

Abstract
Manganese (Mn) is an essential trace element, but chronic overexposure to this metal, either environmentally or occupationally may cause manganism, a disease analogous to Parkinson’s disease. Inhibitors of histone deacetylases, such as valproic acid (VPA) and sodium butyrate (NaB) exert neuroprotective effects in various animal models of neurological disorders. Thus, the present study investigated whether VPA or NaB prevent Mn-induced neurotoxicity by assessing locomotor activities and expression of astrocytic glutamate transporters, glutamate transporter 1 (GLT-1) and glutamate aspartate transporter (GLAST), in C57BL/6 mice. C57BL/6 mice were pretreated with VPA (200 mg/kg, i.p.) or NaB (1200 mg/kg, i.p.) prior to intranasal instillation of Mn (30 mg/kg) continually for 21 days, followed by open-field and rota-rod behavioral tests and analyses of astrocytic glutamate transporters GLT-1 and GLAST protein/mRNA levels. The results showed that Mn significantly decreased locomotor activity as determined by total distance travelled, stereotypic and ambulatory counts. Mn also significantly decreased rota-rod activity reflecting altered motor coordination. Pretreatment with VPA and NaB with Mn reversed the effects of Mn on the locomotor activity and motor coordination. VPA and NaB also attenuated the Mn-induced decrease in GLT-1 and GLAST mRNA and protein levels in the cerebral cortical and cerebellar regions of mice. These results suggest that VPA and NaB exert protective effects against Mn toxicity seem in vitro are also shown in vivo. VPA and NaB pretreatment in mice enhancing astrocytic glutamate transporter GLT-1 expression as well as locomotor activities. Future research endeavors are warranted to determine if the therapeutic potential of VPA and NaB is via common molecular mechanism, namely, inhibition of histone deacetylases.
Keywords: valproic acid, sodium butyrate, glutamate transporters, Manganese, behavior
1. Introduction
Abnormalities in behavioral and locomotor activities are considered as physiological indicators of neurological disorders, such as Parkinson’s disease (PD), epilepsy, Alzheimer’s disease (AD) and autism (Maski et al., 2011). Although the molecular mechanisms of these neurological disorders are still not completely understood, their etiology is based on genetic, environmental or gene × environment interactions (Dauncey, 2012; Peres et al., 2016). Genetic abnormalities leading to neurological diseases are associated with rare gene mutations and irregular gene expression (Dauncey, 2012; Karki et al., 2015; Seifert et al., 2006).
Environmental and idiopathic factors also significantly influence the development and progression of neurodegenerative disorders (Peres et al., 2016; Robison et al., 2012). Previous studies have reported that environmental agents may induce toxicity, which can lead to serious brain injury and damage (Dauncey, 2012; Peres et al., 2016). One of the most studied metal toxicants is manganese (Mn), which is an essential trace element, but chronic high level exposures to this metal may cause a neurological disorder referred to as manganism, with similar clinical features to parkinsonism (Peres et al., 2016), characterized by locomotor dysfunction and behavioral abnormality (Peres et al., 2016; Robison et al., 2012). Upon overexposure, Mn accumulates preferentially in specific brain regions (i.e. globus pallidus), resulting in motor deficits and neurological disorders (Robison et al., 2012). Several therapeutic approaches have been reported for the treatment of manganism, such as levodopa, antioxidants, plant extracts, iron (Fe)-chelating agents and glutathione precursors (Peres et al., 2016).
Glutamate-mediated excitotoxicity is considered one of the critical mechanisms involved in Mn neurotoxicity (Brouillet et al., 1993). The astrocytic glutamate transporters, glutamate aspartate transporter (GLAST) and glutamate transporter 1 (GLT-1) in rodents (excitatory amino acid transporter 1 (EAAT1) and excitatory amino acid transporter 2 (EAAT2) in humans, respectively) are the main transporters to uptake most of glutamate from synaptic clefts after binding to its postsynaptic receptors to transfer glutamate signals. Accordingly, dysfunction of these transporters is directly associated with glutamate excitotoxicity. In fact, several idiopathic neurodegenerative diseases including PD and manganism are associated with excitotoxicity along with a decrease in the function and expression of the glutamate transporter, GLT-1.
Targeting epigenetic regulation, in particular deacetylation of histone molecules using inhibitors of histone deacetylases (HDACs) for the development of neuroprotective and anti-cancer agents has shown significant progress in recent years. Valproic acid (2-n-propylpentanoic acid; VPA) is clinically used as an antiepileptic and anticonvulsant agent exhibiting increasing usage over the years for the treatment of neurological disorders. Interestingly, in addition to its beneficial effect against epilepsy, VPA exerts neuroprotective effects in several animal models of neurodegenerative diseases (Monti and Contestabile, 2009). However, the exact molecular mechanism of its neuroprotective effect remains to be established.
VPA affects cell growth, differentiation and apoptosis, by regulating gene expression at the molecular level, through epigenetic mechanisms as an inhibitor of HDACs (Ximenes et al., 2013). In addition to inhibition of HDACs, VPA can also interfere with multiple regulatory mechanisms such as the glycogen synthase kinase (GSK)3-α and -β, protein kinase B (Akt), extracellular signal-regulated kinases (ERK) and phosphoinositol pathways, tricarboxylic acid (TCA) cycle, and γ-aminobutyric acid (GABA) system. VPA also showed beneficial effects in modulating blood brain barrier (BBB) disruption and brain edema in a rat model of cerebral ischemia (Wang et al., 2011). VPA also exerts anti-inflammatory and antioxidative properties.
Sodium butyrate (NaB), a short chain fatty acid) is an inhibitor of HDACs, and generally derived from the microbial fermentation product of dietary fiber in the colon and thus, inhibits intestinal pathogenic bacteria and maintains gastrointestinal homeostasis (Canani et al., 2011; Kim et al., 2012). NaB, thus, exerts multiple beneficial effects from the intestinal tract to the peripheral tissues (Canani et al., 2011). NaB has been shown to be effective in the prevention and treatment of multiple chronic disorders such as cancer, metabolic syndrome, cardiovascular diseases, and neurodegeneration. Inhibition of HDACs by NaB is related to its epigenetic regulatory effects on gene expression. Due to its modulation on epigenetic mechanisms, NaB has been considered as a specific and efficacious therapeutic strategy for treatment of variety of diseases, including neurodegenerative disorders. In rats, NaB exhibits neuroprotective effects against ischemic stroke by anti-inflammatory effects (Park and Sohrabji, 2016), and antidepressant-like effects (Yamawaki et al., 2012). NaB also exerts neuroprotective effects by anti-apoptotic, -oxidative and -inflammatory effects in a mouse model of cerebral ischemic injury (Sun et al., 2015). NaB improved memory deficits in a mouse model of traumatic brain injury, attributing to increased expression of tight junction-associated proteins such as occludin and ZO-1, and BBB permeability (Li et al., 2016).
Since VPA and NaB exert neuroprotective effects against numerous neurological disorders, not only by HDAC inhibition, but also many other molecular mechanisms, the current study investigated whether VPA and/or NaB attenuate Mn-induced neurotoxicity by assessing neurobehavioral deficits and expression of astrocytic glutamate transporters, GLT-1 and GLAST in mice.
2. Materials and Methods
2.1. Experimental animals
All animal protocols were reviewed and approved by the Meharry Medical College Institutional Animal Care and Use Committee (Nashville, TN). Adult male C57BL/6 mice (6-8 weeks old; weight 18-20 g) were purchased from the Jackson Laboratory and housed for one week prior to study (Bar Harbor, ME). The animals were randomly selected, group-housed (4 mice/cage) and maintained on a 12-h light/dark cycle at 25 ± 2°C and 60‐70% relative humidity with food, water and enrichment available ad libitum in the Animal Care Facility of Meharry Medical College (Nashville, TN).
2.2. Chemicals and reagents
Manganese chloride (MnCl2, ≥ 99% purity), valproic acid (VPA, ≥ 98% purity), and sodium butyrate (NaB, ≥ 98% purity) were obtained from Sigma-Aldrich (St. Louis, MO). EAAT1 (ab416) and EAAT2 (ab41621) antibodies were obtained from Abcam (Cambridge, MA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH; sc-14007) antibody was acquired from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-rabbit IgG-HRP-conjugated (W4018) secondary antibody was obtained from Promega (Madison, WI).
2.3. Experimental procedure
The number of animals needed for this study (n=48; 8 animals/group) was determine by power analysis. Mice were randomly separated into control, VPA, Mn, Mn plus VPA, NaB, Mn plus NaB groups, and initial body-weights were recorded. Male mice were used to alleviate any possible effects of female sex hormone estrogens on the modulation of astrocytic glutamate transporters (Dhandapani and Brann, 2007; Lee et al., 2009). Body-weights were recorded on a weekly basis. Mice were treated once daily for 21 consecutive days with 100 μl of intraperitoneal (i.p.) injection of VPA (200 mg/kg), NaB (1200 mg/kg), or saline (NaCl, 0.9%; control). VPA and NaB were diluted in 0.9% saline. After 30 min, Mn plus VPA, Mn plus NaB, and Mn groups received 2 μl of MnCl2 (30 mg/kg) via intranasal instillation in the left nostril. Likewise, VPA, NaB and control groups received 2 μl of distilled water in the left nostril. Previously published protocols for Mn intranasal instillation (Kim et al., 2012), VPA (Loscher et al., 1993; Zaky et al., 2014) and NaB dosing and route (Ferrante et al., 2003; Langley et al., 2008) were implemented. During the instillation period the mice were placed under isoflurane-induced anesthesia for 3 min pre- and post-instillation to sedate and prevent expulsion of Mn from the nostril, respectively.
2.4. Open-field test
Behavioral data were collected on day 21 in Seamless Open-field Activity Arenas using Activity Monitor 5 software (Med Associates, Fairfax, VT). Each open-field arena was made of clear Plexiglas, measuring 27.3 cm × 27.3 cm × 20.3 cm, and was covered with a Plexiglas lid containing air holes. The following activity measures were calculated directly by the Activity Monitor 5 software: distance traveled, stereotypic counts, and ambulatory counts for each animal in each group. Each animal went through an acclimation period (1 trial for 30 min, 1 day before Mn and/or VPA or NaB treatment). On day 21, control, Mn, VPA, Mn plus VPA, NaB, and Mn plus NaB groups commenced open-field testing to monitor total distance traveled, ambulatory and stereotypic counts. Each mouse was placed in the center of the open-field for observation and the activity measures were recorded for each observed mouse within the 30-min period.
2.5. Rotarod test for motor performance and coordination
Rota-Rod system (Med Associates Inc., Fairfax, VT) was used to measure the time an animal maintains balance on a moving cylinder. Mice were trained for three consecutive days, with one daily session consisting of 3 trials separated by 300 s resting periods. On days 1 and 21, treated mice were placed on the rod and trials were deemed to have started when rod begins start, with speed gradually increasing from 4 rpm to 40 rpm up to 10 min by 0.1 revolutions/sec. The fall latency, a time measurement of how fast a subject falls from the rotarod at various speeds was assessed. If the mouse persisted on the rod for the entire duration, the measurement value was recorded for 650 sec. The mean duration for each mouse was used for comparison.
2.6. Real time RT-PCR analysis
Tissue samples (3 samples/group) were extracted from cerebral cortical and cerebellar regions of C57BL/6 mice used in behavioral experiments. The mRNA levels of GLT-1 and GLAST from these regions were analyzed. Total RNA was extracted from mouse brain tissue using RNeasy® Mini Kit (Qiagen, Valencia, CA). The purified RNA was transcribed to cDNA with high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). For quantitative real-time PCR (qPCR), the following primers were used: for GLT-1, 5′-GCC AAT ACA ACC AAG GCA GTC-3′ (forward) and 5′-TTC ATC CCG TCC TTG AAC TC-3′ (reverse); for GLAST, 5′-GATCGGAAACATGAAGGAGC-3′ (forward) and 5′-CAA GAA GAG GAT GCC CAG AG-3′ (reverse) and for GAPDH, 5′-TCC CTC AAG ATT GTC AGC AA-3′ (forward) and 5′-AGA TCC ACA ACG GAT ACA TT-3′ (reverse). The total reaction volume (25 μl) contained 1μl of cDNA template of each sample 0.4 μM of primers and RT2 SYBR® Green qPCR Master Mix from SABiosciences/Qiagen (Frederick, MD). The qPCR parameters were set for one cycle at 95°C for 10 min, 40 cycles at 95°C for 15 sec and 60°C for 1 min in CFX96 real-time PCR detection system from Bio-Rad (Hercules, CA). GAPDH was used to normalize all samples. Analysis of data was performed using the web-based RT2 PCR Array Profiler Data Analysis version 3.5 (SABiosciences/Qiagen).
2.7. Western blot analysis
For protein analysis, three mice from each treatment group were randomly selected and harvested brain regions (e.g. cerebral cortical and cerebellar regions) were used for protein extraction and further analysis. The protein concentration of homogenized tissue samples [3:1 ratio of radioimmuno precipitation assay (RIPA) buffer with protease inhibitor and tissue sample] was determined by bicinchoninic acid (BCA) assay. Equal amounts of proteins were run on 10% SDS-PAGE followed by western blot analysis. The primary antibody for GLT-1 or GLAST was at 1:5000 dilution, and HRP-conjugated secondary antibody was used at 1:5000 dilution. All blots were developed using a Pierce chemiluminescence detection kit (Rockford, IL), followed by blot imaging and quantification with the Molecular Imager VersaDoc™ MP 4000 System and Image Lab Software version 5.2.1 from Bio-Rad, respectively.
2.8. Statistical analysis
All data were expressed as the mean ± standard error of the mean (SEM). Multiple comparisons analyses were performed using a one-way analysis of variance (ANOVA) or two-way ANOVA, followed by Tukey’s post‐hoc tests using the GraphPad Prism Software version 5.0 (San Diego, CA). F-statistics and degrees of freedom for all analysis were performed, and the detailed F-statistics are described in supplementary tables. A p-value less than 0.05 (p<0.05) was considered statistically significant.
3. Results
3.1. Effect of treatment of Mn, VPA, NaB, or Mn plus VPA or NaB on body weight of mice
Body weights were determined after treatment with saline, Mn, VPA, NaB, and Mn plus VPA or NaB for 21 days. Mn was administered into mice via intranasal instillation (daily, 30 mg/kg/day) (Gianutsos et al., 1997; Henriksson and Tjalve, 2000; Moberly et al., 2012). Saline, VPA (daily, i.p., 200 mg/kg/day) (Kimura et al., 2015; Qian et al., 2010), or NaB (daily, i.p., 1200 mg/kg/day) in respective treatment groups, was pre-treated 30 min before Mn instillation into mice (Kratsman et al., 2016; Malago and Sangu, 2015; Zhong et al., 2014). As shown in Table 1 using a two-way ANOVA, there was no significant change in body weights in any of the treated groups compared to the body weights on day 1. Table 1 shows the average weights of mice in grams (g) on days 1 and 21.
Table 1.
Body weight changes in mice after treated with Mn, VPA, NaB, Mn plus VPA, or Mn plus NaB for 21 days
| Treatment Groups | Day 1 Average Weight (g, n=8) |
Day 21 Average Weight (g, n=8) |
Weight Gain (+)/Loss (−) (g) |
|---|---|---|---|
| Control | 23.04 ± 1.77 | 26.58 ± 1.74 | +3.55 ± 0.94 |
| Mn | 19.54 ± 3.47 | 18.11 ± 2.06 | −1.43 ± 2.02 |
| VPA | 22.58 ± 1.27 | 24.60 ± 1.82 | +2.03 ± 1.16 |
| VPA + Mn | 21.99 ± 0.85 | 22.02 ± 0.99 | +0.03 ± 0.43 |
| NaB | 22.48 ± 1.19 | 23.45 ± 1.65 | +0.97 ± 1.37 |
| NaB + Mn | 21.90 ± 1.73 | 20.07 ± 1.41 | −1.83 ± 0.44 |
Mice were pretreated with VPA (100 μl/200 mg/kg, i.p. daily) or with NaB (100 μl/1200 mg/kg, i.p. daily) 30 min prior to Mn exposure (2 μl/30mg/kg, intranasal instillation, daily) for consecutive 21 days. Body weights were measured throughout the experiment and weight gain recorded on day 1 and day 21. Results represent mean ± S.E.M derived from eight independent experiments and each of their weights are expressed in grams (g). Statistical analysis was performed by two-way ANOVA repeated measures, followed by Tukey’s post-hoc test, (n=8).
3.2. Pretreatment of VPA with Mn reversed the Mn-induced decrease in the locomotor activity in mice
As one of the main pathological effects of Mn toxicity or manganism is PD-like behavioral deficits, we assessed the locomotor activity altered by Mn exposure, and determined if VPA or NaB attenuates Mn effects. As shown in Fig. 1, on day 21 Mn induced a significant decrease in locomotor activities on several parameters such as total distance traveled (1A), ambulatory activity (1B) and stereotypic activity (1C) counts, compared to the control group. During the habituation period, there was no significant change between any animals in each treatment group (data not shown). The results illustrated that 21 days of continuous pretreatment with either VPA or NaB (30 min prior to Mn exposure) attenuated the Mn-induced behavioral deficits. Mn-induced reduction of total distance traveled were significantly reversed by both VPA (F5,42 = 15.83, p<0.01) and NaB (F5,42 = 15.83, p<0.01) (Fig.1A). Additionally, ambulatory counts by VPA (F5,42 = 17.64, p<0.01) and NaB (F5,42 = 17.64, p<0.05) and stereotypic counts were reversed by VPA (F5,42 = 6.545, p<0.01) and NaB (F5,42 = 6.545, p<0.05) (Fig. 1B and 1C), respectively.
Fig. 1. Attenuating effect of VPA and NaB on Mn-induced decrease in locomotor activity in mice.
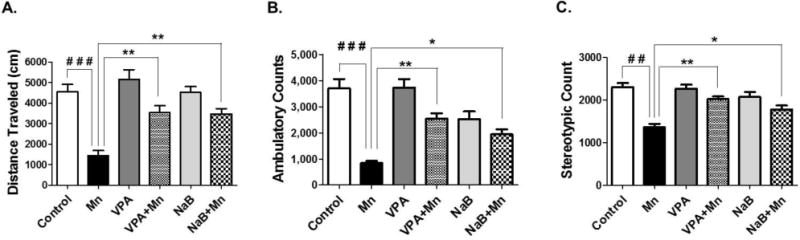
Total distance traveled (1A), ambulatory counts (1B) and stereotypic counts (1C) were measured in C57BL/6 mice. Mice were pretreated with VPA (100 μl/200 mg/kg, i.p. daily) or with NaB (100 μl/1200 mg/kg, i.p. daily) 30 min prior to Mn exposure (2 μl/30mg/kg, intranasal instillation, daily) for 21 days as described in the Methods section. Saline (0.9% NaCl, i.p.) was used as vehicles for VPA and NaB; H2O was used as vehicle for Mn. ###, p<0.001; ##, p<0.01; *p<0.05; **p<0.01 (ANOVA followed by Tukey’s post hoc test; n = 8).
3.3. Effect of VPA and NaB on the Mn-induced decrease of rotarod performance
The rotarod performance test is especially useful to test the effect of experimental drugs or after traumatic brain injury (Mouzon et al., 2012). The length of time that a given animal stays on this rotating rod is a measure of their balance, coordination, physical condition and motor-planning. The Mn group exerted significantly reduced retention times on the bar compared to the control group on days 21 (F5,42 = 7.566, p<0.01, Fig. 2), while pretreatment of VPA and NaB (Fig. 2) with Mn significantly attenuated motor coordination performance compared to the Mn alone group (F5,42 = 7.566, p<0.01 for VPA; F5,42 = 7.566, p<0.05 for NaBu). The VPA or NaB alone group did not change in rotarod performance compared to the control group.
Fig. 2. Attenuating effect of VPA and NaB on Mn-induced decrease in motor coordination by Rota-Rod test in mice.
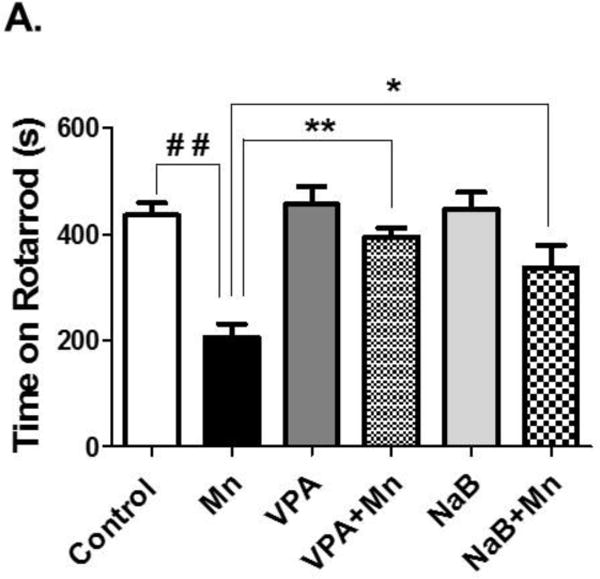
Total time on rota-rod was measured after mice were pretreated with VPA (100 μl/200 mg/kg, i.p. daily) or NaB (100 μl/1200 mg/kg, i.p. daily) 30 min prior to Mn exposure (2 μl/30mg/kg, intranasal instillation, daily) for 21 days as described in the Methods section. Saline (0.9% NaCl, i.p.) was used as a vehicle for VPA and NaB, and H2O was used as a vehicle for Mn. ##, p<0.01; *, p<0.05; **; p<0.01 (ANOVA followed by Tukey’s post hoc test; n = 8).
3.4. VPA and NaB attenuated Mn-induced decrease of GLT-1 mRNA levels in the cerebral cortex and cerebellum of mouse brain
Mn induces glutamate excitotoxicity and dysregulates astrocytic glutamate transporters in the brain (Normandin and Hazell, 2002), and astrocytic glutamate transporters (GLT-1 and GLAST) are critically involved in glutamate toxicity (Rothstein, 1995; Sheldon and Robinson, 2007; Zhang et al., 2016). Since HDAC inhibitors including VPA and NaB have shown to reverse Mn-induced decrease in GLT-1 expression in astrocytes culture system, we investigated if VPA and NaB could reverse the Mn-induced effect. As shown in Fig. 3A and 3B, Mn significantly decreased GLT-1 mRNA levels in the cerebral cortex (F3,8 = 179.5, p<0.001) and cerebellum (F3,8 = 38.72, p<0.001) compared to the control group, whereas pretreatment of VPA with Mn significantly reversed GLT-1 mRNA levels compared to those of the Mn-treated group (F3,8 = 179.5, p<0.001 for cortex and F3,8 = 38.72, p<0.01 for cerebellem). Moreover, co-treatment of NaB with Mn also exerted a similar effect to those of VPA in attenuation of Mn-decreased GLT-1 mRNA levels in the cerebral cortex (F3,8 = 47.63, p<0.001) and cerebellum (F3,8 = 10.28, p<0.01) (Fig. 3C and 3D).
Fig. 3. Effects of VPA and NaB on Mn-induced decrease of GLT-1 mRNA levels in mouse cerebral cortical and cerebellar regions.
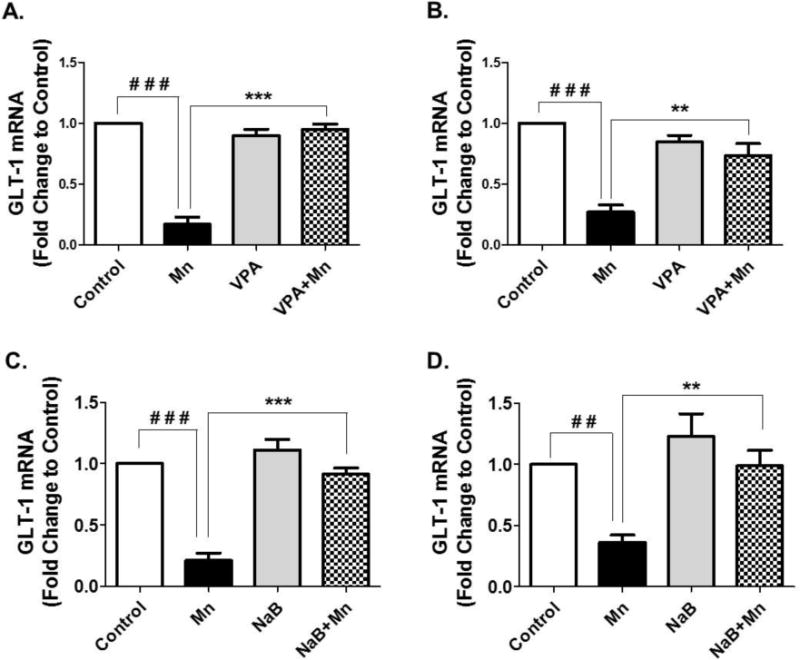
At the end of 21 days of treatment with Mn, VPA, NaB as previously mentioned, tissues from cerebral cortex and cerebellum were prepared and measured for GLT-1 mRNA levels by qPCR. (A, B) GLT-1 mRNA levels in cerebral cortex (4A) and cerebellum (4B) from mouse brain treated with Mn, VPA or Mn plus VPA. (C, D) GLT-1 mRNA levels in cerebral cortex (4C) and cerebellum (4D) from mouse brain treated with Mn, NaB, or Mn plus NaB. ##, p<0.01; ###, p<0.001; **, p<0.01; ***, p<0.001 (ANOVA followed by Tukey’s post hoc test; n = 3).
3.5. VPA and NaB attenuated Mn-induced decrease of GLT-1 protein levels in the cerebral cortex and cerebellum of mouse brain
For GLT-1 to be functional, it has to be in a form of protein, and thus, its protein expression levels are desirable to be tested. The results show a similar trend for GLT-1 protein expression to those of GLT-1 mRNA, as shown in Fig. 4. Mn decreased GLT-1 protein levels significantly in the cerebral cortex and cerebella regions of mice treated for 21 days, while VPA attenuated the Mn-decreased GLT-1 protein levels in cerebral cortex (F3,8 = 10.22, p<0.01) (Fig. 4A) and cerebellum (F3,8 = 40.38, p<0.001) (Fig. 4B). NaB also reversed Mn-induced reduction of GLT-1 protein levels in both cerebral cortex (F3,8 = 31.72, p<0.05) (Fig. 4C) and cerebellum (F3,8 = 14.81, p< 0.01) (Fig. 4D). Neither VPA nor NaB alone altered GLT-1 protein levels significantly compared to the control groups.
Fig. 4. Effects of VPA and NaB on Mn-induced decrease of GLT-1 protein levels in mouse cerebral cortical and cerebellar regions.
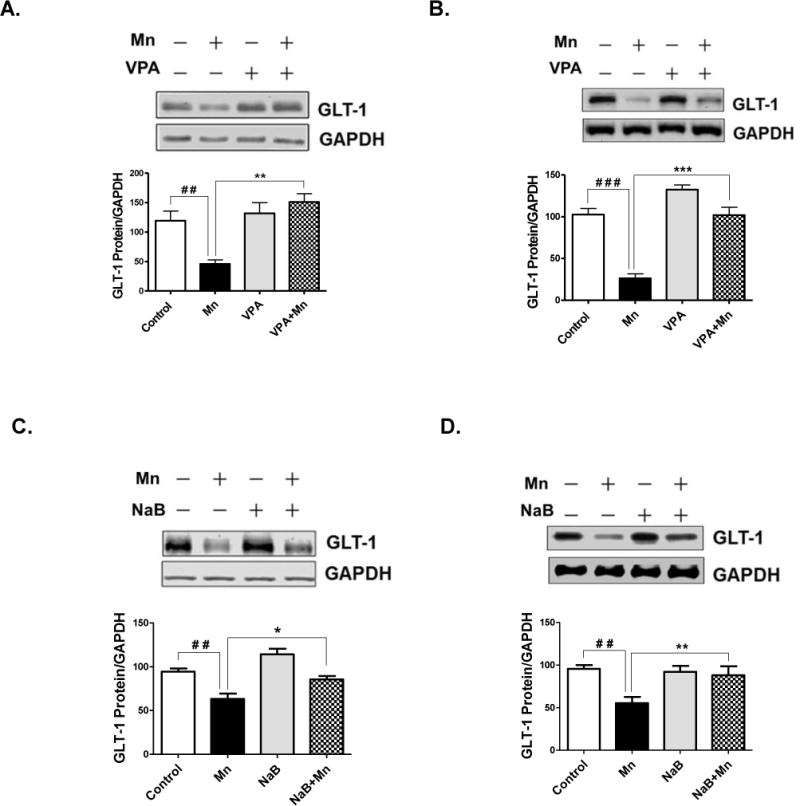
At the end of 21 days of treatment with Mn, VPA, NaB as previously mentioned, tissues from cerebral cortex and cerebellum were prepared and measured for GLT-1 protein levels by western blot analysis. (A, B) GLT-1 protein levels in cerebral cortex (4A) and cerebellum (4B) from mouse brain treated with Mn, VPA or Mn plus VPA. (C, D) GLT-1 protein levels in cerebral cortex (4C) and cerebellum (4D) from mouse brain treated with Mn, NaB, or Mn plus NaB. ##, p<0.01; ###, p<0.001; *, p<0.05; **, p<0.01; ***p<0.001 (ANOVA followed by Tukey’s post hoc test; n = 3).
3.6. VPA and NaB attenuated Mn-induced decrease of GLAST mRNA levels in the cerebral cortex and cerebellum
We also assessed the other main astrocytic glutamate transporter GLAST mRNA levels in cerebral cortex and cerebellum after 21 days of treatment with Mn, VPA, NaB, Mn plus VPA, or Mn plus NaB. While GLT-1 expression is more dominant in the cerebral cortex compared to cerebellum, GLAST is highly expressed in the cerebellar region compared to the cortex (Regan et al., 2007), indicating that these two transporters play a different role in different brain regions. As shown in Fig. 5, Mn significantly decreased GLAST mRNA levels in cerebral cortex (F3,8 = 27.03, p< 0.001) and cerebellum while co-treatment of VPA (30 min prior to Mn exposure) reversed Mn-decreased GLAST mRNA levels in both cerebral cortex (F3,8 = 27.03, p<0.01) (Fig. 5A) and cerebellum (F3,8 = 14.73, p<0.05) (Fig. 5B). NaB also reversed the Mn-induced reduction in GLAST mRNA levels in the cortex (F3,8 = 106.3, p<0.001) (Fig. 5C) and cerebellum (F3,8 = 19.01, p<0.01) (Fig. 5D).
Fig. 5. Effects of VPA and NaB on Mn-induced decrease of GLAST mRNA levels in mouse cerebral cortical and cerebellar regions.
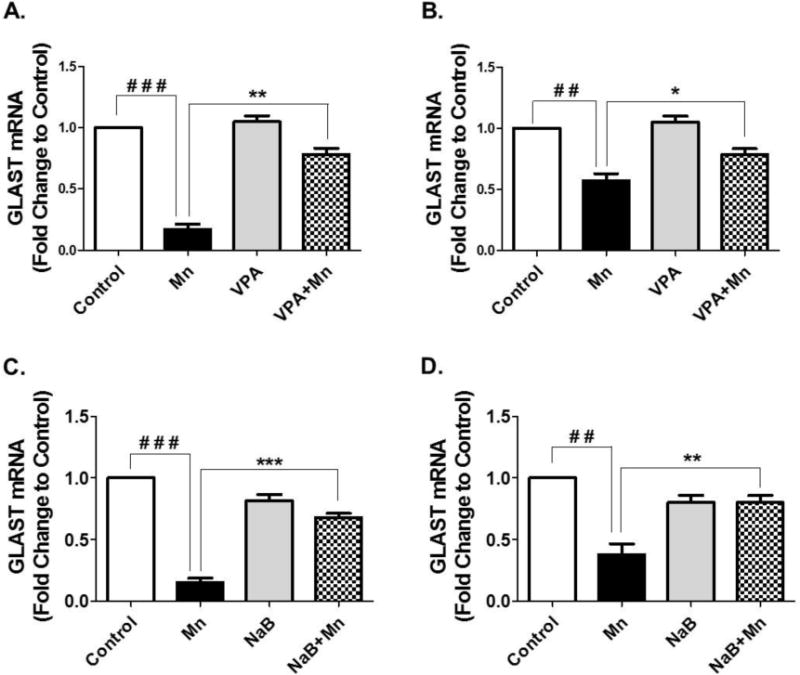
At the end of 21 days of treatment with Mn, VPA, NaB as previously mentioned, tissues from cerebral cortex and cerebellum were prepared and measured for GLAST mRNA levels by qPCR. (A, B) GLAST mRNA levels in cerebral cortex (4A) and cerebellum (4B) from mouse brain treated with Mn, VPA or Mn plus VPA. (C, D) GLAST mRNA levels in cerebral cortex (4C) and cerebellum (4D) from mouse brain treated with Mn, NaB, or Mn plus NaB. ##, p<0.01; ###, p<0.001; *, p<0.05; **; p<0.01 ***, p<0.001 (ANOVA followed by Tukey’s post hoc test; n = 3).
3.7. VPA and NaB attenuated Mn-induced decrease of GLAST protein levels in the cerebral cortex and cerebellum
As shown in Fig. 6, Mn decreased GLAST protein levels significantly, where VPA attenuated Mn-decreased GLAST protein levels in both cerebral cortex (F3,8 = 21.76, p<0.01) (Fig. 6A) and cerebellum (F3,8 = 8.193, p<0.01) (Fig. 6B). NaB also reversed the Mn-induced a decrease of GLAST protein expression levels in both cerebral cortex (F3,8 = 10.18, p<0.01) (Fig. 6C) and cerebellum (F3,8 = 8.193, p<0.05) (Fig. 6D) after 21 days of treatment.
Fig. 6. Effects of VPA and NaB on Mn-induced decrease of GLAST protein levels in mouse cerebral cortical and cerebellar regions.
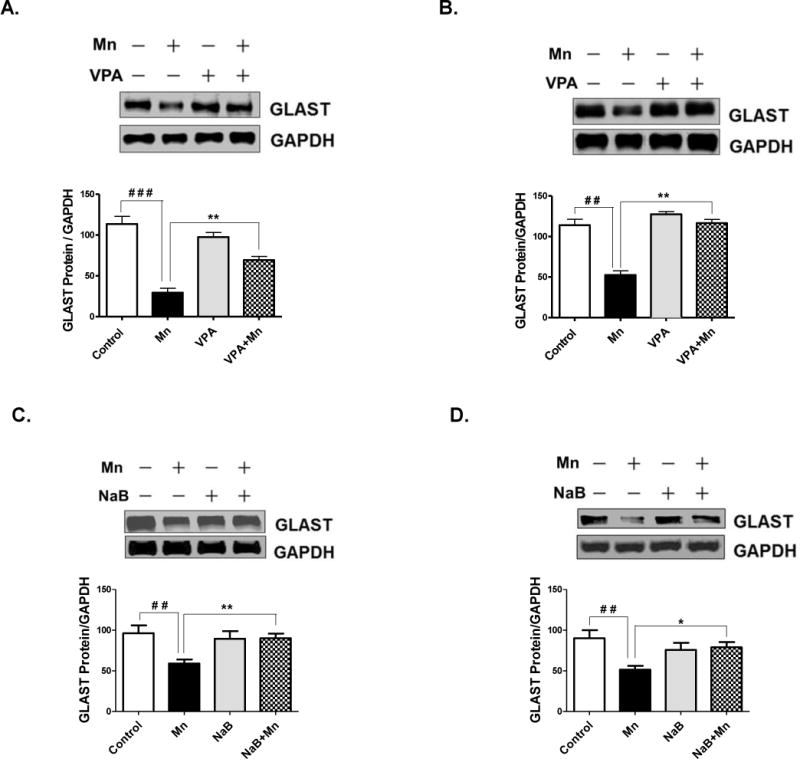
At the end of 21 days of treatment with Mn, VPA, NaB as previously mentioned, tissues from cerebral cortex and cerebellum were prepared and measured for GLAST protein levels by western blot analysis. (A, B) GLAST protein levels in cerebral cortex (4A) and cerebellum (4B) from mouse brain treated with Mn, VPA or Mn plus VPA. (C, D) GLAST protein levels in cerebral cortex (4C) and cerebellum (4D) from mouse brain treated with Mn, NaB, or Mn plus NaB. ##, p<0.01; ###, p<0.001; *, p<0.05; **, p<0.01 (ANOVA followed by Tukey’s post hoc test; n = 3).
4. Discussion
We demonstrate the therapeutic potential of VPA and NaB against Mn-induced behavioral deficits in mice. Moreover, VPA and NaB attenuated the Mn-induced dysregulation of astrocytic glutamate transporters by reversing the Mn-induced decrease in mRNA and protein levels of GLT-1 and GLAST in mouse cerebral cortex and cerebellum. These findings indicate that VPA and NaB may attenuate Mn-induced behavioral deficits, at least in part by reversing expression of GLT-1 and GLAST at the transcriptional level.
The molecular pharmacological commonality of VPA and NaB resides in their ability to inhibit HDACs via epigenetic mechanisms. We did not test the possible role of HDAC inhibition by these compounds in directly attenuating Mn toxicity. However, this attenuation is likely to involve HDAC inhibition, because HDACs regulate expression of GLT-1/GLAST as co-repressors of transcription factor yin yang 1 (YY1), and Mn enhances the interaction of YY1 with HDACs (Karki et al., 2015; Karki et al., 2014). HDAC inhibitors have been previously shown to reverse Mn-induced reduction in GLT-1 promoter activity as well as mRNA and protein levels (Karki et al., 2014). Additional studies on the precise mechanism/s by which VPA and NaB afford neuroprotection against Mn-induced toxicity should be advanced.
We tested motor performance and coordination by open-field and rotarod performance (Curzon et al., 2009). Since the symptoms of Mn toxicity or manganism are similar to those in PD, including tremor, gait, frequent falling and problems with posture and agility (Peres et al., 2016), analysis of Mn-induced reduction of locomotor activity and motor coordination are pertinent to test the effects of VPA and NaB in Mn-induced behavioral deficits. The cerebral cortex is a major component of the neural motor pathway in regulating body movements, and cerebellar regions play a critical role in motor coordination and balance. Therefore, studying these regions to examine any changes at the molecular levels are highly relevant for corroborating the efficacy of VPA and NaB in attenuating the Mn–induced behavioral deficits.
Ambulatory activity represents spontaneous physical activity, which comprises all activity other than formal exercise (Kotz et al., 2012). Animals with reduced motor muscle function have lower ambulatory activity, which is generally associated with decreased horizontal and vertical activities, total distance traveled and movement time (Nagaraju et al., 2000; Rayavarapu et al., 2010; Yu et al., 2013). It has been reported that PD patients showed a decrease in physical ambulatory activities due to motor deficits (Skidmore et al., 2008). The stereotypic activity is believed to be correlated with signs of altered response selection by the basal ganglia (Garner and Mason, 2002). Assessing ambulatory activity associated with Mn toxicity in rodents is highly relevant, because in the initial stages of manganism, neurological symptoms consist of reduced response speed, irritability, mood changes and compulsive behaviors (Roth, 2006). In more advanced stages of the disease, Mn toxicity becomes more prominent in the basal ganglia, resembling the symptoms inherent to idiopathic PD. VPA is an anti-seizure agent, and has long been used clinically to treat epileptic patients with incomplete understanding of mechanism of action involved. VPA is known to modulate γ-aminobutyric acid (GABA) neurotransmission and also inhibit HDACs at the epigenetic levels, warranting further studies to understand molecular mechanisms by which VPA reversed Mn effects on behavioral deficits.
Although both VPA and NaB are well established HDAC inhibitors, they have also been shown to exert neuroprotective effects via other distinct cellular and molecular mechanisms (Chuang et al., 2009). The present study focused on their effects on modulation of astrocytic glutamate transporters GLT-1 and GLAST. Among the five subtypes of glutamate transporters identified in humans, two transporters, namely, EAAT1 (GLT-1 in rodents) and EAAT2 (GLAST in rodents), which are predominantly expressed in astrocytes, effectively remove the majority of glutamate from synaptic clefts. Accordingly, dysregulation of GLT-1 or GLAST leads to glutamate accumulation in the synaptic clefts, ultimately causing excitotoxic neuronal injury and death (Jayakumar and Norenberg, 2016; Karki et al., 2015; Leke and Schousboe, 2016). The results from the present study indicate that altered expression levels of GLT-1 and GLAST by Mn, and the reversing effects of VPA and NaB might be associated with dysregulation of glutamate signaling as well as excitotoxicity in the cerebral cortex and cerebellum, eventually leading to modulation of locomotor activity and motor coordination. Although the molecular mechanism underlying Mn-reduced GLT-1 and GLAST at the transcriptional level remains to be established, YY1 might be the key transcription factor for Mn-reduced expression of GLT-1/GLAST (Karki et al., 2014). Mn increased YY1 expression at the transcription level, and YY1 repressed GLT-1/GLAST with HDACs as co-repressors (Karki et al., 2014). These findings support the notion that inhibition of HDACs might represent a plausible mechanism of VPA/NaB-induced attenuation of Mn-induced motor deficits.
VPA, a conventional anti-epileptic drug, has been shown to improve seizure (Campos et al., 2016), primarily being used to treat epilepsy, bipolar disorder and migraine headaches. VPA-induced antiepileptic effects have been proposed to be due to modulation of levels of brain GABA by raising its levels in cerebral and cerebellar regions, possibly by inhibiting GABA degradative enzymes and transport of T-type calcium ions (Lason et al., 2011; Leke and Schousboe, 2016). However, a growing body of evidence indicates that VPA might be beneficial to treat many other neurologic and/or neurodegenerative diseases such as stroke, traumatic brain injury (TBI), Huntington’s disease (HD), Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), and Fragile X Syndrome (FXS) via their diverse mechanisms of action (Monti et al., 2009). VPA modulates neurotrophic, angiogenic, and anti-apoptotic factors, survival signaling cascades, and oxidative stress pathways as well as HDAC epigenetic mechanisms (Ximenes et al., 2015).
At clinically relevant levels, VPA effectively inhibits HDACs (Gottlicher et al., 2001; Phiel et al., 2001), making it valuable for investigations into the therapeutic role of chromatin remodeling and epigenetic regulation in the CNS disorders. VPA significantly inhibits class I and, to a lesser extent, class II HDACs (Gottlicher et al., 2001). VPA also improves behavioral and cognitive performance, suppresses neurodegeneration and neuroinflammation, and prolongs cell survival in various animal models of CNS disorders such as stroke, PD, HD, AD and ALS (Chiu et al., 2013). Clinically, weight gain and increased appetite in obese patients have been reported as side effects of VPA treatment (Belcastro et al., 2013), but no significant changes in body weight or weight gain were observed in the current study. The protective effects of VPA against Mn-induced motor deficits are likely due to its efficacy in reversing the Mn-reduced effects on GLT-1/GLAST via inhibition of HDACs. VPA upregulates GLAST promotor activity and mRNA levels in chicken Bergmann glial cells (Aguirre et al., 2008), supporting the notion that VPA may exert neuroprotection against Mn-induced toxicity, at least in part, by modulation of histone acetylation and transcription of glutamate transporter activity. Nonetheless, further studies are warranted in order to elucidate the molecular mechanism by which VPA affords protection against Mn-toxicity.
Numerous studies have reported that NaB induces neuroprotection in various animal models of neurological disorders, suggesting a number of molecular mechanisms of action for NaB in its neuroprotection including anti-inflammatory effect (Park and Sohrabji, 2016), anti-oxidative effect, and anti-apoptotic properties (Sun et al., 2015). NaB has also been reported to modulate gene expression that is related to many behavior in the prefrontal cortex (Kratsman et al., 2016). NaB affects genes involved in neuronal excitation or inhibition, particularly promoting the transcription of genes on inhibitory pathways, but the exact mechanisms by which NaB regulates social behavior and neuronal activity on gene expression levels have yet to be established. At the epigenetic levels, NaB is a well-established HDAC inhibitor, suggesting that inhibition of these HDACs is likely involved in the molecular and behavioral effects. In general, it has been reported that HDAC inhibitors have potential to serve as therapeutics to treat learning and memory deficits (Takuma et al., 2014), depression (Wei et al., 2014) and cognition deficits (Graff and Tsai, 2013).
As histone hypoacetylation and transcriptional dysfunction are involved in numerous neurological disorders in both in vivo and in vitro models, particularly, class I and II HDAC inhibitors have shown to protect against neurodegeneration by modulating expression of multiple genes involved in neuroprotection and neurotrophicity (Chuang et al., 2009). Importantly, HDAC inhibitors modulate gene expression including neurotrophin not only in neurons but also in astrocytes. This suggests that astrocytes are also an important target for therapeutic intervention. In agreement with this previous reports, our findings that VPA and NaB attenuated Mn-induced behavioral deficits as well as a decreased expression of astrocytic glutamate transporters GLT-1 and GLAST which are predominantly expressed in astrocytes. Interestingly, VPA and NaB do not exert similar enhancing effects on GLT-1/GLAST between in vivo and in vitro conditions. We have reported that VPA and NaB increased promoter activity of GLT-1 (Karki et al., 2014), and increased the levels of mRNA and protein of GLAST in in vitro astrocytes (Karki et al., 2015). The present study shows that these HDAC inhibitors did not increase significantly expression of GLT-1 and GLAST in mouse brain, while attenuating Mn-induced reduction of expression of these transporters. It is possible that in the in vivo condition, the mouse brain is compromised with the effects of HDAC inhibitors on GLAST/GLT-1 expression to maintain homeostasis. At this point, it is not conclusive whether these HDAC inhibitors attenuate Mn-induced neurotoxicity is solely via inhibition of HDACs or involving additional unknown mechanisms.
In conclusion, we demonstrate that VPA and NaB exert protective effects against Mn-induced decrease of locomotor activity as well as GLT-1/GLAST expression in mice. The VPA- and NaB-induced protective effects likely reflect VPA/NaB’s propensity to increase glutamate transporter activity and glutamate clearance, thus inhibiting glutamate accumulation and excitotoxicity. Therefore, VPA and NaB have the potential to attenuate Mn toxicity as well as other neurodegenerative diseases driven by glutamate excitotoxicity. Further studies are required to establish the optimal doses of VPA and NaB, and identify the molecular targets of VPA and NaB which mediate their efficacy in attenuating the Mn-induced decrease in locomotor activity and GLT-1/GLAST expression in mouse brain.
Supplementary Material
Acknowledgments
The present study has been supported in part by R01 ES024756 (EL), SC1 089630(EL), SC1 CA200519 (DS), R01 ES10563 (MA), 1R03 ES024849 (MA) and 1R21 ES025415 (MA).
Footnotes
Conflicts of interest
There are no conflicts of interest.
References
- Aguirre G, Rosas S, Lopez-Bayghen E, Ortega A. Valproate-dependent transcriptional regulation of GLAST/EAAT1 expression: involvement of Ying-Yang 1. Neurochemistry international. 2008;52(7):1322–1331. doi: 10.1016/j.neuint.2008.01.015. [DOI] [PubMed] [Google Scholar]
- Belcastro V, D’Egidio C, Striano P, Verrotti A. Metabolic and endocrine effects of valproic acid chronic treatment. Epilepsy research. 2013;107(1–2):1–8. doi: 10.1016/j.eplepsyres.2013.08.016. [DOI] [PubMed] [Google Scholar]
- Brouillet EP, Shinobu L, McGarvey U, Hochberg F, Beal MF. Manganese injection into the rat striatum produces excitotoxic lesions by impairing energy metabolism. Experimental neurology. 1993;120(1):89–94. doi: 10.1006/exnr.1993.1042. [DOI] [PubMed] [Google Scholar]
- Campos MS, Ayres LR, Morelo MR, Marques FA, Pereira LR. Efficacy and Tolerability of Antiepileptic Drugs in Patients with Focal Epilepsy: Systematic Review and Network Meta-analyses. Pharmacotherapy. 2016;36(12):1255–1271. doi: 10.1002/phar.1855. [DOI] [PubMed] [Google Scholar]
- Canani RB, Costanzo MD, Leone L, Pedata M, Meli R, Calignano A. Potential beneficial effects of butyrate in intestinal and extraintestinal diseases. World journal of gastroenterology. 2011;17(12):1519–1528. doi: 10.3748/wjg.v17.i12.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CT, Wang Z, Hunsberger JG, Chuang DM. Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacological reviews. 2013;65(1):105–142. doi: 10.1124/pr.111.005512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends in neurosciences. 2009;32(11):591–601. doi: 10.1016/j.tins.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curzon P, Zhang M, Radek RJ, Fox GB. The Behavioral Assessment of Sensorimotor Processes in the Mouse: Acoustic Startle, Sensory Gating, Locomotor Activity, Rotarod, and Beam Walking. In: Buccafusco JJ, editor. Methods of Behavior Analysis in Neuroscience. Boca Raton (FL): 2009. [PubMed] [Google Scholar]
- Dauncey MJ. Recent advances in nutrition, genes and brain health. The Proceedings of the Nutrition Society. 2012;71(4):581–591. doi: 10.1017/S0029665112000237. [DOI] [PubMed] [Google Scholar]
- Dhandapani KM, Brann DW. Role of astrocytes in estrogen-mediated neuroprotection. Exp Gerontol. 2007;42(1–2):70–75. doi: 10.1016/j.exger.2006.06.032. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. Journal of Neuroscience. 2003;23(28):9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner JP, Mason GJ. Evidence for a relationship between cage stereotypies and behavioural disinhibition in laboratory rodents. Behavioural brain research. 2002;136(1):83–92. doi: 10.1016/s0166-4328(02)00111-0. [DOI] [PubMed] [Google Scholar]
- Gianutsos G, Morrow GR, Morris JB. Accumulation of manganese in rat brain following intranasal administration. Fundamental and applied toxicology : official journal of the Society of Toxicology. 1997;37(2):102–105. doi: 10.1006/faat.1997.2306. [DOI] [PubMed] [Google Scholar]
- Gottlicher M, Minucci S, Zhu P, Kramer OH, Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG, Heinzel T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. The EMBO journal. 2001;20(24):6969–6978. doi: 10.1093/emboj/20.24.6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff J, Tsai LH. The potential of HDAC inhibitors as cognitive enhancers. Annual review of pharmacology and toxicology. 2013;53:311–330. doi: 10.1146/annurev-pharmtox-011112-140216. [DOI] [PubMed] [Google Scholar]
- Henriksson J, Tjalve H. Manganese taken up into the CNS via the olfactory pathway in rats affects astrocytes. Toxicological sciences : an official journal of the Society of Toxicology. 2000;55(2):392–398. doi: 10.1093/toxsci/55.2.392. [DOI] [PubMed] [Google Scholar]
- Jayakumar AR, Norenberg MD. Glutamine Synthetase: Role in Neurological Disorders. Advances in neurobiology. 2016;13:327–350. doi: 10.1007/978-3-319-45096-4_13. [DOI] [PubMed] [Google Scholar]
- Karki P, Kim C, Smith K, Son DS, Aschner M, Lee E. Transcriptional Regulation of the Astrocytic Excitatory Amino Acid Transporter 1 (EAAT1) via NF-kappaB and Yin Yang 1 (YY1) The Journal of biological chemistry. 2015;290(39):23725–23737. doi: 10.1074/jbc.M115.649327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki P, Webb A, Smith K, Johnson J, Jr, Lee K, Son DS, Aschner M, Lee E. Yin Yang 1 is a repressor of glutamate transporter EAAT2, and it mediates manganese-induced decrease of EAAT2 expression in astrocytes. Molecular and cellular biology. 2014;34(7):1280–1289. doi: 10.1128/MCB.01176-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Li Y, Buckett PD, Bohlke M, Thompson KJ, Takahashi M, Maher TJ, Wessling-Resnick M. Iron-responsive olfactory uptake of manganese improves motor function deficits associated with iron deficiency. PloS one. 2012;7(3):e33533. doi: 10.1371/journal.pone.0033533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Guo X, Noro T, Harada C, Tanaka K, Namekata K, Harada T. Valproic acid prevents retinal degeneration in a murine model of normal tension glaucoma. Neuroscience letters. 2015;588:108–113. doi: 10.1016/j.neulet.2014.12.054. [DOI] [PubMed] [Google Scholar]
- Kotz C, Nixon J, Butterick T, Perez-Leighton C, Teske J, Billington C. Brain orexin promotes obesity resistance. Annals of the New York Academy of Sciences. 2012;1264:72–86. doi: 10.1111/j.1749-6632.2012.06585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratsman N, Getselter D, Elliott E. Sodium butyrate attenuates social behavior deficits and modifies the transcription of inhibitory/excitatory genes in the frontal cortex of an autism model. Neuropharmacology. 2016;102:136–145. doi: 10.1016/j.neuropharm.2015.11.003. [DOI] [PubMed] [Google Scholar]
- Langley B, D’Annibale MA, Suh K, Ayoub I, Tolhurst A, Bastan B, Yang L, Ko B, Fisher M, Cho S, Beal MF, Ratan RR. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28(1):163–176. doi: 10.1523/JNEUROSCI.3200-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lason W, Dudra-Jastrzebska M, Rejdak K, Czuczwar SJ. Basic mechanisms of antiepileptic drugs and their pharmacokinetic/pharmacodynamic interactions: an update. Pharmacological reports : PR. 2011;63(2):271–292. doi: 10.1016/s1734-1140(11)70497-2. [DOI] [PubMed] [Google Scholar]
- Lee ESY, Sidoryk M, Jiang HY, Yin ZB, Aschner M. Estrogen and tamoxifen reverse manganese-induced glutamate transporter impairment in astrocytes. Journal of Neurochemistry. 2009;110(2):530–544. doi: 10.1111/j.1471-4159.2009.06105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leke R, Schousboe A. The Glutamine Transporters and Their Role in the Glutamate/GABA-Glutamine Cycle. Advances in neurobiology. 2016;13:223–257. doi: 10.1007/978-3-319-45096-4_8. [DOI] [PubMed] [Google Scholar]
- Li H, Sun J, Wang F, Ding G, Chen W, Fang R, Yao Y, Pang M, Lu ZQ, Liu J. Sodium butyrate exerts neuroprotective effects by restoring the blood-brain barrier in traumatic brain injury mice. Brain research. 2016;1642:70–78. doi: 10.1016/j.brainres.2016.03.031. [DOI] [PubMed] [Google Scholar]
- Loscher W, Wahnschaffe U, Honack D, Drews E, Nau H. Effects of valproate and E-2-en-valproate on functional and morphological parameters of rat liver. III. Influence of fasting. Epilepsy research. 1993;16(3):183–194. doi: 10.1016/0920-1211(93)90079-m. [DOI] [PubMed] [Google Scholar]
- Malago JJ, Sangu CL. Intraperitoneal administration of butyrate prevents the severity of acetic acid colitis in rats. Journal of Zhejiang University Science B. 2015;16(3):224–234. doi: 10.1631/jzus.B1400191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maski KP, Jeste SS, Spence SJ. Common neurological co-morbidities in autism spectrum disorders. Current opinion in pediatrics. 2011;23(6):609–615. doi: 10.1097/MOP.0b013e32834c9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moberly AH, Czarnecki LA, Pottackal J, Rubinstein T, Turkel DJ, Kass MD, McGann JP. Intranasal exposure to manganese disrupts neurotransmitter release from glutamatergic synapses in the central nervous system in vivo. Neurotoxicology. 2012;33(5):996–1004. doi: 10.1016/j.neuro.2012.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti B, Contestabile A. Memory-enhancing drugs: a molecular perspective. Mini reviews in medicinal chemistry. 2009;9(7):769–781. doi: 10.2174/138955709788452621. [DOI] [PubMed] [Google Scholar]
- Monti B, Polazzi E, Contestabile A. Biochemical, molecular and epigenetic mechanisms of valproic acid neuroprotection. Current molecular pharmacology. 2009;2(1):95–109. doi: 10.2174/1874467210902010095. [DOI] [PubMed] [Google Scholar]
- Mouzon B, Chaytow H, Crynen G, Bachmeier C, Stewart J, Mullan M, Stewart W, Crawford F. Repetitive mild traumatic brain injury in a mouse model produces learning and memory deficits accompanied by histological changes. Journal of neurotrauma. 2012;29(18):2761–2773. doi: 10.1089/neu.2012.2498. [DOI] [PubMed] [Google Scholar]
- Nagaraju K, Raben N, Loeffler L, Parker T, Rochon PJ, Lee E, Danning C, Wada R, Thompson C, Bahtiyar G, Craft J, Hooft Van Huijsduijnen R, Plotz P. Conditional up-regulation of MHC class I in skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(16):9209–9214. doi: 10.1073/pnas.97.16.9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normandin L, Hazell AS. Manganese neurotoxicity: an update of pathophysiologic mechanisms. Metabolic brain disease. 2002;17(4):375–387. doi: 10.1023/a:1021970120965. [DOI] [PubMed] [Google Scholar]
- Park MJ, Sohrabji F. The histone deacetylase inhibitor, sodium butyrate, exhibits neuroprotective effects for ischemic stroke in middle-aged female rats. Journal of neuroinflammation. 2016;13(1):300. doi: 10.1186/s12974-016-0765-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peres TV, Schettinger MR, Chen P, Carvalho F, Avila DS, Bowman AB, Aschner M. Manganese-induced neurotoxicity: a review of its behavioral consequences and neuroprotective strategies. BMC pharmacology & toxicology. 2016;17(1):57. doi: 10.1186/s40360-016-0099-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. The Journal of biological chemistry. 2001;276(39):36734–36741. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- Qian YR, Lee MJ, Hwang S, Kook JH, Kim JK, Bae CS. Neuroprotection by valproic Acid in mouse models of permanent and transient focal cerebral ischemia. The Korean journal of physiology & pharmacology : official journal of the Korean Physiological Society and the Korean Society of Pharmacology. 2010;14(6):435–440. doi: 10.4196/kjpp.2010.14.6.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayavarapu S, Van der Meulen JH, Gordish-Dressman H, Hoffman EP, Nagaraju K, Knoblach SM. Characterization of dysferlin deficient SJL/J mice to assess preclinical drug efficacy: fasudil exacerbates muscle disease phenotype. PloS one. 2010;5(9):e12981. doi: 10.1371/journal.pone.0012981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan MR, Huang YH, Kim YS, Dykes-Hoberg MI, Jin L, Watkins AM, Bergles DE, Rothstein JD. Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the developing and mature CNS. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27(25):6607–6619. doi: 10.1523/JNEUROSCI.0790-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison G, Zakharova T, Fu S, Jiang W, Fulper R, Barrea R, Marcus MA, Zheng W, Pushkar Y. X-ray fluorescence imaging: a new tool for studying manganese neurotoxicity. PloS one. 2012;7(11):e48899. doi: 10.1371/journal.pone.0048899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth JA. Homeostatic and toxic mechanisms regulating manganese uptake, retention, and elimination. Biological research. 2006;39(1):45–57. doi: 10.4067/s0716-97602006000100006. [DOI] [PubMed] [Google Scholar]
- Rothstein JD. Excitotoxicity and neurodegeneration in amyotrophic lateral sclerosis. Clinical neuroscience. 1995;3(6):348–359. [PubMed] [Google Scholar]
- Seifert G, Schilling K, Steinhauser C. Astrocyte dysfunction in neurological disorders: a molecular perspective. Nature reviews Neuroscience. 2006;7(3):194–206. doi: 10.1038/nrn1870. [DOI] [PubMed] [Google Scholar]
- Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochemistry international. 2007;51(6–7):333–355. doi: 10.1016/j.neuint.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skidmore FM, Mackman CA, Pav B, Shulman LM, Garvan C, Macko RF, Heilman KM. Daily ambulatory activity levels in idiopathic Parkinson disease. Journal of rehabilitation research and development. 2008;45(9):1343–1348. [PubMed] [Google Scholar]
- Sun J, Wang F, Li H, Zhang H, Jin J, Chen W, Pang M, Yu J, He Y, Liu J, Liu C. Neuroprotective Effect of Sodium Butyrate against Cerebral Ischemia/Reperfusion Injury in Mice. BioMed research international. 2015;2015:395895. doi: 10.1155/2015/395895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuma K, Hara Y, Kataoka S, Kawanai T, Maeda Y, Watanabe R, Takano E, Hayata-Takano A, Hashimoto H, Ago Y, Matsuda T. Chronic treatment with valproic acid or sodium butyrate attenuates novel object recognition deficits and hippocampal dendritic spine loss in a mouse model of autism. Pharmacology, biochemistry, and behavior. 2014;126:43–49. doi: 10.1016/j.pbb.2014.08.013. [DOI] [PubMed] [Google Scholar]
- Wang Z, Leng Y, Tsai LK, Leeds P, Chuang DM. Valproic acid attenuates blood-brain barrier disruption in a rat model of transient focal cerebral ischemia: the roles of HDAC and MMP-9 inhibition. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2011;31(1):52–57. doi: 10.1038/jcbfm.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Melas PA, Wegener G, Mathe AA, Lavebratt C. Antidepressant-like effect of sodium butyrate is associated with an increase in TET1 and in 5-hydroxymethylation levels in the Bdnf gene. The international journal of neuropsychopharmacology. 2014;18(2) doi: 10.1093/ijnp/pyu032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ximenes JC, de Oliveira Goncalves D, Siqueira RM, Neves KR, Santos Cerqueira G, Correia AO, Felix FH, Leal LK, de Castro Brito GA, da Graca Naffah-Mazzacorati M, Viana GS. Valproic acid: an anticonvulsant drug with potent antinociceptive and anti-inflammatory properties. Naunyn-Schmiedeberg’s archives of pharmacology. 2013;386(7):575–587. doi: 10.1007/s00210-013-0853-4. [DOI] [PubMed] [Google Scholar]
- Ximenes JC, Neves KR, Leal LK, do Carmo MR, Brito GA, Naffah-Mazzacoratti Mda G, Cavalheiro EA, Viana GS. Valproic Acid Neuroprotection in the 6-OHDA Model of Parkinson’s Disease Is Possibly Related to Its Anti-Inflammatory and HDAC Inhibitory Properties. Journal of neurodegenerative diseases. 2015;2015:313702. doi: 10.1155/2015/313702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamawaki Y, Fuchikami M, Morinobu S, Segawa M, Matsumoto T, Yamawaki S. Antidepressant-like effect of sodium butyrate (HDAC inhibitor) and its molecular mechanism of action in the rat hippocampus. The world journal of biological psychiatry : the official journal of the World Federation of Societies of Biological Psychiatry. 2012;13(6):458–467. doi: 10.3109/15622975.2011.585663. [DOI] [PubMed] [Google Scholar]
- Yu Q, Sali A, Van der Meulen J, Creeden BK, Gordish-Dressman H, Rutkowski A, Rayavarapu S, Uaesoontrachoon K, Huynh T, Nagaraju K, Spurney CF. Omigapil treatment decreases fibrosis and improves respiratory rate in dy(2J) mouse model of congenital muscular dystrophy. PloS one. 2013;8(6):e65468. doi: 10.1371/journal.pone.0065468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaky A, Mahmoud M, Awad D, El Sabaa BM, Kandeel KM, Bassiouny AR. Valproic acid potentiates curcumin-mediated neuroprotection in lipopolysaccharide induced rats. Frontiers in Cellular Neuroscience. 2014;8 doi: 10.3389/fncel.2014.00337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LN, Sun YJ, Wang LX, Gao ZB. Glutamate Transporters/Na(+), K(+)-ATPase Involving in the Neuroprotective Effect as a Potential Regulatory Target of Glutamate Uptake. Molecular neurobiology. 2016;53(2):1124–1131. doi: 10.1007/s12035-014-9071-4. [DOI] [PubMed] [Google Scholar]
- Zhong T, Qing QJ, Yang Y, Zou WY, Ye Z, Yan JQ, Guo QL. Repression of contexual fear memory induced by isoflurane is accompanied by reduction in histone acetylation and rescued by sodium butyrate. British journal of anaesthesia. 2014;113(4):634–643. doi: 10.1093/bja/aeu184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.