

Abstract
Testis-specific serine/threonine kinase 2 (TSSK2) is an important target for reversible male contraception. A high-throughput screen of ~17,000 compounds using a mobility shift assay identified two potent series of inhibitors having a pyrrolopyrimidine or pyrimidine core. The pyrrolopyrimidine 10 (IC50 22 nM; GSK2163632A) and the pyrimidine 17 (IC50 31 nM; ALK inhibitor 1) are the most potent TSSK2 inhibitors in these series, which contain the first sub-100 nanomolar inhibitors of any TSSK isoform reported, except for the broad kinase inhibitor staurosporine. The novel, potent pyrimidine TSSK2 inhibitor compound 19 (IC50 66 nM; 2-[[5-chloro-2-[2-methoxy-4-(1-methylpiperidin-4-yl)anilino]pyrimidin-4-yl]amino]-N-methylbenzenesulfonamide) lacks the potential for metabolic activation. Compound 19 had a rank order potency TSSK1 > TSSK2 > TSSK3 > TSSK6, indicating that potent dual inhibitors of TSSK1/2 can be identified, which may be required for a complete contraceptive effect. The future availability of a TSSK2 crystal structure will facilitate structure-based discovery of selective TSSK inhibitors from these pyrrolopyrimidine and pyrimidine scaffolds.
Keywords: testis-specific serine/threonine kinase, kinase inhibitors, structure–activity relationships, high-throughput screening, male contraception
Sperm beware!
Testis-specific serine/threonine kinase 2 (TSSK2) plays an important role in spermiogenesis and fertilization. A robust mobility shift assay high-throughput screen identified a pyrrolopyrimidine and a pyrimidine series of TSSK2 ATP site inhibitors. A pyrimidine analog lacking a metabolic liability was identified as a potent dual inhibitor of TSSK2 and TSSK1, demonstrating the potential to identify inhibitors of multiple TSSKs for male contraception.
Introduction
Testis-specific serine/threonine kinases (TSSKs) are a family of six kinases in the CAM kinase family expressed in mammalian testis that are involved in spermiogenesis and fertilization. All TSSK isoforms are expressed postmeiotically in spermatids.[1] TSSK1, TSSK2, and TSSK6 are expressed in both human and mouse sperm, whereas TSSK4 is expressed in mouse, but not human, sperm.[1] In contrast, TSSK3 is expressed in testis, but not in sperm of either human or mice.[1] TSSK5 appears to be expressed in mouse,[2] but there is no homologue gene in the human genome based on BLAST searches. TSSK2 is of particular interest due to its critical role in fertility in mice and likely humans as well as the availability of highly active, full-length human protein.[3]
TSSK2 has been localized to various regions of sperm. Hao et al. reported that TSSK2 is localized to the equatorial segment of human sperm,[4] whereas Shang et al. found that TSSK2, TSSK1, and their common endogenous substrate, TSKS, accumulate in a ring-shaped structure around the base of the flagellum and in a cytoplasmic satellite derived from the chromatoid body.[3d] In contrast, Li et al. reported that TSSK2 is expressed mainly in the sperm head in the postacrosomal regions and the acrosome tip,[1] regions involved in sperm-oocyte fusion, where Izumo migrates following the acrosome reaction. TSSK2 is apparently expressed throughout the sperm depending on the stage of spermiogenesis, as it has been localized to the centrioles of post-meiotic spermatids, the tail and acrosomal regions of mouse epididymal sperm, and the midpiece of human spermatozoa.[5]
TSSK2 phosphorylates itself as well as TSKS and SPAG16L. TSSK2 co-immunoprecipitates with TSKS in human sperm[3a] and SPAG16L in mouse testis extracts.[6] Human TSSK2 phosphorylates the N-terminal region of TSKS and displays robust autophosphorylation.[3c] Targeted deletion of TSSK1 and TSSK2 resulted in chimeric mice which fail to form elongated spermatids, possess apoptotic spermatocytes and spermatids, and accumulate numerous round cells in the epididymal lumen, which are likely immature spermatocytes.[3a] TSSK4 knockout mice have reduced fertility[7], whereas deletion of TSSK6[8] and the double TSSK1/TSSK2 knockout[3a] resulted in sterility suggesting that these members of the TSSK family can be targeted with selective kinase inhibitors for male contraception. A triallelic polymorphism of the TSSK6 gene is associated with spermatogenic impairment in humans[9] and two single-nucleotide polymorphisms of the TSSK2 gene are associated with spermatogenesis impairment and may be associated with male idiopathic infertility in humans.[3b]
We recently expressed soluble, full-length human TSSK2 in baculovirus and purified the enzyme by immobilized-metal affinity chromatography (IMAC) followed by gel filtration chromatography.[3c] A mobility shift assay developed using purified, full-length human TSSK2 was used to screen focused compound libraries. Two series of potent inhibitors were identified and their structure-activity relationships are described herein.
Results and Discussion
High-Throughput Screening
A previously described mobility shift assay detecting full-length, human TSSK2 phosphorylation of a synthetic substrate[3c] was used to screen compound libraries for inhibitors. The broad-spectrum kinase inhibitor staurosporine, previously shown to be a potent TSSK2 inhibitor (IC50 20 nM),[3c] was used as a positive control on every screening plate. Vertical validation experiments using alternating columns of full reaction, no enzyme, and an IC50 concentration of staurosporine as positive control yielded Z′ values > 0.85, substantially higher than the generally accepted minimum value of 0.5 for high-throughput screening (HTS). A pilot screen of the Library of Pharmacologically Active Compounds (LOPAC) produced a similar pattern of apparent inhibitors on a repeat test, indicating that the assay was reproducible. About 17,000 compounds were screened, including drug and pharmacologically active collections (Johns Hopkins FDA collection, NIH Clinical Collections 1 and 2, Prestwick Chemical Library, Tocris Screening Collection, and Microsource Spectrum) and kinase inhibitor libraries (SelleckChem kinase inhibitor library, GSK PKIS 1 and 2, Tocris kinase inhibitor toolbox, and a ChemDiv kinase scaffold collection). Of 48 compounds that produced > 20% inhibition (0.3% hit rate), 14 were confirmed as inhibitors in concentration-response experiments conducted with cherry-picked compounds from DMSO stocks. Confirmed hits included several pyrrolopyrimidines from the GSK PKIS and SelleckChem collections and the pyrimidine NVP-TAE684 (hereafter referred to as TAE684) from SelleckChem. Other active compounds included CHIR-124, PF-03814735, and hesperidin (SelleckChem), staurosporine (SelleckChem, Tocris), and the oxadiazol-imidazopyridines SK1392956A and GSK1007102B (GSK PKIS). The commercially available confirmed hits were re-purchased and the GSK PKIS confirmed hits were resupplied by GSK (solid sample) and the University of North Carolina (DMSO stock). Five of these confirmed, resupplied hits inhibited TSSK2 with IC50 values < 100 nM: staurosporine, TAE684, and three pyrrolopyrimidines.
SAR and kinase selectivity of pyrrolopyrimidine TSSK2 inhibitors
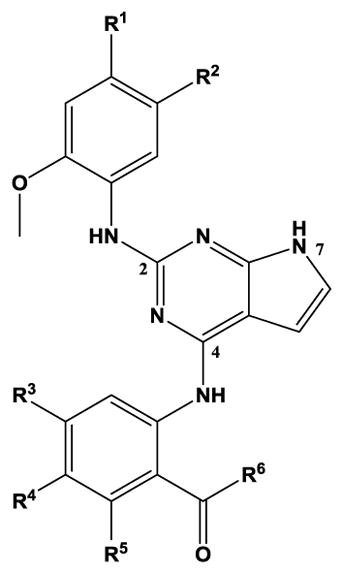
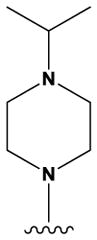
The pyrrolopyrimidines tested exhibited a broad range of potencies for inhibition of TSSK2 activity (Figure 1, Tables 1 and 2), with IC50 values ranging from low double-digit nanomolar to > 10 μM. The structure-activity relationships for substituted 4-anilino-2-(2-methoxyanilino)-7H-pyrrolo[2,3-d]pyrimidine compounds are shown in Table 1. The most active compounds in this series have 6-membered aliphatic rings with one or more basic nitrogen atoms in the R1 position and R6 = -NH2 (carboxamide) as in compounds 1, 2, and 5. Fluorination at R3 and R4 provides similar activity, although the R4, R5 di-fluoro substitution (compound 2) decreases activity 2-fold compared to R4-fluoro (compound 1). Alkylation of the carboxamide (compound 3) or replacement with carboxylic acid (compound 4) decreases activity > 10-fold. Similar activities of compounds 5 (4-(1-propylpiperidin-4-yl) and 6 (4-(-propylpiperidin-3-yl) indicate that the basic nitrogen of the piperidine ring in the 4- and 3-positions provides high activity and that the 1-yl-nitrogen of the piperazine ring is not required for activity. Substitution of the R2 position with a basic nitrogen-containing group affords moderate activity (compound 7), although alkylation of the amide nitrogen (compound 8) reduces potency 50-fold, suggesting the importance of a hydrogen bonding interaction in this region.
Figure 1.

Pyrrolopyrimidine inhibition of human recombinant TSSK2. Concentration-response curves for inhibition of phosphorylation of 5-FAM-GS peptide using the mobility shift assay. Symbols represent the mean ± SEM of at least 3 independent experiments.
Table 1.
Structure-activity relationships for substituted 4-anilino-2-(2-methoxyanilino)-7H-pyrrolo[2,3-d]pyrimidine inhibition of TSSK2 activity
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | R3 | R4 | R5 | R6 | TSSK2 IC50, nM[a] |
| 1 |
|
H | H | F | H | NH2 | 72 ± 10 |
| 2 |
|
H | H | F | F | NH2 | 150 ± 30 |
| 3 |
|
H | H | F | H |
|
1200 ± 200 |
| 4 |
|
H | H | H | F | OH | 2000 ± 200 |
| 5 |
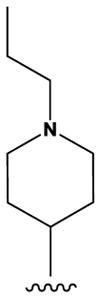
|
H | F | H | H | NH2 | 107 [b] |
| 6 |
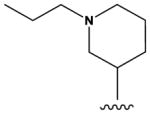
|
H | F | H | H | NH2 | 250 ± 50 |
| 7 | CH3 |
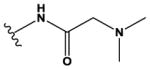
|
H | H | F | NH2 | 280 ± 60 |
| 8 | CH3 |
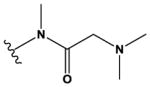
|
H | H | F | NH2 | 14,000 ± 2000 |
| 9 | Cl |
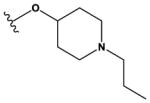
|
H | H | F | NH2 | 1300 ± 200 |
Mean ± SEM of at least 3 independent experiments.
N = 2 due to limited sample availability.
Table 2.
Structure-activity relationships for substituted 2-amino-7H-pyrrolo[2,3-d]pyrimidine inhibition of TSSK2 activity
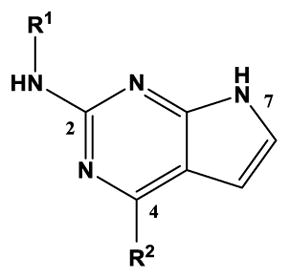
| |||
|---|---|---|---|
| Compound | R1 | R2 | TSSK2 IC50, nM[a] |
| 10 |
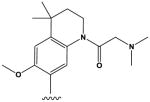
|
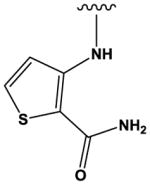
|
22 ± 4 |
| 11 |
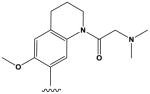
|
|
47 ± 1 |
| 12 |
|
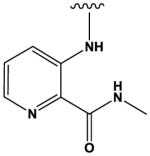
|
247 ± 1 |
| 13 |
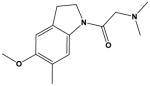
|
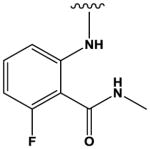
|
1300 ± 200 |
| 14 |
|
|
4800 ± 300 |
| 15 |
|
|
1500 [b] |
| 16 |
|
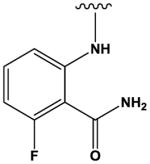
|
6000 ± 200 |
Mean ± SEM of at least 3 independent experiments.
N = 2 due to limited sample availability.
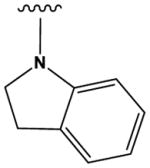
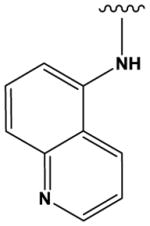
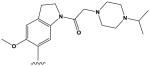
The most potent compounds identified in the 2-amino-7H-pyrrolo[2,3-d]pyrimidine series possess a substituted tetrahydroquinoline at R1 and aminothiophene carboxamide at R2, as in compounds 10 (IC50 = 22 nM) and 11 (IC50 = 47 nM) (Table 2). Methylation of the carboxamide nitrogen atom reduces activity 5-fold (compound 12), and a further 5-fold loss in activity is observed when the tetrahydroquinoline is replaced with the indoline group (compound 13). Other heterocyclic substitutions at R1 and R2 produce low micromolar inhibitors (compounds 14, 15, 16).
The pyrrolopyrimidines included in this study have previously been reported to inhibit other kinases.[10] As many of these compounds were prepared as part of an insulin-like growth factor 1 receptor (IGF-1R) drug discovery program, it is not surprising that they all inhibit the IGF-1R with low or sub-nanomolar IC50 values (Supplemental Table 1).[10a–c, 10e, 10f] Thus, the most potent pyrrolopyrimidine TSSK2 inhibitors identified, compounds 1, 10, and 11, are ≥ 100-fold more potent inhibitors of IGF-1. Many compounds in this series are also reported to inhibit the insulin receptor and anaplastic lymphoma kinase (ALK) with low or sub-nanomolar IC50 values (Supplemental Table 1).[10e, 10f] The potent TSSK2 inhibitors compounds 10 and 11 have been reported to inhibit the G-protein coupled receptor kinases (GRKs) with highest activity against GRK1,[10d] although these compounds are > 5-fold selective for TSSK2 over GRK1. In addition, the weak TSSK2 inhibitor 13 was profiled against 224 kinases and was found to inhibit five additional kinases with IC50 values between 20 and 100 nM. The affinity of compound 13 has also been determined at 78 kinases using the KINOMEScan assay,[11] identifying an additional nine kinases with KD values between 1 and 100 nM. These data suggest that the potent pyrrolopyrimidine TSSK2 inhibitors compounds 1, 10, and 11 are likely to be highly active inhibitors of several kinases in addition to IGF-1, a conclusion supported by profiling data provided by GSK supporting the PKIS collections indicating that these three compounds each inhibit 15 – 20 kinases by > 50% at 100 nM.
SAR and kinase selectivity of pyrimidine TSSK2 inhibitors
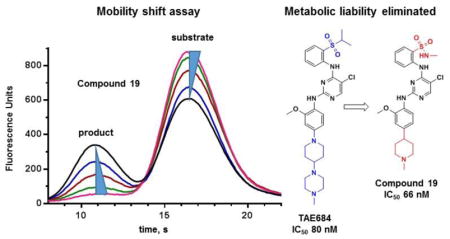
To further evaluate the activity of pyrimidines related to the screening hit TAE684, the TSSK2 inhibitory potency of several commercially available TAE684 analogs was determined. A broad range of potencies were observed in the pyrimidine series, ranging from 31 nM to > 100 μM (Figure 2, Tables 3 and 4). The most potent compounds in this series are 17 (ALK inhibitor 1) and 18 (ALK inhibitor 2), which contain a methylpiperazine A ring (R1 = Me, X = N) and a D ring with R5 = methylsulfonamide. It has previously been shown that piperazine-containing compounds such as 17 and 18 undergo metabolic oxidation to form reactive adducts in the presence of glutathione.[12] Marsilje et al.[12] described multiple reactive metabolic products arising from oxidation of the para-phenylene diamine substructure to a quinonoid structure that then became trapped by glutathione. This metabolic liability was prevented by replacing the piperazine ring with piperidine, an approach that contributed to the discovery of ceritinib, an ALK inhibitor approved by the FDA to treat small cell lung cancer.[12] The discovery of the potent TSSK2 inhibitor 18 prompted the synthesis of compound 19 with the single point substitution of nitrogen at position Y by methine. Replacement of the piperazine in compound 18 with piperidine in compound 19 reduces activity < 2-fold, indicating that potent pyrimidine inhibitors of TSSK2 can be designed without this metabolic liability.
Figure 2.

Pyrimidine inhibition of human recombinant TSSK2. Concentration-response curves for inhibition of phosphorylation of 5-FAM-GS peptide using the mobility shift assay. Symbols represent the mean ± SEM of at least 3 independent experiments.
Table 3.
Structure-activity relationships for substituted N2,N4-diphenyl-2,4-pyrimidinediamine inhibition of TSSK2
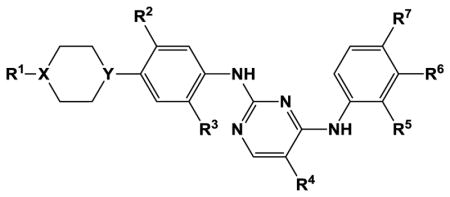
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | R1 | X | Y | R2 | R3 | R4 | R5 | R6 | R7 | TSSK2 IC50, nM[a] |
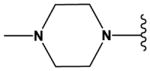
| 17 | Me | N | N | H | OMe | Br |
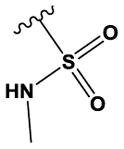
|
H | H | 31 ± 7 |
| 18 | Me | N | N | H | OMe | Cl |
|
H | H | 37 ± 5 |
| 19 | Me | N | CH | H | OMe | Cl |
|
H | H | 66 ± 8 |
| 20 | - | O | N | H | OMe | Cl |
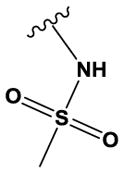
|
H | H | 750 ± 110 |
| 21 | - | O | N | H | OMe | F |
|
H | H | 13,000 ± 2000 |
| 22 |
|
CH | N | H | OMe | Cl |
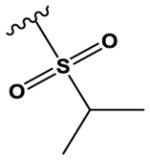
|
H | H | 80 ± 13 |
| 23 | H | N | CH | Me | OiPr | Cl |
|
H | H | 610 ± 30 |
| 24 | N(CH3)2 | CH | N | H | OMe | Cl |
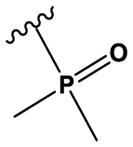
|
H | H | 230 ± 30 |
| 25 |
|
CH | N | H | OMe | Cl |
|
H | H | 510 ± 80 |
| 26 | H | N | N | H | H | Cl |
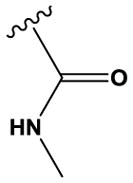
|
H | H | 963 ± 7 |
| 27 | - | O | N | H | OMe | Cl |
|
H | H | 7600 ± 800 |
| 28 | - | O | N | H | H | Cl |
|
H | H | > 100,000 |
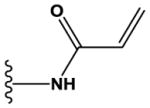
| 29 | H | CH | N | H | H | H | COOH | H | H | > 100,000 |
| 30 | Me | N | N | H | H | Me | H |
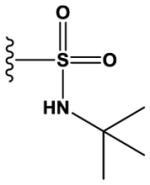
|
H | 6200 ± 900 |
| 31 |
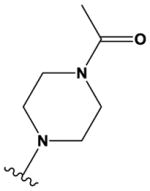
|
N | N | H | OMe | CF3 | H |
|
H | 15,000 ± 1000 |
| 32 |
|
N | N | H | OMe | Cl | OMe | H |
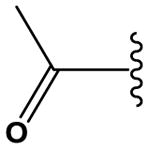
|
62,000 ± 4000 |
| 33 | - | O | N | H | H | F | H | H |
|
> 100,000 |
Mean ± SEM of at least 3 independent experiments.
Table 4.
Structure-activity relationships for pyrimidine core replacements and 4-substituted, 2-anilino-pyrimidine inhibition of TSSK2
| Compound | Structure | TSSK2 IC50, nM[a] |
|---|---|---|
| 34 |
|
690 ± 140 |
| 35 |
|
58 ± 2 |
| 36 |
|
3800 ± 100 |
Mean ± SEM of at least 3 independent experiments.
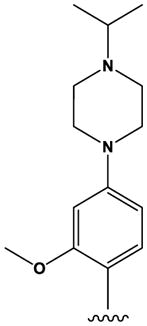
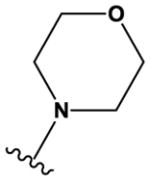
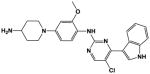
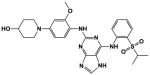
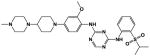
The ‘reverse sulfonamide’ of compound 18 containing morpholine instead of methylpiperazine (compound 20) was 20-fold less active. Compound 21 had a further 17-fold loss in activity, indicating that Cl or Br is preferred over F in the R4 position of the C ring. Substitution of the A ring of compound 18 with methylpiperazine at R1 and replacement of the sulfonamide with iPr-sulfone as in the screening hit compound 22 (TAE684) reduces activity ~2-fold, indicating that a sulfonamide substituted D ring is not required for activity. As sulfonamide–containing drugs can cause hypersensitivity reactions in humans, compounds without a sulfonamide group are preferred. Compounds 23, 24, and 25 with various substitutions at R1, methoxy or i-propoxy at R2, a single nitrogen atom at either X or Y, and either a iPr-sulfone or dimethylphosphine group at R5 are moderately active, indicating that active compounds can be obtained by modifying each of these positions and that a sulfur-containing substituent on the D ring is not required for activity. This conclusion is further supported by compound 26, which contains a methylcarboxamide at R5 and has moderate activity despite suboptimal substitution of the A and B rings, and by compound 34 with indole replacing the D ring (Table 4). Other combinations of substitutions with methylcarboxamide (compounds 27, 28) or carboxyl (compound 29) at R5 or various nitrogen-containing groups at R6 (compounds 30, 31) or R7 (compound 32) resulted in weakly active or inactive compounds. Replacement of the pyrimidine core can produce highly active compounds. Compound 35 (Mps1-IN-3) with a purine rather than pyrimidine core is a potent inhibitor of TSSK2 (Table 4). However, replacement of the pyrimidine core with triazine (compound 36) results in almost 50-fold loss of activity relative to the corresponding pyrimidine (compound 22).
As described for the pyrrolopyrimidines, the pyrimidines included in this study have previously been reported to inhibit other kinases.[12–13] The most potent pyrimidine TSSK2 inhibitors identified (compounds 17, 18) were prepared as part of a focal adhesion kinase (FAK) discovery program at Novartis and have low nanomolar IC50 values for FAK inhibition in patent literature (Supplementary Table 2).[13p] Compound 17 is substantially less potent for inhibiting IGF-1R.[13p] Compounds 17 and 18 are referred to as ‘ALK inhibitor 1’ and ‘ALK inhibitor 2’, respectively, by commercial vendors, although there is apparently no published data for inhibition of ALK by either compound. Considerably more kinase selectivity information has been reported for the potent TSSK2 inhibitors 22 and 35 (Supplementary Table 2). The MPS1 inhibitor 35 is reported to inhibit MPS1 with similar potency[13a] as we observed for TSSK2, with IC50 values of ~50 – 60 nM for these two kinases. This compound also inhibits FAK with similar potency to TSSK2 and MPS1, and inhibits LTK, IGF-1R, insulin receptor, and CLK2 with IC50 values of ~20 – 40 nM (Supplementary Table 2). These selectivity data suggest that appropriate substitutions of the purine core may afford a selective TSSK2 inhibitor. Although compound 22 is a reasonably potent TSSK2 inhibitor (IC50 = 80 nM), it has been reported to have low nanomolar activity at several kinases, including ALK,[12] FLT3,[13b] LRRK2,[13c] and Tie2.[13b]
The potent TSSK2 inhibitor 19 lacking the potential for metabolic activation to thiol-reactive intermediates was profiled in a broad panel of 369 kinases at Reaction Biology Corp. Compound 19 (100 nM) inhibited 62 of 369 kinases to a greater extent than TSSK2 (Fig. 3, Supplementary Table 3). IC50 values determined at four off-target kinases indicated that compound 19 is a sub-nanomolar inhibitor of ALK, FAK, and IRR and a single digit nanomolar inhibitor of FAK consistent with other compounds in this series (Supplementary Table 4). This inhibition profile indicates that compound 19 is a promiscuous kinase inhibitor that would likely have a side effect profile inconsistent with chronic dosing as an elective agent for an indication such as contraception. However, substantial selectivity can be achieved in this pyrimidine series as evidence by the improved selectivity of ceritinib for the target kinase ALK.[12] Interestingly, compound 19 displayed a broad range of potencies for inhibition of different TSSK family members. Despite its synthesis in a program targeting TSSK2, compound 19 is > 40-fold more potent at inhibiting TSSK1 than TSSK2. Compound 19 is 800-fold more potent at TSSK1 than TSSK3 and is almost 100,000-fold selective for TSSK1 over TSSK6. Although the large differences in potency of compound 19 at different TSSK family members is surprising, the IC50 values generally correlate with sequence homology. Compound 19 has highest potency at TSSK1 and TSSK2, which share 72% identity overall and 83% identity in the kinase region.[4] Compound 19 is less potent at TSSK3, which has 47.5 and 49% sequence identity with TSSK1 and TSSK2, respectively.[4] Compound 19 is almost inactive at TSSK6, which has 45 to 49% sequence identity with TSSK1, TSSK2, and TSSK3.[8]
Figure 3. Kinome profiling for compound 19.

Compound 19 (100 nM) was tested for inhibition of 369 wild-type kinases at Reaction Biology Corp. Inhibitory activity is indicated from low (red) to high (blue). TK = tyrosine kinase; TKL = tyrosine kinase-like; STE = sterile-phenotype kinases; CK1 = casein kinase 1; AGC = cAMP-, cGMP-dependent, PKC; CAMK = calcium/calmodulin-dependent protein kinase; CMGC = CDK, MAPK, GSK3, CLK. 315 of 369 kinases are displayed.
Conclusion
Several lines of evidence suggest that TSSK2 is an important target for reversible male contraception, including the finding that double TSSK1/TSSK2 knockout mice are sterile. The availability of highly active, purified, soluble, full-length human recombinant TSSK2 enzyme allowed the discovery of two series of potent TSSK2 inhibitors by HTS having a pyrrolopyrimidine or pyrimidine core. However, these pyrrolopyrimidine and pyrimidine compounds possess higher inhibitory potencies against kinases other than TSSK2, suggesting that identifying selective TSSK inhibitors in these series may be challenging. In particular, the novel, potent TSSK2 inhibitor 19 lacking the potential for metabolic activation was found to be a promiscuous kinase inhibitor that would likely have a side effect profile inconsistent with chronic dosing as an elective agent for an indication such as male contraception. However, compound 19 was found to be a potent inhibitor of both TSSK1 and TSSK2, a profile that may provide a complete loss of fertility by mimicking the phenotype of the double TSSK1/2 knockout mouse. Moreover, compound 19 and related potent TSSK2 inhibitors may be useful as tool compounds to generate X-ray co-crystal structures of TSSK1 and 2 with ligands bound to the ATP sites. These co-crystal structures may also aid in medicinal chemistry lead optimization strategies to identify selective TSSK1/2 inhibitors using these pyrrolopyrimidine and pyrimidine scaffolds.
Experimental Section
Chemical libraries
The screening library consisted of ~17,000 compounds obtained from commercial sources, including the Library of Pharmacologically Active Compounds (LOPAC) (1280) (Sigma, St. Louis, MO), NIH Clinical Collection 1 (446), Prestwick Chemical Library (1120) (Prestwick Chemical, Illkirch-Graffenstaden, France), Tocris Tocriscreen (1120) (Bio-Techne, Minneapolis, MN), Microsource Spectrum Collection (2000) (Gaylordsville, CT), Johns Hopkins FDA Collection (1514) (Generous gift of Drs. Jun Liu and David Sullivan, Johns Hopkins University, Baltimore, MD), Tocris Tocriscreen Kinase Inhibitor Toolbox (80) (Bio-Techne), SelleckChem kinase inhibitor library (277), Published Kinase Inhibitor Set (PKIS) Set 1 (367) and Set 2 (473) (GlaxoSmithKline, Research Triangle Park, NC), and ChemDiv Tyrosine Kinase Targeted Library (8504), (ChemDiv, San Diego, CA). These compounds were stored at ~10 mM in DMSO in 384-well plates.
Compounds
Fourteen pyrrolopyrimidines (1, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 14, 15, and 16) were resupplied by Dr. William Zuercher, GlaxoSmithKline, in ~1 mg amounts. Ten of these pyrrolopyrimidines (1, 3, 4, 5, 6, 8, 9, 12, 14, and 16) were resupplied by Dr. William Zuercher, University of North Carolina, as 1 μL of 10 mM stock solutions. Compound 13 was obtained from SelleckChem (S2703). Compound 2 was obtained from ACES Pharma (131863) (Princeton, NJ). The pyrimidines were obtained as follows: 17 (HY-15357), 25 (HY-12857), 26 (HY-15985), and 31 (HY-15729) (MedChem Express, Monmouth Junction, NJ), 18 (011-15358) (A Chemtek, Worcester, MA), 20 (4534) and 32 (5098) (Bio-Techne), 21 (SML0373) (Sigma-Aldrich), 22 (S1108), 24 (S7000), 27 (S2820), 30 (S2692), 36 (S8054) (SelleckChem), 23 (SYN-1199) (Adipogen Life Sciences, San Diego, CA), 29 (7126239) and 33 (LN00770660) (Lab Network, South Portland, ME), 34 (A8620) (ApexBio, Houston, TX), and 35 (SYN5205) (AK Scientific, Inc., Union City, CA). Compound 28 was re-supplied by Dr. William Zuercher, GlaxoSmithKline. Staurosporine was obtained from Sigma (S4400). All resupplied compounds obtained as solids were dissolved in DMSO at 10 mM.
Chemistry
The synthesis of compound 19 (Scheme 1) was accomplished by the reaction of two commercially available building blocks using the conditions described by Garcia-Echeverria et al.[13p] The purity of all compounds in Tables 1–4 was >95% determined by analytical reverse-phase high performance liquid chromatography (HPLC). General chemical methods, detailed description of the synthesis of compound 19, and purity determination methods can be found in the Supporting Information.
Scheme 1.

Synthesis of compound 19.
Expression and purification of human TSSK2
Soluble human TSSK2 (hTSSK2) was expressed and purified as previously described.[3c] Briefly, the full-length hTSSK2 ORF (358 aa) was amplified by PCR from human testis cDNA, cloned into a plasmid with a His-tag, transposed into a bacmid, and expressed in Sf9 cells. hTSSK2 was purified from the soluble fraction by immobilized metal affinity chromatography (IMAC) on a His binding Ni-NTA column followed by gel filtration. Elution fractions containing partially purified TSSK2 were used for HTS, whereas fractions containing highly purified TSSK2 were used for IC50 determinations.
Mobility Shift Assay
The mobility shift assay (MSA) was conducted as previously described[3c]. Compounds and positive control (staurosporine) dissolved in DMSO were added to low-volume 384-well microplates (Corning #3677) using an Echo 550 dispenser (Labcyte, Sunnyvale, CA). Recombinant hTSSK2 in 5 μL reaction buffer (10 mM MgCl2, 0.015% Brij-35, 4 mM DTT, 100 mM HEPES, pH 7.5) was then added to the plate. The reaction was initiated with the addition of 5 μL of 1.5 μM (final) fluorescently-labeled glycogen synthase peptide substrate (5-FAM-GS) (Anaspec, Fremont, CA) and 5 μM ATP (final) in reaction buffer, the plate was sealed under vacuum, mixed on a plate shaker for 1 min, and incubated for 60 min at 30 °C. The reaction was terminated with the addition of 1 μL of 250 mM EDTA in 100 mM HEPES, pH 7.5. Substrate and product were separated on a Caliper LC3000 (Perkin Elmer) using 5-FAM-GS (2 μM) as a reference marker. The separation buffer consisted of 100 mM HEPES (pH 7.5), 0.015% Brij-35 solution, 0.1% (w/v) Coating Reagent #3 (Perkin Elmer) and 10 mM EDTA. The conversion of substrate to product in the presence of compound was calculated as a percentage of control wells containing the complete reaction mixture after subtraction of background wells containing no enzyme.
High-throughput screening was conducted with 0.84 μg/well partially purified hTSSK2 (specific activity of 0.24 nmol•min−1•mg−1)[3c] and compounds were tested at 10 μM final (0.1 % DMSO). A Multidrop combi (Thermo Fisher Scientific, Waltham, MA) was used for enzyme and substrate additions. Primary screening data were analyzed using ActivityBase (IDBS, Alameda, CA). For concentration-response experiments, 10 ng/well highly purified hTSSK2 (specific activity 9.7 nmol•min−1•mg−1)[3c] and substrate in buffer were added with a multichannel pipet. The final DMSO concentration was 1%, except for compounds resupplied by the University of North Carolina as DMSO stocks (final DMSO 5%). These DMSO concentrations did not reduce control enzyme activity. IC50 values from at least three independent experiments were calculated using Prism (GraphPad, San Diego, CA).
Supplementary Material
Acknowledgments
The authors dedicate this study to Prof. John C. Herr, a devoted scientist, friend, mentor, and colleague, who passed away during the conduct of this study. The PKIS was supplied by GlaxoSmithKline, LLC and the Structural Genomics Consortium under an open access Material Transfer and Trust Agreement: http://www.sgc-unc.org. These studies were supported by NIH-1U01 HD060591, 1U01HD076542, and HHSN275201300017C from the Contraception Research Branch of NICHD and by Schering AG, Berlin.
References
- 1.Li Y, Sosnik J, Brassard L, Reese M, Spiridonov NA, Bates TC, Johnson GR, Anguita J, Visconti PE, Salicioni AM. Mol Hum Reprod. 2011;17(1):42–56. doi: 10.1093/molehr/gaq071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.testis-specific serine/threonine-protein kinase 5 [Mus musculus]: NCBI Reference Sequence: NP_898922.2.
- 3.a) Xu B, Hao Z, Jha KN, Zhang Z, Urekar C, Digilio L, Pulido S, Strauss JF, 3rd, Flickinger CJ, Herr JC. Dev Biol. 2008;319(2):211–222. doi: 10.1016/j.ydbio.2008.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang H, Su D, Yang Y, Zhang W, Liu Y, Bai G, Ma M, Ma Y, Zhang S. J Androl. 2010;31(4):388–392. doi: 10.2164/jandrol.109.008466. [DOI] [PubMed] [Google Scholar]; c) Shetty J, Sinville R, Shumilin IA, Minor W, Zhang J, Hawkinson JE, Georg GI, Flickinger CJ, Herr JC. Protein Expr Purif. 2016;121:88–96. doi: 10.1016/j.pep.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Shang P, Baarends WM, Hoogerbrugge J, Ooms MP, van Cappellen WA, de Jong AA, Dohle GR, van Eenennaam H, Gossen JA, Grootegoed JA. J Cell Sci. 2010;123(Pt 3):331–339. doi: 10.1242/jcs.059949. [DOI] [PubMed] [Google Scholar]
- 4.Hao Z, Jha KN, Kim YH, Vemuganti S, Westbrook VA, Chertihin O, Markgraf K, Flickinger CJ, Coppola M, Herr JC, Visconti PE. Mol Hum Reprod. 2004;10(6):433–444. doi: 10.1093/molehr/gah052. [DOI] [PubMed] [Google Scholar]
- 5.Xu B, Hao Z, Jha KN, Zhang Z, Urekar C, Digilio L, Pulido S, Strauss JF, 3rd, Flickinger CJ, Herr JC. Dev Biol. 2008;319(2):201–210. doi: 10.1016/j.ydbio.2008.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang Z, Shen X, Jones BH, Xu B, Herr JC, Strauss JF., 3rd Biol Reprod. 2008;79(1):75–83. doi: 10.1095/biolreprod.107.066308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang X, Wei Y, Fu G, Li H, Saiyin H, Lin G, Wang Z, Chen S, Yu L. Mol Hum Reprod. 2015;21(2):136–145. doi: 10.1093/molehr/gau097. [DOI] [PubMed] [Google Scholar]
- 8.Spiridonov NA, Wong L, Zerfas PM, Starost MF, Pack SD, Paweletz CP, Johnson GR. Mol Cell Biol. 2005;25(10):4250–4261. doi: 10.1128/MCB.25.10.4250-4261.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su D, Zhang W, Yang Y, Zhang H, Liu YQ, Bai G, Ma YX, Peng Y, Zhang SZ. Asian J Androl. 2010;12(2):234–239. doi: 10.1038/aja.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Chamberlain SD, Redman AM, Patnaik S, Brickhouse K, Chew YC, Deanda F, Gerding R, Lei H, Moorthy G, Patrick M, Stevens KL, Wilson JW, Brad Shotwell J. Bioorg Med Chem Lett. 2009;19(2):373–377. doi: 10.1016/j.bmcl.2008.11.065. [DOI] [PubMed] [Google Scholar]; b) Chamberlain SD, Redman AM, Wilson JW, Deanda F, Shotwell JB, Gerding R, Lei H, Yang B, Stevens KL, Hassell AM, Shewchuk LM, Leesnitzer MA, Smith JL, Sabbatini P, Atkins C, Groy A, Rowand JL, Kumar R, Mook RA, Jr, Moorthy G, Patnaik S. Bioorg Med Chem Lett. 2009;19(2):360–364. doi: 10.1016/j.bmcl.2008.11.077. [DOI] [PubMed] [Google Scholar]; c) Chamberlain SD, Wilson JW, Deanda F, Patnaik S, Redman AM, Yang B, Shewchuk L, Sabbatini P, Leesnitzer MA, Groy A, Atkins C, Gerding R, Hassell AM, Lei H, Mook RA, Jr, Moorthy G, Rowand JL, Stevens KL, Kumar R, Shotwell JB. Bioorg Med Chem Lett. 2009;19(2):469–473. doi: 10.1016/j.bmcl.2008.11.046. [DOI] [PubMed] [Google Scholar]; d) Homan KT, Larimore KM, Elkins JM, Szklarz M, Knapp S, Tesmer JJ. ACS Chem Biol. 2015;10(1):310–319. doi: 10.1021/cb5006323. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Sabbatini P, Korenchuk S, Rowand JL, Groy A, Liu Q, Leperi D, Atkins C, Dumble M, Yang J, Anderson K, Kruger RG, Gontarek RR, Maksimchuk KR, Suravajjala S, Lapierre RR, Shotwell JB, Wilson JW, Chamberlain SD, Rabindran SK, Kumar R. Mol Cancer Ther. 2009;8(10):2811–2820. doi: 10.1158/1535-7163.MCT-09-0423. [DOI] [PubMed] [Google Scholar]; f) Stevens K, Shotwell JB, Redman A, Wilson JW, Lei H, Gerding R, Patnaik S. Preparation of pyrrolopyrimidine derivatives for use as IGF-1R inhibitors. GlaxoSmithKline LLC; USA: 2010. p. 310. [Google Scholar]
- 11.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Nat Biotechnol. 2011;29(11):1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 12.Marsilje TH, Pei W, Chen B, Lu W, Uno T, Jin Y, Jiang T, Kim S, Li N, Warmuth M, Sarkisova Y, Sun F, Steffy A, Pferdekamper AC, Li AG, Joseph SB, Kim Y, Liu B, Tuntland T, Cui X, Gray NS, Steensma R, Wan Y, Jiang J, Chopiuk G, Li J, Gordon WP, Richmond W, Johnson K, Chang J, Groessl T, He YQ, Phimister A, Aycinena A, Lee CC, Bursulaya B, Karanewsky DS, Seidel HM, Harris JL, Michellys PY. J Med Chem. 2013;56(14):5675–5690. doi: 10.1021/jm400402q. [DOI] [PubMed] [Google Scholar]
- 13.a) Tannous BA, Kerami M, Van der Stoop PM, Kwiatkowski N, Wang J, Zhou W, Kessler AF, Lewandrowski G, Hiddingh L, Sol N, Lagerweij T, Wedekind L, Niers JM, Barazas M, Nilsson RJ, Geerts D, De Witt Hamer PC, Hagemann C, Vandertop WP, Van Tellingen O, Noske DP, Gray NS, Wurdinger T. J Natl Cancer Inst. 2013;105(17):1322–1331. doi: 10.1093/jnci/djt168. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Galkin AV, Melnick JS, Kim S, Hood TL, Li N, Li L, Xia G, Steensma R, Chopiuk G, Jiang J, Wan Y, Ding P, Liu Y, Sun F, Schultz PG, Gray NS, Warmuth M. Proc Natl Acad Sci U S A. 2007;104(1):270–275. doi: 10.1073/pnas.0609412103. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang J, Deng X, Choi HG, Alessi DR, Gray NS. Bioorg Med Chem Lett. 2012;22(5):1864–1869. doi: 10.1016/j.bmcl.2012.01.084. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhang L, Holmes IP, Hochgrafe F, Walker SR, Ali NA, Humphrey ES, Wu J, de Silva M, Kersten WJ, Connor T, Falk H, Allan L, Street IP, Bentley JD, Pilling PA, Monahan BJ, Peat TS, Daly RJ. J Proteome Res. 2013;12(7):3104–3116. doi: 10.1021/pr3008495. [DOI] [PubMed] [Google Scholar]; e) Hellwig S, Miduturu CV, Kanda S, Zhang J, Filippakopoulos P, Salah E, Deng X, Choi HG, Zhou W, Hur W, Knapp S, Gray NS, Smithgall TE. Chem Biol. 2012;19(4):529–540. doi: 10.1016/j.chembiol.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Huang WS, Liu S, Zou D, Thomas M, Wang Y, Zhou T, Romero J, Kohlmann A, Li F, Qi J, Cai L, Dwight TA, Xu Y, Xu R, Dodd R, Toms A, Parillon L, Lu X, Anjum R, Zhang S, Wang F, Keats J, Wardwell SD, Ning Y, Xu Q, Moran LE, Mohemmad QK, Jang HG, Clackson T, Narasimhan NI, Rivera VM, Zhu X, Dalgarno D, Shakespeare WC. J Med Chem. 2016;59(10):4948–4964. doi: 10.1021/acs.jmedchem.6b00306. [DOI] [PubMed] [Google Scholar]; g) Feldman HC, Tong M, Wang L, Meza-Acevedo R, Gobillot TA, Lebedev I, Gliedt MJ, Hari SB, Mitra AK, Backes BJ, Papa FR, Seeliger MA, Maly DJ. ACS Chem Biol. 2016;11(8):2195–2205. doi: 10.1021/acschembio.5b00940. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Ramsden N, Perrin J, Ren Z, Lee BD, Zinn N, Dawson VL, Tam D, Bova M, Lang M, Drewes G, Bantscheff M, Bard F, Dawson TM, Hopf C. ACS Chem Biol. 2011;6(10):1021–1028. doi: 10.1021/cb2002413. [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Mori M, Ueno Y, Konagai S, Fushiki H, Shimada I, Kondoh Y, Saito R, Mori K, Shindou N, Soga T, Sakagami H, Furutani T, Doihara H, Kudoh M, Kuromitsu S. Mol Cancer Ther. 2014;13(2):329–340. doi: 10.1158/1535-7163.MCT-13-0395. [DOI] [PubMed] [Google Scholar]; j) Pardanani A, Hood J, Lasho T, Levine RL, Martin MB, Noronha G, Finke C, Mak CC, Mesa R, Zhu H, Soll R, Gilliland DG, Tefferi A. Leukemia. 2007;21(8):1658–1668. doi: 10.1038/sj.leu.2404750. [DOI] [PubMed] [Google Scholar]; k) Ember SW, Zhu JY, Olesen SH, Martin MP, Becker A, Berndt N, Georg GI, Schonbrunn E. ACS Chem Biol. 2014;9(5):1160–1171. doi: 10.1021/cb500072z. [DOI] [PMC free article] [PubMed] [Google Scholar]; l) Liu TJ, LaFortune T, Honda T, Ohmori O, Hatakeyama S, Meyer T, Jackson D, de Groot J, Yung WK. Mol Cancer Ther. 2007;6(4):1357–1367. doi: 10.1158/1535-7163.MCT-06-0476. [DOI] [PubMed] [Google Scholar]; m) Otani H, Yamamoto H, Takaoka M, Sakaguchi M, Soh J, Jida M, Ueno T, Kubo T, Asano H, Tsukuda K, Kiura K, Hatakeyama S, Kawahara E, Naomoto Y, Miyoshi S, Toyooka S. PLoS One. 2015;10(6):e0129838. doi: 10.1371/journal.pone.0129838. [DOI] [PMC free article] [PubMed] [Google Scholar]; n) Walter AO, Sjin RT, Haringsma HJ, Ohashi K, Sun J, Lee K, Dubrovskiy A, Labenski M, Zhu Z, Wang Z, Sheets M, St Martin T, Karp R, van Kalken D, Chaturvedi P, Niu D, Nacht M, Petter RC, Westlin W, Lin K, Jaw-Tsai S, Raponi M, Van Dyke T, Etter J, Weaver Z, Pao W, Singh J, Simmons AD, Harding TC, Allen A. Cancer Discov. 2013;3(12):1404–1415. doi: 10.1158/2159-8290.CD-13-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]; o) Kang CH, Yun JI, Lee K, Lee CO, Lee HK, Yun CS, Hwang JY, Cho SY, Jung H, Kim P, Ha JD, Jeon JH, Choi SU, Jeong HG, Kim HR, Park CH. Biochem Biophys Res Commun. 2015;464(3):762–767. doi: 10.1016/j.bbrc.2015.07.027. [DOI] [PubMed] [Google Scholar]; p) Garcia-echeverria C, Kanazawa T, Kawahara E, Masuya K, Matsuura N, Miyake T, Ohmori O, Umemura I, Steensma R, Chopiuk G, Jiang J, Wan Y, Ding Q, Zhang Q, Gray SN, Karanewsky D. Preparation of 2,4-pyrimidinediamines useful in the treatment of neoplastic diseases, inflammatory and immune system disorders. Novartis A.-G; Switz: Novartis Pharma G.m.b.H.; IRM LLC; 2005. p. 285. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.