

Abstract
Soluble huntingtin exon 1 (Httex1) with expanded polyglutamine (polyQ) engenders neurotoxicity in Huntington’s disease. To uncover the physical basis of this toxicity, we performed structural studies of soluble Httex1 for wild type and mutant polyQ lengths. Nuclear magnetic resonance experiments show evidence for conformational rigidity across the polyQ region. In contrast, hydrogen-deuterium exchange shows absence of backbone amide protection, suggesting negligible persistence of hydrogen bonds. The seemingly conflicting results are explained by all-atom simulations, which show that Httex1 adopts tadpole-like structures with a globular head encompassing the N-terminal amphipathic and polyQ regions and the tail encompassing the C-terminal proline-rich region. The surface area of the globular domain increases monotonically with polyQ length. This stimulates sharp increases in gain-of-function interactions in cells for expanded polyQ, and one of these interactions is with the stress-granule protein Fus. Our results highlight plausible connections between Httex1 structure and routes to neurotoxicity.
Keywords: Huntington’s disease, NMR spectroscopy, molecular simulations, hydrogen-deuterium exchange
Graphical abstract
INTRODUCTION
Huntington’s disease (HD) is caused by mutations in exon 1 of the huntingtin (Htt) gene that expand a CAG trinucleotide repeat sequence, encoding polyglutamine (polyQ), from a normal range of 11 – 25 to lengths beyond 36 [1]. A key feature of HD is the appearance of intracellular aggregates of N-terminal fragments of mutant Htt that encompass the polyQ segment [2]. Ectopic expression of exon 1 in its polyQ-expanded form is sufficient to recapitulate neurological defects and aggregation pathology resembling HD in a variety of animal model contexts [3–5]. Because of this, the Htt exon 1 fragment (Httex1), has been the focus of intense scrutiny in efforts to illuminate the basis of the “gain-of-toxicity” attributable to the polyQ expansion.
There is growing evidence that toxic properties of mutant Htt arise from interactions involving soluble forms of polyQ-expanded Httex1 prior to its aggregation into visible aggregates [6–13]. Yet, an unanswered question persists: how do soluble forms of Httex1 that encompass expanded polyQ tracts give rise to toxicity and cell death? Clearly, detailed investigations of soluble forms of Httex1 are required to guide structure-function studies that connect features within soluble forms of Httex1 to cellular toxicity. Such studies are challenging because of the high aggregation propensity of molecules with polyQ stretches [14]. To overcome problems with aggregation, many researchers have resorted to studying constructs housing short polyQ tracts or using fusion proteins wherein solubilizing folded domains are fused to Httex1 constructs or polyQ peptides of different lengths [15–18]. None of these studies have provided clear insights regarding the connection between expanded polyQ and the toxic attributes of the soluble Httex1 protein. For example, one-dimensional nuclear magnetic resonance (NMR) experiments on short (22Q) versus long (41Q) polyQ sequences as fusions to Glutathione S-transferase yielded no apparent differences in solution structures [19]. Crystals of Httex1 (17Q) fused to maltose binding protein showed the presence of multiple structures indicating that polyQ adopts different conformations, including α-helical structures that extend from the N-terminal end [20]. Yet this study did not report structures of pathogenic polyQ lengths. A recent cryo-electron microscopy (cryo-EM) study reported the structure of full-length Htt complexed with HAP40, which is a Htt binding protein [21]. This remarkable structure, obtained for Htt with a non-pathological polyQ length has several unresolved regions including all of Httex1, which seems to be disordered in agreement with previous reports.
The cryo-EM structure complements recent structural studies of Httex1. Warner et al. utilized a combination of single molecule Förster resonance energy transfer (smFRET) experiments and atomistic simulations to extract information regarding the global conformational properties of Httex1 as a function of polyQ length [22]. The sequences studied spanned wild type (15Q, 23Q) and pathogenic (37Q, 43Q, 49Q) polyQ lengths. The smFRET experiments were performed at concentrations where confounding effects from aggregation could be eliminated. Data gathered using multiple dye pair positions were interpreted using results from atomistic simulations. This combined approach showed that irrespective of polyQ length, monomeric Httex1 adopts tadpole-like architectures defined by a globular head composed of the 17-residue N-terminal amphipathic region (N17) and the polyQ domain, and the C-terminal proline-rich region composing the tail [22]. Simulations reweighted to match the smFRET measurements suggest that Httex1 ensembles lack stable secondary structure – an observation that persists across wild type and pathogenic polyQ lengths. However, an independent assessment of the inferences from simulations could not be obtained from smFRET measurements, because these experiments do not probe details pertaining to local length scales, including secondary structure preferences.
Elucidating the secondary structure features within wild type and expanded forms of Httex1 is important in order to determine whether a preference for specific structural features in expanded polyQ constructs drives toxicity. Antibody-based studies suggest that polyQ targeting antibodies preferentially bind expanded polyQ tracts, however hypotheses as to why have been debated [9, 12, 23, 24]. Some studies suggest that a unique structural epitope exists in expanded polyQ constructs and this structural feature drives toxicity [9, 12]. However, other studies have argued that binding to expanded polyQ constructs results from these constructs having an increased number of binding sites compared to wild type polyQ constructs [23, 24]. Overall, the resolution of antibody-based studies is inadequate to make precise assessments of the impact of polyQ length on the conformational properties of monomeric Httex1.
Our goal was to obtain a structural description of the biologically relevant sequence of Httex1, without fusion domains, and to understand how polyQ expansions alter the structural features of soluble forms of this molecule. For this, we used a combination of hydrogen-deuterium exchange (HDX) with NMR and mass spectrometry and studied two variants: 25Q and 46Q of Httex1. Regardless of polyQ length, Httex1 showed a lack of persistent hydrogen bonding. More detailed studies on Httex1 25Q indicated a lack of persistent secondary structure, and yet the N17 and polyQ domains displayed unusual rigidity. By complementing these solution-phase studies with atomistic simulations we found that the seemingly conflicting features are consistent with Httex1 adopting tadpole-like architectures, where the N17 and polyQ domains form a collapsed, conformationally heterogeneous “head”, as previously determined utilizing smFRET measurements. This tadpole-like topology is preserved for wild type and expanded polyQ lengths. However, the overall size and surface area of the globular polyQ domain grows monotonically with the length of polyQ tracts.
We hypothesized that the growing prominence of the polyQ domain in tadpole-like topologies could engender gain-of-function interactions between soluble Httex1 and components of the cellular proteome. We tested this hypothesis using a proteomic analysis and found that Httex1 with longer polyQ sequences leads to gain-of-function interactions with other proteins. Notable among the gain-of-function interactions are those with the RNA binding protein Fus and other RNA binding proteins.
RESULTS
PolyQ expansion does not alter Httex1 structural (dis)order
We reanalyzed the conformational ensembles that were reweighted to match the smFRET data. The focus of this re-analysis was on the secondary structure propensities of Httex1 as a function of polyQ length. Fig 1a-e shows the per-residue secondary structure propensity for Httex1 constructs containing polyQ tracts of length 15, 23, 37, 43, and 49 residues whereas Fig 1f focuses on the α-helical propensities for the various Httex1 domains. The simulation results predict a lack of persistent secondary structure throughout Httex1 regardless of polyQ length with weak preference for α-helical structures within N17 and the N-terminal portions of polyQ tracts.
Fig 1. Secondary structure propensities from smFRET reweighted ensembles of Httex1 for five polyQ lengths (15, 23, 37, 43, and 49).
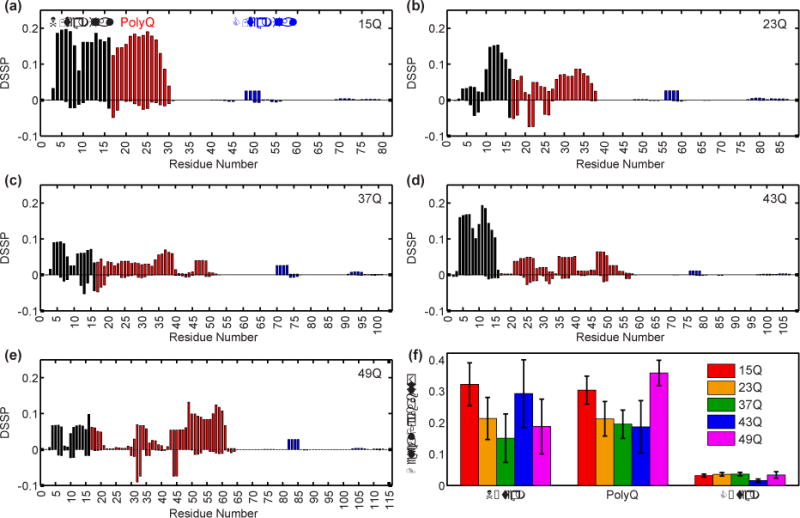
(a-e) Probability that a residue is found in an α-helical stretch of length ≥4 residues (positive) or an extended stretch of ≥2 residues (negative) as defined by the DSSP algorithm [25] for Httex1 of 15Q, 23Q, 37Q, 43Q, and 49Q, respectively. Domains are color-coded as shown. (f) Probability that each region has an α-helical stretch of ≥4 residues for each polyQ length studied.
The use of multiple smFRET measurements yields information on global conformational properties. However, these global conformational features can be consistent with a range of different local conformational/structural properties. Thus, we need additional experimental measurements to test predictions regarding secondary structure propensities calculated from ensembles reweighted to match smFRET data. If the predictions from the simulations are correct, then Httex1 should generally lack stable hydrogen bonds regardless of polyQ length. Hydrogen-deuterium exchange (HDX) combined with NMR spectroscopy or mass spectrometry (MS) serves as a powerful method to detect the presence of stable hydrogen bonds. We focused our attention on two Httex1 variants, namely 25Q (wild-type) and 46Q (pathogenic) (Fig S1; Table S1). The 25Q form of Httex1 lacks persistent protection from exchange by NMR at pH 7.5, suggesting an absence of persistent hydrogen bonds (Fig S2). We could not obtain an NMR spectrum for an expanded Httex1 construct (46Q) for a direct comparison because it aggregated at the concentrations (100 μM) required for NMR. Therefore, we turned to HDX-mass spectrometry (HDX-MS; Fig S3). This approach enabled measurements of global HDX under far more dilute protein concentrations (5 μM) whereby measurements could be performed prior to aggregation. There was no significant difference in HDX between the 25Q and 46Q forms whether the measurements were performed at pH 7.5 or pH 4 (Fig 2b). Treatment with denaturant (8 M urea-d4) also did not change the HDX patterns. Compared to a structured protein, lysozyme, our results indicate that Httex1 is devoid of persistent hydrogen bonding in its soluble form. This is true irrespective of whether the polyQ length is in the normal or pathological range. These findings are consistent with predictions from the computationally derived ensembles that were reweighted to match smFRET data.
Fig 2. Httex1 lacks persistent hydrogen bonding with 25Q or 46Q.
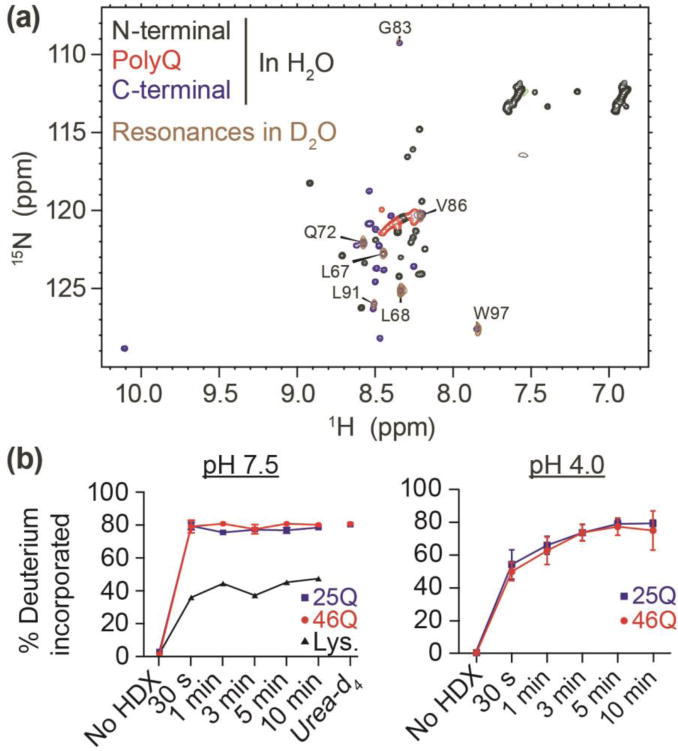
(a) 1H,15N HSQC of Httex1 25Q protein fragment in H2O versus D2O based sodium acetate buffer (150 mM; pH 4). Labelled residues are observed after hydrogen-deuterium exchange. (b) Hydrogen-deuterium exchange of Httex1 25Q and 46Q were measured over 10 minutes using mass spectrometry at both pH 7.5 and pH 4. Exchange plateaued at ~80% (due to back-exchange in the protonated solvent during chromatography separation prior to MS). Httex1 was denatured in urea-d4 (8 M) at pH 7.5, resulting in no further exchange. Hydrogen-deuterium exchange of lysozyme (Lys) was measured as a positive control for protein structure.
Given that the HDX-MS results for both 25Q and 46Q forms imply a lack of stable hydrogen bonding, we focused on the 25Q variant for the remainder of our biophysical assays to probe additional structural details within Httex1. In an effort to rule out weak hydrogen bonding we also investigated HDX combined with NMR of Httex1 25Q at pH 4. Under these conditions, the exchange of a freely exposed peptide group is approximately 3-orders of magnitude slower than at pH 7.5 [26]. For Httex1 25Q, seven backbone amide hydrogens were found to be protected from deuteration at pH 4. These correspond to residues in the C-terminal region L67, L68, Q72, G83, L91, V86, and W97 (Fig 2a). Except for the C-terminal W97 all these residues with protected amide hydrogens were flanked on one or both sides by proline residues. In addition, the side-chain NH2 groups of Gln residues also exchanged rapidly.
Soluble polyQ is devoid of persistent secondary structure but is characterized by the presence of rigid domains
Previous experiments and simulations have suggested that polyQ prefers to adopt an array of collapsed conformations of equivalent thermodynamic preference [22, 27, 28] that arises from water being a poor solvent for polyQ [29]. Collapsed globular conformations minimize the chain-solvent interface between polyQ and aqueous solvent. This behavior is in contrast to that of canonical random coils that prefer expanded conformations to maximize chain-solvent interactions [30]. Additionally, studies based on atomic force microscopy suggest that polyQ globules display considerable mechanical rigidity [31–33]. Importantly, these compact structures have been previously suggested by simulations to be formed via labile combinations of backbone and side-chain hydrogen bonds whereby stable hydrogen bonds do not persist [27, 34–36], an observation that is also true of generic polyamides including polypeptide backbones without any side-chains [37].
Our HDX data are consistent with a lack of persistent hydrogen bonding. This lack of hydrogen bonding may result from conformational heterogeneity within the polyQ domain and throughout Httex1, as predicted by the simulations, or from fully solvated, rod-like conformations with a preference for polyproline II conformations. To understand the specific implications of our HDX data and discern differences, if any, in the sequence-specific conformational preferences of different sequence blocks within Httex1 we turned to high-resolution NMR spectroscopy. Fig 3a shows the near complete non-proline backbone assignment of C, N-labelled-Httex1 (25Q) using triple resonance NMR experiments [38]. Approximately 90% of the Httex1 sequence, excluding the polyQ region and proline residues, was assignable (Fig 3a; Table S2). Although we could not assign individual backbone resonances within the polyQ tract, the side-chain resonances were readily identifiable as overlapping resonances (between 1H of 6.8–7.8 ppm and 15N of 111.5–114 ppm; Fig 3a, boxed). Additionally, the backbone, based on 13Cα–13Cβ peak correlations, formed a resonance arc (between 1H 8.17–8.49 ppm and 15N 120–121.7 ppm; Fig 3A, red). The 13Cα and 13Cβ chemical shifts are sensitive to secondary structure [39]. Analysis of the smoothed ΔCα–ΔCβ values [40] indicated localized regions of distinct secondary structure (values ≥ 1) and regions lacking stable structure but with a tendency towards some α-helical structure throughout the N-terminal and polyQ regions of Httex1 (Fig 3b) [39–41]. This result is consistent with findings that portions of the polyQ region are able to form α-helices [20, 42, 43]. These results are also consistent with predictions that emerge from the computationally derived ensembles that were reweighted to match the smFRET data of Warner et al. [22]. In light of the lack of protection from HDX, the simplest explanation is that the N17 and polyQ stretches fluctuate into and out of α-helical conformations. The C-terminal sequence displayed clear preferences for extended structures (Fig 3b). Overall, these results suggest that Httex1 adopts a heterogeneous ensemble of conformations with localized preferences for specific secondary structure motifs.
Fig 3. Resonance assignment of the Httex1 monomer suggests presence of transient structure.
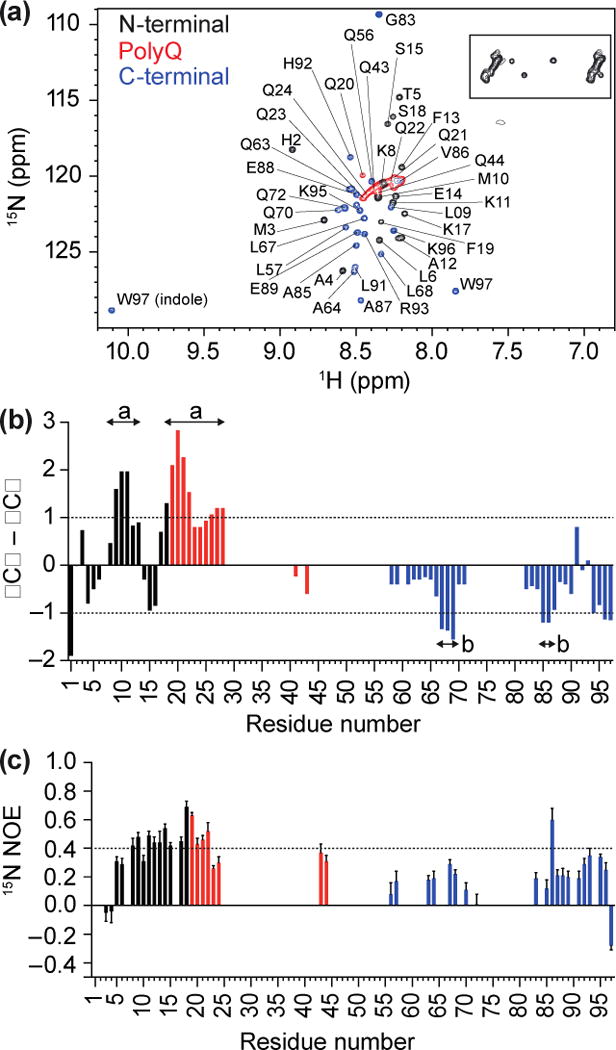
(a) 1H,15N HSQC of Httex1 protein fragment (25Q) in sodium acetate buffer (150 mM; pH 4), recorded at 5 °C, indicating N-terminal, polyQ and C-terminal Httex1 backbone amide peaks, with glutamine side chains (boxed). (b) Analysis of propensity of secondary structure based on ΔCα–ΔCβ values where persistence of positive ΔCα–ΔCβ differences suggest α-helical structures and persistence of negative ΔCα–ΔCβ differences are consistent with β-strands. (c) 15N{1H}-NOE revealed positive NOE values of 0.4 to 0.6 for N-terminal Httex1 25Q and part of the polyQ region.
To investigate the issue of molecular rigidity anticipated from the preference for globular conformations within the polyQ domain, we assessed the backbone heteronuclear Overhauser Enhancements (15N{1H}-NOE) for the 25Q construct. Positive NOE values (> 0.85) indicate a rigid protein backbone (Fig S4). The first four residues within Httex1 have negative NOE values, which is consistent with high mobility of the N-terminus (Fig 3c). The next 13 amino acids N-terminal to the polyQ region as well as the arc of glutamine backbone resonances displayed positive NOE values mostly between 0.4 and 0.6. The C-terminal proline-rich region shows 15N{1H}-NOE values that are < 0.4. These observations suggest that, while the N17 and polyQ tract are not ordered in the canonical sense, there is a persistent preference for conformational rigidity implying low amplitude conformational fluctuations, especially within the N17 and polyQ regions. In comparison, other intrinsically disordered proteins (IDPs) including the synucleins [44, 45], PAGE4 [46], and Ash1 [47] show 15N{1H}-NOE values of 0.1 to 0.4, similar to the proline-rich region of Httex1. Overall, our data point to an interesting duality whereby Httex1 molecules sample conformations in aqueous solutions with labile patterns of intra-molecular hydrogen bonds, although these conformations also involve a coexistence of compact-rigid parts within N17 and the polyQ tract and extended conformations within the proline-rich region.
Atomistic simulations show that tadpole-like architectures for Httex1 explain experimental dualities
To determine whether the tadpole-like architecture of Httex1 is consistent with the dualities observed from HDX and ΔCα–ΔCβ versus 15N{1H}-NOE results, we re-analyzed the Httex1 ensembles determined using smFRET restraints [22]. In addition, we performed new sets of atomistic simulations for the Httex1 25Q and 46Q constructs that correspond to those used for the NMR and HDX experiments. Specifically, the 25Q and 46Q simulations were performed on the sequences used for the NMR and HDX experiments, GHMATLEKLMKAFESLKSF-Qn-P11-QLPQPPPQAQPLLPQPQ-P10-GPAVAEEPLHRPKKW. The previous simulations were performed on the sequences ATLEKLMKAFESLKSF-Qn-P11-QLPQPPPQAQPLLPQPQ-P10-GPAVAEEPLHRP in order to be consistent with the smFRET experiments. Here, n denotes the number of glutamine residues in the polyQ tract. Simulations were performed using the ABSINTH implicit solvation model and force field paradigm [48]. It is important to note that the new simulations were not subjected to any reweighting to match experimental results. Even without reweighting, the back-calculated chemical shifts for Httex1 25Q were generally within error of the experimentally determined chemical shifts (Table S3). The results are summarized in Figs 4–7 and Figs S5-S7. Ensembles of the Httex1 25Q and 46Q constructs globally adopt tadpole-like conformations that are similar to the ensembles derived from reweighting to match the smFRET data (Fig S5). This result is consistent with previous observations that minimal reweighting of the computationally derived ensembles was necessary to match the smFRET measurements [22]. The implication is that simulations based on the unmodified ABSINTH model generate verifiably accurate ensembles for Httex1. Additionally, in accord with the secondary structure propensities quantified for the constructs that were used for smFRET measurements (Fig 1), the computationally derived unweighted ensembles for 25Q and 46Q showed limited persistent secondary structural preferences (Fig 4). A modest preference for α-helical structure extending from the N-terminus through the polyQ domain was observed, and this is consistent with the ΔCα–ΔCβ profile for 25Q. In contrast to both the smFRET reweighted ensembles and the ΔCα–ΔCβ profile for 25Q, the C-terminus shows a preference for α-helical structure. The ABSINTH paradigm only supports a fixed charged state model. The preference for α-helical structure within the C-terminal end arises due to our choice of a protonated state of histidine for the 25Q and 46Q constructs. This choice for the simulations was made in order to be consistent with the NMR and HDX experiments that were performed at pH 4. Httex1 25Q simulations with unprotonated histidine do not show an increased preference for α-helical structure in the C-terminus (Fig S6). This structural discrepancy in the C-terminus makes it clear that improvements need to be made to the ABSINTH model to account for charge state fluctuations.
Fig 4. Secondary structure propensities from ensembles of HDX Httex1 constructs.
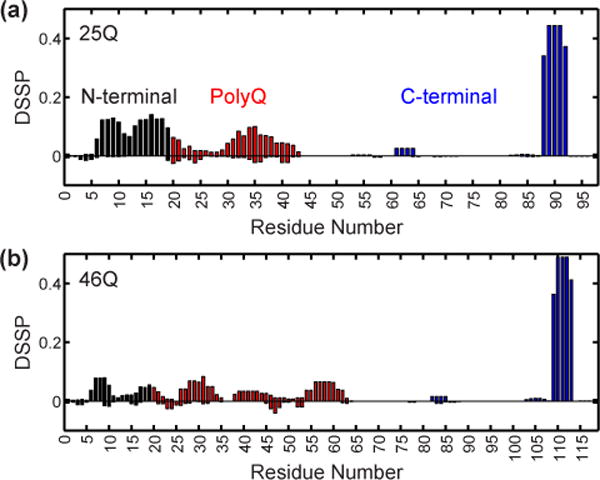
(a-b) Probability a residue is found in an α-helical stretch of length ≥4 residues (positive) or an extended stretch of ≥2 residues (negative) as defined by the DSSP algorithm [25] for Httex1 of 25Q and 46Q, respectively. Domains are color-coded as shown.
Fig 7. Scaling of the polyQ domain as a function of polyQ length and representative snapshots for all Httex1 constructs studied.
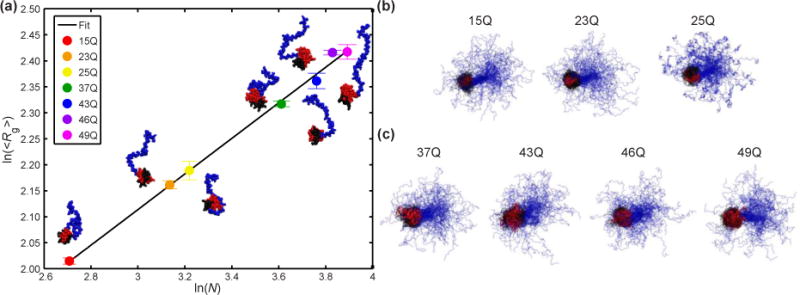
(a) Scaling of the mean size (<Rg>) of the polyQ domain as a function of polyQ length, N. The line corresponds to the best fit to the equation ln(<Rg>)=vln(N)+ln(R0) Here, v=0.34 and R0=2.97 Å. Error bars correspond to the standard error of the mean calculated over five independent simulations. (b) 100 representative snapshots for Httex1 ensembles with wild type polyQ lengths (15, 23, and 25). (c) 100 representative snapshots for Httex1 ensembles with pathogenic polyQ lengths (37, 43, 46, and 49). The N-terminal region is shown in black, the polyQ domain in red, and the C-terminal region in blue. All snapshots are aligned over the polyQ domain and the first 11 residues of the C-terminal region.
As noted above, conformational rigidity can arise due to a preference for low amplitude conformational fluctuations, which we attribute to polyQ having a preference for adopting compact/globular conformations. To test for the presence of low amplitude conformational fluctuations along the Httex1 sequence, which would be consistent with 15N{1H}-NOE values > 0.4, we quantified the degree of local collapse within Httex1 constructs for all polyQ lengths. Local collapse was determined by quantifying the radius of gyration, Rg, over sliding windows of 10 residues in sequence space and normalizing the resultant Rg values by the Rg values obtained from an ideal random coil model (Fig 5). Values less than 1 correspond to regions that are more collapsed than an ideal random coil, whereas values greater than 1 correspond to regions more expanded than an ideal random coil. Consequently, values less than 1 correspond to regions of low amplitude conformational fluctuations. For all polyQ lengths, the N-terminus of N17 shows a minimal amount of local collapse with the normalized dimensions being more expanded or similar to that of an ideal random coil. This is consistent with 15N{1H}-NOE values that are less than 0.4 for the first 6 residues of N17. The rest of N17, as well as the majority of the polyQ domain, is more collapsed than an ideal random coil. Additionally, the two pure polyproline segments (cyan) show local features that are more extended than an ideal random coil, whereas the regions connecting these polyproline segments and C-terminal to the last polyproline segment (blue) behave similarly to an ideal random coil. For comparison, we also plot the local collapse profiles for Ash1 and Ntl9. Ash1 is an IDP and has been shown to have 15N{1H}-NOE values less than 0.2 across the entire sequence, whereas Ntl9 is a folded protein. For Ash1 the local collapse profile is greater than or approximately equal to 1 across the entirety of the sequence. This is consistent with Ash1 adopting expanded conformations as has been shown both experimentally and computationally [47]. For Ntl9, a majority of the sequence adopts local conformations more collapsed than an ideal random coil. Overall, the simulated ensembles are consistent with the HDX, ΔCα–ΔCβ, and 15N{1H}-NOE results.
Fig 5. Local collapse profiles extracted from ensembles of all Httex1 constructs, a representative IDP (Ash1), and a representative folded protein (Ntl9).
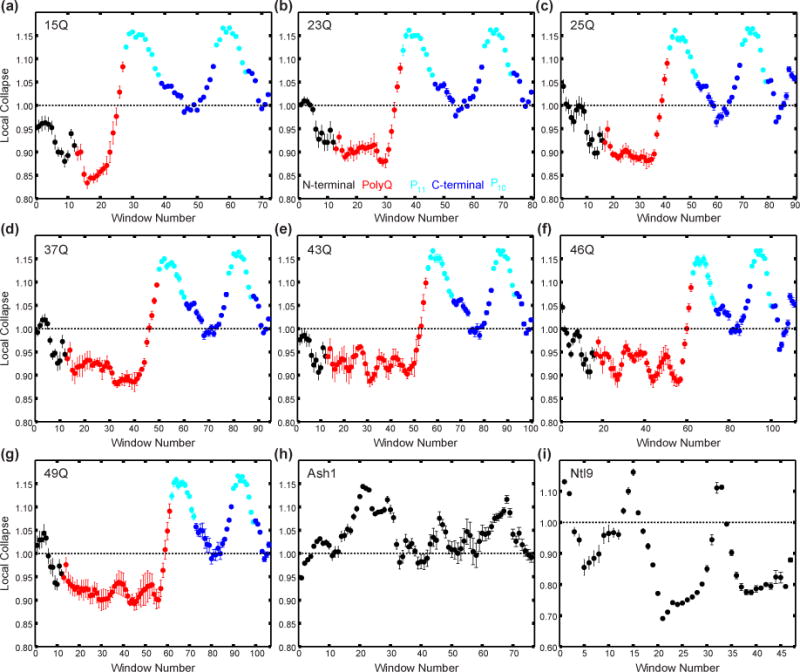
Local collapse is defined by the average radius of gyration, Rg, over sliding windows of ten residues divided by the Rg calculated over the same window from an ideal random coil simulation. Values greater than one imply the local conformational properties are more extended than an ideal random coil, whereas values less than one imply the local conformational properties are more compact that an ideal random coil. (a-g) Local collapse profiles for ensembles of Httex1 15Q, 23Q, 25Q, 37Q, 43Q, 46Q, and 49Q, respectively. Window numbers are colored based on what region the position of the fifth residue resides in. Black corresponds to N-terminal residues, red to polyQ resides, cyan to P11 and P10 residues, and blue to residues within the remainder of the C-terminal region. (h) Local collapse profile for ensembles of Ash1 – an IDP that adopts coil-like conformations. (i) Local collapse profile for ensembles of a representative folded protein (Nt19). Error bars correspond to the standard error of the mean calculated over five independent simulations for the Httex1 constructs, ten independent simulations for the Ash1 construct, and three independent simulations for the Ntl9 construct.
The HDX and ΔCα–ΔCβ results further suggest that Httex1 adopts heterogeneous conformational ensembles. To examine the degree of conformational heterogeneity within Httex1, we quantified the degree of conformational (dis)similarity across ensembles of simulated conformations. Similarity is quantified in terms of an order parameter denoted as Φ where 0 ≤ Φ ≤ 1 [49]. If the degree of conformational heterogeneity within a chain is similar to that of a reference random coil, then the extent of conformational dissimilarity within the ensemble is equivalent to that of the coil ensemble and Φ≈0. Conversely, if a chain adopts a singular, stable structure, then the conformational heterogeneity is low and Φ≈1. Fig 6 shows the Φ values computed for different Httex1 regions. The value of Φ is less than 0.4 for the N-terminal and C-terminal proline-rich regions, as well as full length Httex1, for all polyQ lengths. The C-terminal region is found to be less heterogeneous in the smFRET reweighted ensembles. This is explained by the fact that these ensembles reduce weights for conformations in which the C-terminus interacts with the polyQ domain (Fig S7). Additionally, the value of Φ is less than 0.2 for the polyQ region, irrespective of polyQ length. These numbers point to significant conformational heterogeneity within the polyQ tract. Importantly, the values of Φ are similar for all polyQ lengths for all regions and the full length Httex1. The calculated preference for conformational heterogeneity is consistent with the chemical shifts observed for 25Q, as well as the HDX results for 25Q and 46Q. Furthermore, our results show that sequences with compact domains and a persistent preference for tadpole-like architectures are also compatible with significant conformational heterogeneity. Specifically, the degree of conformational heterogeneity can be similar to or even greater than IDPs that adopt extended conformations (Fig 6).
Fig 6. The degree of similarity, Φ, between simulated ensembles calculated over fragments of Httex1 (N-terminal domain (a), polyQ domain (b), and C-terminal domain (c)) or over full Httex1 (d) for all polyQ lengths.
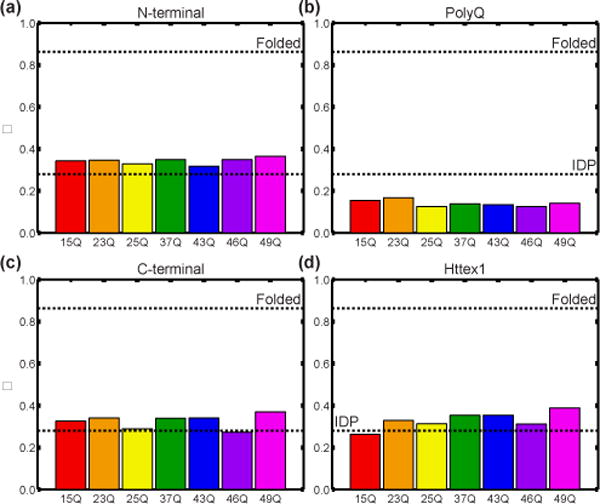
Low Φ values correspond to high degrees of heterogeneity within the simulated ensembles. The upper dashed line corresponds to the Φ-value associated with a reference folded protein (Ntl9), whereas the lower dashed line corresponds to the Φ-value associated with a reference IDP (Ash1).
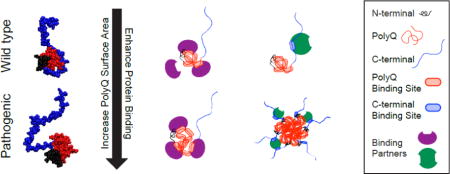
Overall, the combined HDX, ΔCα–ΔCβ, and 15N{1H}-NOE results are consistent with Httex1 adopting tadpole-like topologies that are encoded by distinct blocks of repetitive amino acid sequence blocks. Representative structures from the tadpole-like ensemble are shown in Fig 7b-c. The overall tadpole-like architecture is preserved, as revealed by atomistic simulations, across polyQ lengths. The radii of gyration and surface areas of polyQ globules increase with chain length as N1/3 and N2/3, respectively (Fig 7a) [50]. Here, N is the number of residues within the polyQ tract. Accordingly, with increased polyQ lengths, the globular polyQ tract becomes more prominent within the tadpole-like conformations of Httex1. Therefore, we propose, in accord with previous suggestions [22], that the increased prominence of the globular polyQ tract could lead to a gain of new or enhanced intracellular protein-protein interactions involving the polyQ domain and low complexity domains from other proteins (Fig 8a). Additionally, gain of new or enhanced interactions can arise through multivalent interactions that emerge as a consequence of oligomerization (Fig 8b). In this case, the increased interaction surface of the polyQ domain leads to an increased drive to form homotypic aggregates. Thus, at a given concentration, Httex1 constructs with expanded polyQ lengths can form soluble oligomers, whereas wild type polyQ lengths cannot. These oligomers can then give rise to multivalent interactions that may or may not involve interactions with the polyQ domain, and can lead to enhanced binding. As a first test of this hypothesis, we performed a proteomic analysis to probe changes to the Httex1 interactome as a function of polyQ length.
Fig 8. Proposed models for the generation of new/enhanced interactions due to expansion of the polyQ domain.
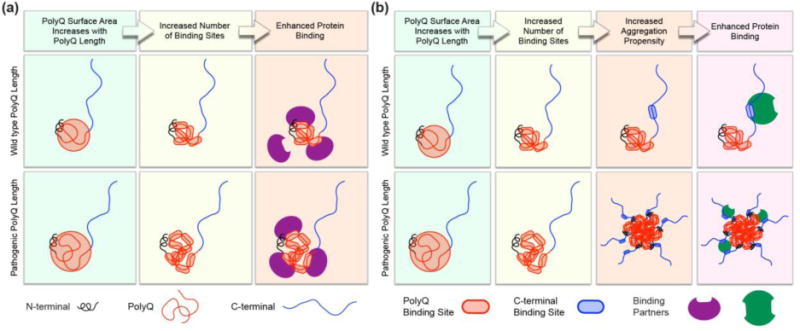
The increased surface area of the polyQ domain upon polyQ expansion can lead to an increased number of binding sites on the polyQ domain. (a) At the monomer level, this can lead to enhanced binding between proteins and the polyQ domain. (b) An increased number of binding sites can also engender an increased drive for homotypic oligomerization. For Httex1 constructs with expanded polyQ tracts, homotypic oligomerization can then lead to gain-of-function interactions that arise from multivalent interactions between the Httex1 oligomer and the binding partner [51]. In this case, these interactions do not have to directly involve the polyQ domain.
PolyQ expansion leads to gain of interactions of soluble Httex1 with the proteome
To probe how polyQ expansion modulates protein-protein interactions in the cellular context, we transiently expressed GFP-tagged Httex1 in mouse neuroblastoma (Neuro2a) cells and immunoprecipitated the soluble pool of Httex1 for proteomic analysis of binding partners after depleting large aggregates by pelleting. Under these conditions, it is estimated that the soluble pool of 46Q Httex1-GFP will comprise of approximately two-thirds monomers (sedimentation coefficient of 2.3 S) and one-third oligomers (140 S) based on our previous sedimentation velocity analysis in the same cell line [52]. By contrast 25Q Httex1-GFP only contains monomers [52]. Using a label-free quantification strategy, we found five proteins to be enriched by more than two-fold (p<0.05) as binding partners of 46Q when compared to the 25Q counterpart (Fig 9; Table S4). These proteins were Fus, Pebp1, Prdx6, Gars, and Hist1h4a. None of these proteins are known to interact with each other as assessed by protein-protein interactions with the v10 of the STRING algorithm [53]. This suggested that the interactions between Httex1 and each of Fus, Pebp1, Prdx6, Gars, and Hist1h4a are independent of one another. One explanation is that they are mediated by novel properties arising from the expanded polyQ tract in 46Q.
Fig 9. PolyQ expansion alters binding partners to soluble Httex1 states in cells.
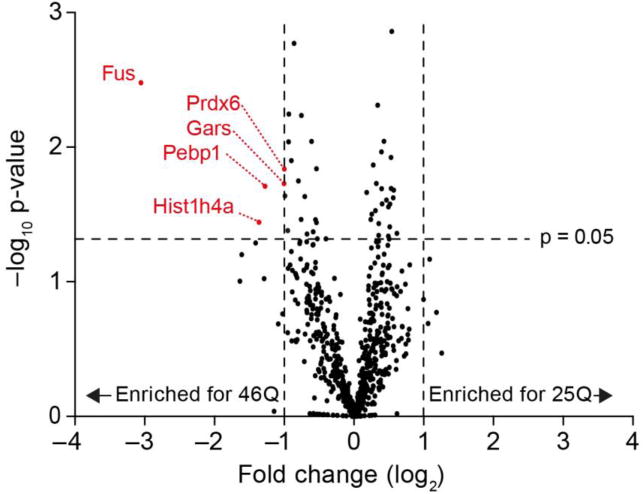
Shown is a Volcano plot of binding partners to soluble Httex1 measured from proteomic analysis of GFP-trap immunoprecipitants from Neuro2a cells transfected with Httex1-GFP. Data were acquired using label free MS/MS methods from n=3 replicates.
The most prominent interaction partner of soluble 46Q Httex1 was Fus, which functions to sequester RNA into stress granules [54, 55]. The low complexity sequence regions within Fus are enriched in polar amino acids, which mediate phase separation that drives Fus into stress granules. The N-terminal low complexity sequence in particular is able to drive liquid-liquid phase separation in the micromolar range and fibril formation that leads to hydrogels in the millimolar range [56]. Our data raise the prospect that soluble Httex1 has a selective affinity to Fus through a phase-separation and co-mixing mechanism between its low complexity domains and the polyQ sequence. Indeed, previous studies showed that proteins with low complexity domains, such as Fus, are preferentially recruited into aggregates formed by Httex1 with expanded polyQ tracts [57].
DISCUSSION
A series of prior studies established that soluble forms of polyQ expanded Httex1 are toxic to cells as quantified in cell survival measurements [7–13]. According to prevailing dogma, encompassed in the so-called “rusty hinge” hypothesis [58], this toxicity should result from a sharp change in either beta-sheet content and/or compactness of the polyQ domain within monomeric Httex1 as the polyQ length crosses the pathological threshold. These insights emerge from experiments where one cannot be certain of the identities of the species being probed [58–60], or from simulations of the atomistic [61, 62] or coarse-grained [61, 62] variety that have not, to date, been substantiated by experimental observations. Specifically, in vitro evidence for the “rusty hinge” hypothesis comes from experiments performed on constructs with solubilizing tags [63] or the interpretation of aggregation assays that assume the soluble phase is purely monomeric [59, 60]. The later assumption has been challenged as soluble spherical aggregates have been observed in vitro [31, 51, 60] and in cells [52, 64, 65], as well as isolated from animal models [66]. Additionally, in cells, a significant decrease in intramolecular FRET efficiency for Httex1 was observed at the pathogenic polyQ threshold length [67]. This result was interpreted as the polyQ domain gaining structure and concomitantly losing its ability to act as a flexible “hinge” to bring N17 and the proline-rich region within close proximity as the polyQ length increased past the pathogenic threshold. However, this change in FRET efficiency was found to be dependent on the presence of a Httex1 binding partner, PACSIN1. This dependence on PACSIN1 suggests that the change in FRET efficiency may be a result of a polyQ length-dependent modulation of protein-protein interactions, rather than an intrinsic change in polyQ structure at the pathogenic polyQ length. Overall, previous results do not provide a direct answer as to whether a sharp structural change occurs upon polyQ expansion.
Here, we have focused on a detailed, direct structural characterization, using a combination of biophysical methods, to uncover the structural features of soluble Httex1 for wild type and pathological polyQ lengths. These studies were designed to investigate the possibility that polyQ expanded Httex1 undergoes a discernible structural change when compared to constructs with wild type polyQ. By measuring HDX-MS we find that there is no clear evidence for changes in solvent protection, to imply the presence of stable beta-sheeted hydrogen bonds, with increased polyQ length. Despite the lack of persistent structure, our analysis of 15N{1H}-NOEs reveals that a large fraction of the N17 and polyQ regions are conformationally rigid, considerably more so than IDPs such as α-synuclein and Ash1 [44–47]. These data point to the presence of compact regions within Httex1 that prevail despite the absence of specific and persistent hydrogen bonding. The apparent duality of conformational rigidity and heterogeneity is reconciled by atomistic simulations, which reveal that Httex1 adopts tadpole-like topologies regardless of polyQ length [22]. In these tadpole-like structures, the globular head is defined by the adsorption of a partially alpha-helical N17 on a globular, albeit disordered polyQ domain whereas the C-terminal proline-rich region adopts semi-flexible, rod-like conformations. The tadpole-like structures explain the impact of polyQ expansions because these expansions increase the prominence of the globular head. Specifically, the surface area and radius of gyration of the globular polyQ region increase as N2/3 and N1/3 respectively with polyQ length N. This points to the prospect of gain-of-function interactions through the polyQ domain that include new heterotypic interactions that are unavailable to Httex1 with wild type polyQ and an increased tendency to aggregate via homotypic associations [31, 68], which would also be weak for Httex1 with wild type polyQ. The latter has been previously hypothesized to lead to cellular toxicity [22, 69] and may explain why reducing Htt levels has clear therapeutic benefits in animal models [70]. Here, we focused on testing the former.
Proteomics analysis reveals gain-of-function interactions whereby certain protein partners interact more prominently with polyQ expanded Httex1 when compared to wild type Httex1. These interaction partners do not share any known functional properties or interaction motifs. We hypothesize that the five proteins viz., Fus, Pebp1, Prdx6, Gars, and Hist1h4a either primarily target the polyQ tract (Fig 8a) or engage in multivalent interactions that emerge as a result of polyQ-dependent oligomerization (Fig 8b) given that the gain of these interactions requires the presence of an expanded polyQ tract within Httex1. However, additional experiments are needed to identify the regions that engage in these gain-of-function interactions and the oligomerization states of Httex1 when such interactions occur. Of the five proteins that we identified as displaying gain-of-function interactions, Fus was enriched the most. This protein is part of RNA stress granules and mutations within Fus are known to be associated with Amyotrophic Lateral Sclerosis [55, 71, 72]. Previous studies showed that the low complexity disordered domain of Fus leads to preferential interactions and recruitment of Fus into aggregates with polyQ expansions [57]. This is consistent with the presence of an increased number of binding sites within the polyQ domain. These sites might promote interactions with the low complexity domain of Fus. We further hypothesize that Httex1 binding partners can modulate the intracellular aggregation and phase behavior of Httex1 through linkage effects [51, 73]. Clearly, what is required is a systematic assessment of the interplay between homotypic interactions that drive aggregation and phase separation of Httex1 [31] and gain-of-function heterotypic interactions with polyQ expanded Httex1 that could either be protective or toxic to cells. This requires assays to characterize the impact of heterotypic interactions on Httex1 phase behavior and a framework to understand the interplay among multiple gain-of-function heterotypic interactions. A recent in vitro study, which queries the impact of profilin on Httex1 aggregation and phase behavior, lays the groundwork for studying how ligand binding can modulate phase boundaries through linkage effects [51]. Similar studies in the presence of each of Fus, Pebp1, Prdx6, Gars, and Hist1h4a and generalizations to assay the effects of multiple ligands simultaneously will be necessarily to obtain an understanding of how heterotypic interactions engender gain-of-function toxicity while also opening the door to protective effects as with profilin.
Overall, our findings converge upon a detailed structural characterization of monomeric Httex1 for wild type and pathological polyQ lengths that expands upon identifying Httex1 constructs as globally adopting tadpole-like architectures [22]. Our results support a model in which Httex1 constructs with wild type and pathogenic polyQ lengths both adopt heterogeneous conformational ensembles that lack stable secondary structure. This model suggests that a sharp, structural change within soluble Httex1 is not the cause for pathogenesis upon polyQ expansion. However, it is conceivable that the barrier to intra- and certainly inter-molecular beta-sheet formation is lowered as polyQ length increases past the pathological threshold. Such a scenario would be consistent with hypotheses that have emerged in the literature [74] and testing such hypotheses will require investigations of the barriers to beta-sheet formation using a combination of non-equilibrium sampling methods and well-crafted experiments. Irrespective of whether or not the barrier to beta-sheet formation is the relevant topological consideration, a viable alternative in vivo could be the simpler scenario whereby the tadpole-like architecture of Httex1, which is characterized by a globular polyQ domain – with an adsorbed N17 – and whose surface area increases monotonically with polyQ length, can engender sharp gain-of-function interactions upon polyQ expansion. We have presented preliminary evidence for these gain-of-function interactions by showing that new interaction partners are gained in cells when compared to the interactome for the wild-type polyQ length. Understanding how these new binding partners modulate Httex1 aggregation and phase behavior and the effects of these binding partners on toxicity are important next steps for understanding Httex1-dependent cellular toxicity.
MATERIALS AND METHODS
Plasmids
For bacterial expression, Httex1 was cloned as a fusion with a TEV protease cleavable his-tagged maltose binding protein (MBP) tag using a pET28 vector (Novagen, Hornsby Australia) using PCR-mediated cloning procedures similar to described [52]. Here, the construct lacked the fluorescent protein tag and had an appended unique tryptophan at the C-terminus (details of sequence in Table S1).
For mammalian expression, cDNA was expressed in pTREX-based vectors (Invitrogen) and encoded Emerald derivative of GFP (EmGFP), Httex1 25Q-EmGFP, 46Q-EmGFP, GFP Y66L, Httex1 25Q-mCherry or Httex1 46Q-mCherry, and mCherry in the pEGFP C1 vector (described in detail in Table S1) [8].
Recombinant Httex1 production
To produce unlabelled or 15N-labelled Httex1, pET28-Httex1 plasmid was transformed into BL21 (DE3; NEB, Ipswich MA) cells using kanamycin as the selection antibiotic. A single colony was picked to create an overnight culture in 10 mL LB at 37 °C in a shaking incubator. 2 mL overnight culture was used to inoculate 500 mL autoinduction media ZM-5052 [75] containing kanamycin (50 μg/mL; Sigma-Aldrich). 15N-labelling was achieved using N-5052 media [75] using 15NH4-Cl (Sigma-Aldrich). Cells were grown to an OD600 ≈ 0.4 at 37 °C in a shaking incubator. The cells were transferred to a 16 °C shaking incubator and incubated a further 35 hours for unlabelled preps and 48 hours for 15N-labelled preps. Cells were harvested by centrifugation (5,000 g; 30 minutes; 4 °C), resuspended in Tris-HCl (100 mM; pH 7.5) with cOmplete protease inhibitor cocktail tablets (Roche) and lysed using a final concentration of 2.5 mg/mL hen egg white lysozyme (Sigma-Aldrich; stock 50 mg/mL lysozyme, 350 mM Tris-HCl, 50% v/v glycerol; pH 7.5) with vigorous shaking and stored at −20 °C for at least 16 hours.
15N-, 13C-labelled Httex1 was produced as previously described [76]. 15NH4Cl and D-[13C] glucose (Sigma-Aldrich) were used as the nitrogen and carbon sources, respectively. Briefly, a 10 mL starter culture was prepared in LB as described above. Modified M9 media [76] (100 mL) supplemented with 14NH4Cl (50 mM), D-[12C] glucose (2 g), and kanamycin (50 μg/mL) inoculated with the starter culture, and this was cultured overnight at 37°C in a shaking incubator. Cells from 50 mL overnight culture were pelleted (2,000 g) and resuspended in modified M9 media containing 15NH4Cl (50 mM), D-[13C] glucose (0.3% w/v), and kanamycin (50 μg/ml). Cells were incubated in a 37 °C shaking incubator until the OD600 reached 0.7–0.8. Cells were induced with IPTG (final concentration 1 mM), and cultured in a shaking incubator at 16 °C until the OD600 plateaued – typically 24 hours post induction. Cells were harvested, lysed and stored as described above.
Proteins were purified first through a 5 mL HisTrap column (GE Healthcare, Australia) using binding buffer (20 mM Tris-HCl, 500 mM NaCl, 5 mM imidazole; pH 7.5) and elution buffer (20 mM Tris-HCl, 150 mM NaCl, 150 mM imidazole; pH 7.5) then a 5 mL MBPTrap column (GE Healthcare; on the GE Healthcare ÄKTA Pure chromatography system).
TEV protease production, purification and cleavage
TEV protease was produced as described [77]. Briefly, BL21 E. coli were transformed with pTEV (S219V) and grown in 2×YT broth with kanamycin as the selection antibiotic. 5 mL of overnight culture was used to inoculate 1 L 2 × YT broth, and the culture was incubated (with shaking) at 37 °C until OD600 ~0.6. IPTG (0.4 mM final concentration) was added for a 4 hour induction at 30 °C. Cells were pelleted (5,000 g; 30 min; 4 °C) and lysed with lysozyme as described above. TEV protease was purified using a HisTrap column (GE Healthcare) using bind buffer (40 mM sodium phosphate, 300 mM NaCl, 25 mM imidazole; pH 6.8) and elution buffer (40 mM sodium phosphate, 150 mM NaCl, 300 mM imidazole; pH 6.8), then stored in 10% glycerol. This protocol produced a final concentration of 3–4 mg/mL TEV protease. Cleavage activity was tested before use.
MBP tagged Httex1 was diluted to a concentration of 20 μM and then cleaved by addition of purified TEV protease (1:1 v/v) for 40 minutes at room temperature. Httex1, which lacks the his-tag, was purified from the other fragments MBP, uncleaved MBP-Httex1 and TEV protease, which all contain his tags, by filtration through a 5 mL HisTrap column equilibrated with a low salt buffer (20 mM Tris, 100 mM NaCl, 25 mM imidazole; pH 7.5). The flow through containing Httex1 was concentrated using an Amicon Ultra centrifugal filter unit (3,000 Da NMWCO cut off; Merck-Millipore, Bayswater Australia). The resultant concentrate was desalted using a PD10 column (Sephadex G-25; GE Healthcare) into sodium acetate buffer (150 mM; pH 4) and adjusted to a final protein concentration of 100 μM as measured by spectrophotometry.
HDX-MS sample preparation
To initiate protein deuteration 5% v/v protein solution (Httex1 Q25, Httex1 Q46, lysozyme; 100 μM) was mixed with 95% deuterated buffer at either pH 7.5 (20 mM Tris-HCl, 100 mM NaCl) or pH 4 (150 mM sodium acetate buffer). H-D exchange was quenched by reducing pH to 2.5 with DCl at five different time points: 30 seconds, 1 minute, 3 minutes, 5 minutes and 10 minutes. Urea-d4 (8 M; Sigma Aldrich) in deuterated buffer (pH 7.5) was used as a control to denature Httex1 proteins for 10 minutes. Each reaction was snap frozen in liquid nitrogen. Proteins were applied to a 2.1 × 10 mm column 300 Å C8 reverse phase column (Agilent Technologies, Mulgrave Australia) on ice in-line to an Agilent esiTOF Mass Spectrometer. Proteins were eluted with a gradient of buffer A: 0.1% v/v formic acid; and B: 95% v/v acetonitrile, 0.1% v/v formic acid in H2O at a flow rate of 0.25 mL/minute, increasing the percentage of B from 5% to 80% v/v over one minute. Mass spectra were deconvoluted using Agilent Mass Hunter, and these deconvoluted masses were compared to total deuterated backbone mass [78].
NMR data acquisition
NMR data for Httex1 25Q were acquired on a 700-MHz Bruker Avance HDTTT spectrometer with a triple resonance cryoprobe. Due to the propensity of Httex1 to aggregate, data were acquired at 5 °C. 3D HNCACB, HNCOCACB, HNCO and HNCACO experiments were acquired using N and C isotopically labelled Httex1. The acquisition parameters for all HSQC involved obtaining 1,024 points for 1H, 128 points for 15N, over 16 scans, with an acquisition time of 80 minutes. Data were processed using NMRPipe [79], and analysed using NMRFAM-SPARKY [80]. Assignments were determined using the PTNE server [81, 82].
Protein (Httex1 25Q) was prepared for NMR as described above. 15N,1H HSQC spectra were acquired on protein in protonated buffer at both pH 7.5 (20 mM Tris-HCl, 100 mM NaCl) and pH 4 (150 mM sodium acetate buffer). The same buffers were prepared using 2H2O. Directly following acquisition of the protonated buffer 15N, 1H HSQC, buffer was exchanged for deuterated buffer using a NAP5 column (GE Healthcare) and another 15N,1H HSQC acquired.
15N{1H}-NOE experiments were acquired in sodium acetate buffer (150 mM; pH 4) at 5 °C with a saturation pulse of 4 seconds and an additional relaxation delay of 5 seconds [83, 84]. Assignments from 3D experiments were superimposed on the 15N{1H}-NOE spectra and the differences in the saturated and reference spectra compiled using Microsoft Excel. Further analyses were performed using relax [85].
The NMR chemical shift data for 15N, NH, 13Cα, 13Cβ and 13C’ of Httex1 25Q have been deposited at the Biological Magnetic Resonance Data Bank with accession code 27161.
Details of atomistic simulations of HDX constructs
Simulations of Httex1 were performed using the CAMPARI simulation package (http://campari.sourceforge.net) utilizing the ABSINTH implicit solvation model and forcefield paradigm [48]. The move set utilized includes translational, pivot, concerted rotation, and sidechain rotation moves, as well as moves that allow for the accurate and efficient sampling of proline rings. Additional details of the implicit solvation model and move sets have been published previously [86, 87]. Five independent simulations were conducted for each construct using parameters from the abs_3.2_opls.prm parameter set. Protein atoms, as well as neutralizing and excess Na+ and Cl− ions, were modeled using atomistic detail. The excess NaCl concentration in the simulations was 5 mM. The specific sequences used were Ace-GHMATLEKLMKAFESLKSF-Qn-P11QLPQPPPQAQPLLPQPQP10-GPAVAEEPLHRPKKW-Nme, in accord with the experimental constructs. Here, n=25 and 46, Ace is the N-terminal acetyl unit, and Nme is the C-terminal N-methyl amide. The protonated version of histidine was used in order to mimic a solution condition of pH 4. Simulations were conducted in spherical droplets with radii of 150 Å. Temperature replica exchange was used to enhance sampling and followed the temperature schedule T=[288 K, 293 K, 298 K, 305 K, 310 K, 315 K, 320 K, 325 K, 335 K, 345 K, 360 K, 375 K, 390 K, 405 K]. Simulations consisted of 6.15×107 total steps of which the first 107 were taken as equilibration steps. Here, a step consists of either a Metropolis Monte Carlo move or a temperature swap. Temperature swap steps were proposed every 5×104 steps. Frames used for analyses were collected every 5×103 steps over the last 5.15×107 simulation steps such that each simulation generated 10,300 frames.
Details of atomistic simulations of smFRET constructs
The details of the simulations reweighted to match smFRET results and the reweighting procedure are given in Warner et al. [22]. In addition to the previously performed three independent simulations per construct, two new independent simulations were conducted for each construct in order to generate additional statistics. Additionally, R0 was taken to be 57 μ. For reference, the specific sequences used were ATLEKLMKAFESLKSF-Qn-P11-QLPQPPPQAQPLLPQPQ-P10-GPAVAEEPLHRP, where n=15, 23, 37, 43, and 49. The N- and C-termini were left uncapped for consistency with the experimental constructs.
Definitions of regions used for analysis of all atomistic simulations
Given the slight variation in sequence between the HDX and smFRET constructs, the following regions were defined as follows and Φ analyses of each region were conducted only over the defined residues: N-term: ATLEKLMKAFESLKSF; polyQ: Qn, C-term: P11-QLPQPPPQAQPLLPQPQ-P10-GPAVAEEPLHRP; and full: ATLEKLMKAFESLKSF-Qn-P11-QLPQPPPQAQPLLPQPQ-P10-GPAVAEEPLHRP.
Proteomics: Sample preparation andMS/MS
Neuro2a cells (1.2 × 107) were plated on a T150 culture flask and transfected with the pT-REx Httex1-25Q-Em or Httex1-46Q-Em vectors using Lipofectamine 2000 according to the manufacturer’s instructions (Thermo Fisher Scientific, Australia). Cells were harvested 48 hours after transfection, transferred to Lo-Bind tubes (Eppendorf, North Ryde Australia) and lysed mechanically by vortexing with silica beads in ATP depleted lysis buffer (10 mM Tris-HCl, 150 mM NaCl, 0.5 mM EDTA, 1 mM PMSF, 7 mM D-glucose, 1 U/mL hexokinase, 20 U/mL benzonase, 1 cOmplete protease inhibitor cocktail tablet per 10 mL buffer; pH 7.5). Lysates were centrifuged to remove protein aggregates (21, 130 g) and matched for equivalent GFP fluorescence (BioRad plate reader, Gladesville Australia). GFP-TrapA beads (ChromoTek, Planegg-Martinsried Germany), pre-equilibrated as per the manufacturer’s instructions with dilution buffer (100 mM Tris-HCl, 150 mM NaCl 1 cOmplete protease inhibitor cocktail tablet per 10 mL; pH 8) were added to the lysate and incubated for 2 hours at 4 °C. The beads were pelleted (2,000 g) and washed thrice in dilution buffer as per manufacturer’s instructions. Beads were resuspended in triethylammonium bicarbonate (Sigma-Aldrich) and snap frozen in liquid nitrogen. Proteins were eluted from the GFP-TrapA beads with 4% w/v SDS, 100 mM DTT, 100 mM Tris-HCl; pH 7.5 at 65 °C for 5 minutes. Beads were washed twice with 100 mM Tris-HCl (pH 7.5) with the beads collected by centrifugation (2,000 g). The wash supernatants were pooled and added to the eluent. Eluted proteins were prepared for LC-MS/MS analysis using a FASP Protein Digestion Kit (Expedeon, San Diego CA) according to the manufacturer’s instructions with two additional wash steps with Urea Sample Solution (Expedeon). Samples were dried using a benchtop CentriVap (Labconco, Kansas City MO) and resuspended in 0.1% v/v formic acid.
Peptides were analysed by LC-MS/MS using the QExactive mass spectrometer (Thermo Fisher Scientific) coupled online with a RSLC nano HPLC (Ultimate 3000, Thermo Fisher Scientific). Samples were loaded on a 100 μm, 2 cm nanoviper pepmap100 trap column in 2% v/v acetonitrile, 0.1% v/v formic acid at a flow rate of 15 μL/minute. Peptides were eluted and separated at a flow rate of 300 μL/minute on RSLC nanocolumn 75 μm × 50 cm, pepmap100 C18, 3 μm 100 Å pore size (Thermo Fisher Scientific) as described [88].
MS/MS data analysis
MS/MS data were analysed using MaxQuant version 1.2.2.5 and the UniProt mouse protein database (accessed August 2013) for peptide identification. Strict bioinformatic criteria were used for the assignment of peptide identity; including the application of a false discovery rate of 0.01 for both proteins and peptides [89, 90] and removal of species contained within a list of common contaminants [91]. Data were further analysed using Perseus version 1.5.6.0 (MaxQuant) to determine significantly enriched species in label free quantification measurements of the various IP experiments. A two-fold increase in relative abundance and a P value < 0.05 (Student’s t-test) were used to determine those enriched peptides/proteins. The mass spectrometry proteomics data have been deposited in PRIDE Archive proteomics data repository [92] with dataset identifier PXD006792.
Supplementary Material
HIGHLIGHTS.
The physical basis for expanded polyglutamine (polyQ) neurotoxicity is unknown
HDX-MS shows a lack of stable hydrogen bonds regardless of polyQ length
Httex1 lacks stable secondary structure but contains conformationally rigid domains
Dualities are explained by tadpole-like structures with a globular polyQ domain
Surface area of polyQ domain increases with length, leading to gain-of-function interactions
Acknowledgments
The US National Institutes of Health supported this work through grant 5R01NS056114 to R.V.P. We thank Oded Kleifeld from the Infection and Immunity Program, Monash Biomedicine Discovery Institute and Department of Biochemistry and Molecular Biology, Monash University for technical assistance with the proteomics work (now at Faculty of Biology, Technion-Israel Institute of Technology, Haifa 3200003). This work builds on a prior collaboration with John Warner IV, Piau Siong Tan, Edward Lemke, and Hilal Lashuel that yielded atomistic descriptions of Httex1 monomers using smFRET and computer simulations [22]. We (K.M.R. and R.V.P) are grateful to Drs. Warner, Lemke, and Lashuel for their assistance and for their insights regarding the conformational ensembles of Httex1.
ABBREVIATIONS
- HD
Huntington’s disease
- Htt
huntingtin
- polyQ
polyglutmaine
- Httex1
huntingtin exon 1
- NMR
nuclear magnetic resonance
- smFRET
single molecule Forster resonance energy transfer
- N17
17-residue N-terminal amphipathic region of Httex1
- HDX
hydrogen-deuterium exchange
- MS
mass spectrometry
- IDPs
intrinsically disordered proteins
- Neuro2a
mouse neuroblastoma cell line
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Walker FO. Huntington’s disease. Lancet (London, England) 2007;369:218–28. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- 2.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–3. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 3.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–48. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 4.von Horsten S, Schmitt I, Nguyen HP, Holzmann C, Schmidt T, Walther T, et al. Transgenic rat model of Huntington’s disease. Hum Mol Genet. 2003;12:617–24. doi: 10.1093/hmg/ddg075. [DOI] [PubMed] [Google Scholar]
- 5.Yang S-H, Cheng P-H, Banta H, Piotrowska-Nitsche K, Yang J-J, Cheng ECH, et al. Towards a transgenic model of Huntington/’s disease in a non-human primate. Nature. 2008;453:921–4. doi: 10.1038/nature06975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leitman J, Ulrich Hartl F, Lederkremer GZ. Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat Commun. 2013;4:2753. doi: 10.1038/ncomms3753. [DOI] [PubMed] [Google Scholar]
- 7.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–10. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 8.Ramdzan YM, Nisbet RM, Miller J, Finkbeiner S, Hill AF, Hatters DM. Conformation sensors that distinguish monomeric proteins from oligomers in live cells. Chem Biol. 2010;17:371–9. doi: 10.1016/j.chembiol.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller J, Arrasate M, Brooks E, Libeu CP, Legleiter J, Hatters D, et al. Identifying polyglutamine protein species in situ that best predict neurodegeneration. Nat Chem Biol. 2011;7:925–34. doi: 10.1038/nchembio.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang QC, Yeh TL, Leyva A, Frank LG, Miller J, Kim YE, et al. A compact beta model of huntingtin toxicity. J Biol Chem. 2011;286:8188–96. doi: 10.1074/jbc.M110.192013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nucifora LG, Burke KA, Feng X, Arbez N, Zhu S, Miller J, et al. Identification of novel potentially toxic oligomers formed in vitro from mammalian-derived expanded huntingtin exon-1 protein. J Biol Chem. 2012;287:16017–28. doi: 10.1074/jbc.M111.252577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters-Libeu C, Miller J, Rutenber E, Newhouse Y, Krishnan P, Cheung K, et al. Disease-associated polyglutamine stretches in monomeric huntingtin adopt a compact structure. J Mol Biol. 2012;421:587–600. doi: 10.1016/j.jmb.2012.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramdzan YM, Trubetskov MM, Ormsby AR, Newcombe EA, Sui X, Tobin MJ, et al. Huntingtin Inclusions Trigger Cellular Quiescence, Deactivate Apoptosis, and Lead to Delayed Necrosis. Cell Rep. 2017;19:919–27. doi: 10.1016/j.celrep.2017.04.029. [DOI] [PubMed] [Google Scholar]
- 14.Wetzel R. Physical chemistry of polyglutamine: intriguing tales of a monotonous sequence. J Mol Biol. 2012;421:466–90. doi: 10.1016/j.jmb.2012.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bugg CW, Isas JM, Fischer T, Patterson PH, Langen R. Structural features and domain organization of huntingtin fibrils. J Biol Chem. 2012;287:31739–46. doi: 10.1074/jbc.M112.353839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darrow MC, Sergeeva OA, Isas JM, Galaz-Montoya JG, King JA, Langen R, et al. Structural Mechanisms of Mutant Huntingtin Aggregation Suppression by the Synthetic Chaperonin-like CCT5 Complex Explained by Cryoelectron Tomography. J Biol Chem. 2015;290:17451–61. doi: 10.1074/jbc.M115.655373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Isas JM, Langen R, Siemer AB. Solid-State Nuclear Magnetic Resonance on the Static and Dynamic Domains of Huntingtin Exon-1 Fibrils. Biochemistry. 2015;54:3942–9. doi: 10.1021/acs.biochem.5b00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baias M, Smith PE, Shen K, Joachimiak LA, Zerko S, Kozminski W, et al. Structure and Dynamics of the Huntingtin Exon-1 N-Terminus: A Solution NMR Perspective. Journal of the American Chemical Society. 2017;139:1168–76. doi: 10.1021/jacs.6b10893. [DOI] [PubMed] [Google Scholar]
- 19.Masino L, Kelly G, Leonard K, Trottier Y, Pastore A. Solution structure of polyglutamine tracts in GST-polyglutamine fusion proteins. FEBS Lett. 2002;513:267–72. doi: 10.1016/s0014-5793(02)02335-9. [DOI] [PubMed] [Google Scholar]
- 20.Kim MW, Chelliah Y, Kim SW, Otwinowski Z, Bezprozvanny I. Secondary structure of Huntingtin amino-terminal region. Structure. 2009;17:1205–12. doi: 10.1016/j.str.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo Q, Bin H, Cheng J, Seefelder M, Engler T, Pfeifer G, et al. The cryo-electron microscopy structure of huntingtin. Nature. 2018;555:117–20. doi: 10.1038/nature25502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Warner JB, IV, Ruff KM, Tan PS, Lemke EA, Pappu RV, Lashuel HA. Monomeric huntingtin exon 1 has similar overall structural features for wild type and pathological polyglutamine lengths. Journal of the American Chemical Society. 2017;139:14456–69. doi: 10.1021/jacs.7b06659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klein FA, Zeder-Lutz G, Cousido-Siah A, Mitschler A, Katz A, Eberling P, et al. Linear and extended: a common polyglutamine conformation recognized by the three antibodies MW1, 1C2 and 3B5H10. Hum Mol Genet. 2013;22:4215–23. doi: 10.1093/hmg/ddt273. [DOI] [PubMed] [Google Scholar]
- 24.Owens GE, New DM, West AP, Jr, Bjorkman PJ. Anti-PolyQ Antibodies Recognize a Short PolyQ Stretch in Both Normal and Mutant Huntingtin Exon 1. J Mol Biol. 2015;427:2507–19. doi: 10.1016/j.jmb.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 26.Englander SW, Kallenbach NR. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q Rev Biophys. 1983:521–655. doi: 10.1017/s0033583500005217. [DOI] [PubMed] [Google Scholar]
- 27.Vitalis A, Wang X, Pappu RV. Atomistic Simulations of the Effects of Polyglutamine Chain Length and Solvent Quality on Conformational Equilibria and Spontaneous Homodimerization. Journal of Molecular Biology. 2008;384:279–97. doi: 10.1016/j.jmb.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lyle N, Das RK, Pappu RV. A quantitative measure for protein conformational heterogeneity. The Journal of Chemical Physics. 2013;139:121907. doi: 10.1063/1.4812791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crick SL, Jayaraman M, Frieden C, Wetzel R, Pappu RV. Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proc Natl Acad Sci U S A. 2006;103:16764–9. doi: 10.1073/pnas.0608175103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das RK, Ruff KM, Pappu RV. Relating sequence encoded information to form and function of intrinsically disordered proteins. Current Opinion in Structural Biology. 2015;32:102–12. doi: 10.1016/j.sbi.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crick SL, Ruff KM, Garai K, Frieden C, Pappu RV. Unmasking the roles of N- and C-terminal flanking sequences from exon 1 of huntingtin as modulators of polyglutamine aggregation. Proc Natl Acad Sci U S A. 2013;110:20075–80. doi: 10.1073/pnas.1320626110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dougan L, Li J, Badilla CL, Berne BJ, Fernandez JM. Single homopolypeptide chains collapse into mechanically rigid conformations. Proc Natl Acad Sci U S A. 2009;106:12605–10. doi: 10.1073/pnas.0900678106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walters RH, Murphy RM. Examining polyglutamine peptide length: a connection between collapsed conformations and increased aggregation. J Mol Biol. 2009;393:978–92. doi: 10.1016/j.jmb.2009.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Vitalis A, Wyczalkowski MA, Pappu RV. Characterizing the conformational ensemble of monomeric polyglutamine. Proteins: Structure, Function, and Bioinformatics. 2006;63:297–311. doi: 10.1002/prot.20761. [DOI] [PubMed] [Google Scholar]
- 35.Vitalis A, Lyle N, Pappu RV. Thermodynamics of <em>β</em>-Sheet Formation in Polyglutamine. Biophysical Journal. 2009;97:303–11. doi: 10.1016/j.bpj.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang H, Vázquez FX, Zhang L, Das P, Toledo-Sherman L, Luan B, et al. Emerging β-Sheet Rich Conformations in Supercompact Huntingtin Exon-1 Mutant Structures. Journal of the American Chemical Society. 2017;139:8820–7. doi: 10.1021/jacs.7b00838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Holehouse AS, Garai K, Lyle N, Vitalis A, Pappu RV. Quantitative Assessments of the Distinct Contributions of Polypeptide Backbone Amides versus Side Chain Groups to Chain Expansion via Chemical Denaturation. Journal of the American Chemical Society. 2015;137:2984–95. doi: 10.1021/ja512062h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gardner KH, Kay LE. The use of 2H, 13C, 15N multidimensional NMR to study the structure and dynamics of proteins. Annu Rev Biophys Biomol Struct. 1998;27:357–406. doi: 10.1146/annurev.biophys.27.1.357. [DOI] [PubMed] [Google Scholar]
- 39.Wishart DS, Sykes BD. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR. 1994;4:171–80. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 40.Metzler WJ, Leiting B, Pryor K, Mueller L, Farmer BT., 2nd The three-dimensional solution structure of the SH2 domain from p55blk kinase. Biochemistry (Mosc) 1996;35:6201–11. doi: 10.1021/bi960157x. [DOI] [PubMed] [Google Scholar]
- 41.Metzler WJ, Constantine KL, Friedrichs MS, Bell AJ, Ernst EG, Lavoie TB, et al. Characterization of the three-dimensional solution structure of human profilin: 1H, 13C, and 15N NMR assignments and global folding pattern. Biochemistry (Mosc) 1993;32:13818–29. doi: 10.1021/bi00213a010. [DOI] [PubMed] [Google Scholar]
- 42.Dlugosz M, Trylska J. Secondary structures of native and pathogenic huntingtin N-terminal fragments. J Phys Chem B. 2011;115:11597–608. doi: 10.1021/jp206373g. [DOI] [PubMed] [Google Scholar]
- 43.Atwal RS, Xia J, Pinchev D, Taylor J, Epand RM, Truant R. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum Mol Genet. 2007;16:2600–15. doi: 10.1093/hmg/ddm217. [DOI] [PubMed] [Google Scholar]
- 44.Bussell R, Eliezer D. Residual structure and dynamics in Parkinson’s disease-associated mutants of alpha-synuclein. J Biol Chem. 2001:45996–6003. doi: 10.1074/jbc.M106777200. [DOI] [PubMed] [Google Scholar]
- 45.Sung YH, Eliezer D. Residual structure, backbone dynamics, and interactions within the synuclein family. J Mol Biol. 2007;372:689–707. doi: 10.1016/j.jmb.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He Y, Chen Y, Mooney SM, Rajagopalan K, Bhargava A, Sacho E, et al. Phosphorylation-induced Conformational Ensemble Switching in an Intrinsically Disordered Cancer/Testis Antigen. J Biol Chem. 2015;290:25090–102. doi: 10.1074/jbc.M115.658583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin EW, Holehouse AS, Grace CR, Hughes A, Pappu RV, Mittag T. Sequence Determinants of the Conformational Properties of an Intrinsically Disordered Protein Prior to and upon Multisite Phosphorylation. Journal of the American Chemical Society. 2016;138:15323–35. doi: 10.1021/jacs.6b10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vitalis A, Pappu RV. ABSINTH: a new continuum solvation model for simulations of polypeptides in aqueous solutions. J Comput Chem. 2009;30:673–99. doi: 10.1002/jcc.21005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lyle N, Das RK, Pappu RV. A quantitative measure for protein conformational heterogeneity. J Chem Phys. 2013;139:121907. doi: 10.1063/1.4812791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vitalis A, Wang X, Pappu RV. Atomistic simulations of the effects of polyglutamine chain length and solvent quality on conformational equilibria and spontaneous homodimerization. J Mol Biol. 2008;384:279–97. doi: 10.1016/j.jmb.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Posey AE, Ruff KM, Harmon TS, Crick SL, Li A, Diamond MI, et al. Profilin reduces aggregation and phase separation of huntingtin N-terminal fragments by preferentially binding to soluble monomers and oligomers. Journal of Biological Chemistry. 2018 doi: 10.1074/jbc.RA117.000357. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olshina MA, Angley LM, Ramdzan YM, Tang J, Bailey MF, Hill AF, et al. Tracking mutant huntingtin aggregation kinetics in cells reveals three major populations that include an invariant oligomer pool. J Biol Chem. 2010;285:21807–16. doi: 10.1074/jbc.M109.084434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447–52. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sama RR, Ward CL, Kaushansky LJ, Lemay N, Ishigaki S, Urano F, et al. FUS/TLS assembles into stress granules and is a prosurvival factor during hyperosmolar stress. J Cell Physiol. 2013;228:2222–31. doi: 10.1002/jcp.24395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li YR, King OD, Shorter J, Gitler AD. Stress granules as crucibles of ALS pathogenesis. Journal of Cell Biology. 2013;201:361–72. doi: 10.1083/jcb.201302044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–67. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wear MP, Kryndushkin D, O’Meally R, Sonnenberg JL, Cole RN, Shewmaker FP. Proteins with Intrinsically Disordered Domains Are Preferentially Recruited to Polyglutamine Aggregates. PLoS One. 2015;10:e0136362. doi: 10.1371/journal.pone.0136362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Caron NS, Desmond CR, Xia J, Truant R. Polyglutamine domain flexibility mediates the proximity between flanking sequences in huntingtin. Proc Natl Acad Sci U S A. 2013;110:14610–5. doi: 10.1073/pnas.1301342110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen S, Ferrone FA, Wetzel R. Huntington’s disease age-of-onset linked to polyglutamine aggregation nucleation. Proc Natl Acad Sci U S A. 2002;99:11884–9. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vitalis A, Pappu RV. Assessing the contribution of heterogeneous distributions of oligomers to aggregation mechanisms of polyglutamine peptides. Biophys Chem. 2011;159:14–23. doi: 10.1016/j.bpc.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen M, Tsai M, Zheng W, Wolynes PG. The Aggregation Free Energy Landscapes of Polyglutamine Repeats. J Am Chem Soc. 2016;138:15197–203. doi: 10.1021/jacs.6b08665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen M, Wolynes PG. Aggregation landscapes of Huntingtin exon 1 protein fragments and the critical repeat length for the onset of Huntington’s disease. Proc Natl Acad Sci U S A. 2017;114:4406–11. doi: 10.1073/pnas.1702237114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nagai Y, Inui T, Popiel HA, Fujikake N, Hasegawa K, Urade Y, et al. A toxic monomeric conformer of the polyglutamine protein. Nat Struct Mol Biol. 2007;14:332–40. doi: 10.1038/nsmb1215. [DOI] [PubMed] [Google Scholar]
- 64.Legleiter J, Mitchell E, Lotz GP, Sapp E, Ng C, DiFiglia M, et al. Mutant huntingtin fragments form oligomers in a polyglutamine length-dependent manner in vitro and in vivo. J Biol Chem. 2010;285:14777–90. doi: 10.1074/jbc.M109.093708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mukai H, Isagawa T, Goyama E, Tanaka S, Bence NF, Tamura A, et al. Formation of morphologically similar globular aggregates from diverse aggregation-prone proteins in mammalian cells. Proc Natl Acad Sci U S A. 2005;102:10887–92. doi: 10.1073/pnas.0409283102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sathasivam K, Lane A, Legleiter J, Warley A, Woodman B, Finkbeiner S, et al. Identical oligomeric and fibrillar structures captured from the brains of R6/2 and knock-in mouse models of Huntington’s disease. Hum Mol Genet. 2010;19:65–78. doi: 10.1093/hmg/ddp467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Caron NS, Desmond CR, Xia J, Truant R. Polyglutamine domain flexibility mediates the proximity between flanking sequences in huntingtin. Proc Natl Acad Sci U S A. 2013;110:14610–5. doi: 10.1073/pnas.1301342110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen S, Berthelier V, Yang W, Wetzel R. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J Mol Biol. 2001;311:173–82. doi: 10.1006/jmbi.2001.4850. [DOI] [PubMed] [Google Scholar]
- 69.Klein FA, Pastore A, Masino L, Zeder-Lutz G, Nierengarten H, Oulad-Abdelghani M, et al. Pathogenic and non-pathogenic polyglutamine tracts have similar structural properties: towards a length-dependent toxicity gradient. J Mol Biol. 2007;371:235–44. doi: 10.1016/j.jmb.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 70.Kordasiewicz HB, Stanek LM, Wancewicz EV, Mazur C, McAlonis MM, Pytel KA, et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron. 2012;74:1031–44. doi: 10.1016/j.neuron.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell. 2015;162:1066–77. doi: 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- 72.Schwartz JC, Cech TR, Parker RR. Biochemical Properties and Biological Functions of FET Proteins. Annual review of biochemistry. 2015;84:355–79. doi: 10.1146/annurev-biochem-060614-034325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wyman J, Gill SJ. Ligand-linked phase changes in a biological system: applications to sickle cell hemoglobin. Proc Natl Acad Sci U S A. 1980;77:5239–42. doi: 10.1073/pnas.77.9.5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kar K, Jayaraman M, Sahoo B, Kodali R, Wetzel R. Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nature structural & molecular biology. 2011;18:328–36. doi: 10.1038/nsmb.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–34. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 76.Cai M, Huang Y, Sakaguchi K, Clore GM, Gronenborn AM, Craigie R. An efficient and cost-effective isotope labeling protocol for proteins expressed in Escherichia coli. J Biomol NMR. 1998;11:97–102. doi: 10.1023/a:1008222131470. [DOI] [PubMed] [Google Scholar]
- 77.Tropea JE, Cherry S, Waugh DS. Expression and Purification of Soluble His6-Tagged TEV Protease. Methods Mol Biol. 2009;498:297–307. doi: 10.1007/978-1-59745-196-3_19. [DOI] [PubMed] [Google Scholar]
- 78.Kavan D, Man P. MSTools-Web based application for visualization and presentation of HXMS data. International Journal of Mass Spectrometry. 2011;302:53–8. [Google Scholar]
- 79.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 80.Lee W, Tonelli M, Markley JL. NMRFAM-SPARKY: enhanced software for biomolecular NMR spectroscopy. Bioinformatics. 2015;31:1325–7. doi: 10.1093/bioinformatics/btu830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bahrami A, Assadi AH, Markley JL, Eghbalnia HR. Probabilistic interaction network of evidence algorithm and its application to complete labeling of peak lists from protein NMR spectroscopy. PLoS Comput Biol. 2009;5:e1000307. doi: 10.1371/journal.pcbi.1000307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee W, Westler WM, Bahrami A, Eghbalnia HR, Markley JL. PINE-SPARKY: graphical interface for evaluating automated probabilistic peak assignments in protein NMR spectroscopy. Bioinformatics. 2009;25:2085–7. doi: 10.1093/bioinformatics/btp345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, et al. Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry (Mosc) 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 84.Sethi A, Bruell S, Patil N, Hossain MA, Scott DJ, Petrie EJ, et al. The complex binding mode of the peptide hormone H2 relaxin to its receptor RXFP1. Nat Commun. 2016;7:11344. doi: 10.1038/ncomms11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.d’Auvergne EJ, Gooley PR. Optimisation of NMR dynamic models II. A new methodology for the dual optimisation of the model-free parameters and the Brownian rotational diffusion tensor. J Biomol NMR. 2008;40:121–33. doi: 10.1007/s10858-007-9213-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Das RK, Crick SL, Pappu RV. N-terminal segments modulate the alpha-helical propensities of the intrinsically disordered basic regions of bZIP proteins. J Mol Biol. 2012;416:287–99. doi: 10.1016/j.jmb.2011.12.043. [DOI] [PubMed] [Google Scholar]
- 87.Radhakrishnan A, Vitalis A, Mao AH, Steffen AT, Pappu RV. Improved atomistic Monte Carlo simulations demonstrate that poly-L-proline adopts heterogeneous ensembles of conformations of semi-rigid segments interrupted by kinks. J Phys Chem B. 2012;116:6862–71. doi: 10.1021/jp212637r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wynne JW, Woon AP, Dudek NL, Croft NP, Ng JH, Baker ML, et al. Characterization of the Antigen Processing Machinery and Endogenous Peptide Presentation of a Bat MHC Class I Molecule. J Immunol. 2016;196:4468–76. doi: 10.4049/jimmunol.1502062. [DOI] [PubMed] [Google Scholar]
- 89.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 90.Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics. 2014;13:2513–26. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–14. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 92.Vizcaíno JA, Côté RG, Csordas A, Dianes JA, Fabregat A, Foster JM, et al. The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013;41:D1063–D9. doi: 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.