

Abstract
Desmosterolosis is a rare multiple congenital anomaly syndrome caused by a defect in the enzyme 3-beta-hydroxysterol delta-24-reductase (DHCR24) in the cholesterol biosynthesis pathway. Defects in this enzyme cause increased level of the cholesterol precursor desmosterol while disrupting development of cholesterol, impacting embryogenesis. A total of 9 cases of desmosterolosis have been reported to date. We report a 20-month-old male from consanguineous parents with multiple congenital anomalies including corpus callosum hypoplasia, facial dysmorphism, cleft palate, pectus deformity, short and wide neck and distal contractures. On analysis of the regions of homozygosity found by microarray, we identified DHCR24 as a candidate gene. Sterol quantitation showed a desmosterol level of 162 μg/mL (nl: 0.82 ± 0.48). Genetic testing confirmed the diagnosis with a homozygous likely pathogenic mutation (p.Glu191Lys) in the DHCR24 gene. Our case expands the known diagnostic spectrum for Desmosterolosis. We suggest considering Desmosterolosis in the differential diagnosis of patients who present with concurrent agenesis of the corpus callosum with white matter atrophy and ventriculomegaly, retromicrognathia with or without cleft palate, hand contractures, and delay of growth and development. Children of consanguineous mattings may be at higher risk for rare recessive disorders and testing for cholesterol synthesis defect should be a consideration for affected children. Initial evaluation can be performed using sterol quantitation, followed by genetic testing.
Keywords: Desmosterol, Inborn errors of metabolism, Cholesterol biosynthesis defect, Congenital anomalies, Consanguinity
1. Introduction
Desmosterolosis (OMIM 602398) is an autosomal recessive disorder that is caused by defects in the DHCR24 gene coding for the enzyme 3-beta-hydroxysterol-delta-24-reductase in the cholesterol biosynthesis pathway (Waterham et al., 2001). This enzyme metabolizes desmosterol to cholesterol as the last step of conversion of lanosterol to cholesterol (Clayton, 1998; FitzPatrick et al., 1998; Kelley and Herman, 2001). Increased levels of desmosterol in plasma, tissue and cultured cells have been reported in desmosterolosis (Waterham et al., 2001).
Cholesterol is vitally important in the body for manufacturing membranes, hormones, cell signaling, and for the induction of SHH-related pathways (Simons and Ehehalt, 2002; Riobo, 2012; Stottmann et al., 2011). Desmosterol and 7-dehydrocholesterol are each the penultimate product in parallel pathways for cholesterol biosynthesis. Smith-Lemli-Opitz syndrome (Opitz, 1999; Irons et al., 1994) results from a block in sterol delta-7-reductase causing accumulation of 7-dehydrocholesterol, and has highly variable expression with well described diagnostic findings including the nearly omnipresent 2–3 toe syndactyly. Desmosterolosis is a lesser-known disorder and is still being characterized (Porter and Herman, 2010).
In 1998, Fitzpatrick et al. reported the first case of desmosterolosis, a female born at 34 weeks gestation who died shortly after birth. Features described included macrocephaly with MRI findings of ventricular dilatation and corpus callosum agenesis, hypoplastic nasal bridge, thick alveolar ridges, gingival nodules, cleft palate, total anomalous pulmonary venous drainage, ambiguous genitalia, short limbs and generalized osteosclerosis. Post-mortem gas chromatography-mass spectrometry in kidney, liver and brain revealed elevated level of desmosterol (FitzPatrick et al., 1998). In 2002, Andersson et al. reported the first living case of desmosterolosis in an infant who presented with severe microcephaly, agenesis of corpus callosum, facial dysmorphic features, clubfoot and persistent patent ductus arteriosus, and a 100-fold increase in the level of desmosterol both in plasma and cultured lymphoblasts (Andersson et al., 2002). Zolotushko et al. (2011) described a consanguineous Israeli Bedouin family with six affected family members of which four were living. All affected individuals described had microcephaly, microretrognathia, psychomotor and growth retardation, hand contractures, underdeveloped corpus callosum and ventriculomegaly (Zolotushko et al., 2011).
Since then, a total of 9 patients with diagnosis of desmosterolosis have been reported in the literature. Dias et al. described an affected sibling pair and summarized all of these cases, focusing on their clinical features and genetic mutations (Dias et al., 2014).
Here we report a 20 month-old baby boy from consanguineous parents with multiple congenital anomalies including global developmental delay, hypoplastic corpus callosum, ventricular dilatation, cleft palate, dysmorphic facial features and hand contractures who was ultimately diagnosed with desmosterolosis.
2. Clinical report
A 20-month old male infant was referred to our clinic for diagnostic workup for cleft palate, hydrocephalus, developmental delay, and exotropia. He was born full term to a 32-year-old healthy father and a 30-year-old healthy mother who had seven previous pregnancies, of which three resulted in miscarriage. Parents were first cousins of Middle Eastern heritage. Pregnancy was complicated by decreased fetal movements and hydrocephalus that was detected on prenatal ultrasound. He was born by spontaneous vaginal delivery with birth weight over 2 kg.
He was noted after birth to have microcephaly, low set ears, mandibular hypoplasia, wide complete cleft palate, small adducted thumbs, long fingers, and talipes equinovarus. Ongoing medical issues include constipation, reactive airway disease, and recurrent otitis media.
On presentation at 20 months, the patient had significant motor and speech delay. He could not sit alone or crawl, but could stand with assistance though could not place his feet flat on the ground. He had frequent drooling. He could babble and speak two words. He was unable to consume solid food, and his diet consisted of soft food and peptide-based formula. He had growth retardation with height of 75.3 cm (< 1%, Z score of −2.97) and weight of 8.77 kg (< 1%, Z-score of −2.2). His head circumference was 48 cm (21st percentile). He had coarse hair. Head shape was asymmetric, with bitemporal narrowing, frontal bossing and a metopic ridge. Facial features include exotropia, mildly down-slanting palpebral fissures, long curled lashes, high nasal bridge, low set and posteriorly rotated ears, retro-micrognathia and cleft palate. He had a short and wide neck, shield-like chest, laterally placed nipples, and small pectus excavatum. He had unilateral cryptorchidism. Extremities had bilateral inward deviation of the thumbs, contracted distal interphalangeal joints with hypoplastic flexion creases, contracted thumbs, overlapping toes, and a hemangioma on the right forearm (see Fig. 1). Neurological examination revealed intact cranial nerves. He did not focus or track objects. Motor testing showed increased tone in the lower limbs and to some extent upper limbs. He had some antigravity movements at the knees and hips. Deep tendon reflexes were brisk at the knees, ankles and biceps and plantar reflexes were extensor.
Fig. 1.

Pictures of the patient at initial presentation i.e. 20 months of age, showing (a) facial dysmorphism, (b) low set and posteriorly rotated ears, retro-micrognathia, (c–d) transverse palmar crease and contractures of the interphalangeal joints.
Family history is depicted in Fig. 2. It demonstrates the parents are first degree cousins. There are no known similarly affected family members.
Fig. 2.
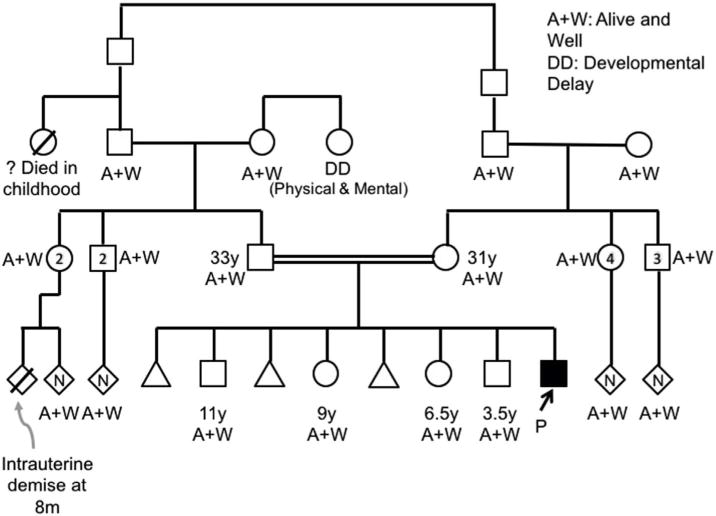
Pedigree of the patient and his family.
Brain magnetic resonance imaging (MRI) demonstrated dilation of the ventricles and hypoplastic corpus callosum (see Fig. 3). Abdominopelvic ultrasound, chest, hip, spine and abdominal x-ray imaging and echocardiography were non-contributory. Chromosomal microarray studies identified no pathogenic variants, but revealed areas of homozygosity comprising greater than 2% of the genome. We conducted a search using the Genomic Oligoarray and SNP Array Evaluation Tool version 3.010 (Wierenga et al., 2013) for autosomal recessive conditions within the areas of homozygosity using search terms “cleft palate” and “corpus callosum hypoplasia.” Of all results matching our query, the diagnosis most consistent with our patient’s presentation was Desmosterolosis. As a next step, sterol quantitation test was performed, which showed a 7-dehydrocholesterol level of 2.82 μg/mL (reference range = 0.04–0.36), a desmosterol level of 162 μg/mL (reference range = 0.82 ± 0.48), and a total cholesterol level of 123 mg/dL (within normal limits). Molecular genetics analysis revealed a homozygous mutation (c.571G > A, p.Glu191Lys) in the DHCR24 gene, which has been previously described in affected siblings and an unrelated individual (Dias et al., 2014). This mutation affects a highly conserved amino acid residue in the flavin adenine dinucleotide-binding domain of the DHCR24 protein (Waterham et al., 2001). This mutation appears to be deleterious on pathogenicity prediction tools.
Fig. 3.
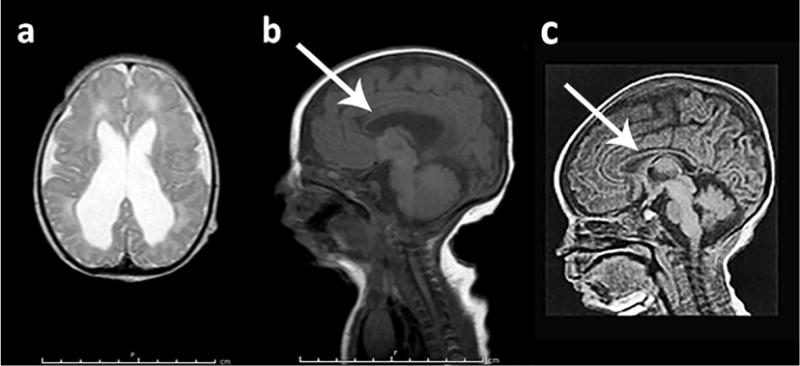
Magnetic resonance imaging (MRI) of brain at day 7 of age. (a) Axial T2 showing hyperintensity of periventricular and frontal area. (b) Sagittal Flair showing thinning of corpus callosum (arrow). (c) Sagittal T1-weighted MRI showing a normal corpus callosum in a 7-day-old male infant.
3. Discussion
Three other cases of desmosterolosis with the same homozygous mutation, c.571G > A (p.Glu191Lys) in DHCR24 gene, have been reported in the literature (Andersson et al., 2002; Dias et al., 2014). Common clinical features include failure to thrive, short stature, intellectual disability, spasticity, distal arthrogryposis, dysgenesis of the corpus callosum and cerebral white matter atrophy. Our proband also presented with these common clinical features and facial dysmorphism. One other patient had a short neck (Dias et al., 2014) as was present in our case. Unique features include metopic ridge, high nasal bridge, and long eyelashes. He had pectus deformity which was not previously reported in cases with this particular mutation. Frontal bossing (FitzPatrick et al., 1998) and prominent forehead (Schaaf et al., 2011) have been reported in previous cases with different mutations. Our patient also had unilateral cryptorchidism. Urogenital anomalies including renal hypoplasia, prominent clitoris and ambiguity of external genitalia have been reported in one case with a different mutation (FitzPatrick et al., 1998). Table 1 summarizes all published cases of Desmosterolosis to date, with their mutation and clinical features.
Table 1.
Summary of clinical features and molecular findings of all reported cases with Desmosterolosis.
| Patient 1 | Patient 2 | Patient 3–6 | Patient 7 | Patient 8 and 9 | Present patient | Frequency n = 10 |
|
|---|---|---|---|---|---|---|---|
| Molecular diagnosis | p.[Y471S] + [N294T; K306N] | p.[E191K] + [E191K] | p.[R103C] + [R103C] | p.[R94H] + [E480K] | p.[E191K] + [E191K] | p.[E191K] + [E191K] | |
| Sex | Female | Male | 2 Female, 2 Male |
Female | Female | Male | |
| Ancestry | European | European | Israeli Bedouin | N/A | Middle Eastern | Middle Eastern | |
| Failure to thrive | N/A | + | + | + | + | + | 9/9 |
| Head circumference | Macrocephaly | Microcephaly | Microcephaly | Relative Macrocephaly | Low normal/Relative Macrocephaly | Relative Macrocephaly | |
| Facial anomalies | |||||||
| Frontal bossing/Prominent forehead | Frontal bossing | − | − | Prominent forehead | − | Frontal bossing | 3/10 |
| Dolichocephaly/bitemporal narrowing | − | − | − | − | + | − | 2/10 |
| Down-slanting palpebral fissures/Epicanthal folds/Telecanthus | − | Down-slanting PF/Epicanthal folds | − | Telecanthus | Down-slanting PF | Down-slanting PF | 5/10 |
| Micrognathia/Microretrognathia | + | + | + | + | 1/2 | + | 9/10 |
| Cleft palate | + | + | − | − | 1/2 | + | 4/10 |
| Abnormal nose | + | − | − | + | + | + | 5/10 |
| Low set/abnormal ears | + | − | − | − | 1/2 | + | 3/10 |
| Neurological anomalies | |||||||
| ID/DD | N/A | + | + | + | + | + | 9/9 |
| Agenesis of corpus callosum (full/partial) | + | + | + | + | + | + | 10/10 |
| Ventriculomegaly | + | − | + | + | + | + | 9/10 |
| Thinning of white matter | N/A | + | + | + | + | − | 8/9 |
| Seizure | N/A | − | 3/4 | N/A | + | − | 5/8 |
| Ophthalmological anomalies | |||||||
| Nystagmus/Strabismus | N/A | − | 3/4 | − | + | + | 6/9 |
| Skeletal anomalies | |||||||
| Short stature | − | + | N/A | N/A | + | + | 4/5 |
| Short/wide neck | − | − | − | − | 1/2 | + | 2/10 |
| Shortening of the limbs | + | − | N/A | + | − | − | 2/6 |
| Short wide ribs | + | − | N/A | − | − | − | 1/6 |
| Pectus deformity | − | − | − | − | − | + | 1/10 |
| Bone mineralization | Osteosclerosis | Normal | N/A | Mild heterogeneity in long bones | 1/2 (Osteopenia) | Normal | |
| Large joints contractures | + | Talipes | N/A | + | 1/2 (Talipes) | Talipes | 5/6 |
| Distal arthrogryposis | − | + | + | + | + | + | 9/10 |
| Spasticity | N/A | + | + | N/A | + | + | 8/8 |
| Syndactyly 2–4 toe | − | − | − | + | − | − | 1/10 |
| Congenital heart disease | TAPVR | PDA | − | − | − | − | 2/10 |
| Urogenital anomalies | Prominent clitoris; Renal hypoplasia | − | − | − | − | Cryptorchidism | 2/10 |
| Other features | Neonatal demise | Cutis aplasia | Diaphragmatic eventration; Congenital hydrocephalus | 2/2 radial head dislocations; 1/2 SNHL; 1/2 Parietal foramina; 1/2 deformed pelvis; 1/2 Sacral cyst |
Abbreviations: N/A = not available, ID = intellectual disability, DD = developmental delay, PF = palpebral fissure, TAPVR = total anomalous pulmonary venous return, PDA = patent ductus arteriosus, SNHL = Sensorineural Hearing Loss. References: Patient 1: FitzPatrick et al., 1998, Patient 2: Andersson et al., 2002, Patients 3–6: Zolotushko et al., 2011, Patient 7: Schaaf et al., 2011, Patient 8–9: Dias et al., 2014.
His hand contractures are in the spectrum of distal arthrogryposis described in other reports. Occipital frontal circumference (OFC) is variable in these patients. Two cases including our patient had relative macrocephaly (Schaaf et al., 2011). Only one previous patient was reported to have macrocephaly (FitzPatrick et al., 1998) (> 97 percentile), 5 other cases had microcephaly (Andersson et al., 2002; Zolotushko et al., 2011) and two were normocephalic (Dias et al., 2014).
Desmosterolosis is a very rare disease with less than a dozen cases reported, and ranging from lethal disease described in a premature newborn to the oldest patient reported at 14 years of age (Dias et al., 2014). Previous reports have also denoted a low genotype-phenotype correlation in this disease, which further hinders identification of affected individuals (Dias et al., 2014; Schaaf et al., 2011). Additional case studies may provide further insight into the clinical and mutation spectrum of this disease.
A few studies have investigated the pathophysiology of the defects in cholesterol biosynthesis using knockout mouse models. Wechsler et al. analyzed mice with targeted disruption of the DHCR24 gene, which had lower survival, poor growth and infertility, but lacked dysmorphology or involvement of body systems other than the testes (Wechsler et al., 2003). In contrast, the DHCR24 knockout mice described by Mirza et al. died within days after birth. These mice had lethal dermopathy and the authors concluded DHCR24 maybe essential for normal skin development (Mirza et al., 2006). The majority of cholesterol biosynthesis disorders cause ichthiosiform dermatitis, which can be attributed to lack of cholesterol in membranes and maybe also toxic effects from the sterol precursors (Elias et al., 2011). However, dermatologic findings have not been reported in Desmosterolosis in humans.
While the symptomatic presentation of these knockout mice differs from what has been seen in reported cases in humans, the presence of marked phenotypic variability in mice models may mirror the variability of severity in affected people. For example, the first reported human case had postnatal death but the subsequent case reports had a non-lethal form.
Of the reported patients, two other families have the same mutation as this proband, one family also of middle-eastern descent and one of European descent. This suggests that, as Dias et al. proposed, only some mutations may be compatible with life, leading to few recurrent mutations even in unrelated families of different ethnic backgrounds (Dias et al., 2014).
In summary, Desmosterolosis is a prenatal onset metabolic disorder of cholesterol synthesis, which presents with multiple congenital anomalies. We describe successful disease gene identification using regions of homozygosity found on microarray. Consanguinity was present in 40% of reported cases. The presence of consanguinity calls for consideration of rare recessive disease investigations, which may include evaluation for sterol disorders. Diagnostic workup for this condition may begin with sterol quantitation, followed by gene sequencing. We suggest including defects of cholesterol synthesis in the differential diagnosis of patients who present with agenesis of the corpus callosum with white matter atrophy with or without ventriculomegaly, retromicrognathia with or without cleft palate, distal arthrogryposis spectrum features, and delay in growth and development. This patient expands the diagnostic spectrum by adding the features of facial dysmorphism, pectus deformity, short, wide neck and cryptorchidism.
Acknowledgments
We thank the family for their interest in this work and willingness to participate in this clinical report.
References
- Andersson HC, Kratz L, Kelley R. Desmosterolosis presenting with multiple congenital anomalies and profound developmental delay. Am J Med Genet. 2002;113:315–319. doi: 10.1002/ajmg.b.10873. [DOI] [PubMed] [Google Scholar]
- Clayton PT. Disorders of cholesterol biosynthesis. Arch Dis Child. 1998;78:185–189. doi: 10.1136/adc.78.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias C, Rupps R, Millar B, et al. Desmosterolosis: an illustration of diagnostic ambiguity of cholesterol synthesis disorders. Orphanet J Rare Dis. 2014;9:94. doi: 10.1186/1750-1172-9-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias PM, Crumrine D, Paller A, Rodriguez-Martin M, Williams ML. Pathogenesis of the cutaneous phenotype in inherited disorders of cholesterol metabolism: therapeutic implications for topical treatment of these disorders. Dermatoendocrinol. 2011;3:100–106. doi: 10.4161/derm.3.2.14831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FitzPatrick DR, Keeling JW, Evans MJ, et al. Clinical phenotype of desmosterolosis. Am J Med Genet. 1998;75:145–152. [PubMed] [Google Scholar]
- Irons M, Elias ER, Tint GS, et al. Abnormal cholesterol metabolism in the smith-lemli-opitz Syndrome: report of clinical and biochemical findings -in four patients and treatment in one patient. Am J Med. 1994;352:347–352. doi: 10.1002/ajmg.1320500409. [DOI] [PubMed] [Google Scholar]
- Kelley RI, Herman GE. Inborn errors of sterol biosynthesis. Annu Rev Genomics Hum Genet. 2001;2:299–341. doi: 10.1146/annurev.genom.2.1.299. [DOI] [PubMed] [Google Scholar]
- Mirza R, Hayasaka S, Takagishi Y, et al. DHCR24 gene knockout mice demonstrate lethal dermopathy with differentiation and maturation defects in the epidermis. J Invest Dermatol. 2006;126:638–647. doi: 10.1038/sj.jid.5700111. [DOI] [PubMed] [Google Scholar]
- Opitz JM. RSH (so-called Smith-Lemli-Opitz) syndrome. Curr Opin Pediatr. 1999;11:353–362. doi: 10.1097/00008480-199908000-00015. [DOI] [PubMed] [Google Scholar]
- Porter FD, Herman GE. Malformation syndromes caused by disorders of cholesterol synthesis. J Lipid Res. 2010;52:6–34. doi: 10.1194/jlr.R009548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riobo NA. Cholesterol and its derivatives in Sonic Hedgehog signaling and cancer. Curr Opin Pharmacol. 2012;12:736–741. doi: 10.1016/j.coph.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf CP, Koster J, Katsonis P, et al. Desmosterolosis-phenotypic and molecular characterization of a third case and review of the literature. Am J Med Genet Part A. 2011;155:1597–1604. doi: 10.1002/ajmg.a.34040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. J Clin Invest. 2002;110:597–603. doi: 10.1172/JCI16390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stottmann RW, Turbe-Doan A, Tran P, et al. Cholesterol metabolism is required for intracellular Hedgehog signal transduction in vivo. PLoS Genet. 2011;7 doi: 10.1371/journal.pgen.1002224. http://dx.doi.org/10.1371/journal.pgen.1002224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterham HR, Koster J, Romeijn GJ, et al. Mutations in the 3beta-hydroxysterol Delta24-reductase gene cause desmosterolosis, an autosomal recessive disorder of cholesterol biosynthesis. Am J Hum Genet. 2001;69:685–694. doi: 10.1086/323473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler A, Brafman A, Shafir M, et al. Generation of viable cholesterol-free mice. Science. 2003;302:2087. doi: 10.1126/science.1090776. [DOI] [PubMed] [Google Scholar]
- Wierenga KJ, Jiang Z, Yang AC, Mulvihill JJ, Tsinoremas NF. A clinical evaluation tool for SNP arrays, especially for autosomal recessive conditions in offspring of consanguineous parents. Genet Med. 2013;15:354–360. doi: 10.1038/gim.2012.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolotushko J, Flusser H, Markus B, et al. The desmosterolosis phenotype: spasticity, microcephaly and micrognathia with agenesis of corpus callosum and loss of white matter. Eur J Hum Genet. 2011;19:942–946. doi: 10.1038/ejhg.2011.74. [DOI] [PMC free article] [PubMed] [Google Scholar]