

Abstract
Paragangliomas are rare neuroendocrine tumors where hypoxia-inducible factor plays a critical role in tumorigenesis. It has been suggested that patients with congenital heart disease, in particular, may have cellular environment and relative hypoxia favorable to the development of these neuroendocrine tumors. Here, we present a case of an 11-year-old child with hypoplastic left heart syndrome previously palliated with Fontan procedure, diagnosed with paraganglioma on surveillance imaging. We present the clinical course, intervention, and outcome as well as review the possible contributory mechanisms. As we continue to improve long-term survival for single ventricle patients, awareness of these tumors during surveillance may be warranted as timely intervention may lead to cure.
Keywords: Congenital heart disease, Fontan, heart transplantation, hypoxia, outcome, paraganglioma
INTRODUCTION
Paragangliomas (PGL) are rare neuroendocrine tumors that arise from the extra-adrenal paraganglionic system, with one-third to one-half of cases being inherited. Genetic susceptibilities resulting in the activation of hypoxia pathways within chromaffin cells have been implicated in the development of these tumors.[1] By extension, chronic hypoxia has been implicated in causation of PGL as demonstrated by reports in patients living at high altitudes and in those suffering from chronic obstructive lung disease.[2] Cyanotic congenital heart disease (CHD) has also been suggested as a risk factor for paraganglioma-pheochromocytoma.[3,4] We present a case of a retroperitoneal paraganglioma in a Fontan patient. We believe chronic hypoxia related to single ventricle palliation to be a trigger for the development of paraganglioma in the above-said patient.
CASE REPORT
An 11-year-old female child with hypoplastic left heart syndrome underwent staged palliation culminating with an extracardiac fenestrated Fontan procedure at 4 years of age. Before the Fontan procedure, she maintained oxygen saturations between 75% and 77%, whereas after the Fontan procedure, she maintained saturations between 92% and 94%. She did well for the subsequent 7 years, but by 12 years of age, there was evidence of Fontan dysfunction. At 12 years of age, a cardiac catheterization was performed which showed evidence of suboptimal Fontan pathophysiology with elevated Fontan pressures of 18 mmHg, hepatic vein wedge pressure was 20 mmHg, and right ventricular end-diastolic pressure was elevated at 16 mmHg.
An echocardiogram at the same time demonstrated moderate-to-severe tricuspid valve regurgitation and moderately decreased right ventricular systolic function.
She underwent an abdominal magnetic resonance imaging (MRI) as a routine screening for Fontan-associated liver disease. MRI was significant for evidence of hepatomegaly, liver congestion, and presence of hypervascular nodules. There was an incidental finding of a 3.7 cm complex, heterogeneous mass that appeared to be arising exophytically from the head/uncinate process of pancreas with the duodenum draped anteriorly over the mass [Figure 1].
Figure 1.
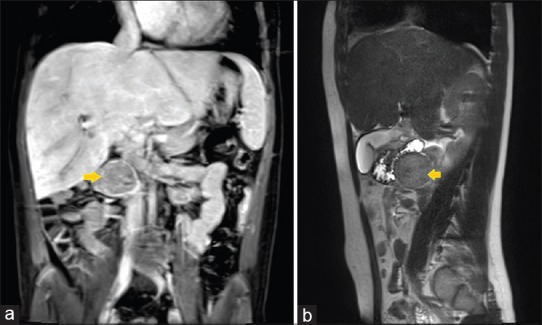
Magnetic resonance imaging of the abdomen showing coronal (a) and sagittal (b) view of the heterogeneous mass (arrows) appearing to arise from the pancreas, close to the right lobe of the liver, and immediately posterior to the duodenum
There was a coincidental history of chronic abdominal discomfort, poor appetite, and poor weight gain over the preceding year. In light of the MRI findings, there was concern that the mass was contributing to some of her symptoms. The main differential diagnosis based on the MRI findings was pseudopapillary tumor of the pancreas. Secretory tumors such as pheochromocytoma were considered less likely due to location and absence of typical signs and symptoms such as hypertension and tachycardia. Diagnostic certainty was necessary to establish heart transplant candidacy. The anatomic location of the mass in continuity or in proximity to the uncinate process of the pancreas warranted planning for potential Whipple procedure. After extensive discussions among the different specialties and the family, a decision was made to proceed with the surgery.
In the operating room, the mass was immediately visible on the right side on abdominal exploration. Neovascularization was seen around the mass and it was found to be moderately adherent to the inferior vena cava. After severing all vascular ties, the mass was found to be discrete from the duodenum and pancreas. It was lifted up from its retroperitoneal location after a generous Kocher maneuver, removed from the body, and sent to pathology. A liver biopsy was also obtained at the same time and sent for pathology.
On gross examination, the mass was pink-red to pink-tan rubbery in consistency. Frozen section was suggestive of paraganglioma. Hematoxylin and Eosin stains showed well-defined nests of cuboidal cell with abundant cytoplasm, separated by highly vascularized fibrous septa [Figure 2a]. Immunostains exhibited synaptophysin positive, chromogranin A positive, and S100 positive for sustentacular cells [Figure 2b]. These findings were consistent with diagnosis of paraganglioma.
Figure 2.
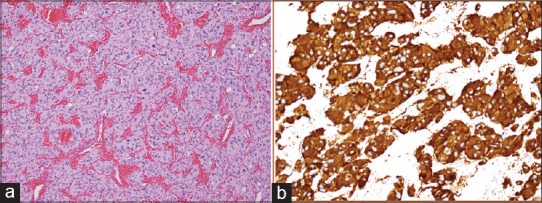
Histopathology of the paraganglioma. (a) H and E stain showing well-defined nests of cuboidal cells with abundant cytoplasm separated by highly vascularized fibrous septa. (b) Immunostaining shows strong positivity to synaptophysin immunostain in the tumor cells
Liver biopsy histopathology showed moderate-to-severe fibrosis with sinusoidal dilatation [Figure 3a]; the Masson's trichrome stain confirmed the degree of fibrosis [Figure 3b]. Findings were consistent with Fontan-associated liver disease.
Figure 3.
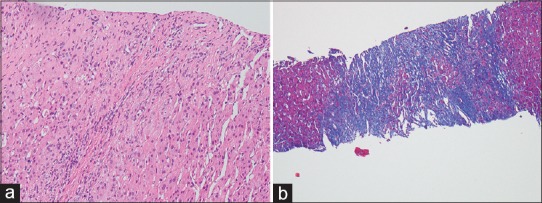
Liver biopsy histopathology demonstrating (a) show hepatic lobules and portal areas with fibrosis with H and E staining while (b) staining with Masson's trichrome stain highlights the extent of bridging fibrosis consistent with Fontan hepatopathy
The patient recovered very well from the procedure and was extubated shortly after arrival at the intensive care unit. Her hemodynamics were stable throughout the procedure as well as recovery. Over the subsequent 2 months, she continued to demonstrate cardiac dysfunction and developed exertional dyspnea. On follow-up, imaging (positron emission scan) as well as endocrine assessment in the form of metanephrine levels was unremarkable.
In the setting of failing Fontan physiology as well as the presence of associated liver disease, the patient was, therefore, evaluated and listed for heart transplantation. In certain patients with Fontan-associated liver disease, including those with significant fibrosis but without ascites, with low (<2) varices, ascites, splenomegaly, and thrombocytopenia scores and low modified end-stage liver disease score, we have opted for single-organ transplantation (heart) rather than dual heart–liver transplantation. She underwent heart transplantation without complications and now 6 months out she is thriving.
DISCUSSION
Patients with Fontan pathophysiology have complex morbidities such as protein-losing enteropathy, associated liver disease, plastic bronchitis, chronic hypoxia, and venous congestion to list a few.[5] This case highlights a rare but potentially important finding of paraganglioma in a Fontan patient.
PGL are rare tumors originating from neural crest cells within the adrenal gland or the sympathetic/parasympathetic chains in the paravertebral regions. Although initially thought to be sporadic tumors, there is increasing evidence that PGL along with pheochromocytoma and carotid body tumors forms a group of neoplasms that may be genetically related.[1] The common thread appears to be relative hypoxia and abnormalities of energy metabolism at the cellular level in cells that are predisposed. Research has implicated genes that cluster in a common cellular pathway that activates hypoxia-inducible factors leading to the proposition of pseudohypoxia hypothesis of PGL development, in which these genetic susceptibilities result in aberrant activation of hypoxia pathways in chromaffin cells resulting in the development of PGL. PGL and related tumors, therefore, are deemed as pseudohypoxic tumors.[1,2] Hypoxia-inducible factors are vital transcription factors the control energy metabolism at the cellular and mitochondrial level.
Normally, oxygen is responsible for degradation of HIF. In the setting of relative hypoxia, there may be an accumulation of HIF causing transcriptional changes to genes leading to tumorigenesis. This mechanism is further validated by the occurrence of high-altitude PGL.[2,6]
Cyanotic CHD as a potential reason for hypoxia-sensitive tumor development was recognized and proposed as early as 1964 by Folger et al. when they reported on the association of cyanotic heart disease with pheochromocytoma.[3] Two of the five cases in this report were pediatric patients and carried a diagnosis of transposition of great arteries. In our literature review, the only other pediatric Fontan patient is reported by Opotowsky et al.[7] Majority of the other patients reported are adults or are reported as autopsy findings.[4,8] Diagnosis continues to be a challenge. It is noteworthy that a significant portion of the early reports were patients diagnosed late in the clinical course, sometimes at autopsy. In the current era, increasing awareness for this rare complication may lead to timely diagnosis. Patients do not have to be cyanotic at the time of diagnosis as demonstrated by Opotowsky et al.[7] We speculate that a period of relative hypoxemia (as in our case, before Fontan) during early stages of single-ventricle palliation may provide adequate stimulation for tumorigenesis. High degree of clinical suspicion is necessary for diagnosis as the symptoms may often be nonspecific in CHD patients. Those with pheochromocytoma may present with signs and symptoms consistent with high catecholamine state such as uncontrolled hypertension and/or tachycardia and tachyarrhythmias. The signs and symptoms may get attributed to the primary cardiac disease rather than an alternative diagnosis. Patients with PGL, however, may be silent for the duration of time and may present with nonspecific symptoms such as abdominal pain as was true in our case. Biochemical testing in the form of blood and urinary catecholamine levels is helpful. Advanced imaging such as computerized tomography scan, MRI or 123I-metaiodobenzylguanidine scintigraphy scan may be necessary to visualize tumors. Fontan patients are increasingly undergoing imaging surveillance for hepatopathy. Being purposeful during these surveillance scans may help us diagnose these tumors at a higher frequency. In the majority of cases, surgical resection when possible is curative as was true in our patient.
CONCLUSION
PGL are rare tumors associated with CHD. These tumors can be symptomatic and have important clinical implications. As we continue to improve long-term survival for single-ventricle patients, attention has shifted to long-term morbidities. Awareness of these tumors during surveillance is, therefore, warranted as timely intervention may lead to cure.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
Authors would like to acknowledge the contributions from Dr. Erwin Schemankewitz, MD (Department of Pathology, Emory University), in proving the pathology images as well as his expertise in diagnosis.
REFERENCES
- 1.Dahia PL. Pheochromocytomas and paragangliomas, genetically diverse and minimalist, all at once! Cancer Cell. 2017;31:159–61. doi: 10.1016/j.ccell.2017.01.009. [DOI] [PubMed] [Google Scholar]
- 2.Cerecer-Gil NY, Figuera LE, Llamas FJ, Lara M, Escamilla JG, Ramos R, et al. Mutation of SDHB is a cause of hypoxia-related high-altitude paraganglioma. Clin Cancer Res. 2010;16:4148–54. doi: 10.1158/1078-0432.CCR-10-0637. [DOI] [PubMed] [Google Scholar]
- 3.Folger GM, Jr, Roberts WC, Mehrizi A, Shah KD, Glancy DL, Carpenter CC, et al. Cyanotic malformations of the heart with pheochromocytoma. A report of five cases. Circulation. 1964;29:750–7. doi: 10.1161/01.cir.29.5.750. [DOI] [PubMed] [Google Scholar]
- 4.Bockelman HW, Arya S, Gilbert EF. Cyanotic congenital heart disease with malignant paraganglioma. Cancer. 1982;50:2513–7. doi: 10.1002/1097-0142(19821201)50:11<2513::aid-cncr2820501143>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 5.Mondésert B, Marcotte F, Mongeon FP, Dore A, Mercier LA, Ibrahim R, et al. Fontan circulation: Success or failure? Can J Cardiol. 2013;29:811–20. doi: 10.1016/j.cjca.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 6.Rodríguez-Cuevas S, López-Garza J, Labastida-Almendaro S. Carotid body tumors in inhabitants of altitudes higher than 2000 meters above sea level. Head Neck. 1998;20:374–8. doi: 10.1002/(sici)1097-0347(199808)20:5<374::aid-hed3>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 7.Opotowsky AR, Moko LE, Ginns J, Rosenbaum M, Greutmann M, Aboulhosn J, et al. Pheochromocytoma and paraganglioma in cyanotic congenital heart disease. J Clin Endocrinol Metab. 2015;100:1325–34. doi: 10.1210/jc.2014-3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reynolds JL, Gilchrist TF. Congenital heart disease and pheochromocytoma. Am J Dis Child. 1966;112:251–5. doi: 10.1001/archpedi.1966.02090120119014. [DOI] [PubMed] [Google Scholar]