red cell agglutination. J Exp Med. 1943;78:407–423. doi: 10.1084/jem.78.5.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koel BF, Mögling R, Chutinimitkul S, Fraaij PL, Burke DF, et al. Identification of amino acid substitutions supporting antigenic change of influenza A(H1N1)pdm09 viruses. J Virol. 2015;89:3763–3775. doi: 10.1128/JVI.02962-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.