

Abstract
Military personnel and athletes exposed to traumatic brain injury may develop chronic traumatic encephalopathy (CTE). Brain pathology in CTE includes intracellular accumulation of abnormally phosphorylated tau proteins (p-tau), the main constituent of neurofibrillary tangles (NFTs). Recently, we found that cholinergic basal forebrain (CBF) neurons within the nucleus basalis of Meynert (nbM), which provide the major cholinergic innervation to the cortex, display an increased number of NFTs across the pathological stages of CTE. However, molecular mechanisms underlying nbM neurodegeneration in the context of CTE pathology remain unknown. Here, we assessed the genetic signature of nbM neurons containing the p-tau pretangle maker pS422 from CTE subjects who came to autopsy and received a neuropathological CTE staging assessment (Stages II, III, and IV) using laser capture microdissection and custom-designed microarray analysis. Quantitative analysis revealed dysregulation of key genes in several gene ontology groups between CTE stages. Specifically, downregulation of the nicotinic cholinergic receptor subunit β-2 gene (CHRNB2), monoaminergic enzymes catechol-O-methyltransferase (COMT) and dopa decarboxylase (DDC), chloride channels CLCN4 and CLCN5, scaffolding protein caveolin 1 (CAV1), cortical development/cytoskeleton element lissencephaly 1 (LIS1), and intracellular signaling cascade member adenylate cyclase 3 (ADCY3) was observed in pS422-immunreactive nbM neurons in CTE patients. By contrast, upregulation of calpain 2 (CAPN2) and microtubule-associated protein 2 (MAP2) transcript levels was found in Stage IV CTE patients. These single-population data in vulnerable neurons indicate alterations in gene expression associated with neurotransmission, signal transduction, the cytoskeleton, cell survival/death signaling, and microtubule dynamics, suggesting novel molecular pathways to target for drug discovery in CTE.
Keywords: : basal forebrain, brain trauma, CTE, expression profiling, genes, TBI
Introduction
Chronic traumatic encephalopathy (CTE), previously termed “dementia pugilistica,” is a neurodegenerative dementia associated with repetitive mild brain trauma. CTE has been identified in American football, ice hockey, rugby, and soccer players; boxers; wrestlers; and military personnel. This concussive dementia is pathologically characterized by the widespread distribution of neurofibrillary tangles (NFT) containing the hyperphosphorylated tau protein (p-tau), a hallmark of Alzheimer's disease (AD), in the neocortex, limbic cortex, and subcortical regions,1–3 including cholinergic basal forebrain (CBF) neurons, located within the nucleus basalis of Meynert (nbM).3–5 These CBF neurons form a continuum of perikarya extending from the septal diagonal band (Ch1–2) caudally throughout the nbM (Ch4),5 are associated with memory and attention dysfunction,6 provide the major source of acetylcholine (Ach) to the cortical mantle,5 and degenerate in AD and traumatic brain injury (TBI).4,7–10 There is a reduction in both the number of choline acetyltransferase (ChAT) positive neurons within the nbM as well as ChAT activity, following TBI.11 In addition, there is also an acute downregulation of the regulator of Ach neurotransmission, the vesicular Ach transporter (vAChT) and acetylcholinesterase (AChE) after TBI.12 Moreover, several studies indicate that the centrally acting United States Food and Drug Administration (FDA)-approved AChE inhibitor, donepezil, improves cognitive function as well as attention after TBI,13 although this remains controversial.14–16 Taken together, these findings suggest that the CBF cortical projection system is damaged after head trauma in humans, and may be impacted significantly in CTE.
Numerous studies have shown that NFT pathology is the best correlate of cognitive decline to date in AD.17 In this regard, we have shown that pretangle p-tau pathology in CBF neurons correlates with cognitive decline during the progression of AD.18 Recently, we reported that nbM neurons progressively develop intracellular tau-immunoreactivity across the CTE pathological stages proposed by McKee and colleagues,1 similar to AD.3,4,18 In particular, there was a significant increase in pretangle phosphorylated pS422 and oligomeric tau proteins within these vulnerable neurons.4 Moreover, the numbers of neurons containing pretangles and tau oligomers correlated with age at symptom onset and age of death in CTE.4 These findings indicate that early tau changes during NFT development contribute to CBF degeneration in CTE. Moreover, it is likely that following an initial brain injury, there are delayed secondary alterations that develop over days, weeks, months, and possibly years, which result in the perturbation of normal homeostatic mechanisms that can activate cell death and impede pro-survival molecular cascades in CBF neurons with the nbM. In this regard, it has been suggested that TBI causes significant gene expression level changes within vulnerable populations of neurons.19,20 Understanding the molecular cellular pathophysiology underlying CTE injury should be considered key to the identification of putative new targets for the treatment of this disorder. However, our current understanding of the molecular mechanisms underlying neurodegeneration post-CTE in the human brain is limited. In this regard, the molecular fingerprint of CBF neurons within the nbM containing p-tau following TBI requires a strategy that will uncover expression alterations at the cellular level.
Although genomic approaches to study the molecular pathogenesis of brain trauma have been applied to homogenized cortical brain tissue from pre-clinical rodent TBI models,20–22 their application to the human condition is under-represented in the literature.23 Over the past several years, we have employed single cell gene expression profiling to evaluate the molecular pathogenesis of human hippocampal and nbM neurons during the onset of AD.24 Evidence from these gene expression studies reveal dysregulation of select classes of genes associated with neurotrophins and their receptors, cholinergic activity, synaptic proteins, and endosomal/lysosomal markers during the progression of AD.25–28 However, whether similar or different gene classes are altered in tangle-bearing nbM neurons in patients with CTE remains unknown. Herein, we assessed, for the first time, the genetic signature of nbM neurons containing the p-tau pretangle maker, pS422, obtained from subjects who came to autopsy and received a neuropathological staging assessment for CTE (Stages II, III, and IV) using a combination of laser capture microdissection (LCM) and custom-designed microarray analysis.29,30
Methods
Subjects
A total of 17 brains obtained from male athletes who played American football and ice hockey as well as from boxers with a history of repetitive mild TBI that we used in a previous study (see Table 1).4 Two of these subjects were also military veterans. Institutional approval for brain donation was obtained through the Boston University School of Medicine. Institutional approval for postmortem clinical record review, interviews with family members, and neuropathological analysis was obtained through Boston University School of Medicine.
Table 1.
Demographics
| Stage | Gender | Race | Sport/act | Age sport began (y) | Years of play | Age at onset of symptoms (y) | Interval between retirement and symptoms (y) | Interval between symptom onset and death (y) | Age at death (decade) |
|---|---|---|---|---|---|---|---|---|---|
| II | M | C | Football | 16 | 15 | 65 | 34 | 5 | 7 |
| M | AA | Football | 11 | 10 | 45 | 24 | 8 | 5 | |
| M | C | Football | 9 | 20 | 28 | −1 | 2 | 3 | |
| M | C | Semi-pro. football | 8 | 18 | 30 | 4 | 32 | 6 | |
| M | C | College football | 6 | 15 | 21 | 0 | 4 | 2 | |
| M | C | HS football, Wrestling, Pole vaulting | 6 | 11 | 20 | 2.5 | 5 | 2 | |
| Mean ± SD | 9.3 ± 3.7 | 14.8 ± 3.8 | 34.8 ± 17.2 | 10.5 ± 14.7 | 9.3 ± 11.2 | 4.1 ± 2.1 | |||
| III | M | AA | Football | 10 | 17 | 28 | 1 | 28 | 5 |
| M | C | Football | 11 | 23 | 35 | 1 | 11 | 4 | |
| M | C | Football | 14 | 17 | 35 | 4 | 32 | 6 | |
| M | C | Football | 10 | 21 | 40 | 9 | 5 | 4 | |
| M | C | Football | 14 | 15 | 24 | −5 | 36 | 6 | |
| Mean ± SD | 11.8 ± 2.0 | 18.6 ± 3.2 | 32.4 ± 6.3 | 2.0 ± 5.0 | 22.4 ± 13.6 | 5.0 ± 1.0 | |||
| IV | M | AA | Boxing | 12 | 19 | 30 | −1 | 43 | 7 |
| M | C | Football | 10 | 30 | 57 | 17 | 22 | 7 | |
| M | C | Football | 16 | 13 | 68 | 39 | 11 | 7 | |
| M | AA | Football | 9 | 22 | 50 | 19 | 12 | 6 | |
| M | AA | Football/Veteran | 14 | 23 | 65 | 28 | 15 | 8 | |
| M | C | Football/Veteran | 15 | 15 | 32 | 2 | 45 | 7 | |
| Mean ± SD | 12.6 ± 2.8 | 20.3 ± 6.1 | 50.3 ± 16.2 | 17.3 ± 15.2 | 24.6 ± 15.4 | 7.0 ± 0.6 |
AA, African American; C, Caucasian; HS, high school; M, male; Semi-pro, semiprofessional.
Clinical evaluation
Cognitive and behavioral changes and clinical status were determined through postmortem interviews with next of kin performed by a neuropsychologist blinded to postmortem neuropathological status.31 To this end, a semistructured telephonic interview was conducted regarding demographics, athletic history, military service, concussion and brain trauma history, medical history (including neurological, psychiatric, and substance use history), family history, social/occupational history, and reported/observed changes in mood, behavior, motor function, cognition, and activities of daily living.31 To evaluate cognitive, mood, and functional changes, modifications of standard measures/interviews were administered to informants to evaluate their perception of the subject in the months or years prior to death. A review of each individual's medical records was performed, and cases with other comorbidities were excluded.
Neuropathological evaluation
Paraffin-embedded sections were processed for neuropathological evaluation according to the procedure of the Veterans Affairs-Boston University–Concussion Legacy Foundation (VA-BU-CLF) Bain Bank as previously described.32 The following stains were applied to the tissue: Luxol fast blue, hematoxylin and eosin, Bielschowsky's silver, p-tau (AT8), α-synuclein, amyloid-β (Aβ), TAR DNA-binding protein (TDP)-43, phosphorylated TDP-43 (pTDP-43), SMI-31 and SMI-34.33 Neuropathological diagnoses were made blinded by a board-certified neuropathologist.
CTE pathological staging
CTE classification was based on the four pathological stages described previously.32 In Stage I, there are isolated perivascular foci of p-tau NFTs, neuropil threads, and astrocytic tangles mostly in the depths of cerebral sulci of the superior, dorsolateral, lateral, and inferior frontal cortices as well as p-tau NFT degeneration in the locus coeruleus, amygdala, entorhinal cortex, hippocampus, medulla, and cingulate gyrus. In Stage II multiple foci of tau pathology at the depths of the sulci mostly in the superior, dorsolateral, lateral, and inferior frontal, anterior inferior and lateral temporal, inferior and superior parietal, insular and septal cortices, and substantia innominata were described. Stage III exhibits NFTs diffusely in the frontal, temporal and parietal cortices, mainly around small vessels and within the depths of sulci. The hippocampus, entorhinal cortex, amygdala, nbM, and locus coeruleus show extensive neurofibrillary pathology as well as frequent NFTs in the hypothalamus, substantia nigra, and dorsal and median raphe nuclei. Stage IV shows a significant loss in brain weight and pronounced frontal and temporal lobe atrophy. At the light microscopic level, there is severe spongiosus of cortical layer II and widespread neuronal loss, including the substantia nigra, patchy widespread myelin loss, extensive glial tangles, and small NFTs in a patchy irregular distribution throughout frontal, temporal, and parietal cortices as well as extensive NFT degeneration in the insula, septa, temporal cortex, amygdala, hippocampus, entorhinal cortex, substantia nigra, and locus coeruleus. In Stage IV, p-tau pathology also involves the cerebellum, medial lemniscus, and inferior olives of the medulla. CTE stage correlates with the progression of clinical symptoms.32
Immunohistochemistry
An initial set of paraffin-embedded blocks containing the nbM was cut at 8 μm, slide mounted, deparaffinized, rehydrated and boiled in a citric acid solution (pH 6) for 5 min, for antigen retrieval.4 After several washes in 0.1M Tris-buffered saline (TBS, pH 7.4), sections were incubated with 3% H2O2 for 30 min at room temperature to eliminate endogenous peroxidase activity, and blocked with 0.1 M TBS containing 0.04% Triton X-100 and 10% normal goat serum (NGS; Vector Labs, Burlingame, CA) for 1 h. Subsequently, sections were incubated in a solution containing polyclonal rabbit antibody directed against the p-tau epitope pS422 (1:500; Thermo Fisher Scientific Pierce, Rockford, IL), a marker of pretangles mixed in a 0.1 M TBS/2% normal goat serum solution overnight at room temperature.18,34 The next day, sections were washed in TBS and then incubated with the secondary biotinylated goat anti-rabbit IgG (1:200; Vector Labs, Burlingame, CA), and processed with avidin-biotin complex reagent (ABC; Vector Labs, Burlingame, CA). Tissue was then developed in 0.05% 3’, 3’-diaminobenzidine (DAB) and 0.005% H2O2 resulting in a brown reaction product. These sections were counter-stained with Cresyl Violet for identification of the nbM neurons. A second set of sections was dual labeled using the abovementioned pS422 tau antibody and a monoclonal antibody directed against p75NTR (1:500; Thermo Scientific, Waltham, MA), an excellent marker of cholinergic nbM neurons in humans to identify non-tangle-bearing cholinergic neurons at the light microscopic level.4,35
Dual-stained neurons were not evaluated for expression level changes, because of difficulties in consistently identifying these cells with the LCM. All slides were dehydrated in a series of graded concentrations of ethanol, (70, 90, 95, and 100%) but not cover-slipped.
Quantitation of the number of pS422 compared with p75NTR neurons in the nbM
Counts of single labeled pS422 nbM neurons were performed within at least two sections using the Nikon Microphot-FXA microscope at 10 × magnification and normalized to the total number of p75NTR positive neurons as previously described.4 Results are presented as percentage of cell counts.
Single-cell gene expression analysis
Single immunolabeled pS422 and p75NTR nbM neurons (a total of 30 neurons per CTE case pooled/per assay) from CTE Stages II (n = 6), III (n = 5), and IV cases (n = 6) were microaspirated by laser capture microdissection (PALM MicroBeam C IP, Carl Zeiss MicroImaging Inc., Thornwood, NY). The amplification of mRNA was performed with terminal continuation (TC) RNA amplification, which preserves the original quantitative relationships among the transcripts.36–38
Microdissected nbM cells were homogenized in Trizol solution (ThermoFisher Scientific, Waltham, MA) and RNAs were reverse transcribed in the presence of the poly d (T) primer (100 ng/mL) and TC primer (100 ng/mL) in 1 × first strand buffer (ThermoFisher Scientific), 500 μM deoxyribonucleotide triphosphate (dNTP)s, 5 mM dithiothreitol (DTT), 20 U of SuperRNase Inhibitor (ThermoFisher Scientific), and 200 U of reverse transcriptase (ThermoFisher Scientific). Single-stranded complementary DNAs (cDNAs) were digested with RNase H and re-annealed with the primers in a thermal cycler: RNase H digestion step at 37°C, 30 min; denaturation step at 95°C, 3 min; and primer reannealing step at 60°C, 5 min. This step generated cDNAs with double-stranded regions at the primer interface. Samples were then purified by column filtration (Montage PCR filters; Millipore, Billerica, MA). RNAs hybridization probes were synthesized by in vitro transcription with the use of 33P incorporation in 40 mmol/L Tris (pH 7.5); 6 mmol/L MgCl2; 10 mmol/L NaCl; 2 mmol/L spermidine; 2.5 mmol/L DTT; 125 μmol/L adenosine triphosphate (ATP), guanosine triphosphate (GTP), and cytidine triphosphate (CTP); 2.5 μmol/L cold uridine triphosphate (UTP); 20 U of RNase inhibitor; 2 kU of T7 RNA polymerase (Epicentre, Madison, WI); and 60 μCi of 33P-UTP (PerkinElmer, Waltham, MA). The labeling reaction was performed at 37°C for 4 h. Radiolabeled terminal continuation RNA probes were hybridized to custom-designed microarrays without further purification. Single-cell gene array expression was run in triplicate for each case.
Custom-designed microarray platforms and data analysis
Platforms consist of 1 μg of linearized cDNA purified from plasmid preparations adhered to high-density nitrocellulose (Hybond XL, GE Healthcare, Piscataway, NJ).26,38,39 cDNAs were verified by sequence analysis and restriction digestion. Approximately 576 cDNAs were used on our custom array platform. Arrays were hybridized for 24 h in a solution consisting of 6 × saline-sodium phosphate-ethylenediaminetetraacetic acid, 5 × Denhardt's solution, 50% formamide, 0.1% sodium dodecyl sulfate (SDS), and denatured salmon sperm DNA (200 μg/mL) at 42°C in a rotisserie oven. Following the hybridization protocol, arrays were washed sequentially in 2 × saline sodium citrate (SSC)/0.1% SDS, 1 × SSC/0.1% SDS, and 0.5 × SSC/0.1% SDS for 15 min each at 37°C. Arrays were placed in a phosphor screen for 24 h and developed on a phosphor imager (Storm 840, GE Healthcare, Piscataway, NJ).
Hybridization signal intensity was determined utilizing ImageQuant software (GE Healthcare). Briefly, each array was compared with negative control arrays utilizing the respective protocols without any starting RNA. Expression of TC amplified RNA bound to each target minus background was expressed as a ratio of the total hybridization signal intensity of the array (a global normalization approach). Global normalization effectively minimizes variation because of differences in the specific activity of the synthesized probe and the absolute quantity of the probe.29
In situ hybridization
In situ hybridization was performed to validate our single cell expression findings. In situ hybridization was performed on 8 μm paraffin-embedded nbM sections from Stage II and Stage IV CTE cases following the manufacturer's instructions. Customized probe of CLCN5 (Cat. No. 300031, Accession No NM_001127899.3; Advanced Cell Diagnostics, Newark, CA) was used for in situ hybridization using the RNAscope 2.5HD Assay-Brown Kit (Cat No. 322300; Advanced Cell Diagnostics, Newark, CA). The probe region covers 1164 bp spanning nucleotides 771–1934. Expression of dihydrodipicolinate reductase mRNA from Bacillus subtilis was used as negative control. Images were obtained using a Nikon Eclipse microscope
Statistical analysis
Demographic, clinical characteristics and cell counts were evaluated across pathological CTE Stages II, III, and IV using a one way analysis of variance (ANOVA) and the Kruskal–Wallis test followed by Holm–Sidak and Dunn's post-hoc test for multiple comparisons as appropriate with statistical significance set at 0.05 (two tailed).
Relative changes in total hybridization signal intensity and in individual mRNAs were analyzed by one way ANOVA with a Holm–Sidak post–hoc method for multiple comparisons. False discovery rates were also estimated as described previously.29,40 Expression levels were analyzed and clustered using bioinformatics and graphics software packages (GeneLinker Gold, Improved Outcomes Inc., Kingston, ON). Expression levels of select transcripts were correlated with clinical pathological variables using a Pearson test. The level of statistical significance was set at p < 0.05.
Results
Case demographics
The demographics of these 17 cases categorized as CTE Stage II, III, or IV1,3 have been previously published4 (see Table 1). Briefly, Stage II cases (n = 6; mean age 44.2 ± 20.0) included three NFL football players, one college football player, one semiprofessional football player, and one high school football player/wrestler/pole vaulter. Stage III cases (n = 5; mean age 54.8 ± 9.3) were all NFL football players. Stage IV cases (n = 6; mean age 75.0 ± 6.8) included five NFL football players, of which two were also military veterans, and a professional boxer. The groups differed by age (p = 0.008), with Stage IV cases significantly older than Stage II cases, but no differences were found when subjects were compared by age to the time at which sport began, years of play, age at symptom onset, time interval between retirement and symptom onset, and interval between symptom onset and death (Table 1). As we reported previously,4 age when sport began was correlated with age at symptoms onset (r = 0.60, p < 0.008), greater interval between symptom onset and death (r = 0.48, p < 0.04), and age at death (r = 0.72, p < 0.0003). Age at symptom onset was related to increased interval between symptom onset and retirement from sport (r = 0.85, p < 0.00001), and with age at death (r = 0.71, p = 0.0005). Moreover, age at death was associated with longer intervals from symptom onset to either retirement from sport (r = 0.52, p = 0.02) or death (r = 0.64, p = 0.003).
Pathological evaluation
The present CTE subjects displayed other pathologies including: 13/18 (72%) with pTDP-43 inclusions, 6/18 (33%) with diffuse Aβ plaques, 3/18 (17%) with neuritic Aβ plaques, and 1/18 (6%) with Lewy body disease. Comorbid pathologies were more common in older subjects with a higher CTE stage.1
Quantitation of the ratio of pS422 compared with p75NTR nbM neurons
Quantitative analysis of pS422 and p75NTR immunolabeled neurons within the nbM revealed that the percentage of pS422-positive neurons significantly increased in Stage IV compared with Stage II, but not Stage III (Kruskal–Wallis with Dunn's post-hoc test, p < 0.001) (Fig. 1).
FIG. 1.

Box plot showing the percentage of pS422-positive relative to total p75NTR -positive nucleus basalis of Meynert (nbM) neurons in chronic traumatic encephalopathy (CTE) Stages II, III, and IV. Percentage of pS422-positive neurons was significantly increased in Stage IV compared with Stage II but not Stage III (Kruskal–Wallis follows Dunn's post-hoc test; *p < 0.01).
Gene expression of pS422 immunopositive nbM neurons
Custom-designed microarrays were used to compare gene expression profiles in pS422 immunopositive nbM neurons among CTE cases displaying neuropathological stages II, III, and IV (Fig. 2). Quantitative analysis revealed that 10/576 total gene transcripts were significantly altered in between CTE stages (Table 2, Fig. 3). Gene ontology category classification included cholinergic and monoaminergic metabolism, endocytosis, intracellular signaling, and cytoskeleton/tau-related biology (Table 2). The nicotinic cholinergic receptor subunit β-2 (CHRNB2) (Fig. 3A; Holm-Sidak post-hoc test, p = 0.03), monoaminergic enzymes catechol-O-methyltransferase (COMT) (Holm–Sidak post-hoc, p = 0.038) and dopa decarboxylase (DDC) (Holm–Sidak post-hoc test, p = 0.005) transcripts were decreased in CTE Stage III compared with Stage II. Expression levels for calpain 2 (CAPN2), a cytoplasmic protease capable of cleaving tau, were downregulated in CTE Stage II compared with Stage III (Fig. 3B; one way ANOVA, Holm–Sidak post–hoc, p < 0.001), and upregulated in CTE Stage IV compared with Stage III (p = 0.004). Microtubule-associated protein 2 (MAP2) mRNA was upregulated in CTE Stage IV compared with Stage II (Fig. 3B; Holm–Sidak post-hoc, p = 0.019). Cell homeostasis- and neurotransmission-related chloride channels CLCN4 and CLCN5 expression levels were downregulated in CTE Stage IV compared with Stage II (Fig. 3C; one way ANOVA, Holm–Sidak post-hoc, p < 0.05), and only CLCN5 was significantly downregulated in CTE Stage IV compared with Stage III (Fig. 3C; Holm–Sidak post-hoc, p < 0.05). The scaffolding protein caveolin 1 (CAV1) and cortical development/cytoskeletal element lissencephaly 1 (LIS1) transcripts were also decreased in CTE Stage IV compared with Stage III (Fig. 3C; Holm–Sidak post-hoc, p < 0.05). The intracellular signaling cascade member adenylate cyclase 3 (ADCY3) was downregulated in CTE Stage IV compared with Stages II (Fig. 3C; Holm–Sidak post-hoc, p < 0.016) and III (Fig. 3C; Holm–Sidak post-hoc, p < 0.006). Table 3 shows examples of 10 randomly chosen genes (β actin [ACTB], glutamate receptor 1 [GRM1], amyloid precursor [A4] protein 1 [APLP1], heat shock protein [DNAJA1], synapsin 1 [SYN1], bcl2-relate protein [BAK1], potassium channel [KCNA1], galanin peptide [GAL], NGFI-A binding protein 1 transcript factor [NAB1] and integrin α 1 [ITGA1]) displaying stable transcript levels in nbM pS422 positive neurons across CTE stages (one way ANOVA, p > 0.05).
FIG. 2.
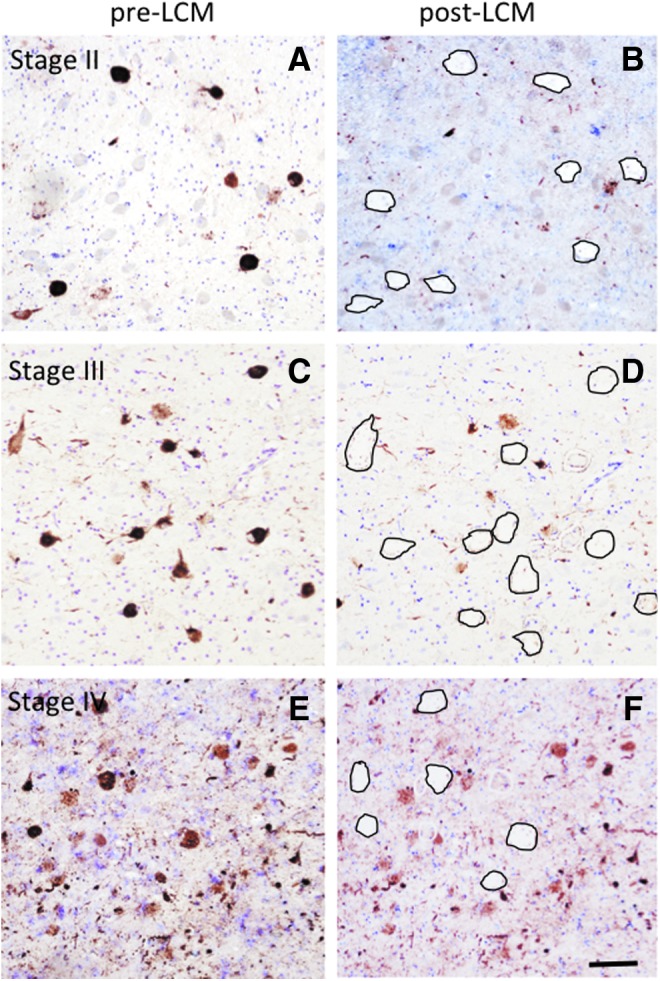
Low power images showing pre (A, C, E) and post (B, D, F) laser capture microdissection (LCM) extraction of pS422-positive nucleus basalis of Meynert (nbM) neurons in a chronic traumatic encephalopathy (CTE) stage II, III, and IV subject. Black ovals outline the location of pS422-positive nbM after LCM retrieval (B, D, E). All sections were counterstained with Cresyl Violet. Scale bar = 50 μm.
Table 2.
Summary of Genes Altered in nbM pS422 Neurons across CTE Stages
| Gene abbreviation | Group | CTE Stage II | CTE Stage III | CTE Stage IV | p value | Pairwise comparison (p value) | Function |
|---|---|---|---|---|---|---|---|
| CLCN4 | CH | 4.11 ± 0.35a | 3.61 ± 0.43 | 3.54 ± 0.27 | 0.026 | II > IV (0.037) | Cell homeostasis |
| CLCN5 | CH | 6.99 ± 0.87 | 6.96 ± 0.94 | 5.7 ± 0.47 | 0.025 | II > IV (0.045) III > IV (0.044) |
Cell homeostasis |
| CAV1 | CYT | 4.21 ± 0.33 | 4.67 ± 0.82 | 3.57 ± 0.46 | 0.018 | III > IV (0.017) | Endocytosis |
| ADCY3 | GP | 1.70 ± 0.07 | 1.75 ± 0.10 | 1.54 ± 0.11 | 0.005 | II > IV (0.016) III > IV (0.006) |
Intracellular signaling |
| LIS1 | DV | 5.20 ± 0.26 | 5.62 ± 0.45 | 4.85 ± 0.38 | 0.012 | III > IV (0.011) | Cortical development cytoskeleton |
| CHRNB2 | CHOL | 3.67 ± 0.59 | 2.87 ± 0.41 | 3.18 ± 0.28 | 0.030 | II > III (0.031) | Neurotransmission |
| COMT | MONO | 3.01 ± 0.19 | 2.61 ± 0.24 | 2.92 ± 0.26 | 0.033 | II > III (0.038) | Neurotransmission |
| DDC | MONO | 4.66 ± 0.30 | 3.82 ± 0.37 | 4.30 ± 0.40 | 0.007 | II > III (0.005) | Neurotransmission |
| MAP2 | CYT | 1.48 ± 0.11 | 1.66 ± 0.15 | 1.73 ± 0.15 | 0.018 | II < IV (0.019) | Cytoskeleton tau |
| CAPN2 | PROT | 5.59 ± 0.38 | 4.50 ± 0.33 | 5.31 ± 0.35 | <0.001 | II > III (<0.001) IV > III (0.004) |
Cell metabolism tau |
Mean ± SD (arbitrary units)
CH, monovalent and divalent channels; CHOL, cholinergic neurotransmission; CTE, chronic traumatic encephalopathy; CYT, cytoskeletal elements; DV, development-related elements; GP, G-protein subunits; MONO, monoamine neurotransmission, nbM, nucleus basalis of Meynert; PROT, protease-related markers.
FIG. 3.

Linear representation showing the mean values of the gene expression level changes for neurotransmission (DDC, CHRNB2, COMT) (A), cytoskeleton/cell metabolism (CAPN2, LIS1, MAP2) (B), and cell homeostasis/endocytosis and intracellular signaling (CLN5, CLN4, CAV1, ADCY3) (C) transcript across CTE stages. Note that MAP2 and CAPN2 mRNA levels were upregulated in CTE Stage IV compared with Stages II (p = 0.019) and III (p = 0.004), respectively, whereas the expression of the other genes was downregulated during the pathological progression of CTE (one way analysis of variance [ANOVA]). Holm–Sidak comparisons indicated by: *II vs. III; #II vs. IV, and $III vs. IV. p < 0.05.
Table 3.
Example of Stable Gene Transcripts in nbM pS422 Positive Neurons across CTE Stages
| Gene abbreviation | Group | CTE Stage II | CTE Stage III | CTE Stage IV | p value | Pairwise comparison (p value) | Function |
|---|---|---|---|---|---|---|---|
| ACTB | CYT | 2.29 ± 0.28a | 2.49 ± 0.22 | 2.51 ± 0.18 | 0.24 | – | Cytoskeleton |
| GRM1 | GLUR | 2.23 ± 0.29 | 2.46 ± 0.21 | 2.43 ± 0.17 | 0.22 | – | Neurotransmission |
| APLP1 | AD | 2.14 ± 0.34 | 2.20 ± 0.35 | 2.09 ± 0.34 | 0.86 | – | Cell metabolism β-amyloid |
| DNAJA1 | SR | 2.50 ± 0.34 | 2.65 ± 0.29 | 2.55 ± 0.26 | 0.69 | – | Cell metabolism |
| SYN1 | SYN | 6.54 ± 1.83 | 6.03 ± 1.65 | 6.16 ± 2.19 | 0.88 | – | Synaptogenesis |
| BAK1 | CD | 2.63 ± 0.39 | 2.74 ± 0.27 | 2.64 ± 0.23 | 0.77 | – | Cell metabolism |
| KCNA1 | CH | 2.54 ± 0.13 | 2.45 ± 0.20 | 2.44 ± 0.13 | 0.52 | – | Cell homeostasis |
| GAL | PEP | 1.86 ± 0.20 | 2.03 ± 0.32 | 1.99 ± 0.39 | 0.63 | – | Neuromodulator |
| NAB1 | TF | 2.86 ± 0.45 | 3.14 ± 0.52 | 2.96 ± 0.40 | 0.56 | – | Cell homeostasis |
| ITGA1 | CYT | 3.76 ± 0.29 | 3.66 ± 0.37 | 3.57 ± 0.33 | 0.64 | – | Cell homeostasis |
Mean ± SD (arbitrary units).
AD, Alzheimer's disease related genes; CD, cell death-related genes; CH, monovalent and divalent channels; CTE, chronic traumatic encephalopathy; CYT, cytoskeletal elements; GLUR, glutamatergic neurotransmission; nbM, nucleus basalis of Meynert; PEP, peptides and peptide receptors; SR, stress response; SYN, synaptic-related markers; TF, transcription factor.
Gene expression in non-tangle bearing p75NTR immunopositive neurons across CTE stages
Evaluation of the expression levels of those transcripts altered in pS422-containing neurons remained stable within non-tangle-bearing p75NTR immunopositive nbM neurons across CTE stages (Fig. 4).
FIG. 4.

Histogram illustrating gene expression for neurotransmission (DDC, CHRNB2, COMT) (A), cytoskeleton/cell metabolism (CAPN2, L1S1, MAP2) (B), and cell homeostasis/endocytosis and intracellular signaling (CLN5, CLN4, CAV1, ADCY3) (C) transcripts in non-tangle-bearing nucleus basalis of Meynert (nbM) p75NTR-positive neurons across chronic traumatic encephalopathy (CTE) stages. Note that mRNA levels were unchanged in non-tangle-bearing neurons across the CTE stages (one way analysis of variance [ANOVA]). p < 0.05.
In situ hybridization
In situ hybridization of the CLCN5 gene was performed to validate our single-cell gene array findings. We found that CLCN5 expression was less abundant in CTE Stage IV compared with Stage II cases (Fig. 5) confirming our findings by custom-designed array analysis.
FIG. 5.
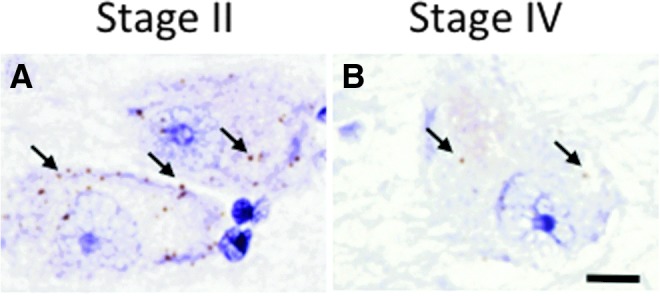
CLCN5 in-situ hybridization images showing differential labeling of nucleus basalis of Meynert (nbM) neurons from chronic traumatic encephalopathy (CTE) Stage II (A) and Stage IV (B) subjects. Note that there are many more CLCN5 mRNA copies (brown dots; arrows) in CTE Stage II than in CTE Stage IV. Sections were counterstained with hematoxylin. Scale bar = 10 μm.
Expression level correlations
Transcript levels of CHRNB2, COMT, and DDC in pS422-immunolabeled neurons showed a strong positive correlation with each other across CTE stages (Fig. 6A, B; Pearson correlation, coefficient [r] > 0.7, p < 0.001). CAV1 correlated positively with ADCY3 (Fig. 6C; r = 0.5, p = 0.03) and LIS1 (Fig. 6D; r = 0.8, p < 0.0001) transcript levels across CTE stages. MAP2 transcript levels showed a weak negative correlation with CAPN2 (r = - 0. 5, p < 0.04), whereas chloride channel CLCN4 and CLCN5 mRNA levels did not correlate with each other across CTE stages. Notably, only increases in MAP2 transcript levels were positively associated with age at which sport began (Fig. 6E; r = 0.5, p = 0.04), whereas CHRNB2 transcripts levels were negatively associated with years played across the CTE stages (Fig. 6F; r = −0.5, p = 0.03).
FIG. 6.

DDC transcript levels correlated positively with COMT (A) and CHRNB2 (B) expression l within the pS422-positive nucleus basalis of Meynert (nbM) across chronic traumatic encephalopathy (CTE) stages. Likewise, CAV1 mRNA levels correlated positively with ADCY3 (C) and LIS1 (D) levels in CTE. MAP2 (E) transcript levels correlated positively with the age at which sport began, whereas CHRNB2 (F) correlated negatively with years of nplay in CTE. Stage II, red circles; Stage III, blue circles; Stage IV, green circles.
Discussion
The present study used single population gene expression profiling to examine the molecular alterations in tau-positive nbM neurons obtained from athletes and military persons who had antemortem brain trauma. Postmortem neuropathological analysis was performed on brain sections immunostained for tau and other pathological hallmarks of neurodegenerative disorders by the Brain Trauma Laboratory at VA Boston Healthcare System.41 Based on the distribution of tau profiles, the cases were categorized as: Stage II, Stage III, or Stage IV.3,41 Single population fingerprinting revealed significant dysregulation of select genes in several relevant gene ontology categories, including neurotransmitter, cell metabolism and homeostasis, and intracellular signaling and cytoskeletal elements within nbM neurons containing the tau pretangle marker pS422 between CTE stages.
Here we report a reduction in the cholinergic neurotransmitter-associated transcript CHRNB2, which encodes the acetylcholine receptor subunit β 2, within nbM neurons in CTE Stage III compared to with Stage II. These findings are consistent with clinical pathological investigations demonstrating cholinergic-related deficits post-TBI in humans, including reduced ChAT activity and immunoreactivity within nbM neurons,12 as well as downregulation of vAChT, and AChE.12,42 Although we did not find alterations in genes related to any of these cholinergic markers, perhaps it is possible that the reduced CHRNB2 expression in CTE Stage III compared with Stage II indicates that the onset of defects in chemical transmission across synapses occurs later in the disease process. Although CHRNB2 has not been linked to TBI previously, studies suggest that CHRNB2 may be a candidate gene for neurodegenerative and neuropsychiatric disorders including schizophrenia43 and AD.44,45 Interestingly, we reported an upregulation of the α7 subunit of the nicotinic receptor (CHRNA7) expression40 and, recently, a downregulation of the CHRNB246 in nbM neurons during the progression of AD. These data suggest that the cholinergic neurotransmission fingerprint of brain trauma resembles, in part, that seen in dementia.
The reduction in COMT and DDC expression within the nbM neurons in CTE Stage III compared with Stage II that was somewhat unexpected, as both genes are mainly associated with dopaminergic metabolism. On the other hand, dopamine plays a role in mood disorders, which are often reported in cases of CTE. Of interest are findings from immunohistochemical studies in primates, including humans, indicating that cells in the nbM may produce dopamine.47–50 Recently, we found a dysregulation of select dopaminergic receptor genes (DRD1 and DRD4) in tau-containing nbM neurons in AD (Tierman and coworkers, in preparation), which is further indication of changes in gene classes related to the dopaminergic metabolism within nbM neurons in CTE and AD.
Expression of CAPN2, a calcium-activated protease implicated in a wide variety of brain-related functions, including the regulation of cell death pathways and synaptic plasticity, was downregulated in CTE Stage II compared with Stage III, and upregulated in Stage IV compared with Stage III.51–54 Numerous in vivo and in vitro investigations support the involvement of calpains in various trauma-induced disorders, including TBI.55–57 Rodent models of cortical brain trauma indicate that various calpain markers are elevated early and then decreased post-TBI. For example, in septohippocampal neuron cultures, mechanical injury induced a persistent elevation in calpain-mediated breakdown products as well as fragmented and beaded dendrites consistent with apoptotic cell death.58 TBI also produces a loss of the cytoskeletal protein MAP2 marked by early changes in dendritic and cellular protein labeling following controlled cortical impact in rodent brain.59–61 We report upregulation of MAP2 expression in the late pathological stage of CTE, suggesting that an increase in MAP2 mRNA levels may be an attempt to compensate for calpain-induced neuronal toxicity.62 Because activated calpain cleaves MAP2, resulting in an alteration in neuronal architecture,54 attenuating calpain abundance and/or activity may modify the molecular pathobiology leading to CTE. Consistent with this hypothesis, targeted gene inactivation of calpain-1 was shown to suppress cortical degeneration in calpin-1 null mice post-TBI.57 Moreover, human tau accumulation inhibits nicotinic acetylcholine receptor α4 by the activation of calpain-2, indicating that intracellular tau causes cholinergic impairments in neuronal cells.63 Perhaps targeted inhibition of calpains may offer a novel therapeutic target not only for tau dysregulation, but also for cholinergic deficits in TBI/CTE.
CAV1 encodes the scaffolding caveolin1 (cav-1) protein found in the caveolae plasma membrane in endocytic processes was downregulated in nbM neurons during CTE progression. Similarly, we reported a downregulation of the endocytic-related CAV2 transcript in nbM neurons in AD.64 These findings suggest that caveloin and endocytic processes in nbM neurons are pathologically compromised post-brain trauma as well as in AD.64 Other studies indicate that cav-1 protein levels in the adult cortex are increased in the endothelium for several days after cold cortical injury,65,66 brain ischemia, and spinal cord injury.67 A similar response was reported following controlled cortical impact in juvenile rats.68,69 Interestingly, astrogliosis and microgliosis are increased in the brains of cav-1 knockout mice post brain insult, suggesting that cav-1 may play a role in the activation of a neuroinflammatory response following brain injury.70,71 Cav-1 knockout mice exhibit enhanced anxiety, impaired spatial memory, and altered cholinergic function similar to findings reported in patients with CTE.72 In addition, a beneficial role for cav-1 delivery has been demonstrated in experiments using cav-1 knockout mice.68 Whether a beneficial affect would be true in humans remains to be determined.
Our findings of a downregulation of ADCY3 expression suggest a defect in the ability of nbM neurons to catalyze the formation of the secondary messenger cyclic adenosine monophosphate (cAMP), which is associated with neuronal survival, and is reduced for many weeks following TBI.73 Restoration of the cAMP pathway improves recovery after brain injury, possibly by stimulating transcription of cell survival genes,74–77 which are reduced in the hippocampus and parietal cortices following fluid-percussion induced brain injury.73 Rolipram, a selective inhibitor of the cAMP-degrading drug phosphodiesterase IV, restored cAMP in the hippocampus, improved neuronal survival compared with vehicle controls, reduced total tau, and improved cognitive performance in a tau mutant mouse.78,79 Perhaps this drug may be useful for the clearance of putative toxic tau isoorms, and should be considered as a treatment approach for human CTE.
The decrease in expression of LIS1 in CTE Stage IV compared with Stage III has not been reported in the pre-clinical or clinical literature. LIS1 is a member of the WD40-repeat family of proteins, and was the first gene identified with neuronal migration defects in the developing and adult brain.80,81 The functional significance of a reduction in LIS1 expression in CTE tau-positive nbM neurons remains to be determined. However, studies indicate a strong relationship between LIS1 and microtubule regulation, microtubule-based motor proteins,82 preservation of normal microtubule network organization,83 neuritic structure, axonal transport, and synaptic function.81,84 Taken together, these findings suggest that the downregulation of the LIS1 gene may be associated with neuronal architecture alterations in advanced CTE.
CLCN4 and CLCN5 are members of the CLCN family of voltage-dependent chloride channel genes.85 Encoded CLCN proteins are mainly localized to the endosomal membrane, and subserve a wide range of cellular functions, including the regulation of electrical excitability of neurons.86 A search of the literature failed to reveal studies showing an interaction between the CLCN4 and CLCN5 genes following brain trauma. Whether the reduction in CLCN5 expression found in the present study is related to mechanisms of cellular apoptosis in nbM neurons in CTE remains to be investigated.
Notable caveats related to the current study are acknowledged. There are currently a limited number of neuropathologically verified CTE cases, effectively limiting the number of basal forebrain samples that we could analyze in this study. The pathological diagnosis of CTE and subsequent staging is performed on paraffin-embedded tissue blocks accrued from donated brains,3 and frozen brain tissue unfortunately is rarely available. A major caveat related to the lack of frozen basal forebrain tissue is the inability to perform quantitative polymerase chain reaction (qPCR) for validation of the current single cell expression data. To address this obstacle, we collaborated with Advanced Cell Diagnostics to generate a customized in situ hybridization probe to independently assess CLCN5 expression. Using this probe, we demonstrated that CLCN5 was less abundant in nbM neurons in CTE Stage IV than in Stage II, validating our custom-designed microarray findings. Another problem is that non-CTE control cases were not available for comparison, as cohorts of age-matched military personnel and/or athletes without TBI/CTE have not been generated to date. Because non-CTE control subjects are not available, we analyzed our data by neuronal chemical phenotype across CTE stages. This scheme allowed only for an analysis of expression profiling in ps422-immunoreactive tau nbM neurons compared and contrasted across CTE stages. Future studies will attempt comparisons of dual labeled neurons when larger cohorts of CTE and control basal forebrain samples are available.
Because this is the first single population expression study of human tau-positive nbM neurons obtained from post-TBI subjects, it is difficult to compare and contrast the current findings with other published reports. For example, a genomic investigation of biopsied frontal and temporal cortical tissue obtained during surgery from TBI patients and homogenized prior to microarray analysis detected 1200 genes, with ∼8% of them showing differential regulation.23 Although there was no overlap between the specific transcripts found in the present study, the broader gene ontology categories showing mRNA alterations displayed similarities, including transcriptional regulation, energy metabolism, and signal transduction. A key feature of the present single population microarray analysis study using specific phospho-tau labeled vulnerable nbM neurons is that it allows the parsing of gene expression signatures from adjacent neuronal and non-neuronal cell types. Further, these findings provide a glimpse into critical mechanistic information at the cellular molecular level related to progressive CTE, which may be used as the infrastructure for finding novel diagnostic and treatment strategies.
Conclusion
In summary, we found that select genes related to cholinergic/monoaminergic neurotransmission, cell survival/death, and signal transduction, among other relevant gene ontology categories, are differentially regulated within nbM neurons during distinct pathological CTE stages. Interestingly, few of the specific transcripts dysregulated in CTE nbM neurons have been reported in single cell array studies of this cell population during the progression of AD.25,87,88 In AD, we found significant changes in a myriad of genes including synaptic, endosomal-lysosomal autophagic, and neurotrophin receptor categories, which is notable, because both AD and CTE contain NFT pathology with in the nbM.4,24–28,88 The differences in altered expression profiles may be related to different mechanisms underlying nbM degeneration in each of these neurological disorders as well as the greater access to AD and age-matched control cases. Age may be a factor as CTE subjects are younger than many AD cases. Neuronal degeneration in CTE results from excessive concussive impacts over time, where AD onset is most likely driven by a confluence of intracellular events and possibly the aging process itself. Trauma also cannot be excluded from AD pathogenesis, although most sporadic AD patients are not professional athletes or military personnel with brain injuries. In conclusion, we provide an initial assessment of tau-containing nbM neurons across CTE stages, and demonstrate select alterations in several genes that may be useful for transcriptional-based drug discovery purposes.
Acknowledgments
This material is based on work supported by the United States Army Medical Research and Material Command and from the United States Department of Veterans Affairs (Chronic Effects of Neurotrauma Consortium) under Award No. W81XWH-13-2-0095. The United States Army Medical Research Acquisition Activity, 820 Chandler Street, Fort Detrick MD 21702-5014 is the awarding and administering acquisition office. Any opinions, findings, conclusions or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the views of the United States government, or the United States Department of Veterans Affairs, and no official endorsement should be inferred. An award from the Department of Veterans Affairs (No. I01 RX001774), National Institute of Neurological Disorders and Stroke, National Institute of Biomedical Imaging and Bioengineering (U01NS086659-01), National Institute of Aging Boston University AD Center (P30AG13846, supplement 0572063345-5), the Veterans Affairs Biorepository (CSP 501), the National Operating Committee on Standards for Athletic Equipment, the Concussion Legacy Foundation, the Andlinger Foundation and the World Wrestling Entertainment and the National Football League supported this work.. The authors gratefully acknowledge the use of the resources and facilities at the Edith Nourse Rogers Memorial Veterans Hospital (Bedford, MA) and Barrow Neurological Institute Department of Neuropathology (Phoenix, AZ).
Author Disclosure Statement
No competing financial interests exist.
References
- 1.McKee A.C., Stern R.A., Nowinski C.J., Stein T.D., Alvarez V.E., Daneshvar D.H., Lee H.S., Wojtowicz S.M., Hall G., Baugh C.M., Riley D.O., Kubilus C.A., Cormier K.A., Jacobs M.A., Martin B.R., Abraham C.R., Ikezu T., Reichard R.R., Wolozin B.L., Budson A.E., Goldstein L.E., Kowall N.W., and Cantu R.C. (2013). The spectrum of disease in chronic traumatic encephalopathy. Brain 136, 43–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stein T.D., Alvarez V.E., and McKee A.C. (2014). Chronic traumatic encephalopathy: a spectrum of neuropathological changes following repetitive brain trauma in athletes and military personnel. Alzheimers Res. Ther. 6, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McKee A.C., Stein T.D., Kiernan P.T., and Alvarez V.E. (2015). The neuropathology of chronic traumatic encephalopathy. Brain Pathol. 25, 350–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mufson E.J., Perez S.E., Nadeem M., Mahady L., Kanaan N.M., Abrahamson E.E., Ikonomovic M.D., Crawford F., Alvarez V., Stein T., and McKee A.C. (2016). Progression of tau pathology within cholinergic nucleus basalis neurons in chronic traumatic encephalopathy: a chronic effects of neurotrauma consortium study. Brain Inj. 30, 1399–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mesulam M.M., Mufson E.J., Levey A.I., and Wainer B.H. (1983). Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J. Comp. Neurol. 214, 170–197 [DOI] [PubMed] [Google Scholar]
- 6.DeKosky S.T., Ikonomovic M.D., Styren S.D., Beckett L., Wisniewski S., Bennett D.A., Cochran E.J., Kordower J.H., and Mufson E.J. (2002). Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann. Neurol. 51, 145–155 [DOI] [PubMed] [Google Scholar]
- 7.Whitehouse P.J., Price D.L., Clark A.W., Coyle J.T., and DeLong M.R. (1981). Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann. Neurol. 10, 122–126 [DOI] [PubMed] [Google Scholar]
- 8.Whitehouse P.J., Price D.L., Struble R.G., Clark A.W., Coyle J.T., and Delon M.R. (1982). Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science 215, 1237–1239 [DOI] [PubMed] [Google Scholar]
- 9.Mufson E.J., Mahady L., Waters D., Counts S.E., Perez S.E., DeKosky S.T., Ginsberg S.D., Ikonomovic M.D., Scheff S.W., and Binder L.I. (2015). Hippocampal plasticity during the progression of Alzheimer's disease. Neuroscience 309, 51–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arciniegas D.B. (2003). The cholinergic hypothesis of cognitive impairment caused by traumatic brain injury. Curr. Psychiatry Rep. 5, 391–399 [DOI] [PubMed] [Google Scholar]
- 11.Murdoch I., Nicoll J.A., Graham D.I., and Dewar D. (2002). Nucleus basalis of Meynert pathology in the human brain after fatal head injury. J. Neurotrauma 19, 279–284 [DOI] [PubMed] [Google Scholar]
- 12.Pike B.R., and Hamm R.J. (1997). Activating the posttraumatic cholinergic system for the treatment of cognitive impairment following traumatic brain injury. Pharmacol. Biochem. Behav. 57, 785–791 [DOI] [PubMed] [Google Scholar]
- 13.Tenovuo O. (2005). Central acetylcholinesterase inhibitors in the treatment of chronic traumatic brain injury-clinical experience in 111 patients. Prog. Neuropsychopharmacol. Biol. Psychiatry 29, 61–67 [DOI] [PubMed] [Google Scholar]
- 14.Masanic C.A., Bayley M.T., VanReekum R., and Simard M. (2001). Open-label study of donepezil in traumatic brain injury. Arch. Phys. Med. Rehabil. 82, 896–901 [DOI] [PubMed] [Google Scholar]
- 15.Walker W., Seel R., Gibellato M., Lew H., Cornis-Pop M., Jena T., and Silver T. (2004). The effects of Donepezil on traumatic brain injury acute rehabilitation outcomes. Brain Inj. 18, 739–750 [DOI] [PubMed] [Google Scholar]
- 16.Kaye N.S., Townsend J.B., 3rd., and Ivins R. (2003). An open-label trial of donepezil (aricept) in the treatment of persons with mild traumatic brain injury. J. Neuropsychiatry Clin. Neurosci. 15, 383–385 [DOI] [PubMed] [Google Scholar]
- 17.Bierer L.M., Hof P.R., Purohit D.P., Carlin L., Schmeidler J., Davis K.L,. and Perl D.P. (1995). Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer's disease. Arch. Neurol. 52, 81–88 [DOI] [PubMed] [Google Scholar]
- 18.Vana L., Kanaan N.M., Ugwu I.C., Wuu J., Mufson E.J., and Binder L.I. (2011). Progression of tau pathology in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer's disease. Am. J. Pathol. 179, 2533–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marciano P.G., Eberwine J.H., Ragupathi R., Saatman K.E., Meaney D.F., and McIntosh T.K. (2002). Expression profiling following traumatic brain injury: a review. Neurochem. Res. 27, 1147–1155 [DOI] [PubMed] [Google Scholar]
- 20.Raghupathi R., McIntosh T.K., and Smith D.H. (1995). Cellular responses to experimental brain injury. Brain Pathol. 5, 437–442 [DOI] [PubMed] [Google Scholar]
- 21.Rojo D.R., Prough D.S., Falduto M.T., Boone D.R., Micci M.A., Kahrig K.M., Crookshanks J.M., Jimenez A., Uchida T., Cowart J.C., Hawkins B.E., Avila M., DeWitt D.S., and Hellmich H.L. (2011). Influence of stochastic gene expression on the cell survival rheostat after traumatic brain injury. PLoS One 6, e23111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Redell J.B., Moore A.N., Grill R.J., Johnson D., Zhao J., Liu Y., and Dash P.K. (2013). Analysis of functional pathways altered after mild traumatic brain injury. J. Neurotrauma 30, 752–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michael D.B., Byers D.M. and Irwin L.N. (2005). Gene expression following traumatic brain injury in humans: analysis by microarray. J. Clin. Neurosci. 12, 284–290 [DOI] [PubMed] [Google Scholar]
- 24.Mufson E.J., Ikonomovic M.D., Counts S.E., Perez S.E., Malek-Ahmadi M., Scheff S.W., and Ginsberg S.D. (2016). Molecular and cellular pathophysiology of preclinical Alzheimer's disease. Behav. Brain. Res. 311, 54–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mufson E.J., Counts S.E., Che S., and Ginsberg S.D. (2006). Neuronal gene expression profiling: uncovering the molecular biology of neurodegenerative disease. Prog. Brain Res. 158, 197–222 [DOI] [PubMed] [Google Scholar]
- 26.Ginsberg S.D., Elarova I., Ruben M., Tan F., Counts S.E., Eberwine J.H., Trojanowski J.Q., Hemby S.E., Mufson E.J., and Che S. (2004). Single-cell gene expression analysis: implications for neurodegenerative and neuropsychiatric disorders. Neurochem. Res. 29, 1053–1064 [DOI] [PubMed] [Google Scholar]
- 27.Ginsberg S.D., Mufson E.J., Alldred M.J., Counts S.E., Wuu J., Nixon R.A., and Che S. (2011). Upregulation of select rab GTPases in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer's disease. J. Chem. Neuroanat. 42, 102–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tiernan C.T., Ginsberg S.D., Guillozet-Bongaarts A.L., Ward S.M., He B., Kanaan N.M., Mufson E.J., Binder L.I., and Counts S.E. (2016). Protein homeostasis gene dysregulation in pretangle-bearing nucleus basalis neurons during the progression of Alzheimer's disease. Neurobiol. Aging 42, 80–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ginsberg S.D. (2008). Transcriptional profiling of small samples in the central nervous system. Methods Mol. Biol. 439, 147–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ginsberg S.D., Alldred M.J., Counts S.E., Cataldo A.M., Neve R.L., Jiang Y., Wuu J., Chao M.V., Mufson E.J., Nixon R.A., and Che S. (2010). Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer's disease progression. Biol. Psychiatry 68, 885–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mez J., Solomon T.M., Daneshvar D.H., Murphy L., Kiernan P.T., Montenigro P.H., Kriegel J., Abdolmohammadi B., Fry B., Babcock K.J., Adams J.W., Bourlas A.P., Papadopoulos Z., McHale L., Ardaugh B.M., Martin B.R., Dixon D., Nowinski C.J., Chaisson C., Alvarez V.E., Tripodis Y., Stein T.D., Goldstein L.E., Katz D.I., Kowall N.W., Cantu R.C., Stern R.A., and McKee A.C. (2015). Assessing clinicopathological correlation in chronic traumatic encephalopathy: rationale and methods for the UNITE study. Alzheimers Res. Ther. 7, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKee A.C., and Daneshvar D.H. (2015). The neuropathology of traumatic brain injury. Handb. Clin. Neurol. 127, 45–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKee A.C., Cantu R.C., Nowinski C.J., Hedley-Whyte E.T., Gavett B.E., Budson A.E., Santini V.E., Lee H.S., Kubilus C.A., and Stern R.A. (2009). Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J. Neuropathol. Exp. Neurol. 68, 709–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guillozet-Bongaarts A.L., Cahill M.E., Cryns V.L., Reynolds M.R., Berry R.W., and Binder L.I. (2006). Pseudophosphorylation of tau at serine 422 inhibits caspase cleavage: in vitro evidence and implications for tangle formation in vivo. J. Neurochem. 97, 1005–1014 [DOI] [PubMed] [Google Scholar]
- 35.Mufson E.J., Bothwell M., Hersh L.B., and Kordower J.H. (1989). Nerve growth factor receptor immunoreactive profiles in the normal, aged human basal forebrain: colocalization with cholinergic neurons. J. Comp. Neurol. 285, 196–217 [DOI] [PubMed] [Google Scholar]
- 36.Alldred M.J., Che S., and Ginsberg S.D. (2009). Terminal continuation (TC) RNA amplification without second strand synthesis. J. Neurosci. Methods 177, 381–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Che S., and Ginsberg S.D. (2004). Amplification of RNA transcripts using terminal continuation. Lab. Invest. 84, 131–137 [DOI] [PubMed] [Google Scholar]
- 38.Ginsberg S.D., and Che S. (2004). Combined histochemical staining, RNA amplification, regional, and single cell cDNA analysis within the hippocampus. Lab. Invest. 84, 952–962 [DOI] [PubMed] [Google Scholar]
- 39.Galvin J.E., and Ginsberg S.D. (2004). Expression profiling and pharmacotherapeutic development in the central nervous system. Alzheimer Dis. Assoc. Disord. 18, 264–269 [PubMed] [Google Scholar]
- 40.Counts S.E., He B., Che S., Ikonomovic M.D., DeKosky S.T., Ginsberg S.D., and Mufson E.J. (2007). Alpha7 nicotinic receptor up-regulation in cholinergic basal forebrain neurons in Alzheimer disease. Arch. Neurol. 64, 1771–1776 [DOI] [PubMed] [Google Scholar]
- 41.McKee A.C., Cairns N.J., Dickson D.W., Folkerth R.D., Keene C.D., Litvan I., Perl D.P., Stein T.D., Vonsattel J.P., Stewart W., Tripodis Y., Crary J.F., Bieniek K.F., Dams-O'Connor K., Alvarez V.E., Gordon W.A., and group T.C. (2016). The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 131, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Donat C.K., Walter B., Deuther-Conrad W., Wenzel B., Nieber K., Bauer R., and Brust P. (2010). Alterations of cholinergic receptors and the vesicular acetylcholine transporter after lateral fluid percussion injury in newborn piglets. Neuropathol. Appl. Neurobiol. 36, 225–236 [DOI] [PubMed] [Google Scholar]
- 43.De Luca V., Voineskos S., Wong, G. and Kennedy J.L. (2006). Genetic interaction between alpha4 and beta2 subunits of high affinity nicotinic receptor: analysis in schizophrenia. Exp. Brain Res. 174, 292–296 [DOI] [PubMed] [Google Scholar]
- 44.Cook L.J., Ho L.W., Taylor A.E., Brayne C., Evans J.G., Xuereb J., Cairns N.J., Pritchard A., Lemmon H., Mann D., St Clair D., Turic D., Hollingworth P., Moore P.J., Jehu L., Archer N., Walter S., Foy C., Edmondson A., Powell J., Lovestone S., Owen M.J., Williams J., Lendon C., and Rubinsztein D.C. (2004). Candidate gene association studies of the alpha 4 (CHRNA4) and beta 2 (CHRNB2) neuronal nicotinic acetylcholine receptor subunit genes in Alzheimer's disease. Neurosci. Lett. 358, 142–146 [DOI] [PubMed] [Google Scholar]
- 45.Steinlein O.K., Stoodt J., de Vos R.A., Steur E.N., Wevers A., Schutz U., and Schroder H. (1999). Mutation screening of the CHRNA4 and CHRNB2 nicotinic cholinergic receptor genes in Alzheimer's disease. Neuroreport 10, 2919–2922 [DOI] [PubMed] [Google Scholar]
- 46.Tiernan C.T., Ginsberg S.D., Guillozet-Bongaarts A.L., Ward S.M., He B., Kanaan N.M., Mufson E.J., Binder L.I., and Counts S.E. (2016). Protein homeostasis gene dysregulation in pretangle-bearing nucleus basalis neurons during the progression of Alzheimer's disease. Neurobiol. Aging 42, 80–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gouras G.K., Rance N.E., Young W.S., 3rd, and Koliatsos V.E. (1992). Tyrosine-hydroxylase-containing neurons in the primate basal forebrain magnocellular complex. Brain Res. 584, 287–293 [DOI] [PubMed] [Google Scholar]
- 48.Wisniowski L., Ridley R.M., Baker H.F., and Fine A. (1992). Tyrosine hydroxylase-immunoreactive neurons in the nucleus basalis of the common marmoset (Callithrix jacchus). J. Comp. Neurol. 325, 379–387 [DOI] [PubMed] [Google Scholar]
- 49.Torres E.M., Rogers D.C., Annett L.E., Sirinathsinghji D.J., and Dunnett S.B. (1993). A novel population of tyrosine hydroxylase immunoreactive neurones in the basal forebrain of the common marmoset (Callithrix jacchus). Neurosci. Lett. 150, 29–32 [DOI] [PubMed] [Google Scholar]
- 50.Ikemoto K., Nagatsu I., Kitahama K., Jouvet A., Nishimura A., Nishi K., Maeda T., and Arai R. (1998). A dopamine-synthesizing cell group demonstrated in the human basal forebrain by dual labeling immunohistochemical technique of tyrosine hydroxylase and aromatic L-amino acid decarboxylase. Neurosci. Lett. 243, 129–132 [DOI] [PubMed] [Google Scholar]
- 51.Goll D.E., Thompson V.F., Li H., Wei W., and Cong J. (2003). The calpain system. Physiol. Rev. 83, 731–801 [DOI] [PubMed] [Google Scholar]
- 52.Wang J., Markesbery W.R., and Lovell M.A. (2006). Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J. Neurochem. 96, 825–832 [DOI] [PubMed] [Google Scholar]
- 53.Wu H.Y., and Lynch D.R. (2006). Calpain and synaptic function. Mol. Neurobiol. 33, 215–236 [DOI] [PubMed] [Google Scholar]
- 54.Saatman K.E., Creed J., and Raghupathi R. (2010). Calpain as a therapeutic target in traumatic brain injury. Neurotherapeutics 7, 31–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saatman K.E., Bozyczko-Coyne D., Marcy V., Siman R., and McIntosh T.K. (1996). Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J. Neuropathol. Exp. Neurol. 55, 850–860 [DOI] [PubMed] [Google Scholar]
- 56.Bralic M., and Stemberga V. (2012). Calpain expression in the brain cortex after traumatic brain injury. Coll. Antropol. 36, 1319–1323 [PubMed] [Google Scholar]
- 57.Yamada K.H., Kozlowski D.A., Seidl S.E., Lance S., Wieschhaus A.J., Sundivakkam P., Tiruppathi C., Chishti I., Herman I.M., Kuchay S.M., and Chishti A.H. (2012). Targeted gene inactivation of calpain-1 suppresses cortical degeneration due to traumatic brain injury and neuronal apoptosis induced by oxidative stress. J. Biol. Chem. 287, 13182–13193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pike B.R., Zhao X., Newcomb J.K., Glenn C.C., Anderson D.K., and Hayes R.L. (2000). Stretch injury causes calpain and caspase-3 activation and necrotic and apoptotic cell death in septo-hippocampal cell cultures. J. Neurotrauma 17, 283–298 [DOI] [PubMed] [Google Scholar]
- 59.Posmantur R.M., Kampfl A., Liu S.J., Heck K., Taft W.C., Clifton G.L., and Hayes R.L. (1996). Cytoskeletal derangements of cortical neuronal processes three hours after traumatic brain injury in rats: an immunofluorescence study. J. Neuropathol. Exp. Neurol. 55, 68–80 [DOI] [PubMed] [Google Scholar]
- 60.Huh J.W., Raghupathi R., Laurer H.L., Helfaer M.A., and Saatman K.E. (2003). Transient loss of microtubule-associated protein 2 immunoreactivity after moderate brain injury in mice. J. Neurotrauma 20, 975–984 [DOI] [PubMed] [Google Scholar]
- 61.Posmantur R.M., Kampfl A., Taft W.C., Bhattacharjee M., Dixon C.E., Bao J., and Hayes R.L. (1996). Diminished microtubule-associated protein 2 (MAP2) immunoreactivity following cortical impact brain injury. J. Neurotrauma 13, 125–137 [DOI] [PubMed] [Google Scholar]
- 62.Kampfl A., Posmantur R.M., Zhao X., Schmutzhard E., Clifton G.L., and Hayes R.L. (1997). Mechanisms of calpain proteolysis following traumatic brain injury: implications for pathology and therapy: implications for pathology and therapy: a review and update. J. Neurotrauma 14, 121–134 [DOI] [PubMed] [Google Scholar]
- 63.Yin Y., Wang Y., Gao D., Ye J., Wang X., Fang L., Wu D., Pi G., Lu C., Zhou X.W., Yang Y., and Wang J.Z. (2016). Accumulation of human full-length tau induces degradation of nicotinic acetylcholine receptor alpha4 via activating calpain-2. Sci. Rep. 6, 27283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Perez S.E., Getova D.P., He B., Counts S.E., Geula C., Desire L., Coutadeur S., Peillon H., Ginsberg S.D., and Mufson E.J. (2012). Rac1b increases with progressive tau pathology within cholinergic nucleus basalis neurons in Alzheimer's disease. Am. J. Pathol. 180, 526–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nag S., Venugopalan R., and Stewart D.J. (2007). Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood–brain barrier breakdown. Acta Neuropathol. 114, 459–469 [DOI] [PubMed] [Google Scholar]
- 66.Nag S., Manias J.L., and Stewart D.J. (2009). Expression of endothelial phosphorylated caveolin-1 is increased in brain injury. Neuropathol. Appl. Neurobiol. 35, 417–426 [DOI] [PubMed] [Google Scholar]
- 67.Shin T. (2007). Increases in the phosphorylated form of caveolin-1 in the spinal cord of rats with clip compression injury. Brain Res. 1141, 228–234 [DOI] [PubMed] [Google Scholar]
- 68.Jasmin J.F., Malhotra S., Singh Dhallu M., Mercier I., Rosenbaum D.M., and Lisanti M.P. (2007). Caveolin-1 deficiency increases cerebral ischemic injury. Circ. Res. 100, 721–729 [DOI] [PubMed] [Google Scholar]
- 69.Badaut J., Ajao D.O., Sorensen D.W., Fukuda A.M., and Pellerin L. (2015). Caveolin expression changes in the neurovascular unit after juvenile traumatic brain injury: signs of blood–brain barrier healing? Neuroscience 285, 215–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Niesman I.R., Zemke N., Fridolfsson H.N., Haushalter K.J., Levy K., Grove A., Schnoor R., Finley J.C., Patel P.M., Roth D.M., Head B.P., and Patel H.H. (2013). Caveolin isoform switching as a molecular, structural, and metabolic regulator of microglia. Mol. Cell Neurosci. 56, 283–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Niesman I.R., Schilling J.M., Shapiro L.A., Kellerhals S.E., Bonds J.A., Kleschevnikov A.M., Cui W., Voong A., Krajewski S., Ali S.S., Roth D.M., Patel H.H., Patel P.M., and Head B.P. (2014). Traumatic brain injury enhances neuroinflammation and lesion volume in caveolin deficient mice. J. Neuroinflammation 11, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gioiosa L., Raggi C., Ricceri L., Jasmin J.F., Frank P.G., Capozza F., Lisanti M.P., Alleva E., Sargiacomo M., and Laviola G. (2008). Altered emotionality, spatial memory and cholinergic function in caveolin-1 knock-out mice. Behav. Brain Res. 188, 255–262 [DOI] [PubMed] [Google Scholar]
- 73.Atkins C.M., Oliva A.A., Jr, Alonso O.F., Pearse D.D., Bramlett H.M., and Dietrich W.D. (2007). Modulation of the cAMP signaling pathway after traumatic brain injury. Exp. Neurol. 208, 145–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kato H., Araki T., Itoyama Y., and Kogure K. (1995). Rolipram, a cyclic AMP-selective phosphodiesterase inhibitor, reduces neuronal damage following cerebral ischemia in the gerbil. Eur. J. Pharmacol. 272, 107–110 [DOI] [PubMed] [Google Scholar]
- 75.Block F., Bozdag I., and Nolden-Koch M. (2001). Inflammation contributes to the postponed ischemic neuronal damage following treatment with a glutamate antagonist in rats. Neurosci. Lett. 298, 103–106 [DOI] [PubMed] [Google Scholar]
- 76.Imanishi T., Sawa A., Ichimaru Y., Miyashiro M., Kato S., Yamamoto T., and Ueki S. (1997). Ameliorating effects of rolipram on experimentally induced impairments of learning and memory in rodents. Eur. J. Pharmacol. 321, 273–278 [DOI] [PubMed] [Google Scholar]
- 77.Mayr B., and Montminy M. (2001). Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2, 599–609 [DOI] [PubMed] [Google Scholar]
- 78.Manganiello V.C., Murata T., Taira M., Belfrage P., and Degerman E. (1995). Diversity in cyclic nucleotide phosphodiesterase isoenzyme families. Arch. Biochem. Biophys. 322, 1–13 [DOI] [PubMed] [Google Scholar]
- 79.Myeku N., Clelland C.L., Emrani S., Kukushkin N.V., Yu W.H., Goldberg A.L. and Duff K.E. (2016). Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 22, 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Reiner O., Carrozzo R., Shen Y., Wehnert M., Faustinella F., Dobyns W.B., Caskey C.T., and Ledbetter D.H. (1993). Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature 364, 717–721 [DOI] [PubMed] [Google Scholar]
- 81.Smith D.S., Niethammer M., Ayala R., Zhou Y., Gambello M.J., Wynshaw-Boris A., and Tsai L.H. (2000). Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat. Cell Biol. 2, 767–775 [DOI] [PubMed] [Google Scholar]
- 82.Reiner O. (2013). LIS1 and DCX: implications for brain development and human disease in relation to microtubules. Scientifica (Cairo) 2013, 393975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jimenez-Mateos E.M., Wandosell F., Reiner O., Avila J., and Gonzalez-Billault C. (2005). Binding of microtubule-associated protein 1B to LIS1 affects the interaction between dynein and LIS1. Biochem. J. 389, 333–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu Z., Steward R., and Luo L. (2000). Drosophila Lis1 is required for neuroblast proliferation, dendritic elaboration and axonal transport. Nat. Cell Biol. 2, 776–783 [DOI] [PubMed] [Google Scholar]
- 85.Jentsch T.J. (2016). VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat. Rev. Mol. Cell Biol. 17, 293–307 [DOI] [PubMed] [Google Scholar]
- 86.Okkenhaug H., Weylandt K.H., Carmena D., Wells D.J., Higgins C.F., and Sardini A. (2006). The human ClC-4 protein, a member of the CLC chloride channel/transporter family, is localized to the endoplasmic reticulum by its N-terminus. FASEB J. 20, 2390–2392 [DOI] [PubMed] [Google Scholar]
- 87.Ginsberg S.D., Che S., Wuu J., Counts S.E., and Mufson E.J. (2006). Down regulation of trk but not p75NTR gene expression in single cholinergic basal forebrain neurons mark the progression of Alzheimer's disease. J. Neurochem. 97, 475–487 [DOI] [PubMed] [Google Scholar]
- 88.Mufson E.J., Counts S.E., and Ginsberg S.D. (2002). Gene expression profiles of cholinergic nucleus basalis neurons in Alzheimer's disease. Neurochem. Res. 27, 1035–1048 [DOI] [PubMed] [Google Scholar]