

Abstract
Adrenal cortical hyperplasia manifests radiologically as a non-malignant growth, or enlargement, of the adrenal glands, specifically the cortex, although the cortex cannot be definitively identified by conventional imaging. Controlled by the pituitary gland, the adrenal cortex drives critical processes, such as the production of cortisol, mineralocorticoid and sex hormones. Any disruption in the multiple enzymes and hormones involved in these pathways may cause serious or life-threatening symptoms, often associated with anatomical changes in the adrenal glands. Diagnosis and treatment of adrenal cortical hyperplasia requires a thorough clinical evaluation. As imaging has become more robust so has its role in the diagnosis and treatment of adrenal conditions. CT has been the primary modality for adrenal imaging owing to reproducibility, temporal and spatial resolution and broad access. MRI serves a complimentary role in adrenal imaging and can be used to further evaluate indeterminate CT findings or serve as an adjunct tool without the use of ionizing radiation. Ultrasound and fluoroscopy (genitography) are most commonly used in children and foetuses to evaluate congenital adrenal hyperplasia. This article will discuss the clinical presentation, laboratory workup and imaging features of adrenal cortical hyperplasia, both congenital and acquired.
Introduction
The adrenal glands are paired glandular neuroendocrine organs within the retroperitoneal perirenal fat, superior to the kidneys.1 They have a rich blood supply, receiving one of the highest rates of blood flow in the body, far in excess of that needed for nutrient delivery.2
The adrenal cortex plays a critical role in homeostasis by controlling the amounts ofcortisol, aldosterone, and androgens, while the adrenal medulla controls “fight or flight” with release of epinephrine and norepinephrine.3
Adrenal cortical hyperplasia is defined radiologically as a non-malignant growth, or enlargement, of the adrenal glands. In a study of 55 patients without known adrenal pathology, Vincent et al found the average maximum width of the normal right adrenal gland to be 0.61 cm, with the average maximum width of each limb measuring 0.28 cm. In the same study, the maximum width of the normal left adrenal gland was found to be 0.79 cm, with average maximum limb widths of 0.33 cm (medial) and 0.30 cm (lateral).4 Hyperplasia is most often smooth, diffuse, bilateral enlargement of the adrenal glands with retained adreniform shape; however, it may be macro- or micronodular, and less commonly unilateral.5 The many subtypes of adrenal cortical hyperplasia can be differentiated by their unique clinical, laboratory and imaging profiles. This article will discuss the pathology and diagnostic workup of the subtypes of adrenal cortical hyperplasia, as well as their pathogenesis, associated imaging findings and presentation. The imaging workup and utility of various modalities, typical imaging features of adrenal cortical hyperplasia and its associations and the correlation of these features with laboratory findings and management will be discussed. Finally, the common mimics of adrenal cortical hyperplasia and their imaging findings will be reviewed.
Pathology and diagnostic workup
A thorough clinical evaluation is paramount in the workup of adrenal cortical hyperplasia and guides subsequent imaging workup, even if hyperplasia is detected incidentally.6 For example, a history of diabetes, proximal muscle fatigue, moon facies and cutaneous striae suggests Cushing syndrome, which may prompt a biochemical evaluation to discern adrenocorticotropic hormone (ACTH)-dependent processes from ACTH-independent processes, by measuring serum ACTH levels. Findings of ACTH-dependent Cushing syndrome necessitate imaging of the pituitary gland as well as the adrenal glands, whereas ACTH-independent processes would typically limit the imaging to only the adrenal glands.6 Conversely, infants born with congenital adrenal hyperplasia (CAH) may present with adrenal insufficiency in the form of salt wasting in infancy, or varying degrees of virilization in infancy or later. These infants are evaluated with an adrenocortical biochemical profile, and possibly genetic testing, often followed by ultrasound of the adrenal glands.7
Imaging workup
CT is the most common imaging tool for the evaluation of adrenal pathology as the adrenal glands can be well visualized in nearly all patients when proper technique is used.1,8 Advantages of CT include availability, speed, reproducibility and spatial plus temporal resolution. However, CT requires the use of ionizing radiation and the soft tissue contrast is inferior to that with MRI.7 While MRI can be especially useful, such as in the detection of intracellular lipid, disadvantages of MRI include increased cost, potential contraindications such as cardiac pacemakers and the potential need for sedation in young children and patients with claustrophobia.7,8 CT and MRI often demonstrate hyperplastic adrenal glands to be enlarged while having otherwise similar characteristics to normal adrenal glands, with a few exceptions such as non-contrast CT attenuation, which can be reduced in some cases.8 Like normal adrenal glands, enlarged hypertrophic adrenal glands are well delineated on T2 weighted fat suppression images as hyperintense to the surrounding fat.1 Although most adrenal glands are well visualized with non-contrast CT and MRI, the addition of intravenous contrast has improved diagnostic accuracy.1
Ultrasound is the modality of choice in children and foetuses with advantages including low cost, the lack of ionizing radiation and broad availability. It is most often used in congenital adrenal hyperplasia, where the adrenal glands may appear enlarged and cerebriform. Ultrasound is, however, operator dependent and limited by patient body habitus.7
Subtypes of adrenal cortical hyperplasia
Adrenal cortical hyperplasia can be divided into three broad categories: ACTH-dependent, ACTH-independent, and CAH. Additionally, several processes can mimic adrenal cortical hyperplasia.
Cushing syndrome
Cushing syndrome, caused by prolonged exposure of tissues to high levels of cortisol, represents a constellation of symptoms including central obesity, muscle fatigue/atrophy, hirsutism, infertility, osteoporosis, moon facies, dorsocervical and supraclavicular fat pads and wide purple striae, among others. It is most often iatrogenic from administration of exogenous glucocorticoids, in which case the need for further diagnostics and imaging is obviated.9 However, approximately 13 new cases per million per year are caused by endogenous ACTH overproduction from a pituitary tumour (Cushing disease), ectopic ACTH production from a benign or malignant extrapituitary tumour, or ACTH-independent processes secondary to adrenocortical neoplasia.9
Diagnosis of Cushing syndrome is established by measuring 24 h urine-free cortisol, dexamethasone suppression tests and/or or measuring late night salivary cortisol.9 The distinction between ACTH-dependent and ACTH-independent processes must be made with measurement of plasma concentrations of ACTH.9
Morning plasma ACTH levels less than 5 pg ml−1 indicate primary adrenal pathology (ACTH-independent), and morning plasma ACTH levels > 15 pg ml−1 indicate ACTH-dependent disease. Morning plasma levels between 5 and 15 pg ml−1 are equivocal.10 The low-dose dexamethasone suppression test aids in determining whether Cushing syndrome exists; then the high-dose dexamethasone suppression test helps to differentiate between pituitary and ectopic production of ACTH.
Cushing disease
Cushing disease, resulting from a pituitary corticotroph adenoma, and rarely carcinoma, makes up 80–85% of endogenous Cushing syndrome cases.11 True Cushing disease can be diagnosed with a two-step dexamethasone suppression test. First, the low-dose test does not suppress morning cortisol levels. Second, the high-dose test does suppress morning cortisol levels, indicating a pituitary source of ACTH.
The adrenal glands are typically bilaterally smooth, enlarged and retain their adreniform shape. However, nodularity is thought to follow indolent long-term ACTH production and is seen in up to 40% of patients with ACTH-dependent adrenal cortical hyperplasia (both central and ectopic).11 The adrenal glands can appear normal in up to 30% of patients with confirmed Cushing disease (Figure 1).9
Figure 1.

A 20-year-old female with recent weight gain, menstrual irregularity, headache, worsening acne and dry skin, as well as hypercortisolism, which suppresses with high-dose dexamethasone suppression test. Axial and coronal contrast-enhanced T1 weighted images of a hypoenhancing microadenoma in the floor of the anterior pituitary gland (a and b) (arrows). Coronal contrast-enhanced CT image of the abdomen shows normal adrenal glands (arrows) (c).
Most visible pituitary microadenomas (<1 cm) are seen as focal areas of pituitary hypo-enhancement, and may show subtle remodelling of the sella floor and deformity of the gland contour on T1 weighted MRI. Dynamic sequencing in the 1–2 min after contrast enhancement has refined the diagnostic utility of MRI for pituitary microadenomas.12 Less commonly, pituitary macroadenomas, which are >1 cm, are identified. With macroadenomas, assessment of extrasellar extension, including chiasmatic compression and cavernous sinus involvement is imperative.11
Curiously, the pituitary gland can appear normal on imaging in more than 50% of cases of proven Cushing disease (Figure 2).11 In these cases, bilateral inferior petrosal sinus sampling (BIPSS) with corticotropin-releasing hormone stimulation may be useful to confirm a pituitary source of ACTH, with sensitivity and specificity of 88–100% and 67–100% respectively.13 Bilateral inferior petrosal sinus sampling is less accurate at predicting laterality, with a positive predictive value of only 50–70%.13 Additionally, pituitary microadenomas can appear incidentally in up to 10% of the population, without Cushing disease, indicating that isolated pituitary findings do not always confirm a diagnosis of Cushing disease.9 However, Yogi-Moren et al found that a pituitary tumour ≥6 mm is highly suggestive of Cushing disease.14
Figure 2.
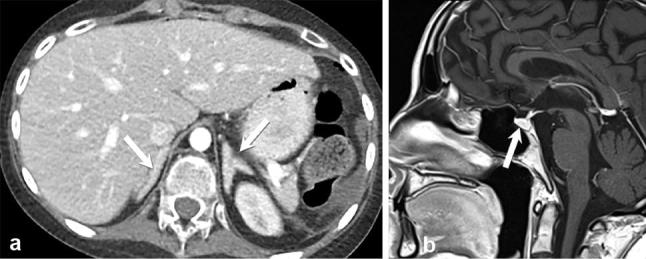
Cushing disease in a 13-year-old female with 3-month history of worsening acne, hirsutism, fatigue, headache and muscle weakness. Contrast-enhanced axial CT image (a) shows bilateral hypertrophied adrenal glands (arrows). Sagittal T1 weighted image of the brain demonstrates a normal-appearing pituitary gland (arrow) (b).
Ectopic ACTH production
Up to 10–15% of ACTH-dependent adrenal cortical hyperplasia is due to an ectopic source of ACTH, most commonly as part of a paraneoplastic process.9,11 In most of these cases, cortisol is not suppressed with either low- or high-dose dexamethasone suppression. Again, the adrenal glands may be bilaterally enlarged, smooth, yet retain their adreniform shape. However, long-term stimulation is thought to result in nodularity, and is more commonly seen with ectopic ACTH production.9 Additionally, adrenal limb width shows a positive correlation with the circulating cortisol level, with larger adrenal glands producing higher levels of cortisol.11
The most common neoplastic source of exogenous ACTH production is the lung (48%), with bronchial carcinoid tumours making up the majority (30%), followed by small cell lung cancer (18%).9 Similarly, these lung tumours made up 44.4% of all cases of neoplasm-related ectopic ACTH causing Cushing syndrome in a large study conducted by Ejaz et al.15 Other sources of ectopic ACTH include neuroendocrine tumours of the thymus, bowel, pancreas (Figure 3), medullary carcinoma of the thyroid, pheochromocytomas and mesotheliomas.9 Occasionally, in up to 12–20%, the source of ectopic ACTH production is not discovered.9
Figure 3.
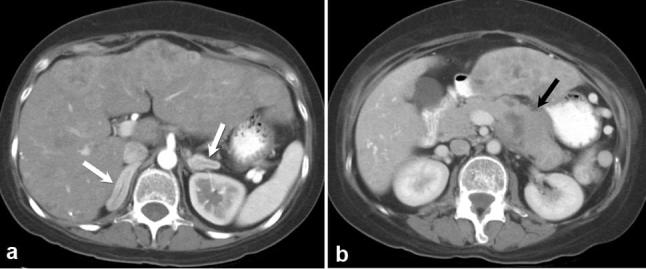
A 33-year-old female with severe Cushing syndrome. Axial contrast-enhanced CT images of the abdomen (a) show diffusely enlarged adrenal glands (white arrows) secondary to ectopic ACTH production in the setting of a metastatic neuroendocrine carcinoma of the pancreas (black arrow) (b).
ACTH-independent cushing syndrome
ACTH-independent hypercortisolism accounts for 15–20% of Cushing syndrome and always arises from primary adrenal disease.16 Although this is most commonly a unilateral process such as a functioning adenoma or carcinoma, bilateral diffuse adrenal enlargement may be seen with primary pigmented nodular adrenal disease (PPNAD) or ACTH-independent macronodular adrenal hyperplasia (AIMAH).16
Diagnosis of ACTH-independent Cushing syndrome includes clinical features of hypercortisolism, absence of serum cortisol diurnal rhythm, elevated late-night cortisol levels and incomplete suppression of cortisol production with low-dose dexamethasone suppression test.16 This article will focus on the bilateral processes of PPNAD and AIMAH.
Primary pigmented nodular adrenocortical disease (PPNAD)
PPNAD, also known as primary pigmented adrenal nodular dysplasia, bilateral micronodular adenomatosis and micronodular adrenal disease, is a rare cause of Cushing syndrome, and is considered a benign disease.16 It can be seen in isolation, or associated with Carney complex (>90% of recorded cases), which is characterized by cardiac myxomas, cutaneous myxomas, mammary fibroadenomatosis, spotty mucocutaneous pigmentation, primary pigmented nodular adrenocortical disease, testicular tumours, or a pituitary adenoma that secretes growth hormone.16–18 Symptoms of hypercortisolism are mild, so patients often present long after onset of symptoms, often in the second decade of life, although an age range of 4–44 years has been recorded in the literature.17 PPNAD is inherited as an autosomal dominant trait and there appears to be a female predominance.17 Normal to hyperplastic bilateral adrenal glands with multiple small nodules may be seen on cross-sectional imaging (Figure 4).16,17 The recommended curative treatment is bilateral adrenalectomy.16
Figure 4.

A 15-year-old girl with surgically proven primary pigmented nodular adrenal dysplasia in the context of Carney complex, treated with bilateral adrenalectomy. Axial contrast-enhanced CT image of the abdomen (a), coronal T2 weighted image of the abdomen (b) and magnified coronal T2 weighted image isolating the right adrenal gland (c) show normal-sized adrenal glands with multiple subtle small nodules (arrows).
ACTH-independent macronodular adrenal hyperplasia (AIMAH)
AIMAH is an even more rare cause of ACTH-independent Cushing syndrome, characterized by a male predominance and later age of onset (average age 48 years).16,19 The adrenal glands characteristically have a large mass of cortical tissue and multiple bilateral nodules measuring up to 5 cm each (Figure 5).16,19 Combined adrenal gland weight of more than 300 g has been noted, compared with a normal range of 8–12 g.19 Histology demonstrates large, yellow macronodules in all patients with AIMAH.19 The treatment of choice is bilateral adrenalectomy and well-controlled glucocorticoid replacement, after which most patients return to normal diurnal ACTH rhythm.19
Figure 5.
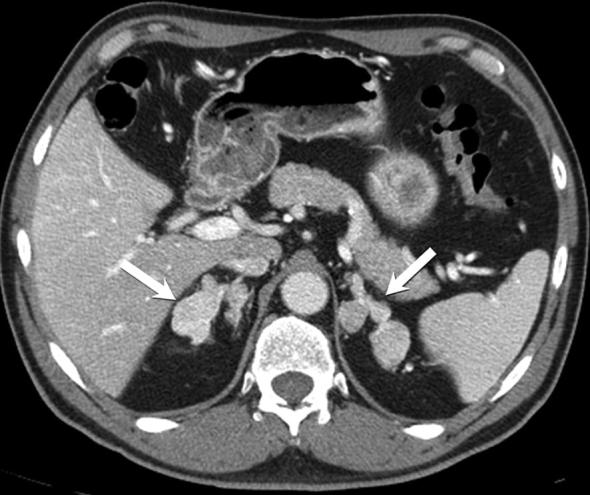
A 39-year-old male with surgically proven ACTH-independent macronodular adrenal hyperplasia. Axial contrast-enhanced CT demonstrating multiple large nodules replacing both adrenal glands, with retained adreniform shape (arrows).
Incidentally, a subset of patients with AIMAH shows ectopic expression and/or increased sensitivity to gastric inhibitory peptide (food-dependent hypercortisolism), vasopressin receptors and beta-adrenergic receptors.19 Screening for abnormal receptors has been recommended, but their presence does not necessarily affect therapy.19
Congenital adrenal hyperplasia
Congenital adrenal hyperplasia is due to an autosomal recessive disorder, which impairs steroidogenesis, resulting in various degrees of mineralocorticoid and cortisol deficiency.20 The negative feedback inhibition of ACTH by the pituitary is consequently interrupted, leading to hyperplasia of the adrenal cortex and increased production of steroid precursors in the pathway, most of which are converted to androgens.21,22 It most often presents in the perinatal period with ambiguous genitalia in females and salt wasting in males, or milder forms can present later with virilization at puberty.20,22 In all forms of congenital adrenal hyperplasia, there is a defect in one of the enzymatic steps required to synthesize cortisol from cholesterol. The overwhelming majority of cases, up to 95%, are caused by deficiency of the 21-hydroxylase enzyme.20
Congenital adrenal hyperplasia: classical 21-hydroxylase deficiency
This is the most severe form of CAH, with total or near total deficiency of 21-hydroxylase enzyme, and total inability to produce cortisol. Female infants born with classical CAH present in the perinatal period with ambiguous external genitalia (Figures 6 and 7), and are occasionally defined as male gender.22 Males born with this form of CAH are born with normal external genitalia, but soon develop isosexual precocious puberty.22 Decreased production of aldosterone in most of these patients, causing salt wasting, leads to hyponatremia, hyperkalemia and hypotension.22 These electrolyte abnormalities can cause life-threatening adrenal crisis in the first days to weeks of life in up to 75% of affected infants.20,22 Classical CAH patients are at higher risk for testicular adrenal rest tumours (Figure 8), and less commonly ovarian adrenal rest tumours, owing to the increased ACTH secretion.20,23 The early detection of these tumors is essential to help prevent fertility impairment and organ damage.20
Figure 6.
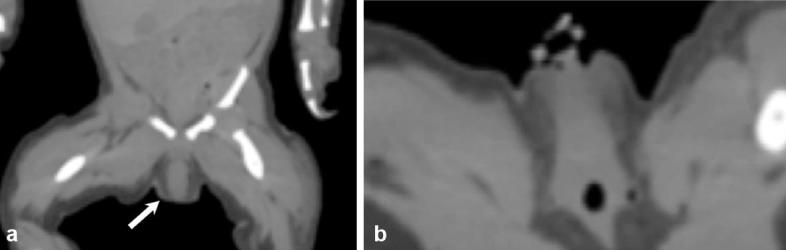
Newborn female infant with ambiguous genitalia. Coronal (a) and axial non-contrast CT (b) show ambiguously female genitalia with an enlarged clitoris (arrow).
Figure 7.

Images of a female infant born with ambiguous genitalia, who was thought to be male at birth. Subsequent ultrasound showed a uterus, vagina and ovaries, and no testicles. Enlarged cerebriform adrenal glands (arrows) were noted on ultrasound (a and b). Persistent urogenital sinus (arrow) seen on fluoroscopy (genitography) (c) owing to in utero androgen exposure.
Figure 8.

A 27-year-old male with classical congenital adrenal hyperplasia. Contrast-enhanced axial and coronal CT ( a and b) show a macronodular appearance of the adrenal glands (arrows). Colour Doppler ultrasound (c) shows an eccentrically located, ill-defined hypoechoic testicular mass with posterior shadowing (arrow), representing a testicular adrenal rest tumour.
Fluoroscopy (genitography) and voiding cystography are useful tools for confirming gender in cases of external genital ambiguity and surgical planning for feminization procedures in females.20 Genitography can show the urethra, the level of the external sphincter, the presence or absence of a vagina, the urethrovaginal confluence and the cervical impression of the uterus. The addition of voiding cystography adds the ability to show upper urogenital abnormalities, which are common in 21–80% of patients, usually females, with congenital adrenal hyperplasia.20
The imaging modality of choice for perinatal and paediatric adrenal glands is ultrasound, with its ease of availability and lack of ionizing radiation. Adrenal glands in patients with classical CAH are often enlarged (one limb >4 mm) and cerebriform.20
Simple virilizing congenital adrenal hyperplasia
Simple virilizing CAH makes up 25% of cases of 21-hydroxylase deficiency, and is characterized by a partial deficiency of the 21-hydroxylase enzyme, resulting in milder manifestations. However, female infants can still be born with external genitalia so ambiguous that they are thought to be male on initial examination.21 The increased ACTH produces normal or near normal cortisol levels, increased aldosterone secretion and therefore less salt losing tendency. Additionally, these patients may show less severe masculinization, with premature adrenarche and pubarchy.21 Poor control of the disease in males has been associated with small testes and infertility, with reduced sperm count. This is likely because of the peripheral aromatization of excess androgens to estrogens, which leads to suppression of pituitary gonadotropins.21 There is also an increased risk for testicular adrenal rest tumours in these patients.20,23
Non-classic or late onset form of congenital adrenal hyperplasia
An even milder deficiency of 21-hydroxylase enzyme, with normal levels of cortisol and aldosterone, this is the most common human autosomal recessive disease worldwide. It is most common in Ashkenazi Jews, with a frequency as high as 1/27.21 Symptoms are generally more related to androgen excess than mineralocorticoid deficiency. Premature pubarche (before age 8 in girls and before age 9 in boys), tall stature, accelerated linear growth velocity and advanced skeletal maturation are common features.7 The adrenal glands are usually normal in appearance, and there is no salt wasting or genital ambiguity at birth.20,21 Other features include masculinization, abnormal menses and polycystic ovaries in girls, as well as an increased incidence of gonadal rest tumors in both sexes (primarily testicular).7,20
Non-classical CAH due to 11-B hydroxylase or 3-B hydroxylase dehydrogenase deficiency can have the same presentation, but is extremely rare.7
Congenital adrenal hyperplasia with steroidogenic acute regulatory (StAR) mutation
Congenital lipoid adrenal hyperplasia is caused by mutations in the steroid acute regulatory protein, and represents the most dangerous form of CAH.24 It is most common in Japanese, Korean and Palestinian Arab populations. All affected patients are born with female external genitalia regardless of karyotype, and with low amounts of all steroids as a result of a severe defect in conversion of cholesterol to pregnenolone by the adrenal glands and gonads.24 Lipid deposits within the adrenal cortex may not be seen on imaging.24
Congenital adrenal hyperplasia—spectrum of imaging associations
Gonadal adrenal rest tumours are caused by migrated adrenal steroid-producing cells, which have descended into the gonads and retain ACTH responsiveness.7 They are more commonly seen in the testes than in the ovaries , are most often bilateral and can be the presenting sign of non-classical/late onset CAH.23 On ultrasound, gonadal rest tumours are usually <2 cm with no sound attenuation, but can be >2 cm heterogeneous or hyperechoic, ill-defined, eccentrically located nodules and may demonstrate posterior shoadowing.20 Early detection is essential in order to preserve fertility.7
Subclinical brain white matter involvement in CAH is seen as multiple T2 hyperintense white matter foci, and may be due to hormonal imbalance during brain development or to corticosteroid treatments.25 Bergamachi et al found a suggestion of association of CAH with demyelinating diseases.25
Mimics of adrenal cortical hyperplasia
Several abnormalities may result in diffuse adrenal enlargement, with apparent maintenance of adreniform shape. These mimics can resemble cortical hyperplasia.
Neurofibromatosis (NF1)
Neurofibromatosis (specifically NF1) can cause bilateral adrenal cortical hyperplasia, which is most commonly macronodular (Figure 9). There is evidence that biallelic loss of NF1 is associated with autonomous adrenal cortical hyperactivity including female-specific overproduction of corticosterone and aldosterone, as well as female-specific StAR hyperphosphorylation.26 Notably, effects are specific to the adrenal cortex and not to the medulla.26 Up to 16% of NF1 patients have hypertension, and up to 5% of NF1 patients have findings of precocious puberty.26 However, no Cushing-like phenotypes are documented in NF1 patients.26
Figure 9.

Axial and coronal contrast-enhanced CT images (a and b) show bilateral enlarged hypodense, nodular adrenal glands (white arrows) with a single calcification (black arrow) in a patient with neurofibromatosis Type 1. Coronal contrast-enhanced T1 weighted image (c) in 15 min delayed phase demonstrates delayed enhancement of these nodules.
Lipomatous metaplasia
Adrenal lipomatous metaplasia is characterized by the presence of small oval foci of macroscopic lipid within the adrenal cortex, with maintained adreniform shape of the gland.27 Figure 10 shows a patient with presumed bilateral adrenal lipomatous metaplasia, not biopsy proven owing to the seemingly common lack of symptomatic adrenal hyperactivity.27 Lipomatous metaplasia of the adrenal glands has also been reported in association with symptomatic cortical hyperplasia or neoplasms.28
Figure 10.
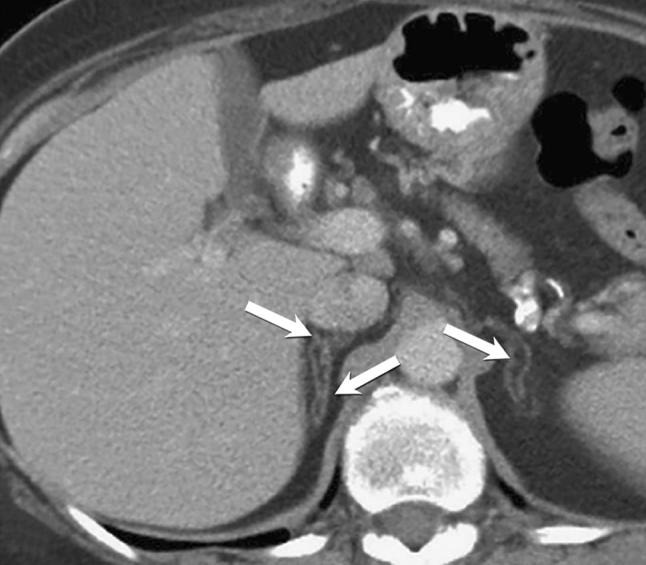
A 64-year-old female with incidentally noted asymptomatic adrenal lipomatous metaplasia found on staging scan for oesophageal cancer. Axial contrast-enhanced CT demonstrates oval foci of macroscopic lipid occupying the limbs of both adrenal glands (arrows), while the overall adreniform shape is maintained.
Adrenal lymphoma
Primary adrenal lymphoma, although very rare, is typically highly symptomatic, aggressive, metabolically hyperactive high-grade lymphoma, which most commonly affects older males.29 The most common subtype is large B-cell non-Hodgkin’s lymphoma, which makes up 87% of cases.29 Patients often suffer from adrenal insufficiency, but more commonly present with B-symptoms initially.29 On imaging, adrenal lymphoma usually presents as large bilateral adrenal masses, which are heterogeneously hypoechoic on ultrasound. They are hypodense with slight to moderate enhancement on CT, and show high fludeoxyglucose avidity because they are hypermetabolic (Figure 11).29
Figure 11.

A 35-year-old male with non-Hodgkin’s lymphoma. Axial, contrast-enhanced CT (a and b) shows bilateral hypodense, heterogeneously enhancing adrenal masses (arrows). Axial positron emission tomography/CT (c) shows marked avidity of both adrenal glands (arrows).
Adrenal Haematoma
Adrenal haemorrhage can be traumatic or non-traumatic, unilateral or bilateral and rarely causes symptoms of adrenal insufficiency.30 Traumatic adrenal haemorrhage usually results from blunt trauma to the abdomen. Non-traumatic adrenal haemorrhage may be related to a large number of causes, some of which include stress (from surgery, burns, sepsis, hypotension), coagulopathy or underlying mass.30 Acute primary adrenal insufficiency resulting from massive bilateral adrenal haemorrhage is rare, but life threatening.30
On CT imaging, adrenal haematomas are usually round or oval, with higher attenuation when acute. They become more hypodense in the centre with age, developing a characteristic “tram-track” appearance (Figure 12). They are often associated with periadrenal fat stranding, and can be associated with periadrenal haemorrhage into the perinephric space.30 On ultrasound, early haematomas appear solid, with diffuse or heterogeneous echogenicity, and are avascular on Doppler.30 With liquefaction, a central anechoic region develops, and eventually the entire haematoma becomes anechoic. Calcifications can begin to appear as early as 1–2 weeks.30 MR imaging is often used to age adrenal haematomas as each evolutionary stage shows different characteristics.30 Acute haematomas (up to 7 days) are slightly T1 hypointense and markedly T2 hypointense owing to a high concentration of intracellular deoxyhaemoglobin.30 Subacute haematomas (7 days to 7 weeks) are hyperintense on both T1 and T2 weighted images owing to the presence of free haemoglobin and serum respectively.30 Finally, chronic haematomas (older than 7 weeks) develop a hypointense rim on T1 and T2 imaging owing to haemosiderin deposition and development of a fibrous capsule.30
Figure 12.
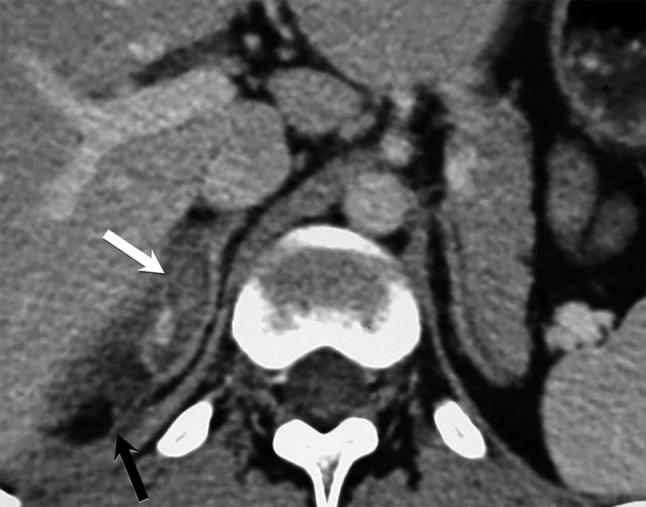
Axial contrast-enhanced CT showing “tram-track” appearance (white arrow), typical for adrenal haematoma with surrounding periadrenal fat stranding (black arrow).
Infectious processes
Adrenal infections can be caused by a multitude of pathogens including viruses, bacteria, fungi and parasites, yet represent one of the most underdiagnosed processes affecting the adrenal glands.31 Adrenal infections are most often bilateral and most commonly result in Addisonian symptoms. They can affect the appearance of the glands and mimic adrenal cortical hyperplasia on imaging (Figure 13) . One of the reasons that ante mortem diagnosis is rare is that 80–90% of the gland must be destroyed before Addisonian symptoms appear. Additionally, these symptoms are often non-specific, such as fatigue, anorexia, hypotension, hyperkalemia, hypernatremia and hypoglycemia.31
Figure 13.

Axial and coronal contrast enhanced CT images (a and b) demonstrate bilateral enlarged adrenal glands (arrows) due to biopsy proven histoplasmosis infection. Coronal PET CT (c) shows FDG avidity of the infected and inflamed adrenal glands (arrows).
Immunocompromised individuals, particularly those affected with human immunodeficiency virus, are the most susceptible to adrenal infection, with cytomegalovirus representing the most significant player in adrenal destruction associated with human immunodeficiency virus.31 Mycobacterium tuberculosis is the most common bacterial infection associated with destruction of the adrenal glands. It reaches the adrenal glands haematogenously, and can be present for up to 10 years before symptoms are seen.32 Fungal infection of the adrenal glands is most often seen in individuals with depressed cell-mediated immunity, and usually after disseminated infection. Parasitic infection of the adrenal glands is extremely rare and almost always requires residence in an endemic area.31
Conclusion
Adrenal cortical hyperplasia is most often characterized by smooth, diffuse enlargement of the adrenal glands, with preservation of adreniform shape. Various nuanced appearances, including macro- or micronodularity, may be seen depending on the underlying aetiology. The most important guide in differential diagnosis is an in-depth clinical evaluation. Imaging serves a critical role in the evaluation of these adrenal conditions and compliments clinical examination and laboratory findings.
Acknowledgment
We would like to thank Dr. Steven G. Waguespack, Professor of Endocrine Neoplasia and Hormonal Disorders, The University of Texas MD Anderson Cancer Center, for his enormous support and providing excellent clinical input and cases to our article.
Contributor Information
Agrons Michelle M, Email: Michelle.Agrons@bcm.edu.
Corey T Jensen, Email: CJensen@mdanderson.org.
Mouhammed Amir Habra, Email: MAHabra@mdanderson.org.
Christine O Menias, Email: Michelle.Agrons@bcm.edu.
Akram M Shaaban, Email: akram.shaaban@hsc.utah.edu.
Nicolaus A Wagner-Bartak, Email: NWagner@mdanderson.org.
Alicia M Roman-Colon, Email: CJensen@mdanderson.org.
Khaled M Elsayes, Email: kmelsayes@mdanderson.org.
References
- 1.Goldman SM, Kenney PJ. Computed Body Tomography with MRI Correlation. Vol. 1 Philadelphia, WA: Lippincot Williams & Wilkins; 2006. 1311 1311–75. [Google Scholar]
- 2.Breslow MJ. Regulation of adrenal medullary and cortical blood flow. Am J Physiol 1992; 262(5 Pt 2): 1317–30. [DOI] [PubMed] [Google Scholar]
- 3.Vincent JM, Morrison ID, Armstrong P, Reznek RH. The size of normal adrenal glands on computed tomography. Clin Radiol 1994; 49: 453–5. DOI: 10.1016/S0009-9260(05)81739-8 [DOI] [PubMed] [Google Scholar]
- 4.Elsayes KM, Mukundan G, Narra VR, Lewis JS, Shirkhoda A, Farooki A, et al. Adrenal masses: mr imaging features with pathologic correlation. Radiographics 2004; 24(Suppl 1): S73–S86. DOI: 10.1148/rg.24si045514 [DOI] [PubMed] [Google Scholar]
- 5.Zeiger MA, Thompson GB, Duh QY, Hamrahian AH, Angelos P, Elaraj D, et al. American Association of Clinical endocrinologists and American Association of Endocrine Surgeons Medical guidelines for the management of Adrenal incidentalomas: executive summary of recommendations. Endocr Pract 2009; 15: 450–3. DOI: 10.4158/EP.15.5.450 [DOI] [PubMed] [Google Scholar]
- 6.Teixeira SR, Elias PC, Andrade MT, Melo AF, Elias Junior J, Elias J Jr. The role of imaging in congenital adrenal hyperplasia. Arq Bras Endocrinol Metabol 2014; 58: 701–8. DOI: 10.1590/0004-2730000003371 [DOI] [PubMed] [Google Scholar]
- 7.Elsayes K, Caoili EM. Adrenal imaging: a practical guide to diagnostic workup and spectrum of imaging findings. Appl Radiol 2011; 40: 14–19. [Google Scholar]
- 8.Sahdev A, Reznek RH, Evanson J, Grossman AB. Imaging in Cushing's syndrome. Arq Bras Endocrinol Metabol 2007; 51: 1319–28. DOI: 10.1590/S0004-27302007000800018 [DOI] [PubMed] [Google Scholar]
- 9.Lila A, Jagtap V, Menon P, Sarathi V, Bandgar T, Shah N. Cushing′s syndrome: Stepwise approach to diagnosis. Indian J Endocrinol Metab 2011; 15(Suppl 8): 317–S321. DOI: 10.4103/2230-8210.86974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sohaib SA, Hanson JA, Newell-Price JD, Trainer PJ, Monson JP, Grossman AB, et al. CT appearance of the adrenal glands in adrenocorticotrophic hormone-dependent cushing's syndrome. AJR Am J Roentgenol 1999; 172: 997–1002. DOI: 10.2214/ajr.172.4.10587135 [DOI] [PubMed] [Google Scholar]
- 11.Chaudhary V, Bano S. Imaging of the pituitary: recent advances. Indian J Endocrinol Metab 2011; 15(Suppl 3): 216–S223. DOI: 10.4103/2230-8210.84871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zampetti B, Grossrubatscher E, Dalino Ciaramella P, Boccardi E, Loli P. Bilateral inferior petrosal sinus sampling. Endocr Connect 2016; 5: R12–R25. DOI: 10.1530/EC-16-0029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yogi-Morren D, Habra MA, Faiman C, Bena J, Hatipoglu B, Kennedy L, et al. Pituitary mri findings in patients with Pituitary and Ectopic Acth-dependent cushing syndrome: does a 6-mm Pituitary tumor size Cut-off value exclude Ectopic acth syndrome? Endocr Pract 2015; 21: 1098–103. DOI: 10.4158/EP15662.OR [DOI] [PubMed] [Google Scholar]
- 14.Ejaz S, Vassilopoulou-Sellin R, Busaidy NL, Hu MI, Waguespack SG, Jimenez C, et al. Cushing syndrome secondary to ectopic adrenocorticotropic hormone secretion. Cancer 2011; 117: 4381–9. DOI: 10.1002/cncr.26029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rockall AG, Babar SA, Sohaib SA, Isidori AM, Diaz-Cano S, Monson JP, et al. CT and MR imaging of the adrenal glands in ACTH-independent cushing syndrome. Radiographics 2004; 24: 435–52. DOI: 10.1148/rg.242035092 [DOI] [PubMed] [Google Scholar]
- 16.Manipadam MT, Abraham R, Sen S, Simon A. Primary pigmented nodular adrenocortical disease. J Indian Assoc Pediatr Surg 2011; 16: 160–2. DOI: 10.4103/0971-9261.86881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bain J. Carney's complex. Mayo Clin Proc 1986; 61: 508 DOI: 10.1016/S0025-6196(12)61989-2 [DOI] [PubMed] [Google Scholar]
- 18.Doppman JL, Chrousos GP, Papanicolaou DA, Stratakis CA, Alexander HR, Nieman LK. Adrenocorticotropin-independent macronodular adrenal Hyperplasia: an uncommon cause of primary adrenal hypercortisolism. Radiology 2000; 216: 797–802. DOI: 10.1148/radiology.216.3.r00au40797 [DOI] [PubMed] [Google Scholar]
- 19.New MI, Wilson RC. Steroid disorders in children: congenital adrenal Hyperplasia and apparent mineralocorticoid excess. Proc Natl Acad Sci U S A 1999; 96: 12790–7. DOI: 10.1073/pnas.96.22.12790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chung EM, Biko DM, Schroeder JW, Cube R, Conran RM. From the radiologic pathology archives: precocious puberty: radiologic-pathologic correlation. Radiographics 2012; 32: 2071–99. DOI: 10.1148/rg.327125146 [DOI] [PubMed] [Google Scholar]
- 21.Witchel SF, Azziz R. Nonclassic congenital adrenal hyperplasia. Int J Pediatr Endocrinol 2010; 2010: 625105 DOI: 10.1186/1687-9856-2010-625105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stikkelbroeck NM, Hermus AR, Schouten D, Suliman HM, Jager GJ, Braat DD, et al. Prevalence of ovarian adrenal rest tumours and polycystic ovaries in females with congenital adrenal hyperplasia: results of ultrasonography and MR imaging. Eur Radiol 2004; 14: 1802–6. DOI: 10.1007/s00330-004-2329-x [DOI] [PubMed] [Google Scholar]
- 23.Bose HS, Sato S, Aisenberg J, Shalev SA, Matsuo N, Miller WL. Mutations in the steroidogenic acute regulatory protein (StAR) in six patients with congenital lipoid adrenal hyperplasia. J Clin Endocrinol Metab 2000; 85: 3636–9. DOI: 10.1210/jc.85.10.3636 [DOI] [PubMed] [Google Scholar]
- 24.Bergamaschi R, Livieri C, Uggetti C, Candeloro E, Egitto MG, Pichiecchio A, et al. Brain white matter impairment in congenital adrenal hyperplasia. Arch Neurol 2006; 63: 413–6. DOI: 10.1001/archneur.63.3.413 [DOI] [PubMed] [Google Scholar]
- 25.Kobus K, Hartl D, Ott CE, Osswald M, Huebner A, von der Hagen M, et al. Double NF1 inactivation affects adrenocortical function in NF1Prx1 mice and a human patient. PLoS One 2015; 10: e0119030 DOI: 10.1371/journal.pone.0119030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elsayes KM, Korobkin MT, Neiderman BJ, Caoili EM. Lipomatous adrenal metaplasia: computed tomography findings in 2 presumed cases. J Comput Assist Tomogr 2009; 33: 715–6. DOI: 10.1097/RCT.0b013e3181934407 [DOI] [PubMed] [Google Scholar]
- 27.Lack EE. Tumors of the adrenal gland and extra-adrenal paraganglia : Atlas of Tumor Pathology. 3rd series Washington, DC: Armed Forces Institute of Pathology; 1997. 102 102–4. [Google Scholar]
- 28.Rashidi A, Fisher SI. Primary adrenal lymphoma: a systematic review. Ann Hematol 2013; 92: 1583–93. DOI: 10.1007/s00277-013-1812-3 [DOI] [PubMed] [Google Scholar]
- 29.Kawashima A, Sandler CM, Ernst RD, Takahashi N, Roubidoux MA, Goldman SM, et al. Imaging of nontraumatic hemorrhage of the adrenal gland. Radiographics 1999; 19: 949–63. DOI: 10.1148/radiographics.19.4.g99jl13949 [DOI] [PubMed] [Google Scholar]
- 30.Paolo WF, Nosanchuk JD. Adrenal infections. Int J Infect Dis 2006; 10: 343–53. DOI: 10.1016/j.ijid.2005.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kelestimur F. The endocrinology of adrenal tuberculosis: the effects of tuberculosis on the hypothalamo-pituitary-adrenal Axis and adrenocortical function. J Endocrinol Invest 2004; 27: 380–6. DOI: 10.1007/BF03351067 [DOI] [PubMed] [Google Scholar]
- 32. Mayo-Smith WW, Boland GW, Noto RB, Lee MJ. State-of-the-art adrenal imaging. Radiographics 2001; 21: 995–1012. DOI: 10.1148/radiographics.21.4.g01jl21995 [DOI] [PubMed] [Google Scholar]