

Abstract
Objective
To evaluate the safety and efficacy of crenezumab in patients with mild to moderate Alzheimer disease (AD).
Methods
In this phase 2 trial, 431 patients with mild to moderate AD 50 to 80 years of age were randomized 2:1 (crenezumab:placebo). Patients received low-dose subcutaneous crenezumab 300 mg or placebo every 2 weeks (n = 184) or high-dose intravenous crenezumab 15 mg/kg or placebo every 4 weeks (n = 247) for 68 weeks. Primary outcome measures were change in Alzheimer's Disease Assessment Scale–Cognitive Subscale (ADAS-Cog12) and Clinical Dementia Rating–Sum of Boxes scores from baseline to week 73.
Results
The primary and secondary endpoints were not met. In an exploratory post hoc analysis, a reduction in decline on the ADAS-Cog12 was observed in the high-dose group. Separation from the placebo group on the ADAS-Cog12 was greatest in the milder subsets of AD patients and reached statistical significance in the group with Mini-Mental State Examination scores of 22 to 26. In both groups, there was a significant increase in CSF β-amyloid1-42 levels that correlated with crenezumab CSF levels. The overall rate of adverse events was balanced between groups. One case of amyloid-related imaging abnormalities indicative of vasogenic edema or effusions was reported.
Conclusions
Although prespecified criteria for testing treatment effects were not met, these data suggest a potential treatment effect in patients with mild AD treated with high-dose crenezumab. Together with the safety profile for crenezumab, these data support the exploration of crenezumab treatment at even higher doses in patients with early AD.
Clinicaltrials.gov identifier
Classification of evidence
This study provides Class II evidence that, for people with AD, crenezumab does not significantly improve cognition or function at 18 months. The study is rated Class II because <80% of enrolled patients completed the study.
Alzheimer disease (AD) is the most common form of dementia1 and is characterized by deposition of amyloid plaques in the brain composed primarily of β-amyloid (Aβ) peptides.2 Aβ peptides may accumulate as soluble monomers and aggregate as oligomers and insoluble fibrils,1 but soluble oligomers are suggested to be a major driver of neurotoxicity.3–5
Crenezumab, a fully humanized immunoglobulin isotype G4 monoclonal antibody, binds to monomers and aggregated forms of Aβ with a 10-fold–higher affinity for oligomers.6 The immunoglobulin isotype G4 backbone confers reduced activation of Fc-gamma receptors (FcγRs) and minimizes the FcγR-mediated inflammatory activation of microglia, hypothesized to contribute to neurotoxicity,7,8 while preserving FcγR-mediated microglial phagocytosis and removal of Aβ oligomers.6
Amyloid-related imaging abnormalities (ARIA) indicative of vasogenic edema or effusions (ARIA-E) and microhemorrhage and siderosis (ARIA-H) have been reported recently with monoclonal antibodies that bind aggregated forms of Aβ and have immunoglobulin isotype G1 backbones with fully FcγR-mediated effector function, limiting the dose levels that could be safely administered.9–11 Crenezumab was designed on the basis of the hypothesis that an antibody with reduced effector function would have a lower risk of inducing ARIA-E/H.12,13
Methods
Primary research question
This phase 2, multicenter, randomized, double-blind, placebo-controlled, parallel-group study was designed to evaluate the safety and efficacy of crenezumab in patients with mild to moderate AD that was conducted from April 25, 2011, to February 18, 2014, at 72 sites in North America and Europe. Class II evidence is provided here.
Standard protocol approvals, registrations, and participant consents
The study protocol was approved by the local institutional review board at each site. Written informed consent was obtained from each patient (or legally authorized representative) before entry into the study (ClinicalTrials.gov identifier NCT01343966). The study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Consolidated Guidelines on Good Clinical Practice.
Patients
Eligible patients were 50 to 80 years old, met the criteria for mild to moderate probable AD according to the National Institute of Neurologic and Communicative Disorders and Stroke—Alzheimer's Disease and Related Disorders Association criteria14 and had a Mini-Mental State Examination (MMSE) score of 18 to 26 points.15 Additional inclusion criteria were a Geriatric Depression Scale score of <6, a Clinical Dementia Rating–Sum of Boxes (CDR-SB) score of ≥0.5,16–18 and an Alzheimer's Disease Assessment Scale–Cognitive Subscale (ADAS-Cog) Delayed Word Recall score of ≥5.19 Treatment with approved AD drugs such as acetylcholinesterase inhibitors or memantine was permitted if initiated ≥3 months and stabilized ≥2 months before randomization.
Study design and treatment
The study was conducted in 2 overlapping parts (figure e-1, links.lww.com/WNL/A461). Patients were randomly assigned in a 2:1 ratio to receive crenezumab 300 mg SC every 2 weeks (the low-dose cohort) or placebo in part 1 and to crenezumab 15 mg/kg IV every 4 weeks (the high-dose cohort) or placebo in part 2. Randomization into the 2 parts was independent and sequential. Enrollment in part 2 of the study began only when randomization in part 1 and the safety run-in (described below) were complete. Randomization was managed by a central IxRS vendor using dynamic hierarchical randomization based on 3 factors: APOE ε4 genotype, MMSE score (<22 vs ≥22), and study site.
To assess the potential for using a higher dose of crenezumab compared to phase 1, part 2 of the phase 2 study was preceded by a safety run-in period that was conducted in parallel with part 1 and consisted of at least 2 monthly IV administrations of 15 mg/kg crenezumab in 13 patients (11 active: 2 placebo). On the basis of the available safety data, the 15-mg/kg dose was selected for the high-dose IV cohort of the study. Patients from this cohort were not included in the primary efficacy analysis.
Outcomes
The primary efficacy outcome measures were changes in the 12-item ADAS-Cog (ADAS-Cog12) and CDR-SB scores from baseline to week 73.20,21 The secondary efficacy outcome measure, the Alzheimer's Disease Cooperative Study–Activities of Daily Living (ADCS-ADL) score, was analyzed in the same manner as the primary efficacy outcome measures.22
To assess the exploratory outcome of crenezumab pharmacokinetics, blood samples were collected to measure serum crenezumab concentrations and analyzed with a validated ELISA (limit of detection 50 ng/mL).
CSF collection was conducted as an optional procedure at week 1 (day 1/baseline) and before study drug administration at week 69 (steady state). CSF crenezumab concentrations were analyzed with a validated ELISA (limit of detection 12.5 ng/mL). CSF Aβ1-42 was measured with the Elecsys Aβ1-42 immunoassay under development by Roche Diagnostics.23 CSF tau and phosphorylated-tau 181 were measured with INNOTEST ELISAs (Fujirebio, Tokyo, Japan).
MRI assessments were performed by a central imaging reader (NeuroRx, Montreal, Quebec, Canada) and analyzed longitudinally and included the following scans for safety assessments and volumetric measurements: a high-resolution T1-weighted structural scan, a T2-weighted gradient-recalled echo, and a T2-weighted fluid-attenuated inversion recovery. Ventricular, whole-brain, and hippocampal volumes were measured from the T1 MRI.
Safety monitoring
Safety was assessed from reports of adverse events (AEs), serious AEs (SAEs), and AEs of special interest. Safety assessments included clinical laboratory testing, clinical examinations, ECG, and brain MRI. All safety data were assessed by an unblinded Internal Safety Monitoring Committee on a regular basis.
Blood samples were collected to test for the presence of antitherapeutic antibody in serum.
Population and statistical analysis
The study was designed to enroll ≈180 patients each in both the low- and high-dose cohorts; in each part, 60 patients would be enrolled in the placebo arm and 120 patients would be enrolled in the crenezumab arm. Assuming a mean decline of 6 points for change in ADAS-Cog12, an SD of 9 points, and 30% dropout, this sample size would provide 80% power to detect a true treatment delta of 3.6 (60% reduction relative to placebo) when testing at the 2-sided 0.2 level. In addition, assuming a mean decline of 2.4 points for CDR-SB, an SD of 3 points, and 30% dropout, this sample size would provide 80% power to detect a true treatment delta of 1.2 (50% reduction relative to placebo) when testing at the 2-sided 0.2 level.
The efficacy analysis was based on the modified intent-to-treat population, which included all patients who were randomized and had both a baseline measurement and at least 1 postbaseline measurement for that endpoint. Patients were grouped according to the treatment assigned at randomization. In the high-dose 15 mg/kg IV cohort, 2 patients receiving crenezumab were excluded from the safety evaluable population as a result of 2 dosing deviations and 2 patients leaving the study before the first dose, leaving 165 crenezumab and 82 placebo patients.
Cohorts from part 1 (low-dose 300 mg SC cohort) and part 2 (high-dose 15 mg/kg IV cohort) were analyzed separately, reflecting their independent and sequential randomization. Three subpopulations determined by baseline MMSE score were analyzed: MMSE score of 18 to 26 (all patients), MMSE score of 20 to 26 (mild AD), and MMSE score of 22 to 26 (very mild AD). The subpopulations with MMSE scores of 18 to 26 and 20 to 26 were prespecified, while the post hoc subpopulation with MMSE scores of 22 to 26 was based on stratification criteria used at study randomization. Efficacy endpoints with >1 postbaseline outcome measure were analyzed with mixed-effect model repeat measurement for the change scores from the baseline to postbaseline time points. An unstructured variance-covariance matrix for within-patient errors was used.
Efficacy endpoints with 1 postbaseline outcome measure were analyzed with analysis of covariance on the change scores from baseline to the postbaseline time point.
The mixed-effect model repeat measurement and analysis of covariance models for efficacy endpoints were used to estimate mean declines for each treatment group and time point, the least-squares mean treatment difference between the crenezumab and placebo groups at each time point, the 95% CIs for the mean treatment deltas, and the corresponding p values. The CIs and p values were not adjusted for multiplicity in this study.
The safety analysis was performed on all randomized patients who received at least 1 dose of study drug during the study. All analyses were performed with SAS version 9.2 (SAS Institute Inc, Cary, NC). The pharmacokinetics analysis was performed on all patients randomized to active treatment who received at least 1 dose of study drug and provided at least 1 valid pharmacokinetics assessment.
Data availability
We provide qualified researchers access to individual patient-level data through the clinical study data request platform (clincalstudydatarequest.com). Further details of Roche's Data Sharing Policy are available here (clinicalstudydatarequest.com/Study-Sponsors-Roche-Details.aspx).
Results
Participant disposition
The disposition of the enrolled participants is summarized in figure e-2 (links.lww.com/WNL/A461). In total, 431 patients received at least 1 dose of crenezumab or placebo.
The 300 mg SC crenezumab cohort consisted of 184 patients, while 247 patients were allocated to the 15 mg/kg IV cohort. The percentage of patients who had a week 73 assessment was similar across the cohorts and treatment arms. Patients within both parts 1 and 2 of the study had balanced baseline characteristics with no significant differences (table 1).
Table 1.
Baseline demographics and disease characteristics

Efficacy
None of the efficacy outcome measures showed statistically significant differences in any of the prespecified analysis subpopulations.
Changes from baseline over time in scores on the ADAS-Cog12 in patients with mild to moderate AD (MMSE score 18–26) showed no statistically significant difference at week 73 between active treatment and placebo in the 300 mg SC cohort (0.04-point difference; table e-1, links.lww.com/WNL/A462). Similarly, the 15 mg/kg IV crenezumab cohort showed no statistically significant difference on the ADAS-Cog12 between crenezumab treatment and placebo at week 73 (1.78-point difference). In the prespecified subgroup analysis in patients with mild AD (MMSE score 20–26), no effect was observed on the ADAS-Cog12 for the 300 mg SC crenezumab cohort (figure 1B and table e-1). Prespecified subgroup analysis of the 15 mg/kg IV crenezumab cohort (figure 1D and table 2) showed a greater reduction of cognitive decline in patients with a baseline MMSE score of 20 to 26 (2.4-point difference in ADAS-Cog12 score).
Figure 1. ADAS-Cog12 scores.

Mean change from baseline to week 73 in (A and D) mild to moderate (MMSE score 18–26), (B and E) mild (MMSE score 20–26), and (C and F) milder (MMSE score 22–26) populations. (A–C) Low-dose 300 mg SC cohort. (D–F) High-dose 15 mg/kg IV cohort. Error bars show SE of the least-squares mean. AD = Alzheimer disease; ADAS-Cog12 = 12-point Alzheimer's Disease Assessment Scale–Cognitive Subscale; BL = baseline; Cr = crenezumab; Diff = difference; MMSE = Mini-Mental State Examination; %Red = percentage reduction; Pl = placebo; SC = subcutaneous; SE = standard error.
Table 2.
Primary, secondary, and exploratory clinical outcomes in high-dose 15 mg/kg IV cohort at week 73 (placebo vs crenezumab; mITT population)

At week 73, no statistically significant drug-placebo group separations were seen on the CDR-SB in the patients with mild to moderate (0.69-point difference) and mild (0.71-point difference) AD who received crenezumab in the 300 mg SC cohort (table e-1, links.lww.com/WNL/A462). Likewise, no treatment effect was seen in any prespecified patient population receiving 15 mg/kg IV crenezumab (table 2).
The secondary efficacy endpoint evaluated the change in ADCS-ADL score from baseline to week 73. No statistically significant differences were observed between crenezumab and placebo in the 300 mg SC cohort. At week 73, a difference of −1.42 points for the mild to moderate AD population was observed. A difference of −2.78 points in the prespecified mild AD (MMSE score 20–26) population was also observed (table e-2, links.lww.com/WNL/A462). In the 15 mg/kg IV cohort, there was a reduction of 0.51 points for the mild to moderate (MMSE score 18–26) population and 2.18 points in the mild (MMSE score 20–26) AD population (table 2).
In the exploratory post hoc subgroup analysis of patients with very mild AD (MMSE score 22–26), no effect was observed on the ADAS-Cog12 for the low-dose 300 mg SC crenezumab cohort (figure 1, B and C and table e-1, links.lww.com/WNL/A462). However, patients receiving 15 mg/kg IV crenezumab (figure 1F and table 2) with a baseline MMSE score of 22 to 26 had a 3.44-point difference. No treatment effect on the CDR-SB was seen in patients with very mild AD with a baseline MMSE score of 22 to 26 in either cohort (table e-1 and table 2). No change in ADCS-ADL score was seen from baseline to week 73; patients with MMSE scores of 22 to 26 in the 300 mg SC crenezumab cohort demonstrated a difference of –1.10 points compared with placebo (table e-1). In the 15 mg/kg IV cohort, there was a reduction of 0.12 points in the very mild (MMSE score 22–26) population (table 2).
For the 15 mg/kg IV dose cohort, the population with a baseline MMSE score of 22 to 26 was the first post hoc subpopulation based on MMSE to be examined. Subsequently, even milder populations defined by an MMSE score of 24 to 26 were also analyzed, as were those with an MMSE score of 26 alone (table e-2, links.lww.com/WNL/A462). These results indicate that the observed percentage reduction relative to placebo consistently increases for ADAS-Cog12 and generally increases for CDR-SB as the subgroups become milder. For ADAS-Cog12, the percentage reduction in the high-dose 15 mg/kg IV cohort ranges from 16.8% for the subgroup with a baseline MMSE score of 18 to 26 to 53.0% for the subgroup with a baseline MMSE score of 26. For CDR-SB, the percent reduction is 3.1% for the subgroup with a baseline MMSE score of 18 to 26 and 54.4% for the subgroup with a baseline MMSE score of 26. No beneficial treatment effects were observed for ADCS-ADL except in the mildest subgroup with a baseline MMSE score of 26, in whom a 42.4% reduction relative to placebo was observed (table e-2).
Crenezumab pharmacokinetics
Crenezumab serum trough concentrations at steady state were 69.2 (29.6) µg/mL after 300 mg SC every 2 weeks dosing and 118 (71.3) µg/mL after 15 mg/kg IV every 4 weeks dosing.
Crenezumab mean steady-state trough concentrations in CSF were 0.19 (SD 0.14) μg/mL and 0.25 (SD 0.12) μg/mL in the 300 mg SC and the 15 mg/kg IV cohort, respectively (figure e-3, links.lww.com/WNL/A461). The proportion of crenezumab detected in the CSF in relation to the serum concentration was similar between doses and routes of administration, with a mean ratio of CSF to serum of 0.28% (SD 0.19%) and 0.29% (SD 0.16%) in the 300 mg SC and 15 mg/kg IV cohorts, respectively.
Biomarker outcomes
No treatment effect was observed in an exploratory volumetric MRI analysis of hippocampal volume, ventricular volume, and whole-brain volume (figure e-4, links.lww.com/WNL/A461).
A statistically significant difference in mean change from baseline in CSF Aβ1-42 levels between the crenezumab and placebo groups of −120.16 pg/mL (unadjusted p = 0.017) (300 mg SC cohort) and −170.50 pg/mL (unadjusted p = 0.022) (15 mg/kg IV cohorts), respectively, was detected (figure e-5A and e-5D, links.lww.com/WNL/A461). No consistent drug-placebo difference was observed in CSF tau or phosphorylated tau levels in either dose group (figure e-5B, e-5C, e-5E, and e-5F). Time-matched CSF crenezumab levels and CSF Aβ1–42 changes showed no clear correlation (figure 2).
Figure 2. CSF Aβ1-42 and crenezumab correlation analysis.
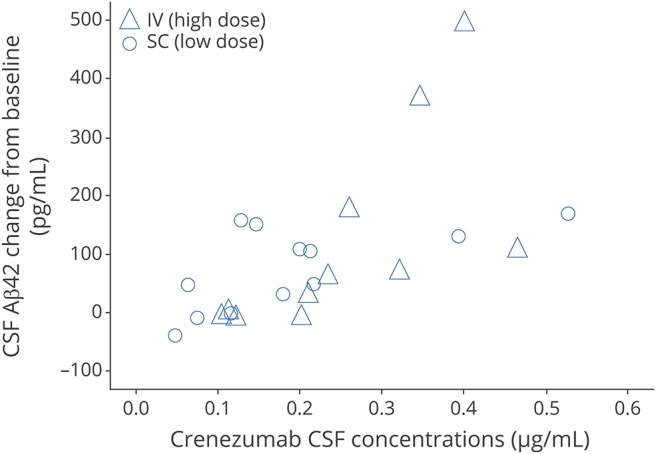
Correlation analysis of change in CSF Aβ1-42 from baseline and crenezumab concentrations in patients receiving low-dose 300 mg SC (circles) and those receiving high-dose 15 mg/kg IV (triangles). Aβ = β-amyloid.
Safety
The overall rate of AEs was balanced between the active and placebo arms of the study and between the 2 cohorts. The majority of reported AEs were grade 1 or 2 in severity (table 3). An imbalance in the frequency of SAEs was observed that was driven mostly by reports of serious pneumonia, syncope, and Alzheimer-type dementia. An imbalance in the rates of SAEs and non-SAEs of pneumonia was observed; however, there was no evidence of an exposure-response relationship within each cohort. An imbalance in fatal events was observed, with 3 deaths (1.0%) in the crenezumab arms compared with no deaths in the placebo arms. All cases were considered by the investigators to be unrelated to the treatment and were within the expected rates for this population.24
Table 3.
Summary of AEs

The frequency of AEs grade 3 or greater was balanced between the placebo and crenezumab treatment arms across both the 300 mg SC and 15 mg/kg IV cohorts. In the 15 mg/kg IV cohort, more AEs of grade 3 or greater were observed in the crenezumab arm (17.0%, 28 patients) relative to the placebo arm (13.4%, 11 patients). This imbalance was driven mainly by cardiovascular events (25% of grade 3 or greater AEs, 7 patients in the crenezumab arm; 18% of grade 3 or greater AEs, 2 patients in the placebo arm), with no clear causal relationship to the study drug, and events (e.g., syncope and atrial fibrillation) were as expected for this patient population.24 The incidences of AEs leading to discontinuation were balanced between the placebo (4.2%, 6 patients) and crenezumab (3.3%, 10 patients) groups, and no clear pattern for discontinuation was observed.
The incidence of new ARIA-H was balanced between the low-dose and high-dose cohorts (table e-3, links.lww.com/WNL/A462). A single case of asymptomatic ARIA-E was observed in an APOE ε4 homozygous female patient in the crenezumab 15 mg/kg IV cohort. A single case of asymptomatic macrohemorrhage was detected in 1 patient receiving crenezumab in the 15 mg/kg IV cohort; the patient was discontinued from study treatment at the time of diagnosis (week 35).
Injection- or infusion-related AEs were balanced across treatment arms, and there was no evidence of clinically relevant immunogenicity in patients receiving crenezumab.
Discussion
This proof-of-concept study in mild to moderate AD found no difference between 2 dose levels of crenezumab and placebo on the coprimary endpoint (ADAS-Cog12 and CDR-SB scores). In post hoc exploratory analysis of patients who received the higher dose of 15 mg/kg IV crenezumab, consistently increasing percentage reductions relative to placebo for the ADAS-Cog12 and CDR-SB endpoints were observed in progressively milder subgroups. These post hoc analyses need confirmation in follow-up studies, but they suggest that earlier treatment and/or higher doses may improve the beneficial effect of crenezumab.
A dose-dependent increase (1.3–1.7 fold) in crenezumab trough concentrations in serum and CSF was observed. The CSF/serum ratio was ≈0.3% for both cohorts, suggesting dose-proportional penetration into the CNS. The current study does not reveal the exposure driver for efficacy because dose is confounded by route of administration (IV vs SC) and schedule (every 4 vs every 2 weeks). However, the 15 mg/kg IV dose maintains concentrations above the 300 mg SC dose throughout the dosing interval, supporting testing doses that provide higher concentrations.
The increase in CSF Aβ1-42 observed in this study suggests crenezumab achieved target engagement in the brain. There were, however, no changes in CSF total tau or phosphorylated tau. The increase in CSF Aβ1-42 associated with crenezumab treatment could reflect increased input of Aβ1-42 into CSF, decreased clearance of CSF Aβ1-42 out of CSF, or a shift in CSF Aβ content toward more species that are detected by the Aβ1-42 assay. Understanding the relationship between the pharmacodynamic effects on CSF Aβ and amyloid PET and their correlation with cognitive change requires larger follow-up studies.
The incidence of ARIA-E was low with crenezumab and within the background rates of ARIA-E in the mild to moderate AD population (0.2%–0.4%), as observed in similar studies9,25 and lower than observed with other anti-Aβ monoclonal antibody treatments that showed rates of ARIA-E up to 55% among APOE ε4 homozygotes.9,26–28 In addition, the incidence of ARIA-H was 14.6% in patients receiving placebo and 10.3% in patients receiving crenezumab, suggesting that the drug did not increase the number of new microhemorrhages in this trial.
The overall rate of AEs was balanced in crenezumab- and placebo-treated patients. Although the rate of pneumonia was imbalanced with a higher percentage of cases in crenezumab-treated patients, the frequency in crenezumab-treated patients was within the range that is expected in an elderly population (2.5%–4.4%).29 In addition, there was a dose-independent imbalance in the rate of urinary tract infections in crenezumab-treated patients. While dose independence does not rule out a causal relationship to treatment, the lower rate of urinary tract infections in the high-dose cohort (10.3%) than in the placebo group (12.2%) suggests that this is unlikely. The higher rate of headaches and falls was also dose independent and, on the basis of individual case assessment, considered unrelated to study drug. The 2 ongoing phase 3 studies will provide more precise and detailed data on the safety profile of crenezumab. Together, these data suggest that crenezumab was generally well tolerated.
Although this trial did not meet the primary endpoints, post hoc analysis of the effects of crenezumab in patients with very mild AD in the high-dose 15 mg/kg IV cohort suggests the utility of treating AD earlier and with higher doses of crenezumab. Because the safety profile observed in this trial suggested generally good tolerability, allowing the evaluation of higher dose levels, two similar crenezumab phase 3 studies (A Study of Crenezumab Versus Placebo to Evaluate the Efficacy and Safety in Participants With Prodromal to Mild Alzheimer's Disease [CREAD, NCT02670083; CREAD2, NCT03114657]) are ongoing in patients in the prodromal to mild phase (MMSE score 22–30) using a higher dose of crenezumab intended to drive efficacy while maintaining tolerability.
Acknowledgment
The authors thank all the patients with AD and their caregivers who participated in these studies. They acknowledge the following individuals for their assistance with the study: Pravina Kittredge, Shane Smith, Erin Elman, Mira Blendstrup, Amy Sullivan, and Tracy Smith from Clinical Operations, and Hiral Raval from Statistical Programming. Editorial support in drafting the manuscript by incorporating technical editing, copyediting, and word processing was provided by Jennifer Smith of MediTech Media Ltd.
Glossary
- Aβ
β-amyloid
- AD
Alzheimer disease
- ADAS-Cog
Alzheimer's Disease Assessment Scale–Cognitive Subscale
- ADCS-ADL
Alzheimer's Disease Cooperative Study–Activities of Daily Living
- AE
adverse event
- ARIA
amyloid-related imaging abnormalities
- ARIA-E
amyloid-related imaging abnormalities indicative of vasogenic edema or effusions
- ARIA-H
amyloid-related imaging abnormalities indicative of microhemorrhage and siderosis
- CDR-SB
Clinical Dementia Rating–Sum of Boxes
- CREAD
A Study of Crenezumab Versus Placebo to Evaluate the Efficacy and Safety in Participants With Prodromal to Mild Alzheimer's Disease
- FcγR
Fc-gamma receptor
- MMSE
Mini-Mental State Examination
- SAE
serious adverse event
Footnotes
Author contributions
J.L.C., M.W., D.C., and R.P. designed the study. J.L.C., M.W., M.F., A.Q., R.N.F., and R.P. contributed to the writing of the first draft, Methods section, and background of the report. J.L.C., S.C., M.B., and C.H.v.D. were involved in data collection and study execution. W.C., M.W., M.F., C.R., F.B., A.Q., L.A.H., D.C., D.M., and C.H. provided guidance on and performed statistical analysis and data interpretation. All authors critically reviewed and edited the manuscript.
Study funding
This study was funded by Genentech, Inc.
Disclosure
J. Cummings has received in-kind research support from Avid Radiopharmaceuticals and Teva Pharmaceuticals and has provided consultation to AbbVie, Acadia, Actinogen, ADAMAS, Alzheon, Anavex, Avanir, Biogen-Idec, Biotie, Boehringer-Ingelheim, Chase, Eisai, Forum, GE Healthcare, Genentech, Grifols, Intracellular Therapies, Lilly, Lundbeck, MedAvante, Merck, Neurotrope, Novartis, Nutricia, Otsuka, QR Pharma, Resverlogix, Roche, Servier, Suven, Takeda, and Toyoma companies. S. Cohen’s institution received a clinical research grant for the conduct the study at the investigative site. C. van Dyck has received grant support from Genentech, Inc for the completion of the study, honoraria and consultancy fees from Roche Pharmaceuticals and AbbVie, and grant support from Roche Pharmaceuticals, Eli Lilly, Biogen Idec, Eisai, Inc, Pfizer, Inc, Merck, TauRx, Toyama Chemical Co, and Forum Pharmaceuticals. M. Brody institution received a clinical research grant for the conduct the study at the investigative site. C. Curtis reports no disclosures relevant to the manuscript. W. Cho, M. Ward, and M. Friesenhahn are current employees of Genentech (a member of the Roche group) and own stock or stock options in Roche. C. Rabe is a current employee of Roche Diagnostics and owns stock or stock options in Roche. F. Brunstein, A. Quartino, L. Honigberg, R. Fuji, D. Clayton, and D. Mortensen are current employees of Genentech (a member of the Roche group) and own stock or stock options in Roche. C. Ho and R. Paul are former employees of Genentech (a member of the Roche group) and owns stock or stock options in Roche. Go to Neurology.org/N for full disclosures.
References
- 1.Alzheimer's Association Report. Alzheimer's disease facts and figures. Alzheimers Dement 2017;13:325–373. [Google Scholar]
- 2.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002;297:353–356. [DOI] [PubMed] [Google Scholar]
- 3.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol 2007;8:101–112. [DOI] [PubMed] [Google Scholar]
- 4.Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat Neurosci 2012;15:349. [DOI] [PubMed] [Google Scholar]
- 5.Wang ZX, Tan L, Liu J, Yu JT. The essential role of soluble Aβ oligomers in Alzheimer's disease. Mol Neurobiol 2016;53:1905. [DOI] [PubMed] [Google Scholar]
- 6.Adolfsson O, Pihlgren M, Toni N, et al. An effector-reduced anti-β-amyloid (Aβ) antibody with unique binding properties promotes neuroprotection and glial engulfment of Aβ. J Neurosci 2012;32:9677–9689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol 2015;14:388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xing B, Bachstetter AD, Van Eldik LJ. Microglial p38alpha MAPK is critical for LPS-induced neuron degeneration, through a mechanism involving TNFalpha. Mol Neurodegener 2011;6:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuller JP, Stavenhagen JB, Teeling JL. New roles for Fc receptors in neurodegeneration: the impact on Immunotherapy for Alzheimer's disease. Front Neurosci 2014;8:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salloway S, Sperling R, Fox NC, et al. Two Phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer's disease. N Engl J Med 2014;370:322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lasser R, Ostrowitzki S, Scheltens P, et al. Efficacy and safety of gantenerumab in prodromal Alzheimer's disease: results from SCarlet RoAD: a global, multicenter trial. Alzheimers Dement 2015;11:P331–P332. [Google Scholar]
- 12.Wilcock DM, Alamed J, Gottschall PE, et al. Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci 2006;26:5340–5346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ultsch M, Li B, Maurer T, et al. Structure of crenezumab complex with Aβ shows loss of β-hairpin. Sci Rep 2016;6:39374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 15.Folstein MF, Folstein SE, McHugh PR. “Mini-Mental State": a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 16.Sheikh RL, Yesavage JA. Geriatric Depression Scale (GDS): recent evidence and development of a shorter version. Clin Gerontol 1986;5:165–173. [Google Scholar]
- 17.O'Bryant SE, Waring SC, Cullum CM, et al. Staging dementia using Clinical Dementia Rating Scale Sum of Boxes scores: a Texas Alzheimer's Research Consortium study. Arch Neurol 2008;65:1091–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berg L, Miller JP, Baty J, Rubin EH, Morris JC, Figiel G. Mild senile dementia of the Alzheimer type, 4: evaluation of intervention. Ann Neurol 1992;31:242–249. [DOI] [PubMed] [Google Scholar]
- 19.Sano M, Raman R, Emond J, et al. Adding delayed recall to the Alzheimer Disease Assessment Scale is useful in studies of mild cognitive impairment but not Alzheimer disease. Alzheimer Dis Assoc Disord 2011;25:122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohs RC, Knopman D, Petersen RC, et al. Development of cognitive instruments for use in clinical trials of antidementia drugs: additions to the Alzheimer's Disease Assessment Scale that broaden its scope: the Alzheimer's Disease Cooperative Study. Alzheimer Dis Assoc Disord 1997;11(suppl 2):S13–S21. [PubMed] [Google Scholar]
- 21.Hughes CP, Berg L, Danziger WL, Coben LA, Martin RL. A new clinical scale for the staging of dementia. Br J Psychiatry 1982;140:566–572. [DOI] [PubMed] [Google Scholar]
- 22.Galasko D, Bennett D, Sano M, et al. An inventory to assess activities of daily living for clinical trials in Alzheimer's disease: the Alzheimer's Disease Cooperative Study. Alzheimer Dis Assoc Disord 1997;11(suppl 2):S33–S39. [PubMed] [Google Scholar]
- 23.Bittner T, Zetterberg H, Teunissen CE, et al. Technical performance of a novel, fully automated electrochemiluminescence immunoassay for the quantitation of beta-amyloid (1-42) in human cerebrospinal fluid. Alzheimers Dement 2016;12:517–526. [DOI] [PubMed] [Google Scholar]
- 24.Henley DB, Sundell KL, Sethuraman G, Schneider LS. Adverse events and dropouts in Alzheimer's disease studies: what can we learn? Alzheimers Dement 2015;11:24–31. [DOI] [PubMed] [Google Scholar]
- 25.Doody RS, Farlow M, Aisen PS. Phase 3 trials of solanezumab and bapineuzumab for Alzheimer's disease. N Engl J Med 2014;370:1460. [DOI] [PubMed] [Google Scholar]
- 26.Sevigny J, Chiao P, Bussiere T, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature 2016;537:50–56. [DOI] [PubMed] [Google Scholar]
- 27.Sevigny J, Chiao P, Williams L, Miao X, O'Gorman J. Randomized, double-blind, phase 1b study of BIIB037, an anti-amyloid beta monocloncal antibody, in patients with prodromal or mild Alzheimer's disease. Neurodegener Dis 2015;15(suppl 1):311. [Google Scholar]
- 28.Lasser R, Ostrowitzki S, Scheltens P, et al. Efficacy and safety of gantenerumab from the phase 3 SCarlet RoAD trial, a study of gantenerumab in patients with prodromal AD. JPAD 2015;2:275. [Google Scholar]
- 29.Chong CP, Street PR. Pneumonia in the elderly: a review of the epidemiology, pathogenesis, microbiology, and clinical features. South Med J 2008;101:1141–1145; quiz 1132, 1179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
We provide qualified researchers access to individual patient-level data through the clinical study data request platform (clincalstudydatarequest.com). Further details of Roche's Data Sharing Policy are available here (clinicalstudydatarequest.com/Study-Sponsors-Roche-Details.aspx).