

Abstract
Purpose.
Without therapeutic intervention, corneal neovascularization rapidly compromises visual acuity, and is a leading cause of blindness. Artesunate was reported to inhibit angiogenesis in tumors, although, the effects of artesunate on nontumor angiogenesis have not been investigated. This study was designed to investigate the effect of artesunate on corneal neovascularization and delineate its underlying mechanism of action.
Methods.
Rats with alkali-burned corneas were treated with artesunate for 11 days. Corneal neovascularization was evaluated by measuring the length and area of corneal vasculature in the rats. Apoptotic cells were stained with AnnexinV and propidine iodide (PI), and measured with flow cytometry analysis. Apoptosis-related and p38 mitogen-activated protein kinases (p38MAPK) signaling were evaluated by Western blot analysis.
Results.
Artesunate significantly inhibited corneal neovascularization and inflammation via specifically inducing apoptosis of vascular endothelial cells. In vascular endothelial cells, artesunate increased the Bax/Bcl-2 ratio, reduced mitochondrial membrane potential, stimulated release of cytochrome C, and cleavage of caspase 9 and 3, suggesting that the mitochondrial apoptotic pathway was involved. Artesunate activated p38MAPK, and specific p38MAPK inhibitors suppressed artesunate-induced apoptosis in endothelial cells. Reactive oxygen species (ROS) levels were increased by artesunate. N-acetyl-L-cysteine blocked p38MAPK activation and protected endothelial cells from artesunate-induced apoptosis. Ferrous salt increased ROS levels and elevated the cytotoxic effect of artesunate on endothelial cells, while the iron chelating agent deferoxamine decreased ROS levels and artesunate-induced apoptosis. Artesunate had no effect on expression of Fas, Fas Ligand, or caspase 8 cleavage.
Conclusions.
These results suggest that artesunate induces apoptosis of endothelial cells via an iron/ROS-dependent p38MAPK-mitochondrial pathway.
Keywords: artemisinin, corneal neovascularization, free radicals, apoptosis
Artesunate significantly inhibited corneal neovascularization by specially inducing endothelial cell apoptosis via iron/reactive oxygen species–dependent p38MAPK-mitocholdrial pathway, which provides the evidence that artesunate may have therapeutic potential for corneal neovascularization.
Introduction
Corneal neovascularization (CNV) consists of the formation of new vasculature in the avascular cornea, and plays a pathologic role in most corneal diseases.1,2 CNV can be induced by inflammatory, infectious, degenerative, or traumatic eye disorders and is a high risk factor for rejection after corneal transplantation.3 CNV is a major cause of blindness due to lack of sufficient antiangiogenic treatment options.1
Angiogenesis, the growth of new blood vessels from preexisting vessels, occurs physiologically during human growth and development. Aberrant angiogenesis plays a significant pathologic role in cancer, rheumatoid arthritis, diabetic complications, and diseases of the eye.4 Angiogenesis is a complex, multistep process, but proliferation and migration of activated endothelial cells comprise the critical first steps, and both processes are, therefore, attractive therapeutic targets for angiogenesis.5
Artemisinin (ART), the active component of Artemisia annua L, was first identified in 1972, and is a sesquiterpene lactone compound with an endoperoxide bridge, which is essential for its antimalaria activity.6,7 ART has low solubility in oil and water, and most semisynthetic ART derivatives are oil soluble, but only artesunate (ARS) is soluble in water.7 Because ART and its derivatives have a rapid and potent therapeutic effect on the malaria parasite without significant side effects in patients, they are recommended by the World Health Organization as the first-line antimalarial treatment for Plasmodium falciparum malaria.8
In the past decade, ART and its bioactive derivatives have been demonstrated to have an anticancer effect.9 Clinical studies also identified that ARS or artemether, combined with other treatments, decreased primary tumor volume, improved patient quality of life, and increased the mean survival time.10,11 Several putative anticancer mechanisms of ART compounds have been investigated including: inhibition of proliferation, apoptosis, oncogene and tumor suppressor gene regulation, modulation of chemoresistance, and antiangiogenic effects.12 It has been reported that ART and its derivatives have a direct cytotoxic effect on vascular endothelial cells, and are able to inhibit proliferation and migration, and induce apoptosis, thus, blocking tube formation in vitro.13 However, the underlying molecular mechanism has not been well defined. Moreover, the effects of ART and its derivatives on nontumor angiogenesis were unknown prior to the following studies.
ARS, a safe, inexpensive, and soluble derivative of ART, appears to be an ideal antiangiogenic candidate. Therefore, the present study was designed to investigate the effect and mechanism of ARS on CNV.
Materials and Methods
Chemicals and Reagents
ARS was obtained from Guilin Pharmaceutical Co., Ltd. (Guangxi, China). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT), N-acetyl-L-cysteine (NAC), desferrioxamine (DFOM), gelatin, epidermal growth factor (EGF), and anti-β actin antibody were acquired from Sigma-Aldrich (St. Louis, MO). Carboxy-H2DCFDA and DiOC2(3) Assay Kits were bought from Molecular Probes (Eugene, OR). Annexin V/propidine iodide (PI) Apoptosis Detection Kit was from KeyGEN (Nanjing, Jiangsu, China). M199, RPMI1640, and fetal bovine serum (FBS) were purchased from GIBCO (GibcoBRL, Gaithersburg, MD). Endothelial cell growth supplement (ECGS) was obtained from BD Bioscience (Franklin Lakes, NJ). SB203580 and SB202190 were bought from Calbiochem (Gibbstown, NJ). Caspase 3, caspase 9, Bax, p38 mitogen-activated protein kinases (p38MAPK), t-p38MAPK, COX IV, and KDR/Flk-1 antibodies were purchased from Cell Signaling Technology (Beverly, MA). Cytochrome C and Bcl-2 antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). HRP-conjugated goat antirabbit secondary antibody and horseradish peroxidase (HRP)-conjugated goat antimouse secondary antibody were acquired from Vector (Burlingame, CA). Mitochondria isolation and electrochemiluminescence (ECL) detection kits were bought from Applygen Technologies, Inc. (Beijing, China). CD31 antibody was acquired from Thermo Fisher Scientific, Inc. (Fremont, CA). TUNEL staining kit was obtained through Roche (Indianapolis, IN).
Animals
Female Sprague-Dawley rats (150–230 g) were purchased from the Laboratory Animal Center of Guangdong Province (Guangzhou, Guangdong, China). Animals were maintained in temperature-controlled conditions under a light/dark photo cycle with food and water supplied ad libitum, and treated in strict agreement with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Corneal Alkali Burn Model
Sprague-Dawley rats were anesthetized by intraperitoneal injection of 3% pentobarbital solution at 45 mg/kg. A round filter paper (4 mm in diameter) was soaked in 1 N NaOH, and applied on the central corneal surface for 40 seconds. The ocular surface was rinsed with 30 mL of sterile normal saline. Rats were randomly divided into three groups, and treated with eye drops containing with 25 μM ARS (n = 11), PBS, (negative control) (n = 10), and 0.1% dexamethasone solution (Dex, positive control) (n = 8), respectively, for 11 days, 4 times/d directly following the operation.
Qualification of Corneal Neovascularization
Rats were anesthetized and examined under a slit-lamp microscope every other day after injury. The inflammatory index was analyzed as previously described.14 The length and clocks of CNV were measured, and the areas of neovascularization were calculated by the following formula:
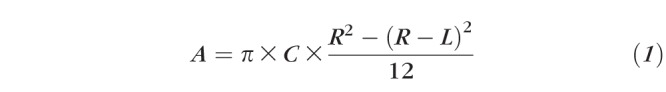 |
where C is the clocks of CNV, L is the length of CNV, and R is the measured radius of the rat cornea, R is equal to 3.5 mm.
Cell Culture
Human umbilical vein endothelial cells (HUVECs) were freshly isolated from human umbilical cords obtained from Guangzhou Women and Children's Medical Center (Guangzhou, Guangdong, China), and identified as described previously.15 All experiments were carried out between cell passages two to six.
Human dermal microvascular endothelial cells (HDMECs) were purchased from ATCC (Washington, DC) and cultured in vascular special medium and supplementary growth kit as recommended. All experiments were carried out in passage three or four.
Rat corneal epithelial cells (RCEpiCs) were isolated in primary culture from Sprague-Dawley rats. Rats were sacrificed and corneas were dissected and cut into five to eight pieces, and then placed in a 2% gelatin-coated 60-mm plate with the epithelial side faced down. The plates were cultured in a humidified incubator with 5% CO2 at 37°C. Culture medium was replaced every 2 days. RPMI 1640 (GibcoBRL) medium was used as the culture medium of RCEpiCs, and was supplemented with 10% FBS, 10 ng/mL EGF, 100 U/mL streptomycin and 100 U/mL penicillin. All experiments were carried out between cell passages one to two.
Human corneal epithelial cells (HCEpiCs) were kindly provided by Zhi-cong Wang in Zhongshan Ophthalmic Center of Sun Yat-sen University (Guangzhou, Guangdong, China). Cells were cultured in Dulbecco's modified Eagle's Medium (DMEM; GibcoBRL) supplemented with 10% FBS, 10 ng/mL EGF, 200 nmol/mL L-Glutamine, 10 μg/mL insulin, 5 μg/mL dexamethasone, 5.5 μg/mL transferrin, 100 U/mL streptomycin, and 100 U/mL penicillin.
Cell Viability Assay
Cells were treated with increasing concentrations of ARS for 48 hours. Cell viability was measured using the MTT assay according to the manufacturer's protocol. Data represented absorbance and were expressed as percentages of negative controls. The inhibitory effects of ARS were expressed as half maximal inhibitory concentration (IC50) values, which were determined from three independent studies.
Cell Apoptosis Assay
HUVECs were treated with ARS for 24 hours. Apoptotic cells were double-stained with AnnexinV and PI (KeyGEN) according to the manufacturer's recommendation and analyzed by flow cytometry.
Determination of Mitochondrial Membrane Potential
After treatment with 0, 25, or 100 μM ARS for 12 hours, HUVECs were stained with fluorescent dye DiOC2(3) and the mitochondrial membrane potential (Ψm) was quantified by flow cytometry according to the manufacturer's instructions.
ROS Measurement
HUVECs were treated with or without ARS for indicated times, and subsequently incubated with 5 μM Carboxy-H2DCFDA for another 30 minutes in dark and analyzed by flow cytometry at an excitation 492 nm and an emission 527 nm.
Isolation of Mitochondria
Cells were harvested by centrifugation at 300g for 5 minutes, and mitochondria were extracted using a mitochondria isolation kit according to the manufacturer's protocol. Mitochondrial pellets were resuspended in SDS lysis buffer.
Cytoplasmic Fraction Extraction
Cytoplasmic fraction was prepared as described previously.16 Briefly, cells were gently broken with glass homogenizers, and then centrifuged at 14,000 rpm for 20 minutes. The cytoplasmic protein was isolated from the supernatant.
Western Blot Analysis
Approximately 60 μg protein of each group was analyzed by Western blot analysis using specific primary antibodies. HRP-conjugated secondary antibodies were used to visualize the protein. Bands were visualized by using an ECL detection kit. The same membrane was stripped and reprobed with an antibody specific to β-actin as the protein loading control of total cell lysates and cytoplasmic extraction, or to COX IV as the control for mitochondrial extractions.
TUNEL and Anti-CD31 Double Staining
Frozen rat corneas were cut into 10-μm thick sections. Slides were air-dried for 30 minutes, fixed in cold acetone for 30 minutes, blocked with 10% normal goat serum for 30 minutes in room temperature, and, subsequently, incubated with anti-CD31 antibody overnight at 4°C, followed by incubation with goat antirabbit immunoglobulin G (Ig G) conjugated fluorescein for 1 hour at room temperature.
TUNEL staining was applied to slides prelabeled with anti-CD31 antibody. TUNEL staining was performed according to the manufacturer's instructions. Slides were viewed by fluorescence microscope equipped with appropriate filters.
Statistical Analysis
All data were expressed as mean ± SEM. SPSS 13.0 software (SPSS, Inc., Chicago, IL) was used for Student's t-test in all statistical analyses. A P value of less than 0.05 was considered statistically significant.
Results
ARS Inhibits Alkali-Induced Corneal Neovascularization
To investigate the effect of ARS on CNV, the length of the longest new vessel in alkali-burned cornea was monitored by slip lamp examination on the first, second, third, fifth, seventh, ninth, and eleventh day after the operation, and areas of CNV were calculated. Decreased neovascularization was identified in ARS-treated groups compared with the PBS-treated group (Figs. 1A, 1B). Hematoxylin and eosin (HE) staining revealed that many messy large vessels and inflammatory cells present in the PBS-treated group and less and smaller vessels and inflammation reactions were present in ARS-treated corneas (Fig. 1A). Meanwhile, our data demonstrated that CNV initiation and progression were decreased in ARS-treated groups (Fig. 1B). Moreover, ARS and Dex decreased the inflammation index in alkali-burned cornea (Fig. 1C). Taken together, these data suggested that ARS significantly ameliorated neovascularization and inflammation in alkali-burned cornea.
Figure 1.

Effects of ARS on alkali-induced rat CNV. (A) Top panels: Representative corneal photograph of PBS-treated group and ARS-treated group at PD11. Bottom panels: Representative HE staining of PBS-treated group and ARS-treated group at PD11 (asterisks show vessels in cornea). Original magnification ×200. (B, C) Statistical analysis of total CNV area and average inflammation index. (D) Representative image of CD31-TUNEL double staining in alkali-burned cornea. Green is TUNEL positive staining, and red is CD31 positive staining. **P < 0.01.
CD31 and TUNEL double staining were performed to determine whether endothelial cells were undergoing apoptosis in alkali-wounded cornea. There were more vessels in the corneal epithelium and stroma in the PBS-treated group relative to the ARS-treated group. ARS significantly increased apoptosis of vascular endothelial cells in alkali-burned cornea (Fig. 1D). Taken together, these results suggested that ARS suppressed corneal neovascularization by inducing vascular endothelial cell apoptosis.
ARS Has Selective Inhibitory Effects on Vascular Endothelial Cells
ARS significantly inhibited proliferation of HUVECs and HDMECs in a concentration-dependent manner with an IC50 of 25 μM (Fig. 2A). ARS exerted inhibitory effects on the proliferation of RCEpiCs and HCEpiCs up to the concentration of 200 μM (Fig. 2B). These results exhibited that ARS selectively induced apoptosis in vascular endothelial cells.
Figure 2.

Effects of ARS on HUVECs proliferation and apoptosis. (A) HDMECs and HUVECs were treated with ARS at indicated concentrations for 48 hours. The viable cells were quantified by MTT assay. (B) RCEpiCs and HCEpiCs were treated with ARS at different concentrations for 48 hours. The viable cells were quantified by MTT. (C) After incubation with ARS for 24 hours, apoptotic HUVECs were stained with AnnexinV and PI, and analyzed by flow cytometry. (D) Quantitative analysis of apoptosis in endothelial cell induced by ARS. n = 3, * and ** denote P < 0.05 and P < 0.01, respectively.
Furthermore, the percentages of apoptotic HUVECs treated with PBS, 25 μM colchicines (positive control), and different concentrations of ARS (12.5 μM ∼ 100 μM) for 24 hours were 5.33 ± 0.612%, 38.98 ± 1.590%, 10.88 ± 1.036%, 21.7 ± 0.936%, 34.48 ± 1.216%, and 47.41 ± 1.180%, respectively (Figs. 2C, 2D), suggesting that ARS promoted endothelial cells death at least in part by inducing apoptosis.
ARS Induces Apoptosis of Endothelial Cells Through the Mitochondrial Pathway
ARS increased mitochondrial Bax, while simultaneously decreasing mitochondrial Bcl-2 (Fig. 3A). Consistent with elevation of the mitochondrial Bax/Bcl2 ratio, ARS promoted cytochrome C release in a concentration-dependent fashion (Fig. 3B).
Figure 3.

Effect of ARS on the expression of Bax, Bcl-2, and cytochrome C. HUVEC cells were treated with ARS for 12 hours. (A) Mitochondrial Bax and Bcl-2 levels were determined by Western blot analysis, and quantified with densitometry. (B) Cytosolic cytochrome C was detected and quantified. n = 3. * and ** denote P < 0.05 and P < 0.01, respectively.
To identify the effect of ARS on mitochondrial membrane potential (Ψm), HUVECs were stained with DiOC2(3) and the fluorescence intensity was qualified by flow cytometry. DiOC2(3) is identified to specifically assemble in mitochondria at a low working concentration, which can be used to indicate the membrane potential changes. In brief, when mitochondrial membrane potential is disrupted, less DiOC2(3) binds to the mitochondrial membrane, and the fluorescence changes from red to green. As mitochondrial membrane potential disruption is aggravated, the red fluorescence will be attenuated. In the PBS-treated group, there was a considerable quantity of red-emitting mitochondria. However, in the 25 μM ARS-treated group, the amount of red-emitting mitochondria was decreased, and almost no red-emitting mitochondria were detected in the 100 μM ARS-treated group (Fig. 4), suggesting that ARS significantly decreased mitochondrial membrane potential in a concentration-dependent manner. Moreover, ARS stimulated cleavage of caspase 3 and 9 in both HUVECs and HDMECs (Fig. 5, Supplementary Fig. S2), while Fas and Fas ligand expression and caspase 8 cleavage were unchanged (Fig. 5, Supplementary Fig. S3). Furthermore, we found that cleaved caspase 9 and 3 were increased in ARS-treated rat corneas (Supplementary Fig. S1A). Taken together, these data suggested that ARS induced apoptosis of endothelial cells via the intrinsic mitochondrial pathway but not the extrinsic death receptor pathway.
Figure 4.

ARS decreased mitochondrial membrane potentials in HUVECs. HUVECs, treated with ARS for 12 hours, mitochondrial membrane potentials were measured by DiOC2(3) staining. (A) Representative confocal microscope images. Original magnification ×600. (B) Quantitative analysis of red/green intensity ratio by flow cytometry analysis. n = 3. ** denotes P < 0.01.
Figure 5.

Effect of ARS on the cleavage of caspases in HUVECs. HUVEC cells were treated with ARS for 24 hours. Total and cleaved caspase 9, caspase 8, and caspase 3 were detected by Western blot analysis.
ARS Induces Apoptosis by Activating p38 MAPK
The p38MAPK pathway is activated by cellular stress, and is heavily involved in drug-induced apoptosis. We found ARS activated p38MAPK in a time- and concentration-dependent manner (Figs. 6A, 6B). To examine whether p38MAPK was involved in ARS-induced apoptosis, HUVECs were pretreated with two specific p38MAPK inhibitors, SB202190 (10 μM) and SB203580 (10 μM), respectively. Both inhibitors blocked ARS-stimulated p38MAPK activation in endothelial cells (Fig. 6C). Furthermore, the two inhibitors decreased caspase 9 cleavage (Fig. 6D), and decreased the ARS-induced apoptotic ratio of HUVECs (Fig. 6E). These data indicated that ARS induced apoptosis of endothelial cells by triggering p38MAPK activation.
Figure 6.

ARS induced p38MAPK activation in HUVECs. ARS activated p38MAPK in time-dependent manner (A) and concentration-dependent manner (B). Specific p38MAPK inhibitors SB202190 and SB203580 inhibited ARS-activated p38MAPK (C). SB202190 and SB203580 protected cells from ARS-induced apoptosis (D) and (E). n = 3. ** denotes P < 0.01.
Cytotoxicity of ARS is Dependent Upon Generation of Reactive Oxygen Species
It is known that generation of free radicals is one of the main mechanisms of ART derivatives' antimalarial activity, and we hypothesized that it might also play a role in CNV inhibition. We applied the fluorescent probe Carboxy-H2DCFDA to qualify the reactive oxygen species (ROS) level in ARS-treated endothelial cells. We found that ROS were elevated as early as 10 minutes after 25 μM ARS treatment, reached the maximum level at 4 hours, and persisted to 24 hours (Fig. 7A). ARS induced ROS generation in a concentration-dependent manner (Fig. 7B). However, ARS unexpectedly decreased ROS levels in RCEpiCs (Fig. 7C), suggesting a paradoxical effect.
Figure 7.

ARS specifically induced ROS accumulation in HUVECs. ROS level was determined by Carboxy-H2DCFDA staining and analyzed by flow cytometry. ARS induced ROS accumulation in a time-dependent manner (A) and a concentration-dependent manner (B). ARS changed ROS level in RCEpiCs after 2 hours of treatment (C). n = 3. * and ** denote P < 0.05 and P < 0.01, respectively.
To further examine whether ROS were involved in the ARS-induced HUVEC apoptosis, the ROS scavenger NAC was employed. We found that NAC inhibited ARS-induced ROS generation and phosphorylation of p38MAPK (Figs. 8A, 8B). Moreover, NAC decreased ARS-induced cleavage of caspase 9 and protected HUVECs from apoptosis (Figs. 8C, 8D). Taken together, these results suggested that ROS are essential for ARS-induced apoptosis of endothelial cells.
Figure 8.

ARS induced formation of ROS in HUVECs. (A) HUVECs were pretreated with NAC for 3 hours and co-incubated with ARS for additional 4 hours. ROS formation was measured by flow cytometry. (B) After cotreated with ARS and NAC for 8 hours, phosphorylation of p38MAPK was detected by Western blot analysis and the bands were semiquantified with densitometry in HUVECs. (C) HUVECs were co-incubated with ARS for 24 hours. Cleavage level of caspase 9 was determined by Western blot analysis. (D) Apoptotic HUVECs were quantified by flow cytometry. n = 3. ** and ## denote P < 0.01, respectively. *: versus control group, #: versus ARS group.
ARS-Induced ROS Formation Is Iron-Dependent
Ferrous iron plays an essential role in the antimalarial and antitumor activities of ART and its derivatives. To identify whether the antiangiogenic effect of ARS is iron-dependent, we used FeSO4 or the iron chelating agent DFOM in vitro. HUVECs were pretreated with 10 μM FeSO4 or 25 μM DFOM for 3 hours, and then cotreated with ARS for the indicated time points. We found that accumulation of ARS-induced ROS was enhanced by FeSO4, and abrogated by DFOM (Fig. 9A). Furthermore, FeSO4 enhanced ARS-induced caspase 9 cleavage and elevated the apoptotic ratio of endothelial cells after ARS treatment, and DFOM inhibited ARS-induced caspase 9 cleavage and apoptosis (Figs. 9B, 9C). These results suggested that ROS formation was iron-dependent in endothelial cells.
Figure 9.

ARS-induced ROS accumulation and apoptosis are iron-dependent. FeSO4 and DFOM were used to explore the effect of iron on ARS-induced apoptosis in HUVECs. Cells were pretreated with FeSO4 or DFOM for 3 hours and co-incubated with ARS for indicated times. (A) ROS were stained with Carboxy-H2DCFDA and quantified by flow cytometry. (B) The cleaved fragment of caspase 9 was determined by Western blot analysis. (C) Apoptotic HUVECs was measured by flow cytometry. n = 3. ** and ## denote P < 0.01, respectively. *: versus control group, #: versus ARS group.
Discussion
Recent studies reported that ART and its derivatives can inhibit tumor angiogenesis by inducing vascular endothelial cells apoptosis.17 Therefore, we hypothesized that ART may also inhibit form of nontumor angiogenesis, such as CNV, which is a leading cause of blindness.1 The rat alkali burn-induced CNV model was employed to assess the therapeutic effect of ARS in this study. Our results demonstrated for the first time that ARS could notably inhibit CNV.
Currently, several antiangiogenic agents for CNV treatment have been identified, but all remain unsatisfactory due to undesired side effects.1,2 In this study, we used Dex, a synthetic glucocorticoid, as the positive control in vivo. Corticosteroids are the most commonly used and effective drugs in the therapy of ocular inflammation. Our recent results showed that Dex can inhibit angiogenesis mainly by down-regulation of VEGF.18 Therefore, Dex was set as an effective control group in this study to evaluate the antiinflammation and antiangiogenic effects of ARS. However, many adverse effects identified in clinical treatment and in preclinical research limit the use of corticosteroids, including drug-induced ocular hypertension and glaucoma, tear-film instability, epithelial toxicity, crystalline keratopathy, decreased wound strength, orbital fat atrophy, ptosis, and limitation of ocular movement.19 It has been reported that Dex retarded corneal epithelial healing in rabbit alkali-burning corneal wounding and carried a 7% to 8% risk of producing a significant rise of IOP, which may result in corneal glaucoma.20–22 Consistent with the mentioned side effects of Dex, our results showed that 0.1% Dex resulted in corneal ulcer, although it could significantly inhibit corneal inflammation and revascularization. By CD31-TUNEL double staining, we found ARS can significantly induce apoptosis of vascular endothelial cells in vivo. Moreover, ARS specifically inhibited proliferation and induced apoptosis of endothelial cells in vitro, while corneal epithelial cells were unaffected. The selective inhibition of endothelial cells may be responsible for the decreased rate of adverse effects. Furthermore, we use fluorescein sodium to stain and quantify the area of epithelial defects in damaged corneas on the first day after operation, according to the previous report.23 The data showed ARS treatment can decrease corneal defect area compared with PBS treatment group (Supplementary Fig. S1B), which at least suggested ARS inhibited corneal inflammation and neovascularization without significant side effects on corneal wound healing. These results suggested that ARS could be an ideal agent for CNV treatment as it is highly effective, presents few adverse effects, and is inexpensive. We chose the concentration of ARS in the animal experiments according to the IC50 of ARS on proliferation of vascular endothelial cells, which is approximately 25 μM (9.6 mg/L). It was reported that ARS had a maximal concentration (Cmax) of 35.6 μM (13.7 mg/L) in patients who suffered from vivax malaria and were treated with intravenous injection of ARS.24 As artemisin and derivatives have an elimination half-life time, another clinical study demonstrated that the range of Cmax of ARS was from 2.7 μM (1.02 mg/L) to 426.6 μM (164 mg/L) in Ugandan adults with severe malaria.25 Therefore, the concentration of ARS in our animal work is not over the Cmax. To achieve an effective concentration of ARS in the cornea, we chose local administration of artesuante by using eye drops in this study. We made artesuante eye drops and treated the animals for 11 days immediately following the operation and 4 times per day until our experiment endpoint. The results suggested that eye drop may be the best way to deliver ARS in patients with CNV.
Vascular endothelial cells are the main target for antiangiogenic therapeutics.5 Besides the apoptosis induction effect of ARS, we detected expression levels of VEGF receptor 2 (KDR/Flk-1) and VEGF secretion levels in HUVECs and HDMECs, respectively. We found ARS down-regulated expression of KDR/Flk-1 in both endothelial cells in a concentration-dependent manner, which is consisted to previous report13 (Supplementary Figs. S5A, S5B). However, ARS has no effect on VEGF secretion in endothelial cells (Supplementary Fig. S5C). These results suggested that ARS could inhibit corneal neovascularization by both inducing endothelial cell apoptosis and disrupting the VEGF signaling pathway. In our study, ARS inhibited cell proliferation and induced cell apoptosis in HUVECs and HDMECs. Cells undergo apoptosis via the intrinsic mitochondrial pathway or the extrinsic death receptor pathway.26 Previous studies have reported that ART and its derivatives induced apoptosis of several cancer cell lines via the mitochondrial pathway, and the death receptor pathway in some cell types.17,27
In our study, ARS increased the mitochondrial Bax/Bcl-2 ratio, reduced mitochondrial membrane potential, stimulated cytochrome C release, and caspase 3 and 9 cleavage. Contrastingly, ARS had no effects on the expression of Fas and Fas-ligand and cleavage of caspase 8, which suggested that the mitochondrial pathway, rather than the death receptor pathway, was activated in ARS-induced endothelial cell apoptosis. Similar results were reported in Jurkat cells, suggesting that the effect is specific to the ART derivative and cell type.28,29 Absence of Fas-associating protein with a novel death domain or caspase 8 activation did not alter apoptotic rates in dihydroartemisinin-treated Jurkat cells, while overexpression of dominant-negative caspase 9 or antiapoptotic proteins Bcl-xL and Bcl-2 largely decreased the cytotoxicity of dihydroartemisinin, which demonstrated selective intrinsic apoptotic pathway was involved in ART compounds-induced apoptosis.28,29 However, the underlying mechanism by which ART activates the mitochondrial apoptosis pathway in endothelial cells was previously poorly understood. Therefore, in this study, we sought to identify which signaling pathway was upstream of mitochondrial apoptosis, and demonstrated that ROS and p38 MAPK were implicated in ARS-induced endothelial cell apoptosis and played essential roles.
The formation of free radicals is strongly associated with the antimalarial and antitumor activities of ART and its derivatives.6 In malaria, when ART and its derivatives bind to hemin or ferrous heme, the peroxide bridge is cleaved, and carbon-centered radicals are formed, which rapidly convert to other free radicals. ART and derivative compounds kill malaria parasites by generating free radicals to alkylate proteins and interrupt malaria parasite metabolism.6 In cancer cells, several studies have shown that ROS generation is the essential factor of ART compound-induced apoptosis.7 In this study, we observed that the ROS level in endothelial cells was enhanced by ARS, and that the ROS scavenger NAC blocked ARS-induced endothelial cell apoptosis, suggesting an essential role of ROS generation. In contrast, ROS accumulation in corneal epithelial cells was lower indicating that ARS-induced ROS generation was cell type-specific. The differential induction of ROS in different cell types may be responsible for the selective apoptosis induction effect of ARS.
Ferrous iron plays an essential role in the antimalarial and antitumor activities of ART and its derivatives. In cancer studies, ferrous salt and the transferrin receptor promoted the inhibitory effect of ART and its derivatives on tumor cells and tumor tissues.30–33 Furthermore the iron chelating agent DFOM blocked the inhibitory effect of ART and compounds on tumor cells, suggesting that iron-dependent ROS generation was responsible for ART-induced apoptosis.27,30 We also observed that ferrous salt enhanced ARS-mediated ROS generation and toxicity in endothelial cells. These results suggested that ARS-induced endothelial cell apoptosis is mediated by iron-dependent ROS generation.
Several studies have shown that the p38MAPK pathway is essential for intrinsic mitochondrial apoptosis.34,35 It has been reported that activation of p38MAPK increases the Bax/Bcl-2 ratio, decreases mitochondrial membrane potential, promotes release of cytochrome C, and initiates cleavage of caspases sequentially.36 In our study, we demonstrated that ARS induced p38 MAPK activation in a concentration- and time-dependent manner. p38MAPK inhibitors decreased the apoptotic rate of ARS-treated HUVECs, suggesting that p38 MAPK plays a central role in ARS-mediated HUVEC apoptosis. Conversely, ARS decreased the phosphorylation levels of p42/44 MAPK and c-Jun N-terminal kinases (JNK) (Supplementary Fig. S4). The same types of changes of MAPK family members were observed in dihydroartemisinin-treated HL60 cells.37 These results suggested that p38MAPK activation might play a more important role in the pro-apoptotic activity of ART and its derivatives. The changes in p42/44 MAPK and JNK activation and their role in ARS-induced apoptosis need to be further evaluated. Moreover, NAC blocks the phosphorylation of p38MAPK mediated by ARS, indicating that ARS-induced ROS formation is the upstream event of p38MAPK activation.
In conclusion, this study provides evidence for the first time that ARS inhibits CNV by specifically inducing endothelial cell apoptosis. Furthermore, we demonstrated that ARS induced endothelial cell apoptosis through an iron/ROS-dependent p38MAPK-mitocholdrial pathway. Together, our findings suggest that ARS may have therapeutic potential for CNV treatment, and elucidate its mechanism of action in this disease model.
Acknowledgments
The authors thank Elizabeth Moran from the Department of Cell Biology, University of Oklahoma for her editing, which has greatly improved the manuscript.
Supported by National Nature Science Foundation of China Grants 30971208, 30973449, 81070746 81001014, 81172163, 81272338, 81272515, and 81200706; National Key Sci-Tech Special Project of China Grants 2009ZX09103-642 and 2013ZX09102-053; Program for Doctoral Station in University Grants 20100171110049, 20100470955, 2011M501364, and 20120171110053; Key Project of Nature Science Foundation of Guangdong Province, China, Grant 10251008901000009; Key Sci-Tech Research Project of Guangdong Province, China, Grant 2011B031200006; Key Sci-Tech Research Project of Guangzhou Municipality, China, Grants 2007Z3-E5041, 2011Y1-00017-8, and 12A52061519; and Program for Young Teacher in University Grants 09YKPY73 and 10YKPY28.
Disclosure: R. Cheng, None; C. Li, None; C. Li, None; L. Wei, None; L. Li, None; Y. Zhang, None; Y. Yao, None; X. Gu, None; W. Cai, None; Z. Yang, None; J. Ma, None; X. Yang, None; G. Gao, None
Footnotes
CR and LC contributed equally to the work presented here and should therefore be regarded as equivalent authors.
References
- 1.Chang JH, Gabison EE, Kato T, Azar DT. Corneal neovascularization. 2001;12:242–249. doi: 10.1097/00055735-200108000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Casey R, Li WW. Factors controlling ocular angiogenesis. 1997;124:521–529. doi: 10.1016/s0002-9394(14)70868-2. [DOI] [PubMed] [Google Scholar]
- 3.Price MO, Thompson RW Jr, Price FW., Jr. Risk factors for various causes of failure in initial corneal grafts. 2003;121:1087–1092. doi: 10.1001/archopht.121.8.1087. [DOI] [PubMed] [Google Scholar]
- 4.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 5.Folkman J. Angiogenesis. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 6.Klayman DL. Qinghaosu (artemisinin): an antimalarial drug from China. 1985;228:1049–1055. doi: 10.1126/science.3887571. [DOI] [PubMed] [Google Scholar]
- 7.Golenser J, Waknine JH, Krugliak M, Hunt NH, Grau GE. Current perspectives on the mechanism of action of artemisinins. 2006;36:1427–1441. doi: 10.1016/j.ijpara.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 8.White NJ. Qinghaosu (artemisinin): the price of success. 2008;320:330–334. doi: 10.1126/science.1155165. [DOI] [PubMed] [Google Scholar]
- 9.Efferth T. Molecular pharmacology and pharmacogenomics of artemisinin and its derivatives in cancer cells. 2006;7:407–421. doi: 10.2174/138945006776359412. [DOI] [PubMed] [Google Scholar]
- 10.Berger TG, Dieckmann D, Efferth T, et al. Artesunate in the treatment of metastatic uveal melanoma–first experiences. 2005;14:1599–1603. [PubMed] [Google Scholar]
- 11.Singh NP, Panwar VK. Case report of a pituitary macroadenoma treated with artemether. 2006;5:391–394. doi: 10.1177/1534735406295311. [DOI] [PubMed] [Google Scholar]
- 12.Efferth T. Willmar Schwabe Award 2006: antiplasmodial and antitumor activity of artemisinin–from bench to bedside. 2007;73:299–309. doi: 10.1055/s-2007-967138. [DOI] [PubMed] [Google Scholar]
- 13.Chen HH, Zhou HJ, Wu GD, Lou XE. Inhibitory effects of artesunate on angiogenesis and on expressions of vascular endothelial growth factor and VEGF receptor KDR/flk-1. 2004;71:1–9. doi: 10.1159/000076256. [DOI] [PubMed] [Google Scholar]
- 14.Laria C, Alio JL, Ruiz-Moreno JM. Combined non-steroidal therapy in experimental corneal injury. 1997;29:145–153. doi: 10.1159/000268009. [DOI] [PubMed] [Google Scholar]
- 15.Yang H, Cheng R, Liu G, et al. PEDF inhibits growth of retinoblastoma by anti-angiogenic activity. 2009;100:2419–2425. doi: 10.1111/j.1349-7006.2009.01332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan J, Quintas-Cardama A, Kantarjian HM, et al. EXEL-0862, a novel tyrosine kinase inhibitor, induces apoptosis in vitro and ex vivo in human mast cells expressing the KIT D816V mutation. 2007;109:315–322. doi: 10.1182/blood-2006-04-013805. [DOI] [PubMed] [Google Scholar]
- 17. Firestone GL, Sundar SN. Anticancer activities of artemisinin and its bioactive derivatives 2009. 11 e32. [DOI] [PubMed] [Google Scholar]
- 18.Li C, Li L, Cheng R, et al. Acidic/neutral amino acid residues substitution in NH2 terminal of plasminogen kringle 5 exerts enhanced effects on corneal neovascularization. 2013;32:680–688. doi: 10.1097/ICO.0b013e3182781ec9. [DOI] [PubMed] [Google Scholar]
- 19.McGhee CN, Dean S, Danesh-Meyer H. Locally administered ocular corticosteroids: benefits and risks. 2002;25:33–55. doi: 10.2165/00002018-200225010-00004. [DOI] [PubMed] [Google Scholar]
- 20.Chung JH, Kang YG, Kim HJ. Effect of 0.1% dexamethasone on epithelial healing in experimental corneal alkali wounds: morphological changes during the repair process. 1998;236:537–545. doi: 10.1007/s004170050118. [DOI] [PubMed] [Google Scholar]
- 21.Gelatt KN, Mackay EO. The ocular hypertensive effects of topical 0.1% dexamethasone in beagles with inherited glaucoma. 1998;14:57–66. doi: 10.1089/jop.1998.14.57. [DOI] [PubMed] [Google Scholar]
- 22.Holland EJ, Bartlett JD, Paterno MR, Usner DW, Comstock TL. Effects of loteprednol/tobramycin versus dexamethasone/tobramycin on intraocular pressure in healthy volunteers. 2008;27:50–55. doi: 10.1097/ICO.0b013e31815873c7. [DOI] [PubMed] [Google Scholar]
- 23.Han Y, Shao Y, Lin Z, et al. Netrin-1 simultaneously suppresses corneal inflammation and neovascularization. 2012;53:1285–1295. doi: 10.1167/iovs.11-8722. [DOI] [PubMed] [Google Scholar]
- 24.Batty KT, Le AT, Ilett KF, et al. A pharmacokinetic and pharmacodynamic study of artesunate for vivax malaria. 1998;59:823–827. doi: 10.4269/ajtmh.1998.59.823. [DOI] [PubMed] [Google Scholar]
- 25. Byakika-Kibwika P, Lamorde M, Mayito J, et al. Pharmacokinetics and pharmacodynamics of intravenous artesunate during severe malaria treatment in Ugandan adults 2012. 11 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hengartner MO. The biochemistry of apoptosis. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 27.Disbrow GL, Baege AC, Kierpiec KA, et al. Dihydroartemisinin is cytotoxic to papillomavirus-expressing epithelial cells in vitro and in vivo. 2005;65:10854–10861. doi: 10.1158/0008-5472.CAN-05-1216. [DOI] [PubMed] [Google Scholar]
- 28.Mercer AE, Maggs JL, Sun XM, et al. Evidence for the involvement of carbon-centered radicals in the induction of apoptotic cell death by artemisinin compounds. 2007;282:9372–9382. doi: 10.1074/jbc.M610375200. [DOI] [PubMed] [Google Scholar]
- 29.Handrick R, Ontikatze T, Bauer KD, et al. Dihydroartemisinin induces apoptosis by a Bak-dependent intrinsic pathway. 2010;9:2497–2510. doi: 10.1158/1535-7163.MCT-10-0051. [DOI] [PubMed] [Google Scholar]
- 30.Efferth T, Benakis A, Romero MR, et al. Enhancement of cytotoxicity of artemisinins toward cancer cells by ferrous iron. 2004;37:998–1009. doi: 10.1016/j.freeradbiomed.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 31.Sadava D, Phillips T, Lin C, Kane SE. Transferrin overcomes drug resistance to artemisinin in human small-cell lung carcinoma cells. 2002;179:151–156. doi: 10.1016/s0304-3835(02)00005-8. [DOI] [PubMed] [Google Scholar]
- 32.Lai H, Singh NP. Selective cancer cell cytotoxicity from exposure to dihydroartemisinin and holotransferrin. 1995;91:41–46. doi: 10.1016/0304-3835(94)03716-v. [DOI] [PubMed] [Google Scholar]
- 33.Lai H, Sasaki T, Singh NP, Messay A. Effects of artemisinin-tagged holotransferrin on cancer cells. 2005;76:1267–1279. doi: 10.1016/j.lfs.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 34.Chang L, Karin M. Mammalian MAP kinase signalling cascades. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 35.Cowan KJ, Storey KB. Mitogen-activated protein kinases: new signaling pathways functioning in cellular responses to environmental stress. 2003;206:1107–1115. doi: 10.1242/jeb.00220. [DOI] [PubMed] [Google Scholar]
- 36.Chen YJ, Liu WH, Kao PH, Wang JJ, Chang LS. Involvement of p38 MAPK- and JNK-modulated expression of Bcl-2 and Bax in Naja nigricollis CMS-9-induced apoptosis of human leukemia K562 cells. 2010;55:1306–1316. doi: 10.1016/j.toxicon.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 37.Lu JJ, Meng LH, Cai YJ, et al. Dihydroartemisinin induces apoptosis in HL-60 leukemia cells dependent of iron and p38 mitogen-activated protein kinase activation but independent of reactive oxygen species. 2008;7:1017–1023. doi: 10.4161/cbt.7.7.6035. [DOI] [PubMed] [Google Scholar]