

Abstract
Phosphothreonine (pThr)-embedded peptide catalysts are found to mediate the reductive amination of 3-amidocyclohexanones with divergent selectivity. Choice of peptide sequence can be used to alter the diastereoselectivity to favor either cis-product or trans-product, which are obtained in up to 93:7 er. NMR studies and DFT calculations are reported and indicate that both pathways rely on secondary interactions between substrate and catalyst to achieve selectivity. Furthermore, catalysts appear to accomplish a parallel kinetic resolution of the substrates. The facility for phosphopeptides to tune reactivity and access multiple products in reductive aminations may translate to the diversification of complex substrates, such as natural products, at numerous reactive sites.
Graphical Abstract
Introduction
Synthetic chemists have often relied on “privileged scaffolds”, in which one catalytic architecture can obtain high selectivities over diverse sets of reactions.1 Chiral phosphoric acids (CPAs, Figure 1A) represent a highly successful example of this paradigm, and CPAs relying on rigid, C2-symmetric scaffolds, such as BINOL, have become one of the most widespread and prodigious classes of asymmetric catalysts.2–5 Alternatively, enzymes have evolved to overcome the challenges associated with selective catalysis by retaining specific, catalytically active, and “privileged” residues and three-dimensional “folds,” which are then effective for a broad range of biochemical transformations.6–9 Changes to the vast number of potential amino acid sequences can facilitate subtle or drastic alterations to protein structure, molding enzymes to fit particular substrate classes, tailored to achieve site-selective modifications of individual complex molecules, and even tuned to access different types of reactivity from the same catalytic residues.10,11 Given the growing challenges faced by synthetic chemists today, exploring the development of new catalytic archetypes is advantageous, particularly ones that are tunable to selectively catalyze reactions on specific, complex substrates.
Figure 1.

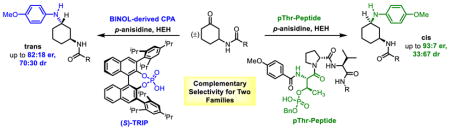
(A) Example BINOL-derived C2-symmetric chiral phosphoric acid (CPA) and pThr-embedded phosphopeptide. (B) Enantioselective transfer hydrogenaticn of 8-aminoquinoiines with pThr-containing peptide P1. (C) Regie- and Enantioselective Baeyer–Vllliger oxidation of 3-amidocyclohexanones with aspartic acid-derlved peptide 6. (D) The focus of this report, the stereoselective reductive amination of 3-amldocyclohexanones.
One such approach to asymmetric catalysis involves mimicking the breadth and selectivity of enzymes by appending minimal and distinct peptide sequences (normally <4 residues) to a conserved catalytic residue.12 The diverse peptide sequences fold in different ways, resulting in a wide variety of accessible conformations, which allow for tuning of the catalytically active residue’s reactivity and selectivity.13 Simultaneously, the functional group-rich nature of peptides chains can also offer secondary points of contact—in a sense, outer sphere interactions—to bind to substrates, differentiating diastereomeric transition state energies, and leading to good levels of selectivity.15
We envisioned that Brønsted acid catalysis could be a useful format for peptide-based catalysis, as non-C2-symmetric CPA scaffolds have been underexplored in this regime and could be broadly applicable, given the breadth of CPA-catalyzed chemistry. As phosphorylated amino acids, such as phosphothreonine, are ubiquitous in nature,14–18 we wondered whether these could be adopted in phosphoric acid catalyzed reactions. The incorporation of the pThr-monomer into the peptide chain is achieved using a HATU-mediated peptide coupling of commercially available Fmoc-pThr(Bn)-OH to various sequences, resulting in phosphopeptides with high yields and purity (Figure 1A).19
Our studies in phosphopeptide-mediated transformations began with transfer hydrogenation. BINOL-derived CPAs have previously been used in the reductions of numerous imines,20–24 enamines,25 and heterocycles,26–29 normally in the presence of Hantzsch ester (HEH) as the hydride source.30–33 Dihydropyridine HEH mimics the activity of NAD(P)H, one of nature’s most prolific hydride sources,34,35 and it was particularly compelling to combine this reductant with the bioinspired pThr-residue. As such, the transfer hydrogenation of 8-aminoquinolines (2) was pursued, and enantioselectivities as high as 94:6 er are achieved (Figure 1B).36 Notably, selectivities are equivalent to BINOL-derived CPAs, despite the increased flexibility of the phosphopeptides. The effectiveness of a non-C2-symmetric CPA-scaffold represents a fundamental departure in strategy for CPA catalyst development, as the pThr-embedded peptides appear to achieve selectivity via secondary interactions between catalyst and substrates, as opposed to forming a small, sterically hindered binding pocket. This success not only offers credence to the idea that phosphopeptides and BINOL-derived catalysts could serve as a complementary system, but also could spark curiosity in previously underexplored in vivo functions of pThr and its potential to serve as a Brønsted acid in nature. As we have expanded our purview into this area, we wished to assess the generality of pThr-containing peptides for confronting an unusual, stereochemically complicated and functional group-rich scaffold that would differ greatly from 2, and perhaps be representative of functionality found in natural products, as a model for site-selective modifications.37 In order to address this challenge, we assembled >200 unique phosphopeptides for evaluation.
Results and Discussion
Catalyst optimization
Ketones are a ubiquitous in natural products,38 and reductive aminations have previously been reported using BINOL-derived CPAs on a range of ketones.32,39,40 In a parallel line of research, our group has pursued the aspartyl-peptide-catalyzed Baeyer–Villiger oxidation (BVO) of 3-amidocyclohexanones (4, Figure 1C), with a similar goal of achieving peptide-based, divergent stereocontrol.41 The presence of a directing group on this substrate class enhances its potential to engage in secondary interactions with peptide catalysts. Accordingly, we wondered if these 3-ami-docyclohexanones could be amenable to selective reductive aminations, catalyzed by the pThr/HEH system (Figure 1D). Reactions of 4 have the potential to produce both trans- and cis-products, offering the possibility for pThr-embedded catalysts and BINOL-derived CPAs to exhibit divergent selectivities and access to different products. As such, our studies on the reductive amination of 3-amidocyclohexanones are presented herein.
The intrinsic diastereoselectivity for the reductive amination of substrate 4a with p-anisidine, in the presence of HEH, was assessed with diphenyl phosphate, a simple achiral phosphoric acid. A 69:31 dr was observed in favor of trans-7a (Table 1, entry 1), establishing this value as an intrinsic dr to which all subsequent results could be compared. Reductive amination in the presence of P1, which achieves up to 94:6 er in the transfer hydrogenation of 8-aminoquinolines, resulted in racemic product (Table 1, entry 2). Continued screening with the phosphopeptide library revealed thatP2 induces a 79:21 er for cis-7a (Table 1, entry 3), suggestive of a significant opportunity to enhance catalyst-substrate interactions. Additional screening indicated that 1,1-disubstitued residues at the i+2 position (see Figure 1A for “i+n” nomenclature)42 were unsuccessful at achieving er, and mono-substituted amino acids were next pursued. The incorporation of residues with heteroatoms for additional points of contact with substrates resulted in diminished enantioselectivity for cis-7a (Table 1, entries 4,5; er = 61:39 and 66:34) Varying the i+2 position revealed a subtle preference for larger side chains. Furthermore, an additional methylene spacer between the bulky group and the backbone results in lower er for cis-7a [Chg to Cha (Table 1, entries 6,7; er = 76:24 and 72:28), Phg to Phe (Table 1, entries 8,9; er = 74:26 and 70:30), and Ile to Leu (Table 1, entries 11,12; er = 79:21 and 77:23)]. Overall, catalysts with Tle (P2) and Val (P12) exhibited the highest selectivities (up to 83:17 er, Table 1, entry 13) and thus catalysts with these residues at the i+2 position were chosen as sequences to examine further. Of note, this catalyst proved to shift the dr in favor of the cis-product, as trans-7a/cis-7a now reached 58:42.
Table 1.

|
Reported results are the average of at minimum two trials,
Conversion to product (conv.) was determined by UPLC/MS.
Enantiomeric ratios (er) and diastereomeric ratios (dr) were determined by HPLC.
Abbreviations are presented in the supporting information.
Varying the stereochemistry at the i+1 and i+2 positions resulted in diminished er. L-stereochemistry at the i+1 position reduced the er for cis-7a to 53:47 (Table 1, entry 15). Additionally, P13, which contains D-stereochemistry at the i+2 position, retained some enantioselectivity for cis-7a (72:28), but not trans-7a (59:41, Table 1, entry 14).
Given these results, and in light of the fact that further evaluation of the i+2 and i+3 positions did not improve selectivities, we hypothesized that the catalytic residue itself is critical for binding to imine 8a. Hence, alterations closer to the N-terminus might induce enhanced selectivities. As such, the stereochemistry of the pThr residue was examined. Interestingly, only the naturally occurring Thr-stereochemistry was compatible with good selectivity, as variation of the Thr β-stereocenter (i.e., use of allo-pThr or DpThr) resulted in low enantioselectivty (Table 1, entries 16,17; er = 58:42 and 57:43). The importance of a β-methyl group was demonstrated by replacing pThr with pSer, resulting in 59:41 er for cis-7a (Table 1, entry 18).
Furthermore, substituted amido-proline catalysts P18 and P19 were examined due to the potential for offering additional points of contact for substrates binding adjacent to the pThr-residue43. Intriguingly, incorporation of a trans-amidoproline directing group overturns the enantioselectivity to favor the opposite enantiomer of cis-7a in 39:61 er (Table 1, entry 19). Alternatively, the cis-amidoproline directing group simply yields cis-7a with slightly diminished er (Table 1, entry 20; er = 71:29).
Encouraged by these variations in observed enantioselectivity with alterations near to the pThr residue, the N-terminal protecting group of the peptide was varied (Table 1, entries 21–26). Utilization of electron rich benzamide protecting groups, such as p-MeO-Bz (P24) results in selectivities as high as 91:9 er for cis-7a, and with a dr of 51:49 (Table 1, entry 25).
With these results in hand, given the lack of substantial variation in selectivities with alterations to the i+2 and i+3 positions, a truncation study on the peptide was performed, in which each residue is sequentially deleted (Table 1, entries 27–31). The C-terminus was capped both as a methyl ester, in analogy to the hit sequence, and also as a methyl amide. Moderate levels of er were maintained upon truncation, with even a minimally protected pThr-monomer yielding 75:25 er of cis-7a (Table 1, entry 31). While Fmoc-protected P27 results in even higher er (Table 1, entry 28; er = 85:15) than Fmoc-protected tetramer P12 (Table 1, entry 13; er = 83:17), these catalysts are both less selective than the elaborated tetramers. In all cases, the methyl esters performed better than the methyl amides. This indicates that the peptide conformer that leads to high levels of selectivity in this reaction does not rely on the i to i+3 β-turn, which requires an NH bond on the i+2/i+3 amide and is generally enhanced by the presence of a C-terminal amide.
Based on these observations, we designed a catalyst incorporated all of the key attributes identified in the above studies. A minimal screen was performed to establish the best pairing of the N-terminal functional group within a truncated tripeptide catalyst. Thus, P31 resulted in the highest cis-7a-selective dr, 45:55 (Table 1, entry 32). Finally, Ser(tBu) as the i+3 residue was found to slightly tune enantioselectivities for cis-7a to 93:7 (Table 1, entry 31). This selectivity represented the maximum enantioselectivity we had observed to this point, peptide P32 was identified as the hit catalyst to study additional aspects of the chemistry.
We also note some results with catalysts of dramatically different structure. These include peptide sequences that are vastly different from the hit catalyst, and also a comparative result with a venerable C2-symmetric CPA (S)-TRIP (1), which is utilized in a myriad of transformations.2 In order to target divergent selectivity with alternative peptide-based catalysts, we examined a pThr-based catalyst with the appended sequence of peptide 6, previously utilized in the BVO of 3-ami-docyclohexanones (4),41 an analogous substrate to the corresponding imine undergoing reduction in the present work. As such, peptide P33 was synthesized, wherein pThr replaces the aspartic acid catalytic residue of 6. P33 results in an entirely different selectivity profile, instead favoring trans-7a with 74:26 dr and 75:25 er (Table 1, entry 34). This result highlights how the diversity of peptide sequences appended to a distinct catalytic residue enables access to multiple products with complementary selectivity. Minimal optimization of P33 resulted in the observation of i+4 Phe-containing P34 as a slightly better performing catalyst (Table 1, entry 35, trans-7a er = 82:18, cis-7a er = 78:22). Finally, the results with catalysts P32 and P34 were compared to (S)-TRIP (1). In this case, catalyst 1a favors the formation of trans-7a with 79:21 dr and 71:29 er (Table 1, entry 34) under the conditions we employed in our study, which were not further optimized. P32 and 1 are complementary catalysts, allowing access to both diastereomers of 7a with good levels of selectivity.
Tuning of reaction conditions
While enantioselectivities as high as 93:7 can be achieved with this substrate class, conversions were normally low with 24 h reaction times. Product 7a could be obtained in higher conversion with longer reaction times, as 96 h resulted in approximately twofold increase in conversion (Table 2). The lower reactivity of this system is ascribed both to difficulties associated formation of imine 8a, in addition to catalyst inhibition induced by the basicity of the secondary amine product 7a and the pyridine byproduct of HEH.
Table 2.
Conditions Optimization.a
| ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | catalyst | time | note | ⊢erc⊣ | drd (trans:cis) | conv.b (%) | ||
| frans-7a | cis-7a | 4a | ||||||
| 1 | 9 | 96 | - | - | - | - | 61:39 | 52 |
| 2 | P32 | 96 | - | 76:24 | 91:9 | 63:37 | 38:62 | 65 |
| 3 | P34 | 96 | - | 81:19 | 80:20 | 47:53 | 60:40 | 35 |
| 4 | 1 | 96 | - | 72:28 | 47:53 | 12:88 | 71:29 | 75 |
|
| ||||||||
| 5 | 9 | 24 | NaHCO3 washe | - | - | - | 55:45 | 15 |
| 6 | P32 | 24 | NaHCO3 washe | 78:22 | 92:8 | 59:41 | 33:67 | 42 |
| 7 | P34 | 24 | NaHCO3 washe | 81:19 | 78:22 | 51:49 | 54:46 | 12 |
| 8 | 1 | 24 | NaHCO3 washe | 82:18 | 30:70 | 36:64 | 70:30 | 41 |
Reported results are the average of at minimum two trials,
Conversion to product (conv.) was determined by UPLC/MS.
Enantiomeric ratios (er) were determined by HPLC.
Diastereomeric ratio (dr) was determined by UPLC/MS.
An aqueous wash with NaHCO3 (saturated) was performed prior to analysis.
Additional optimization of conditions was performed using catalyst P32, and it was found that an aqueous wash with a saturated NaHCO3 solution prior to flash chromatography modestly perturbed the dr for these reductive aminations. With diphenyl phosphate as catalyst, the intrinsic diastereoselectivity was still found to favor trans-7a with 55:45 dr with these new conditions (Table 2, entry 5). P32 and P34 gave 33:67 and 54:46 dr respectively, increased amounts of the cis diastereomer, while maintaining similar er levels for both cis-4a and trans-4a (Table 2, entries 6–7). Alternatively, (S)-TRIP produced a similar dr value (70:30) with increased er values for both cis and trans product (82:18 and 70:30 respectively, Table 2, entry 8). We presume that catalysts may induce a secondary resolution of products through selective protonation. Peptides appear to be matched to protonate cis product preferentially, enhancing dr for cis-4a.44 Alternatively, BINOL-derived 1 instead selectively protonates the favored enantiomers of cis-4a and trans-4a, resulting in an increase in observed er for both diastereomers with an additional base wash to breakup this salt.
Assessment of directing group scope
Under optimized conditions, catalyst P32 tolerates alterations of the N-protecting group of 4 (Table 3). In addition to the previously utilized phenyl acetamide (4a, Table 3, entries 1–2), P32 was able to process benzamide-protected 4b to cis-7b with an er of 82:18, with slight perturbation of dr favoring the cis product (Table 3, entries 3–4, dr changes from 62:38 with 9 to 51:49 with P32). Cbz-protected 4c was also tolerated, and cis-7c could be obtained with an er of 86:14 and a dr of 35:65, which is more cis-selective in comparison to reactions employing (PhO)2P(O)OH (9) as catalyst (Table 3, entries 5–6, dr = 56:44). Finally, sulfonamide 4d provides the most drastic change from 4a, and is converted to cis-7d with 89:11 er and 49:51 dr (Table 3, entry 8). The intrinsic dr with achiral catalyst 9 was 68:32, once again revealing that the peptides induce significant perturbations (Table 3, entry 7).
Table 3.
Variation of 3-amido Protecting Group.a
| ||||||
|---|---|---|---|---|---|---|
| entry | substrate | catalyst | ⊢e.r.c⊣ | d.r.d (trans:cis) | conv.b (%) | |
| trans-7 | cis-7 | |||||
| 1 |
4a
|
9 | - | - | 61:39 | 52 |
| 2 | P32 | 76:24 | 91:9 | 38:62 | 65 | |
|
| ||||||
| 3 |
4b
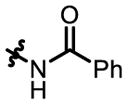
|
9 | - | - | 62:38 | 65 |
| 4 | P32 | 68:32 | 82:18 | 51:49 | 52 | |
| 5 |
4c
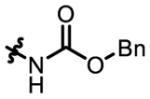
|
9 | - | - | 56:44 | 38 |
| 6 | P32 | 61:39 | 86:14 | 35:65 | 59 | |
| 7 |
4d
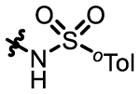
|
9 | - | - | 68:32e | 73 |
| 8 | P32 | 72:28 | 89:11 | 49:51e | 40 | |
Reported results are the average of at minimum two trials,
Conversion to product (conv.) was determined by UPLC/MS.
Enantiomeric ratios (e.r.) were determined by HPLC.
Diastereomeric ratio (dr) was determined by UPLC/MS.
Dr was determined by HPLC.
Mechanism-driven experiments
The observed diastereo- and enantioselectivity for reactions of substrates like 4 offer convenient metrics to gauge the effectiveness of the catalysts in a complex parallel kinetic resolution (see Figure 2).45.46 As ketone 4a is racemic, the catalyst must react with two enantiomers [(R)-4a and (S)-4a]. The reductive amination of each of these enantiomers therefore results in both a cis-product and a trans-product with conserved absolute stereochemistry; the overall transformation thus consists of two diastereoselective reactions. Therefore, in sum, the selectivities of these reactions can be reduced to three ratios: catalyst selectivity for (1) the reaction of (R)-4a versus (S)-4a [(kR1+kR2)/(kS1+kS2)], (2) the formation of (R)-trans-7a versus (R)-cis-7a, and (3) the formation of (S)-trans-7a versus (S)-cis-7a.
Figure 2.

(A) Percentages of each stereoisomer of 7a formed out of 100. (B) Relative rates of reactivity of each enantiomer of 4a, and relative rates of formation of each stereoisomer of 7a.
Analysis of these different rates revealed a difference in the capacity of the pThr peptide-based catalysts and either diphenyl phosphate 9 or (S)-TRIP (1) to process functionalized substrates. The crude product ratios (out of 100) are shown in Figure 2A and the relevant rates for each catalyst are shown in Figure 2B. While peptides P32 and P34 exhibit characteristics of a parallel kinetic resolution, reacting with both enantiomers of 4a at similar rates and converting each to a different diastereomer of 7a, (S)-TRIP (1) reveals a preference to mediate the reductive amination of (S)-4a operating closer to a kinetic resolution paradigm. Furthermore, while 1 only yields good selectivity for (S)-4a (1 : 3.59 ratio), which is matched with the catalyst, it is notable that P32 is able to carry out the reductive amination of both (R)-4a and (S)-4a at high levels of selectivity (1 : 6.00 and 1 : 7.12 ratio respectively). These results show the substantially different mechanisms of actions of these phosphopeptides and the BINOL-derived CPAs, and encourages the investigation of these catalytic systems as complementary scaffolds.
Consideration of the mechanistic basis of stereochemical outcomes
We next sought to understand the catalytically active conformation of P32, and how the peptide interacts with the imines en route to reaction products. The absolute configuration of enantiomerically pure product cis-7d was determined by X-ray crystallography, and crystal structures of (±)-cis-7d and (±)-trans-7d were also solved (see Supporting Information for more details), which is critical for understanding possible catalyst-substrate interactions. However, identifying either the lowest energy, or reactive conformation of the reacting cycloheximine 8 is non-trivial. Thus, we sought to assess computationally the relative energies of both the E- and Z-imines, with the 3-amido group in both the axial and equatorial orientation. DFT calculations were performed on the B3LYP/6-311+G(d,p) level to optimize all structures, and single point energy calculations were performed with M06-2×/6-311++G(2d,3p).547–49 These calculations were performed in conjunction with the IEF-PCM solvation model.50,51 As previously described for cyclohexane 10,52 as a control, the amido group was favored in the equatorial position by ΔG298K = 1.06 kcal/mol as expected (Figure 3A). Addition of the ketone moiety (4a), however, the favorability of the equatorial orientation was negligible, indicating both conformations are accessible in solution (Figure 3B). This could be the result of removing one 1,3-diaxial interaction between the 3-amido group and an axial C–H, in addition to 4a-eq having a substantially larger dipole moment than 4a-ax (Δ = 3.31 debye).
Figure 3.

(A) Calculation of A-value for cyclohexane 10. (B) Calculation of the relative energies of chair conformations of ketone 4a. (C) Calculation of the equilibrium ratios of four conformations of imine 8a. All calculations performed using M06-2×/6-311++G(2d,3p)//B3LYP/6-311+G(d,p) and an IEF-PCM solvation model in PhMe. Dipoles are reported in Debye.
We next applied this computation technique to the four potential conformations of 8a, and for ease of calculation, it was assumed that P32 does not selectively catalyze the imine formation, and that all imine isomers are in equilibrium. E-8a-ax was found to be the most stable imine by a small margin; however, all imines are energetically accessible in the reaction mixture (Figure 3C). We hypothesized that the directing group in an equatorial position could be more favorable with regard to steric considerations. Due to the orientation of the benzamide group, it is possible that an attractive CH–π interaction with the anisidine arene results in some stabilization (Figure 4A). E-8a-eq, alternatively, cannot geometrically access this CH–π interaction (Figure 4B). Peptide catalysts are therefore able to navigate a complex mixture of equilibrating imines (both E and Z, and axial and equatorial isomers, for both enantiomers of 4a; potentially producing eight imines), yielding orthogonally protected 1,3-diamine products with high selectivities.
Figure 4.

Calculated, lowest energy structure of (A) E-8a-ax and (B) E-8a-eq.
With some understanding of the substrate conformational profile in hand, we turned our attention to studying the conformational issues associated with the catalysts by NMR spectroscopy. One challenge associated with NMR analysis of the phosphopeptides is their propensity to exhibit multiple conformations and tautomers.40 Indeed, the pThr residue itself can theoretically adopt a number of intramolecular H-bonding interactions between the phosphoric acid proton, the phosphate O- atoms, and any of the amides on the peptides, in addition to the number of secondary structures the peptide backbone can adopt. Catalyst P25, which shows comparable selectivities to hit catalyst P32 (see Table 1, entries 26 and 33) was chosen for this study as the 1H NMR stretches were well-resolved.
Upon assignment of the full proton NMR spectrum of P25, 2D NOESY experiments were conducted. At similar concentration to peptide-mediated reactions (6.0 mM), 14 inter-residue NOE correlations were observed and are shown in Figure 5A. NOEs, shown in green, orange, and blue, point to a preferred orientation for the pThr residue. The three magenta NOE contacts in Figure 5B suggest contacts between the NHVal and the protons of the α-C–H of Thr, and C–H on the bottom face of Pro. These contacts are possible if this N–H bond has rotated underneath the Pro residue. This places NHVal in close proximity to both C=OThr (green), and the C=OPG (blue), and could indicate competitive formation of a NHVal–C=OThr γ-turn and a β-turn involving the NHVal–C=OPG. (This possible turn is notable in that it is one residue shifted from the proverbial C=OThr–NHLeu β-turn). This hypothesis is further validated with the observation of NOE contacts between a furanoyl C–H bond and the Val methine C–H (Figure 5B, red). This possible conformation is consistent with results of the catalytic reactions involving truncation or changes to the i+3 position. This residue, in the observed solution conformation, may only partially assist to orient the NHi+2. The incorporation of more electron-rich N-terminal protecting groups, such as p-MeO-Bz or furanoyl-groups, could enforce this turn by either favoring the more Lewis basic C=OPG to act as an intramolecular H-bond acceptor, or by disfavoring NHThr from serving as an intramolecular H-bond donor.
Figure 5.

(A) 1H–1H NOESY NMR of P25 revealed 14 inter-residue NOE correlations. Red: evidence of NHval–C=OThr γ-turn NHval–C=OPG β-turn. Magenta: flexibility of NHval to rotate beneath Pro. Orange: relating to ψ–angle of pThr. Blue: relating to side-chain dihedral angle of pThr. Green: correlation between phosphoric acid and NHThr, (B) Proposed structure of P25, resulting from an equilbirum between a γ-turn and β-turn. (C–D) Mechanistic models for formation of cis- and trans- products, which incorporate the above hybrid γ/β-turn. Putative H-bonding interactions are shown in magenta.
With these considerations in mind, mechanistic hypotheses for formation of both trans-product and cis-product may be advanced, and are presented in Figures 5C and 5D. First, a β-turn between NHVal–C=OPG is proposed as the secondary structure for peptide catalysts, in accord with our experimental observations. As DFT calculations indicated the accessibility of multiple chair conformations of 8a in solution, we envision that E-8a-eq could interact most productively, in a Curtin-Hammett fashion, with P32 as shown. Given the importance of the 3-amido directing group, it is likely that the basic imine lone pair is oriented on the same side as the directing group, implying that E-imines may be favored over Z-imines, facilitating secondary points of contact. While E-8a-eq is not the lowest energy conformation of 8a, it is energetically accessible, and presents a sterically accessible environment upon protonation by P32. E-8a-ax, of course, could also be processed, but our analysis includes the intuitive assumption that reactions of the axial conformations should be highly cis-selective, which we do not observe. Thus, returning to models involving E-8a-eq, the NHThr is oriented directly towards the phenyl acetamide’s C=O to engage in an H-Bond (Figures 5C and 5D). Intriguingly, this interaction is possible for both enantiomers of 8a, albeit presenting the opposite imine face towards the bifunctionally activated HEH. This results in one enantiomer of 8a being processed to the trans-product, and one to cis-product, in accord with results under the optimized conditions. Hence, the high selectivities in this parallel kinetic resolution could be the result of flexibility in the peptide structure to accommodate two dissimilar substrates in the same binding pocket.
Conclusion
In conclusion, we have reported the application of pThr-embedded peptides to the reductive amination of 3-amidocy-clohexanones. The phosphopeptides tolerate a range of N-protecting groups on the substrates and are able to overturn the inherent trans selectivity of this reaction to achieve up to 93:7 er and 35:65 dr for cis-product. Hit catalyst, P32, adopts an intriguing secondary structure and is believed to engage in secondary interactions with both enantiomers of starting material, and processes each to a different diastereomer of product through a parallel kinetic resolution. We are hopeful that the study of the pThr-catalyzed reductive aminations of small molecules will translate to advances in the site-selective modification of ketone-containing natural products.
EXPERIMENTAL SECTION
General Experimental Methods
Room temperature is defined as 21–23 °C. All reagents were purchased from commercial sources and used as received unless otherwise noted. Solvents used for reactions and for azeotropic drying, such as toluene (PhMe), methylene chloride (CH2Cl2), N,N-dimethylformamide (DMF), and tetrahydrofuran (THF), were obtained from a Seca Solvent Purification System by Glass Countour, in which the solvents were dried over alumina and dispended under an atmosphere of argon. For all other purposes, solvents were used as received from commercial sources unless otherwise noted.
1H NMR spectra were recorded on 400 MHz, 500 MHz, or 600 MHz Agilent spectrometers at ambient temperature. Samples were prepared in chloroform-d (CDCl3), dimethyl sulfoxide-d6 (d6-DMSO), methanol-d4 (CD3OD), or benzene-d6 (C6D6). 1H NMR data are reported as chemical shifts with multiplicity, coupling constants (J) in Hz, and integrations. Proton chemical shifts are reported in ppm (δ) and referenced to tetramethylsilane (TMS) or residual solvent (CHCl3, δ 7.26 ppm, DMSO δ 2.50 ppm, CD3OH δ 3.31 ppm, C6H6 δ 7.16 ppm).57 Multiplicity is reported as follows: singlet (s), broad singlet (bs), doublet (d), broad doublet (bd), doublet of doublets (dd), broad triplet (bt), doublet of doublet of doublets (ddd), doublet of doublet of doublet of doublets (dddd), doublet of doublet of doublet of triplets (dddt), doublet of doublet of triplets (ddt), doublet of triplets (dt), doublet of triplet of doublets (dtd), doublet of triplets of triplets (dtt), doublet of pentets (dp), triplet (t), triplet of doublets (td), triplet of triplets (tt), triplet of doublet of doublets (tdd), triplet of doublet of triplets (tdt), triplet of triplet of doublets (ttd), quartet (q), multiplet (m), and overlapping multiplets (comp). 13C NMR spectra were recorded on 500 (126) MHz or 600 (151) MHz Agilent spectrometers with complete proton decoupling at ambient temperature, unless otherwise noted. Carbon chemical shifts are reported in ppm (δ) and referenced to tetramethylsilane (TMS) or solvent (CDCl3, δ 77.16 ppm, d6-DMSO δ 39.52 ppm, CD3OD δ 49.00 ppm, C6D6 δ 128.06 ppm).53 31P NMR spectra were recorded on 500 MHz Agilent spectrometers with complete proton decoupling at ambient temperature.
Low resolution mass spectrometry (MS) was acquired on a Waters SQD2 UPLC-MS equipped with an electrospray ionization (ESI) detector, a QToF mass spectrometer, and a photodiode array detector, with Waters ACQUITY UPLC BEH C18 (1.7 μm, 2.1 × 50 mm) and Waters CORTECS UPLC C18 (1.6 μm, 3.0 × 50 mm) columns. High resolution mass spectrometry (HRMS) was conducted by the Mass Spectrometry Laboratory at the University of Illinois at Urbana-Champaign using a Waters Synapt G2-Si instrument equipped with a QToF mass spectrometer, an ESI detector. Elemental Analysis (EA) was conducted by Robinson Microlit Laboratories (Ledgewood, NJ).
Infrared spectra were obtained using a Nicolet ATR/FT-IR spectrometer, and νmax (cm−1) were partially recorded in accordance with convention. Optical rotations were recorded on a Perkin Elmer Polarimeter 341 at the sodium D line (1.0 dm path length) at 20 °C. Analytical thin-layer chromatography (TLC) was performed using EMD Millipore silica gel 60 F254 precoated plates (0.25 mm thickness). The developed plates were visualized by a UV lamp and/or potassium permanganate (KMnO4) stain.
Normal-phase column chromatography was performed with either silica gel 60 Å (32–63 microns) or with a Biotage Isolera One flash purification system equipped with 15 g, 30 g, 60 g, or 120 g SNAP Ultra HP-Sphere 25 μm columns using an appropriate linear gradient of EtOAc/Hexanes. Reversed-phase column chromatography as performed with a Biotage Isolera One flash purification system equipped with 30 g, 60 g, or 120 g SNAP KP-C18-HS or SNAP Ultra-C18 columns with an appropriate gradient of MeCN/H2O.
Enantiomeric ratio (er) values were acquired using an Agilent 1100 series analytical chiral HPLC equipped with a photodiode array detector (210 nm, 230 nm, 250 nm, and 254 nm) with Chiralpak IC, IB, OD-H, and AD-H columns (5 μm particle size, 4.5 × 250 mm).
Representative Synthesis and Characterization of Peptide P12 (For P1–P31, General Procedure #1)
See Scheme S1. Peptide Coupling #1: To a flame dried 100 mL round bottom flask equipped with a stir bar was added HCl•Leu-OMe (1.36 g, 7.5 mmol, 1.0 equiv.), Boc-Val-OH (1.63 g, 7.5 mmol, 1.0 equiv.), EDC•HCl (1.63 g, 8.3 mmol, 1.1 equiv.), and HOBt•H2O (1.63 g, 8.3 mmol, 1.1 equiv.). To the solid mixture was added CH2Cl2 (30 mL), followed by Hunig’s Base (958 μL, 16.5 mmol, 2.2 equiv.). The resulting solution was stirred overnight for 12–18 h, upon which the reaction was transferred to a separatory funnel and the organics were washed with NaHCO3 (30 mL, saturated aqueous), 10% citric acid (50 mL, aqueous), and brine (50 mL, saturated, aqueous). The organics were collected, dried over Na2SO4, filtered, and concentrated in vacuo to yield 2.63 g of Boc-Val-Leu-OMe as a white solid (98% yield), the identity of which was confirmed by UPLC-MS. MS (ESI-QToF) m/z: [M+Na]+ Calcd for C17H32N2O5Na 367.22; Found 367.22.
Deprotection #1
A 250 mL round bottom flask was charged with Boc-Val-Leu-OMe (2.36 g, 7.5 mmol, 1.0 equiv.) and a stirbar, and then capped with a septum pierced with a Teflon cannula leading to a solution of NaHCO3 (saturated, aqueous). HCl (7.5 mL, 4N in 1,4-dioxane) was then added, and the resulting solution was stirred vigorously for 1 h. After completion of the reaction, Nitrogen gas was vigorously bubbled through the solution using a needle until all solvent was removed, revealing HCl•H-Val-Leu-OMe as a crude white solid. The solid was dried under vacuum for a 1 h and was used in the next step without further characterization or purification.
Peptide Coupling #2
To the 250 mL round bottom flask containing HCl•H-Val-Leu-OMe was added Boc-DPro-OH (1.60 g, 7.5 mmol, 1.0 equiv.), EDC•HCl (1.63 g, 8.3 mmol, 1.1 equiv.), and HOBt•H2O (1.63 g, 8.3 mmol, 1.1 equiv.). To the solid mixture was added CH2Cl2 (30 mL), followed by Hunig’s Base (958 μL, 16.5 mmol, 2.2 equiv.). The resulting solution was stirred overnight for 12–18 h, upon which the reaction was transferred to a separatory funnel and the organics were washed with NaHCO3 (30 mL, saturated, aqueous), 10% citric acid (50 mL, aqueous), and brine (50 mL, saturated aqueous). The organics were collected, dried over Na2SO4, filtered, and concentrated in vacuo to yield 3.26 g of Boc-DPro-Val-Leu-OMe as a white solid (98% yield), the identity of which was confirmed by UPLC-MS. MS (ESI-QToF) m/z: [M+Na]+ Calcd for C22H39N3O6Na 464.27; Found 464.37.
Deprotection #2
A 250 mL round bottom flask was charged with Boc-DPro-Val-Leu-OMe (593 mg, 1.34 mmol, 1.1 equiv.) and a stirbar, and then capped with a septum pierced with a Teflon cannula leading to a solution of NaHCO3 (saturated, aqueous). HCl (4.0 mL, 4N in 1,4-dioxane) was then added, and the resulting solution was stirred vigorously for 1 h. After completion of the reaction, Nitrogen gas was vigorously bubbled through the solution using a needle until all solvent was removed, revealing HCl•H-DPro-Val-Leu-OMe as a crude white solid. The solid was dried under vacuum overnight for 24 and was used in the next step without further characterization or purification.
Peptide Coupling #3
To the 25 mL round bottom flask containing HCl•H-DPro-Val-Leu-OMe was added Fmoc-pThr(Bn)-OH (625 mg, 1.22 mmol, 1.0 equiv.). After cooling the solid mixture was to 10 °C using a brine/ice bath, CH2Cl2 (7 mL) was added. To the mixture was added 4-methylmorpholine (NMM, 470 μL, 4.27 mmol, 3.5 equiv.), followed by HATU (557 mg, 1.47 mmol, 1.2 equiv.). The round bottom was capped with a septum and placed under a balloon of Ar and the reaction was stirred for 18 h. Upon completion of the reaction, the solution was transferred to a separatory funnel, diluted with CH2Cl2 (10 mL), and washed with 10% citric acid (25 mL, aqueous). The organic layer was separated and the aqueous layer extracted with CH2Cl2 (25 mL). The combined organics were washed with brine (50 mL, saturated aqueous), dried over Na2SO4, filtered, and concentrated in vacuo to yield a pale yellow solid. The crude product was purified via automatic reverse phase flash chromatography with a gradient of 10% to 100% MeCN/H2O with a 0.1% formic acid buffer solution, resulting in 874 mg P12 as a white solid (86% yield).
Representative Synthesis of Peptide P26 (Procedure #2)
See Scheme S2. The same procedure as shown in Peptide Coupling #1 was followed, instead using methylamine hydrochloride (1.0 equiv.) as the amine source with Boc-Val-OH as the carboxylic acid. These couplings were performed on 2.5 mmol scale (wrt S2).
Synthesis of Peptide P30 (Procedure #3)
See Scheme S3. The same procedure as shown in Peptide Coupling #3 was followed, instead using dimethylamine hydrochloride (1.1 equiv.) as the amine source and Fmoc-pThr(Bn)-OH as the carboxylic acid. This coupling was performed on 1.00 mmol scale (wrt S6).
Fmoc-allo-pThr(Bn)-OH (for P15) and Cbz-pThr(Bn)-OH (for P21) were synthesized according to literature precedent.54
Representative Synthesis of Fmoc-pThr(Bn)-DPro-Val-Ser(tBu)-OMe (precursor to P32) by Solid Phase Synthesis (for P32–P34, General Procedure #4)
See Scheme S4. Peptide Coupling #1: A 20 mL solid phase synthesis tube was charged with HCl•Ser(tBu)-2-Cl-Trt-resin (1.1 meq/g, 1.00 g, 1.10 mmol, 1.0 equiv.) and 20 mL CH2Cl2. The vessel was rotated for 30 min to swell the resin and drained upon completion. Simultaneously, a 20 mL vial was charged with Fmoc-Val-OH (1.12 g, 3.30 mmol, 3.0 equiv.), HCTU (2.28 g, 5.50 mmol, 5.0 equiv.), Cl-HOBt (930 mg, 5.50 mmol, 5.0 equiv.) followed by 17 mL NMP and then NMM (1.20 mL, 11.0 mmol, 10.0 equiv.) was next added. This solution was added to the resin (rinsing the vial with 1 mL NMP). The vessel was rotated to homogeneity for 5 h. Upon completion of the reaction, the vessel was drained and washed with DMF (20 mL × 3) and CH2Cl2 (20 mL × 3), agitating to homogeneity each time. Upon draining the final wash, the resin was taken onto the next step without any further purification.
Deprotection #1
The resin was suspended in 20% v/v piperidine/DMF and rotated for 20 min. Upon draining the reaction vessel, the resin was washed with DMF (20 mL × 3) and CH2Cl2 (20 mL × 3), agitating to homogeneity each time. Upon draining the final wash, the resin was taken onto the next step without any further purification.
Peptide Coupling #2
The same reaction conditions as in Peptide Coupling #1 were utilized, except using Fmoc-DPro-OH (1.86 g, 5.50 mmol, 5.0 equiv.) as the amino acid. The reaction was also rotated overnight for 12 h.
Cleavage
The resin was suspended in 20 mL 4:1:1 CH2Cl2/trifluroethanol/AcOH and rotated for 30 min. Upon completion of the reaction, the vessel was drained and the supernatant collected. The resin was further washed with CH2Cl2 (20 mL × 3) agitating to homogeneity each time, and the combined organics were concentrated in vacuo to yield 287 mg Fmoc-DPro-Val-Ser(tBu)-OH as a white solid (45% yield, 4 steps). The cleaved peptide was taken onto the next step without any further purification
Esterification
To a 250 mL round bottom flask containing Fmoc-DPro-Val-Ser(tBu)-OH (287 mg, 0.495 mmol, 1.0 equiv.) was added EDC•HCl (190 mg, 0.990, 2.0 equiv.), HOBt•H2O (152 mg, 0.990 mmol, 2.0 equiv.), and 7.0 mL MeOH. The reaction was stirred overnight. After 18 h, the reaction was concentrated in vacuo and redissolved in 30 mL CH2Cl2. The organics were washed with NaHCO3 (30 mL, saturated, aqueous), 10% citric acid (30 mL, aqueous), and brine (30 mL, saturated, aqueous). The organics were dried over Na2SO4, filtered, and concentrated in vacuo. The crude solid was purified via automatic reverse phase flash chromatography, eluting with a gradient of 40% to 100% MeCN/H2O to yield Fmoc-DPro-Val-Ser(tBu)-OMe as a white solid.
Deprotection #2
To a flame dried 20 mL vial was added Fmoc-DPro-Val-Ser(tBu)-OMe (388 mg, 0.823 mmol, 1.1 equiv.) and 3.6 mL 1:1 di-ethylamine/CH2Cl2. The reaction for stirred for 30 min and then concentrated in vacuo. The crude solid was dried in vacuo overnight.
Peptide Coupling #3
To the 20 mL vial was added Fmoc-pThr(Bn)-OH (383 mg, 0.748 mmol, 1.0 equiv.). After cooling the solid mixture was to −10 °C using a brine/ice bath, CH2Cl2 (4.8 mL) was added. To the mixture was added NMM (470 μL, 4.27 mmol, 3.5 equiv.), followed by HATU (557 mg, 1.47 mmol, 1.2 equiv.). The vial was flushed with Ar, capped, and stirred for 18 h. Upon completion of the reaction, the solution was transferred to a separatory funnel, diluted with CH2Cl2 (10 mL), and washed with 10% citric acid (25 mL, aqueous). The organic layer was separated and the aqueous layer extracted with CH2Cl2 (25 mL). The combined organics were washed with brine (50 mL, saturated aqueous), dried over Na2SO4, filtered, and concentrated in vacuo to yield a pale yellow solid. The crude product was purified via automatic reverse phase flash chromatography with a gradient of 10% to 100% MeCN/H2O with a 0.1% formic acid buffer solution, resulting in 440 mg peptide as a white solid (69% yield).
Variation of N-Terminal Protecting Group (General Procedure #5)
See Scheme S5-A. This procedure was used for peptides P22 (utilizing p-nitrobenzoic acid as the carboxylic acid), P23 (benzoic acid), P24 (p-methoxybenzoic acid), and P25 (2-furanoic acid). To an oven dried 8 mL scintillation vial was added Fmoc-pThr(Bn)-DPro-Val-Leu-OMe (P12, 200 mg, 0.240 mmol, 1.0 equiv.), followed by 1.2 mL of a 1:1 solution of diethylamine and CH2Cl2. The solution was stirred for 1 h and then concentrated in vacuo. The crude white solid was dried under vacuum overnight for 24 h and was used in the next step without further analysis or purification. To the 8 mL scintillation vial containing Et2NH2•H-pThr(Bn)-DPro-Val-Leu-OMe was added relevant carboxylic acid (0.288 mmol, 1.2 equiv.), followed by CH2Cl2 (2.0 mL). The solution was cooled to −10 °C using a brine/ice bath. To the mixture was added NMM (66 μL, 0.600 mmol, 2.5 equiv.), followed by HATU (118 mg, 0.311 mmol, 1.3 equiv.). The vial was flushed with Ar, capped with a teflon cap, and the reaction was stirred for 18 h. Upon completion of the reaction, the solution was transferred to a separatory funnel, diluted with CH2Cl2 (5.0 mL), and washed with 10% citric acid (15 mL, aqueous). The organic layer was separated and the aqueous layer extracted with CH2Cl2 (15 mL). The combined organics were washed with brine (30 mL, saturated aqueous), dried over Na2SO4, filtered, and concentrated in vacuo to yield a pale yellow or white solid. The crude product was purified via automatic reverse phase flash chromatography with a gradient of 10% to 100% MeCN/H2O with a 0.1% formic acid buffer solution, resulting in product.
Acylation of N-deprotected Phosphopeptide for P20 (Procedure #6)
See Scheme S5-B. To an oven dried 8 mL scintillation vial was added Fmoc-pThr(Bn)-DPro-Val-Leu-OMe (P12, 200 mg, 0.240 mmol, 1.0 equiv.), followed by 1.2 mL of a 1:1 solution of diethylamine and CH2Cl2. The solution was stirred for 1 h and then concentrated in vacuo. The crude white solid was dried under vacuum overnight for 24 h and was used in the next step without further analysis or purification. To the 8 mL scintillation vial containing Et2NH2•H-pThr(Bn)-DPro-Val-Leu-OMe was added CH2Cl2 (2.0 mL). The solution was cooled to −10 °C using a brine/ice bath, followed by addition of acetic anhydride (340 μL, 0.360 mmol, 1.50 equiv.). The vial was flushed with Ar, capped with a teflon cap, and the reaction was stirred for 18 h. Upon completion of the reaction, the solution was transferred to a separatory funnel, diluted with CH2Cl2 (5.0 mL), and washed with 10% citric acid (15 mL, aqueous). The organic layer was separated and the aqueous layer extracted with CH2Cl2 (15 mL). The combined organics were washed with brine (30 mL, saturated aqueous), dried over Na2SO4, filtered, and concentrated in vacuo to yield a pale yellow or white solid. The crude product was purified via automatic reverse phase flash chromatography with a gradient of 10% to 100% MeCN/H2O with a 0.1% formic acid buffer solution, resulting in product.
Synthesis of 4-Amidoproline Substituted Peptides (Procedures #7 and #8)
See Schemes S6 and S7. 1-(tert-butyl) 2-methyl (2R,4S)-4-azidopyrrolidine-1,2-dicarboxylate (trans-11) and 1-(tert-butyl) 2-methyl (2R,4R)-4-azidopyrrolidine-1,2-dicarboxylate (cis-11) were synthesized according to literature precedent.55,56
1-(tert-butyl) 2-methyl (2R,4S)-4-(3-phenylureido)pyrrolidine-1,2-di-carboxylate (trans-12) was synthesized according to literature precedent.61 A 100 mL round bottom flask was charged with trans-11 (550 mg, 2.00 mmol, 1.00 equiv.), followed by 10 mL MeOH. To this solution was added Pd/C (60 mg). The vessel was fitted with a septum and placed under a balloon of H2. The reaction was stirred at 40 °C for 12 h. Upon completion, the reaction was passed through a plug of celite and concentrated in vacuo. The crude product was utilized in the next step without further purification assuming quantitative yield. To a 100 mL round bottom flask containing trans-12 (489 mg, 2.00 mmol, 1.0 equiv.) was added 15 mL THF. The vessel was fitted with a septum and placed under an Ar balloon. Phenyl isocyanate (240 μL, 2.20 mmol, 1.1 equiv.) was next added, and the reaction was stirred at RT for 12 h. Upon completion, the solution was washed with 10% citric acid (20 mL, aqueous) and brine (20 mL, saturated, aqueous). The organics were dried over Na2SO4, filtered, concentrated in vacuo to yield crude product. The product was purified via automatic normal phase flash chromatography, eluting with a gradient of 30% to 100% EtOAc/Hex, followed by automatic normal phase flash chromatography, eluting with a gradient of 20% to 100% MeCN/H2O to yield 253 mg pure, white solid (35% yield, 2 steps). 1H NMR (500 MHz, CDCl3): δ 8.31 (d, J = 14.0 Hz, 1H), 7.33 (d, J = 7.4 Hz, 2H), 7.23–7.14 (m, 2H), 6.86 (t, J = 7.4 Hz, 1H), 6.46 (dd, J = 10.3, 6.9 Hz, 1H), 4.28–4.13 (m, 2H), 3.64 (s, 3H), 3.60–3.50 (m, 1H), 3.22–3.10 (m, 1H), 2.49–2.44 (m, 2H), 2.23–2.00 (m, 2H), 1.34 (s, 9H); 13C NMR (151 MHz, d6-DMSO): δ 173.0, 172.5, 154.8, 153.5, 152.8, 140.1, 128.7, 121.3, 117.7, 79.2, 57.5, 57.2, 52.0, 51.6, 48.2, 47.66, 36.01, 34.97, 28.04, 27.87. HRMS (ESI-QToF) m/z [M+H]+ Calcd for C18H26N3O5 364.1872; Found364.1855. IR: (cm−1, neat): 3330, 2979, 1746, 1698, 1648, 1598, 1548, 1499, 1394, 1313, 1238, 1203, 1177, 1154, 1125, 890, 854, 750; [α]D20 23.1° (c = 0.0060 g/mL, MeOH); TLC: Rƒ (1:2 Hexanes/EtOAc) 0.42, visualized with UV light.
(2R,4S)-1-(tert-butoxycarbonyl)-4-(3-phenylureido)pyrrolidine-2-carboxylic acid (trans-13) was synthesized according to literature precedent.16 A flame dried 20 mL scintillation vial was charged with trans-12 (140 mg, 0.385 mmol, 1.0 equiv.), 4.0 mL THF, and 1.5 mL H2O. The vial was cooled to 0 °C and LiOH (35.0 mg, 0.826 mmol, 2.0 equiv.) was added. The reaction was warmed to RT and allowed to stir for 2h. Upon completion of the reaction, the solution was washed with 10% citric acid (20 mL, aqueous). The aqueous layer was extracted with CH2Cl2 (30 mL × 2). The combined organics were dried over Na2SO4, filtered, and concentrated in vacuo to reveal 135 mg pure, white solid without need for further purification (<99% yield). 1H NMR (400 MHz, CD3OD): δ 7.31 (d, J = 7.8 Hz, 2H), 7.22 (t, J = 7.8 Hz, 2H), 6.95 (t, J = 7.3 Hz, 1H), 4.42–4.24 (comp, 2H), 3.77–3.68 (m, 1H), 3.36–3.31 (m, 1H), 2.31–2.18 (m, 2H), 1.44 (s, 9H); 13C NMR (151 MHz, d6-DMSO, mixture of rotamers): δ 174.0, 173.5, 171.3, 154.9, 154.8, 154.7, 153.5, 153.1, 140.2, 140.1, 128.7, 121.3, 117.7, 117.6, 79.1, 57.6, 57.3, 52.0, 51.6, 48.2, 48.1, 47.6, 47.6, 47.6, 47.5, 36.2, 36.1, 35.2, 35.1, 28.1, 28.0; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C17H24N3O5 350.1716; Found 350.1703; IR (cm−1, neat): 2970, 1733, 1649, 1622, 1472, 1427, 1366, 1313, 1203, 1160, 1142, 1002, 909, 894, 854, 801, 764, 740, 696. [α]D20 13.8° (c = 0.0098 g/mL, MeOH); TLC: Rf (9:1 CH2Cl2/MeOH) 0.13, visualized with UV light.
1-(tert-butyl) 2-methyl (2R,4R)-4-(3-phenylureido)pyrrolidine-1,2-di-carboxylate (cis-12) was synthesized according to literature precedent.61 A flame dried 100 mL round bottom flask was charged with cis-11 (550 mg, 2.00 mmol, 1.00 equiv.), followed by 10 mL MeOH. To this solution was added Pd/C (60 mg). The vessel was fitted with a septum and placed under a balloon of H2. The reaction was stirred at 40 °C until complete. The reaction was passed through a plug of celite and concentrated in vacuo. The crude product was utilized in the next step without further purification assuming quantitative yield. To a 100 mL round bottom flask containing trans-12 (489 mg, 2.00 mmol, 1.0 equiv.) was added mL CH2Cl2. The vessel was fit with a septum and placed under an atmosphere of Ar. Phenyl isocyanate (326 μL, 3.00 mmol, 1.50 equiv.) was then added slowly, and the resulting solution was stirred overnight for 18 h. Upon completion of the reaction, the solution was washed with 10% citric acid (60 mL, aqueous), followed by brine (60 mL, saturated, aqueous). The organics were dried over Na2SO4, filtered, and concentrated in vacuo. The resulting crude material was first purified by automatic normal phase flash chromatography eluting with a gradient of 25% to 100% EtOAc/Hex. Additional purification using automatic reverse phase flash chromatography, eluting with a gradient of 25% to 100% MeCN/H2O was required, and yielded 430 mg white solid (59 % yield). 1H NMR (600 MHz, CDCl3): δ 7.36–7.28 (comp, 4H), 7.05 (t, J = 6.7 Hz, 1H), 6.64 (d, J = 34.4 Hz, 1H), 5.82 (d, J = 69.1 Hz, 1H), 4.57 (s, 1H), 4.31 (ddd, J = 49.6, 9.7, 2.7 Hz, 1H), 3.80–3.57 (comp, 4H), 3.51 (dd, J = 46.8, 11.5 Hz, 1H), 2.49 (dddd, J = 24.8, 13.8, 9.8, 6.1 Hz, 1H), 1.99 (dd, J = 30.2, 13.8 Hz, 1H), 1.43 (s, 9H); 13C NMR (151 MHz, CDCl3, mixture of rotamers): δ 175.4, 174.9, 155.0, 154.5, 153.8, 138.6, 129.3, 129.1, 123.7, 123.7, 120.6, 120.3, 80.8, 58.0, 57.8, 53.9, 53.1, 52.8, 52.5, 49.9, 48.9, 37.2, 36.0, 28.5, 28.4; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C18H26N3O5 364.1872; Found 364.1867; IR (cm−1, neat): 2978, 1646, 1598, 1468, 1393, 1247, 1158, 1129, 1001, 899, 854, 753, 693. [α]D20 14.6° (c = 0.010 g/mL, MeOH); TLC: Rf (1:2 Hexanes/EtOAc) 0.46, visualized with UV light.
(2R,4R)-1-(tert-butoxycarbonyl)-4-(3-phenylureido)pyrrolidine-2-carboxylic acid (cis-13) was synthesized according to literature precedent.16 A flame dried 20 mL scintillation vial was charged with cis-12 (150 mg, 0.413 mmol, 1.0 equiv.), 4.0 mL THF, and 1.5 mL H2O. The vial was cooled to 0 °C and LiOH (35.0 mg, 0.826 mmol, 2.0 equiv.) was added. The reaction was warmed to RT and allowed to stir for 2.5 h. Upon completion of the reaction, the solution was washed with 10% citric acid (20 mL, aqueous). The aqueous layer was extracted with CH2Cl2 (30 mL × 2). The combined organics were dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was partially purified by automatic reverse phase flash chromatography, eluting with a gradient of 25% to 100% MeCN/H2O, to reveal 127 mg of mostly pure white solid that was taken onto the next step without further purification (88% yield). 1H NMR (400 MHz, CD3OD): δ 7.38–7.29 (m, 2H), 7.20 (t, J = 7.8 Hz, 2H), 6.93 (t, J = 7.3 Hz, 1H), 4.38–4.25 (m, 1H), 4.23–4.14 (m, 1H), 3.77–3.67 (m, 1H), 3.30–3.27 (m, 1H), 2.61–2.42 (m, 1H), 2.03–1.89 (m, 1H), 1.42 (s, 9H); HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C17H24N3O5 350.1716; Found 350.1706.
Synthesis of pThr-Embedded Peptides
Fmoc-pThr(Bn)-DPro-Acpc-Met-OMe (P1)
Synthesized using General Procedure #1 on 0.600 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 502 mg white solid (63% yield); 31P NMR (162 MHz, CDCl3) δ −0.20; MS (ESI-QToF) m/z: [M+H]+ Calcd for C41H50N4O11PS 837.2934; Found 837.2916.
Fmoc-pThr(Bn)-DPro-Tle-Leu-OMe (P2)
Synthesized using General Procedure #1 on 0.606 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 221 mg white solid (43% yield); 31P NMR (162 MHz, CDCl3): δ −1.75; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C44H58N4O11P 849.3840; Found 849.3821.
Fmoc-pThr(Bn)-DPro-Trp-Leu-OMe (P3)
Synthesized using General Procedure #1 on 0.780 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 296 mg white solid (51% yield); 31P NMR (162 MHz, CDCl3): δ −1.43; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C49H57N5O11P 922.3792; Found 922.3770.
Fmoc-pThr(Bn)-DPro-Thr(Me)-Leu-OMe (P4)
Synthesized using General Procedure #1 on 0.648 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 228 mg white solid (38% yield); 31P NMR (202 MHz, CDCl3): δ −1.13; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C42H54N4O12P 837.3476; Found 837.3466.
Fmoc-pThr(Bn)-DPro-Chg-Leu-OMe (P5)
Synthesized using General Procedure #1 on 0.648 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 407 mg white solid (71% yield); 31P NMR (162 MHz, CDCl3): δ −1.32; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C46H60N4O11P 875.3996; Found 875.3979.
Fmoc-pThr(Bn)-DPro-Cha-Leu-OMe (P6)
Synthesized using General Procedure #1 on 0.648 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 379 mg white solid (66% yield); 31P NMR (162 MHz, CDCl3): δ −0.70; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C47H62N4O11P 889.4153; Found 889.4127.
Fmoc-pThr(Bn)-DPro-Phg-Leu-OMe (P7)
Synthesized using General Procedure #1 on 0.648 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 347 mg white solid (62% yield); 31P NMR (202 MHz, CDCl3): δ −0.40; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C46H54N4O11P 869.3527; Found 869.3510.
Fmoc-pThr(Bn)-DPro-Phe-Leu-OMe (P8)
Synthesized using General Procedure #1 on 0.648 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 210 mg white solid (37% yield); 31P NMR (162 MHz, CDCl3): δ −1.30; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C47H56N4O11P 883.3683; Found 883.3661.
Fmoc-pThr(Bn)-DPro-Ala-Leu-OMe (P9)
Synthesized using General Procedure #1 on 0.648 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 309 mg white solid (59% yield); 31P NMR (162 MHz, CDCl3): δ −1.25; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C41H52N4O11P 807.3370; Found 807.3362.
Fmoc-pThr(Bn)-DPro-Ile-Leu-OMe (P10)
Synthesized using General Procedure #1 on 0.648 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 424 mg white solid (77% yield); 31P NMR (162 MHz, CDCl3): δ −0.33; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C44H58N4O11P 849.3840; Found 849.3827.
Fmoc-pThr(Bn)-DPro-Leu-Leu-OMe (P11)
Synthesized using General Procedure #1 on 0.648 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 406 mg white solid (56% yield); 31P NMR (162 MHz, CDCl3): δ −0.68; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C44H58N4O11P 849.3840; Found 849.3829.
Fmoc-pThr(Bn)-DPro-Val-Leu-OMe (P12)
Synthesized using General Procedure #1 on 1.22 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 874 mg white solid (86% yield); 31P NMR (162 MHz, CDCl3): δ −0.79; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C43H56N4O11P 835.3683; Found 835.3671.
Fmoc-pThr(Bn)-DPro-DTle-Leu-OMe (P13)
Synthesized using General Procedure #1 on 0.648 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 223 mg white solid (41% yield); 31P NMR (162 MHz, CDCl3): δ −1.44; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C44H58N4O11P 849.3840; Found 849.3833.
Fmoc-pThr(Bn)-Pro-DTle-Leu-OMe (P14)
Synthesized using General Procedure #1 on 0.648 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 378 mg white solid (69% yield); 31P NMR (162 MHz, CDCl3): δ −2.66; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C44H58N4O11P 849.3840; Found 849.3826.
Fmoc-allo-pThr(Bn)-DPro-Val-Leu-OMe (P15)
Synthesized using General Procedures #1 on 0.293 mmol scale wrt Fmoc-allo-pThr(Bn)-OH. Yield: 210 mg white solid (86% yield); 31P NMR (162 MHz, CDCl3): δ −0.74; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C43H56N4O11P 835.3683; Found 835.3671.
Fmoc-DpThr(Bn)-DPro-Val-Leu-OMe (P16)
Synthesized using General Procedure #1 on 0.196 mmol scale wrt Fmoc-DpThr(Bn)-OH. Yield: 108 mg white solid (66% yield); 31P NMR (162 MHz, CDCl3): δ −2.01; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C43H56N4O11P 835.3683; Found 835.3678.
Fmoc-pSer(Bn)-DPro-Val-Leu-OMe (P17)
Synthesized using General Procedure #1 on 0.606 mmol scale wrt Fmoc-pSer(Bn)-OH. Yield: not reported; 31P NMR (162 MHz, CDCl3): δ −1.59; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C42H54N4O11P 821.3527; Found 821.3520.
Fmoc-pThr(Bn)-DPro(4-trans-Ph-urea)-Val-Leu-OMe (P18)
Synthesized using General Procedures #1 and #7 on 0.614 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 440 mg white solid (73% yield); 31P NMR (162 MHz, CDCl3): δ −1.81; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C51H64N6O12P 983.4320; Found 983.4301.
Fmoc-pThr(Bn)-DPro(4-cis-Ph-urea)-Val-Leu-OMe (P19)
Synthesized using General Procedures #1 and #8 on 0.244 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 188 mg white solid (78% yield); 31P NMR (202 MHz, CDCl3): δ −0.60; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C51H64N6O12P 983.4320; Found 983.4320.
Ac-pThr(Bn)-DPro-Val-Leu-OMe (P20)
Synthesized using General Procedures #1 and #6 on 0.240 mmol scale wrt P11. Yield: 56.5 mg white solid (36% yield); 31P NMR (202 MHz, CDCl3): δ −1.68; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C30H48N4O10P 655.3108; Found 655.3109.
Cbz-pThr(Bn)-DPro-Val-Leu-OMe (P21)
Synthesized using General Procedures #1 on 0.228 mmol scale wrt P11. Yield not reported; 31P NMR (202 MHz, CDCl3): δ −1.84; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C36H52N4O11P 747.3370; Found 747.3364.
p-O2N-Bz-pThr(Bn)-DPro-Val-Leu-OMe (P22)
Synthesized using General Procedures #1 and #5 on 0.400 mmol scale wrt P11. Yield: 72.2 mg white solid (39% yield); 31P NMR (162 MHz, CDCl3) δ −0.54; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C35H49N5O12P 762.3115; Found 762.3118.
Bz-pThr(Bn)-DPro-Val-Leu-OMe (P23)
Synthesized using General Procedures #1 and #5 on 0.400 mmol scale wrt P11. Yield: 143 mg white solid (50% yield); 31P NMR (162 MHz, CDCl3): δ 0.01; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C35H50N4O10P 717.3265; Found 717.3254.
p-MeO-Bz-pThr(Bn)-DPro-Val-Leu-OMe (P24)
Synthesized using General Procedures #1 and #5 on 0.240 mmol scale wrt P11. Yield: 150 mg white solid (84% yield); 31P NMR (162 MHz, CDCl3): δ −1.05; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C36H52N4O11P 747.3370; Found 747.3365.
Furanoyl-pThr(Bn)-DPro-Val-Leu-OMe (P25)
Synthesized using General Procedures #1 and #5 on 0.400 mmol scale wrt P11. Yield: 136 mg white solid (48% yield); 1H NMR (600 MHz, Benzene-d6): δ 8.70 (bs, 1H), 7.82 (bd, J = 8.6 Hz, 1H), 7.49 (s, 1H), 7.31–7.27 (m, 2H), 7.19 (d, J = 3.5 Hz, 1H), 7.11–7.06 (m, 2H), 7.06–7.00 (m, 2H), 5.93 (dd, J = 3.5, 1.7 Hz, 1H), 5.15–5.05 (comp, 3H), 4.90 (td, J = 8.6, 5.8 Hz, 1H), 4.82 (t, J = 6.1 Hz, 1H), 4.68 (d, J = 8.3 Hz, 1H), 4.53 (t, J = 8.5 Hz, 1H), 3.89 (bt, J = 7.7 Hz, 1H), 3.30 (s, 3H), 3.20–3.12 (m, 1H), 2.94 (bs, 1H), 2.44–2.37 (m, 1H), 2.17 (dt, J = 13.8, 4.8 Hz, 1H), 1.99 (q, J = 10.5, 9.7 Hz, 1H), 1.87–1.80 (m, 1H), 1.80–1.73 (m, 0H), 1.73–1.64 (m, 2H), 1.50 (ddt, J = 12.3, 9.2, 5.2 Hz, 1H), 1.33 (d, J = 6.2 Hz, 3H), 1.06 (d, J = 6.7 Hz, 3H), 0.96 (d, J = 6.7 Hz, 3H), 0.93 (d, J = 6.6 Hz, 3H), 0.89 (d, J = 6.6 Hz, 3H). 31P NMR (162 MHz, CDCl3): δ −0.58; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C33H48N4O11P 707.3057; Found 707.3046.
Fmoc-pThr(Bn)-DPro-Val-NHMe (P26)
Synthesized using General Procedures #1 and #2 on 1.47 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 997 mg white solid (94% yield); 31P NMR (162 MHz, CDCl3): δ −1.16; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C37H46N4O9P 721.3002; Found 721.2991.
Fmoc-pThr(Bn)-DPro-Val-OMe (P27)
Synthesized using General Procedure #1 on 2.77 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 1.63 g white solid (82% yield); 31P NMR (162 MHz, CDCl3): δ −0.86; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C37H45N3O10P 722.2843; Found 722.2836.
Fmoc-pThr(Bn)-DPro-NHMe (P28)
Synthesized using General Procedures #1 and #2 on 2.16 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 1.07 g white solid (80% yield); 31P NMR (162 MHz, CDCl3): δ −0.41; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C32H37N3O8P 622.2318; Found 622.2316.
Fmoc-pThr(Bn)-DPro-OMe (P29)
Synthesized using General Procedure #1 on 0.250 mmol scale wrt Fmoc-pThr(Bn)-OH (32% yield); 31P NMR (202 MHz, CDCl3): δ −0.06; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C32H36N2O9P 623.2158; Found 623.2150.
Fmoc-pThr(Bn)-NHMe (P30)
Synthesized using General Procedures #3 on 1.00 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 353 mg white solid (66% yield); 31P NMR (162 MHz, CDCl3): δ −1.51; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C28H32N2O7P 539.1947; Found 539.1945.
p-MeO-Bz-pThr(Bn)-DPro-Val-OMe (P31)
Synthesized using General Procedures #1 and #5 on 0.240 mmol scale wrt P25. Yield: 62.6 mg white solid (41% yield); 31P NMR (162 MHz, CDCl3): δ −0.37; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C30H41N3O10P 634.2530; Found 634.2526.
p-MeO-Bz-pThr(Bn)-DPro-Val-Ser(tBu)-OMe (P32)
Synthesized using General Procedures #4 and #5 on 0.173 mmol scale wrt Fmoc-pThr(Bn)-DPro-Val-Ser(tBu)-OMe. Yield: 440 mg white solid (69% yield); 31P NMR (162 MHz, CDCl3): δ −0.27; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C37H54N4O12P 777.3476; Found 777.3488.
Fmoc-pThr(Bn)-Pro-DLys(Boc)-DPro-Tyr(tBu)-OMe (P33)
Synthesized using General Procedure #4 on 0.433 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 339 mg white solid (67% yield); 31P NMR (162 MHz, CDCl3): δ −2.26; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C61H80N6O15P 1167.5419; Found 1167.5402.
Fmoc-pThr(Bn)-Pro-DLys(Boc)-DPro-Phe-OMe (P34)
Synthesized using General Procedure #4 on 0.621 mmol scale wrt Fmoc-pThr(Bn)-OH. Yield: 429 mg white solid (57% yield); 31P NMR (162 MHz, CDCl3): δ −2.13; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C57H72N6O14P 1095.4844; Found 1095.4833.
Synthesis of 3-Amidocyclohexanone Substrates (4)
Tert-butyl (3-oxocyclohexyl)carbamate (14) was synthesized according to literature precedent (Procedure #9, see Scheme S8.).56 A flame dried 100 mL round bottom flask was charged with tert-butyl carbamate (11.7 g, 100 mmol, 1.0 equiv.) and bismuth nitrate pentahydrate (9.70 g, 20.0 mmol, 0.20 equiv.), followed by addition of cyclohexenone (9.68 mL, 100 mmol, 1.0 equiv.). The neat solution was stirred vigorously until formation of a pale yellow solid inhibited stirring. The crude solid was suspended in CH2Cl2 and partially purified by flash chromatography, eluting with a gradient of 2:1 to 1:4 Hex/EtOAc, revealing 14.5 g of pale yellow, mostly pure solid (68% yield), which was used in the next step without further purification. 1H NMR (400 MHz, CDCl3): δ 4.68–4.31 (m, 1H), 3.86 (s, 1H), 2.63 (ddt, J = 14.1, 4.6, 1.6 Hz, 1H), 2.32 (dddd, J = 13.6, 6.7, 3.0, 1.4 Hz, 1H), 2.25–2.15 (m, 2H), 2.07–1.87 (m, 2H), 1.73–1.47 (m, 2H), 1.39 (m, 9H); TLC: Rf (1:1 Hex/EtOAc) 0.47, visualized using KMNO4 stain.
Variation of N-Protecting Group of 3-Amidocylohexanones (General Procedure #10)
See Scheme S9. To a 250 mL round bottom flask containing 14 (1.0 equiv.) was added CH2Cl2 (1.0 mL/mmol 14), followed by trifluoroacetic acid (1.0 mL/mmol 14). The flask was fitted with a septum pierced with a cannula leading to a solution of NaHCO3 (saturated, aqueous). The solution was allowed to stir for 1 h. Upon completion of the deprotection, N2 was blown into the solution via a needle inlet. After 1 h, the remaining solution was concentrated in vacuo, using a base trap connected to the round bottom filled with KOH to neutralize any volatile TFA. After drying under reduced pressure for a few hours, the vessel was charged with CH2Cl2 (6.0 mL/mmol 14) and cooled to 0 °C with an ice bath. Electrophile (1.2 equiv.) was then added and the flask was fitted with a septum and pierced with an argon balloon. After this, Hünig’s base (2.4 equiv.) was added, and the reaction was allowed to stir overnight, slowly warming to rt. Upon completion of the reaction, the solution was transferred to a separatory funnel and washed with NaHCO3 (6.0 mL/mmol 14, saturated, aqueous) and 10% citric acid (6.0 mL/mmol 14, aqueous), dried over Na2SO4, filtered, and concentrated in vacuo. The crude material was purified via flash chromatography.
N-(3-oxocyclohexyl)-2-phenylacetamide (4a)
General Procedure #10 was used on 7.00 mmol scale (wrt 14). Phenylacetyl chloride (1.12 mL, 8.44 mmol, 1.2 equiv.) was used as the electrophile. The crude material was purified via flash chromatography, using a gradient eluent of 2:1 to 1:1 Hex/EtOAc, yielding 625 mg 4a as a white solid (38% yield). 1H NMR (400 MHz, CDCl3): δ 7.36–7.19 (comp, 5H), 5.37 (bd, J = 7.5 Hz, 1H), 4.19 (dddd, J = 13.1, 8.8, 6.9, 4.0 Hz, 1H), 3.53 (s, 3H), 2.65–2.56 (m, 1H), 2.37–2.28 (m, 1H), 2.23–2.09 (m, 2H), 2.04–1.95 (m, 1H), 1.88–1.77 (m, 1H), 1.68 (dddd, J = 15.2, 13.7, 6.8, 3.5 Hz, 1H), 1.54 (dtd, J = 13.2, 9.5, 3.7 Hz, 1H); 13C NMR (151 MHz, CDCl3): δ 208.5, 170.2, 134.6, 129.3, 129.1, 127.5, 48.5, 47.4, 43.8, 40.8, 30.6, 22.0; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C14H18NO2 232.1338; Found 232.1335; IR: (cm−1, neat) 3255, 3080, 2934, 1714, 1635, 1557, 1495, 1454, 1421, 1345, 1267, 1221, 1174, 1032.4, 978, 903, 717; TLC: Rf (1:4 Hexanes/EtOAc) 0.32, visualized with a KMnO4 stain; HPLC: Chiralpak IC column, 1.0 mL/min, 15% EtOH/Hex, 254 nm and 210 nm, peaks observed at 16.2 and 22.8 min.
N-(3-oxocyclohexyl)benzamide (4b)
General Procedure #10 was used on 14.0 mmol scale (wrt 14). Benzoyl chloride (2.20 mL, 16.8 mmol, 1.2 equiv.) was used as the electrophile. The crude material was purified via flash chromatography, using a gradient eluent of 4:1 to 0:1 Hex/EtOAc, yielding 1.27 g of mostly pure 4b as a beige solid. The mixture was further purified using automatic normal phase flash chromatography, eluting with a gradient of 0 to 100% EtOAc/Hex to yield 992 mg 4b as a beige solid (33% yield). 1H NMR (400 MHz, CDCl3): δ 7.73–7.68 (m, 2H), 7.52–7.42 (m, 1H), 7.38 (t, J = 7.5 Hz, 1H), 6.31 (d, J = 8.4 Hz, 1H), 4.46–4.34 (m, 1H), 2.75 (ddt, J = 14.1, 4.8, 1.6 Hz, 1H), 2.43–2.32 (m, 2H), 2.26 (dddd, J = 16.3, 8.9, 5.6, 1.4 Hz, 1H), 2.19–2.10 (m, 1H), 2.04–1.94 (m, 1H), 1.83–1.69 (m, 2H); 13C NMR (151 MHz, CDCl3): δ 208.8, 166.8, 134.2, 131.6, 128.6, 126.9, 49.0, 47.6, 40.9, 30.8, 22.2; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C13H16NO2 218.1181; Found 218.1182; IR: (cm−1, neat): 3323, 2960, 2937, 1709, 1634, 1580, 1525, 1490, 1452, 1325, 1311, 1290, 1270, 1221, 1151, 1070, 1051, 1029, 802, 721; TLC: Rf (1:1 Hexanes/EtOAc) 0.2, visualized with a KMnO4 stain.
Benzyl (3-oxocyclohexyl)carbamate (4c)
General Procedure #10 was used on 17.8 mmol scale (wrt 14). Benzyl chloroformate (3.0 mL, 21.4 mmol, 1.2 equiv.) was used as the electrophile. The crude material was purified via flash chromatography, using a gradient eluent of 4:1 to 1:1 Hex/EtOAc, yielding 3.33 g 4c as a white solid (35% yield). 1H NMR (600 MHz, CDCl3): δ 7.32–7.23 (comp, 5H), 5.44 (bd, J = 8.5 Hz, 1H), 5.10 (s, 2.5H, major conformer), 5.02 (d, J = 2.3 Hz, 0.5H, minor conformer), 3.89 (s, 1H), 2.65–2.55 (m, 1H), 2.33–2.09 (comp, 3H), 2.05–1.95 (m, 1H), 1.94–1.85 (m, 1H), 1.65–1.47 (comp, 2H); 13C NMR (151 MHz, CDCl3): δ 209.1, 157.1, 155.4, 136.4, 128.5, 128.5, 128.1, 128.1, 128.0, 66.7, 50.1, 47.8, 40.7, 30.9, 21.8; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C14H18NO3 248.1287; Found 248.1282; IR: (cm−1, neat): 3332, 2949, 1715, 1686, 1528, 1453, 1284, 1266, 1246, 1215, 1142, 1083, 1034, 1024, 971, 844, 764, 726; TLC: Rf (1:1 Hexanes/EtOAc) 0.52, visualized with a KMnO4 stain.
4-methyl-N-(3-oxocyclohexyl)benzenesulfonamide (4d)
General Procedure #10 was used on 8.90 mmol scale (wrt 14). p-Toluenesulfonyl chloride (2.04 g, 10.7 mmol, 1.2 equiv.) was used as the electrophile. The crude material was purified via flash chromatography, using a gradient eluent of 2:1 to 1:1 Hex/EtOAc, yielding 821 mg 4d as a white solid (35% yield). 1H NMR (400 MHz, CDCl3): δ 7.75 (d, J = 8.3 Hz, 2H), 7.31 (d, J = 7.8 Hz, 2H), 4.90 (bd, J = 7.4 Hz, 1H), 3.59–3.49 (m, 1H), 2.50 (ddt, J = 14.2, 4.8, 1.7 Hz, 1H), 2.45 (s, 3H), 2.36–2.26 (m, 1H), 2.26–2.14 (comp, 2H), 2.06–1.90 (m, 2H), 1.70–1.54 (comp, 2H); 13C NMR (151 MHz, CDCl3): δ 208.0, 143.7, 137.5, 129.9, 126.9, 52.4, 48.5, 40.6, 31.9, 21.7, 21.5; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C13H18NO3S 268.1007; Found 268.0999; IR: (cm−1, neat): 3232, 2957, 1701, 1596, 1446, 1327, 1230, 1150, 1112, 1086, 1022, 1010, 906, 818, 694;; TLC: Rf (1:1 Hexanes/EtOAc) 0.42, visualized with a KMnO4 stain.
Synthesis and Isolation of Racemic 1,3-Diamine Products 7 (General Procedure #11)
To a flame dried 20 mL scintillation vial was added 4 (0.400 mmol, 1.0 equiv.), p-anisidine (62.0 mg, 0.500 mmol, 1.25 equiv.), Hantzsch ester (152 mg, 0.600 mmol, 1.5 equiv.), diphenyl phosphate (50.0 mg, 0.200 mmol, 0.50 equiv.), and calcium sulfate (4.0 g, 100 g/mmol 2), followed by 10.0 mL solvent (see below). The vial’s headspace was flushed with Ar, capped with a Teflon cap, and stirred for a designated amount of time at either RT or 40 °C. The crude mixtures were next filtered through a fine fritted funnel to remove the CaSO4, washing with additional CH2Cl2 and EtOAc. The solution was concentrated in vacuo, redissolved in a minimal amount of CH2Cl2 and purified via automatic normal phase flash chromatography (specific conditions listed below). Further purification was required via preparative HPLC, using a Waters SymmetricPrep C8 7 μm (19 × 300 mm) column. Gradient conditions with H2O (0.1% formic acid) and MeCN (0.1% formic acid) were utilized as follows: Held at 15% MeCN for 5 min, ramped to 20% MeCN for 5 min, ramped to 22% MeCN for 30 min, ramped to 35% MeCN for 10 min, ramped to 95% MeCN for 5 min, ramped to 5% MeCN for 1 min, held at 5% MeCN for 5 min. The relevant fractions were collected and concentrated in vacuo. The crude material was then redissolved in 20 mL CH2Cl2 and washed with NaHCO3 (20 mL, saturated, aqueous). The aqueous layer was extracted with 30 mL CH2Cl2. The combined organics were dried over Na2SO4, filtered, and concentrated in vacuo.
Trans-3-((4-methoxyphenyl)amino)cyclohexyl)-2-phenylacetamide (trans-7a)
See Scheme S10. General Procedure #11 was used, with CHCl3 as the solvent, 40 °C as the temperature, and for 96 h. Five individual reactions were performed in tandem and combined prior to purification. The crude material was purified by automatic normal phase flash chromatography, eluting with a gradient of 40% to 100% EtOAc/Hex, yielding 524 mg of a mixture of trans-7a, cis-7a, and 4a. The three compounds were further separated by preparative HPLC to yield 79.9 mg light brown solid (11% combined yield). 1H NMR (600 MHz, CDCl3): δ 7.33–7.30 (comp, 2H), 7.29 –7.23 (m, 1H), 7.21 (d, J = 7.1 Hz, 2H), 6.74 (d, J = 8.8 Hz, 2H), 6.52 (d, J = 8.8 Hz, 2H), 5.38 (bd, J = 8.3 Hz, 1H), 3.87 (tdt, J = 11.7, 8.1, 4.0 Hz, 1H), 3.72 (s, 3H), 3.51 (s, 2H), 3.22 (tt, J = 10.8, 3.8 Hz, 1H), 2.94 (s, 1H), 2.26 (bd, J = 12.5 Hz, 1H), 2.01 (d, J = 13.0 Hz, 1H), 1.89 (d, J = 11.2 Hz, 1H), 1.75 (dp, J = 14.7, 3.7 Hz, 1H), 1.44–1.34 (m, 1H), 1.01–0.92 (m, 2H), 0.83 (q, J = 11.4 Hz, 1H); 13C NMR: (151 MHz, CDCl3): δ 170.0, 152.2, 140.9, 134.9, 129.4, 129.0, 127.3, 115.0, 114.9, 55.8, 51.9, 47.3, 43.9, 39.7, 32.8, 32.4, 22.6; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C21H27N2O2 339.2073; Found 339.2063; IR: (cm−1, neat): 3365, 3321, 2934, 2865, 1646, 1528, 1511, 1461, 1353, 1240, 1182, 1035, 964, 893, 819, 762, 709; Mp: 124–125 °C; TLC: Rf (1:4 Hexanes/EtOAc) 0.60, visualized with UV light. HPLC: Chiralpak IC, 1.0 mL/min, 15% EtOH/Hex, 254 nm and 210 nm, peaks observed at 10.5 and 11.8 min.
Cis-3-((4-methoxyphenyl)amino)cyclohexyl)-2-phenylacetamide (cis-7a)
See Scheme S10. General Procedure #11 was used, with CHCl3 as the solvent, 40 °C as the temperature, and for 96 h. Five individual reactions were performed in tandem and combined prior to purification. The crude material was purified by automatic normal phase flash chromatography, eluting with a gradient of 40% to 100% EtOAc/Hex, yielding 524 mg of a mixture of trans-7a, cis-7a, and 4a. The three compounds were further separated by preparative HPLC to yield 49.8 mg light brown solid (7% combined yield). 1H NMR (600 MHz, CDCl3): δ 7.36 (t, J = 7.5 Hz, 2H), 7.30 (t, J = 7.4 Hz, 1H), 7.25 (d, J = 6.8 Hz, 2H), 6.73 (d, J = 8.8 Hz, 2H), 6.44 (d, J = 8.7 Hz, 2H), 5.50 (d, J = 7.8 Hz, 1H), 4.16–4.08 (m, 1H), 3.72 (s, 3H), 3.55 (s, 2H), 3.28 (s, 1H), 3.17 (s, 1H), 1.74–1.53 (m, 5H), 1.43–1.29 (m, 3H); 13C NMR (151 MHz, CDCl3): δ 170.2, 152.0, 141.1, 135.1, 129.3, 129.1, 127.4, 114.9, 114.6, 55.8, 48.7, 44.8, 44.0, 37.0, 31.1, 31.0, 19.9; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C21H27N2O2 339.2073; Found 339.2068; IR: (cm−1, neat): 3212, 2931, 2856, 1635, 1541, 1513, 1456, 1352, 1230, 1175, 1033, 821, 726; Mp: 116–118 °C; TLC: Rf (1:4 Hexanes/EtOAc) 0.60, visualized with UV light; HPLC: Chiralpak IC, 1.0 mL/min, 15% EtOH/Hex, 254 nm and 210 nm, peaks observed at 14.5 and 19.9 min.
Trans-N-(3-((4-methoxyphenyl)amino)cyclohexyl)benzamide (trans-7b)
See Scheme S11. General Procedure #11 was used, with PhMe as the solvent, at room temperature, and for 144 h. The crude material was purified by automatic normal phase flash chromatography, eluting with a gradient of 5% to 100% EtOAc/Hex, yielding a mixture of trans-7b, cis-7b and 4b. The three compounds were further separated by preparative HPLC to yield 25.0 mg beige solid (19% yield). 1H NMR (400 MHz, CDCl3): δ 7.75 (d, J = 6.9 Hz, 2H), 7.53–7.46 (m, 1H), 7.43 (t, J = 7.5 Hz, 2H), 6.79–6.75 (m, 2H), 6.63–6.56 (m, 2H), 6.11 (bd, J = 7.7 Hz, 1H), 4.41–4.31 (m, 1H), 3.73 (s, 3H), 3.67–3.58 (m, 1H), 1.99–1.81 (comp, 3H), 1.75 (ddt, J = 13.6, 8.8, 3.9 Hz, 2H), 1.66–1.55 (m, 2H), 1.54–1.43 (m, 1H); 13C NMR (151 MHz, CDCl3): δ 167.0, 152.2, 141.2, 135.0, 131.5, 128.7, 127.0, 115.1, 114.9, 55.9, 49.2, 45.3, 37.3, 31.9, 30.7, 20.1; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C20H25N2O2 325.1916; Found 325.1908; Mp: 118–124 °C; TLC: Rf (1:2 Hexanes/EtOAc) 0.60, visualized with UV light; HPLC: Chiralpak IC, 1.0 mL/min, 20% iPrOH/Hex, 254 nm, peaks observed at 20.4 and 32.7 min.
Cis-N-(3-((4-methoxyphenyl)amino)cyclohexyl)benzamide (cis-7b)
See Scheme S11. General Procedure #11 was used, with PhMe as the solvent, at room temperature, and for 144 h. The crude material was purified by automatic normal phase flash chromatography, eluting with a gradient of 5% to 100% EtOAc/Hex, yielding a mixture of trans-7b, cis-7b and 4b. The three compounds were further separated by preparative HPLC to yield 14.8 mg beige solid (11% yield). 1H NMR (600 MHz, Chloroform-d): δ 7.74–7.69 (m, 2H), 7.50–7.45 (m, 1H), 7.43–7.39 (m, 2H), 6.80–6.76 (m, 2H), 6.65–6.60 (m, 2H), 6.25 (s, 1H), 4.12 (dddt, J = 14.9, 11.3, 8.1, 3.8 Hz, 1H), 3.74 (s, 3H), 3.36 (ddt, J = 10.6, 7.7, 3.8 Hz, 1H), 2.45 (d, J = 11.8 Hz, 1H), 2.15–2.04 (m, 2H), 1.87 (dt, J = 13.9, 4.0 Hz, 1H), 1.51 (dddd, J = 15.9, 14.2, 7.9, 3.6 Hz, 1H), 1.30–1.21 (m, 1H), 1.21–1.10 (comp, 2H); 13C NMR (151 MHz, CDCl): δ 166.7, 152.7, 140.8, 134.9, 131.5, 128.7, 127.0, 115.6, 115.1, 55.9, 52.3, 47.8, 39.6, 33.0, 32.7, 22.6; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C20H25N2O2 325.1916; Found 325.1913; Mp: 144–147 °C; TLC: Rf (1:2 Hexanes/EtOAc) 0.70, visualized with UV light; HPLC: Chiralpak OD-H column, 1.0 mL/min, 20% iPrOH/Hex, 254 nm, peaks observed at 20.9 and 55.2 min.
Trans -Benzyl (3-((4-methoxyphenyl)amino)cyclohexyl)carbamate (trans-7c)
See Scheme S12. General Procedure #11 was used, with CH2Cl2 as the solvent, at room temperature, and for 120 h. The crude material was purified by automatic normal phase flash chromatography, eluting with a gradient of 15% to 100% EtOAc/Hex, yielding a mixture of trans-7c, cis-7c and 4c. The three compounds were further separated by preparative HPLC to yield 20.9 mg yellow oil (14% yield). 1H NMR (400 MHz, CDCl3): δ 7.42–7.28 (comp, 5H), 6.77 (d, J = 8.8 Hz, 2H), 6.57 (d, J = 8.6 Hz, 2H), 5.10 (s, 2H), 4.79 (bd, J = 7.9 Hz, 1H), 3.94 (s, 1H), 3.74 (s, 3H), 3.51 (s, 1H), 3.32 (bs, 1H), 1.88–1.60 (comp, 5H), 1.60–1.36 (comp, 3H); 13C NMR (151 MHz, CDCl3): δ 155.7, 152.3, 141.1, 136.6, 128.7, 128.3, 128.3, 115.1, 115.0, 66.8, 55.9, 49.0, 46.6, 37.5, 31.8, 31.1, 10.0; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C21H27N2O3 355.2022; Found 355.2018; TLC: Rf (2:1 Hexanes/EtOAc) 0.30, visualized with UV light; HPLC: Chiralpak IC column, 1.0 mL/min, 10% EtOH/Hex, 254 nm, peaks observed at 14.1 and 17.0 min.
Cis-Benzyl (3-((4-methoxyphenyl)amino)cyclohexyl)carbamate (cis-7c)
See Scheme S12. General Procedure #11 was used, with CH2Cl2 as the solvent, at room temperature, and for 120 h. The crude material was purified by automatic normal phase flash chromatography, eluting with a gradient of 15% to 100% EtOAc/Hex, yielding a mixture of trans-7c, cis-7c and 7c. The three compounds were further separated by preparative HPLC to yield 3.5 mg yellow oil (2% yield). 1H NMR (400 MHz, CDCl3): δ 7.39–7.29 (comp, 5H), 6.78 (d, J = 8.9 Hz, 2H), 6.68 (d, J = 8.4 Hz, 2H), 5.08 (s, 2H), 4.67 (d, J = 8.1 Hz, 1H), 4.01 (s, 1H), 3.75 (s, 3H), 3.58 (d, J = 12.3 Hz, 1H), 3.24 (s, 1H), 2.40 (d, J = 12.1 Hz, 1H), 2.12–1.96 (comp, 2H), 1.82 (dt, J = 14.1, 3.6 Hz, 1H), 1.48–1.34 (m, 1H), 1.14–0.94 (comp, 3H); 13C NMR (151 MHz, CDCl3): δ 163.5, 155.6, 153.4, 136.6, 128.7, 128.3, 128.3, 116.7, 115.1, 66.8, 55.9, 53.5, 49.3, 40.0, 33.0, 32.4, 22.9; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C21H27N2O3 355.2022; Found 355.2012; TLC: Rf (2:1 Hexanes/EtOAc) 0.23, visualized with UV light; HPLC: Chiralpak IC column, 1.0 mL/min, 10% EtOH/Hex, 254 nm, peaks observed at 20.1 and 26.6 min.
Trans-N-(3-((4-methoxyphenyl)amino)cyclohexyl)-4-methylben-zenesulfonamide (trans-7d) with CH2Cl2 as the solvent, at room temperature, and for 96 h. See Scheme S13. General Procedure #11 was used. The crude material was purified by automatic normal phase flash chromatography, eluting with a gradient of 10% to 100% EtOAc/Hex, yielding a peak with trans-7d, and a second peak with cis-7d and 4d. Both mixtures were further purified with preparative HPLC to yield 47.7 mg colorless oil (33% yield). 1H NMR (600 MHz, CDCl3): δ 7.74 (d, J = 7.9 Hz, 2H), 7.20 (d, J = 7.9 Hz, 2H), 6.71 (d, J = 8.3 Hz, 2H), 6.47 (d, J = 8.3 Hz, 2H), 5.32 (d, J = 7.7 Hz, 1H), 3.74 (s, 3H), 3.51 (s, 1H), 3.46 (tt, J = 7.8, 3.7 Hz, 1H), 3.02 (s, 1H), 2.38 (s, 3H), 1.82–1.66 (comp, 2H), 1.64–1.51 (comp, 3H), 1.51–1.35 (comp, 2H), 1.35–1.23 (comp, 1H); 13C NMR (151 MHz, CDCl3): δ 152.1, 143.3, 141.1, 137.9, 129.8, 126.9, 114.9, 114.9, 55.9, 49.4, 48.3, 37.7, 32.2, 31.4, 21.6, 19.8; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C20H27N2O3S 375.1742; Found 375.1732; IR: (cm−1, neat): 3393, 3266, 2940, 1511, 1439, 1312, 1234, 1155, 1073, 1046, 936, 886, 812, 660; TLC: Rf (2:1 Hexanes/EtOAc) 0.32, visualized with UV light; HPLC: Chiralpak AD-H column, 1.0 mL/min, 20% EtOH/Hex, 254 nm, peaks observed at 33.2 and 39.1 min. [α]D20 12.8° (c = 0.0058 g/mL). See supporting information for crystal structure of (±)-trans-7d.
Cis-N-(3-((4-methoxyphenyl)amino)cyclohexyl)-4-methylbenzene-sulfonamide (cis-7d) with CH2Cl2 as the solvent, at room temperature, and for 96 h. See Scheme S13. General Procedure #11 was used. The crude material was purified by automatic normal phase flash chromatography, eluting with a gradient of 10% to 100% EtOAc/Hex, yielding a peak with trans-7d, and a second peak with cis-7d and 4d. Both mixtures were further purified with preparative HPLC to yield 19.0 mg beige solid (13% yield). 1H NMR (600 MHz, CDCl3): δ 7.71 (d, J = 8.2 Hz, 2H), 7.26–7.23 (m, 2H), 6.73 (d, J = 8.9 Hz, 2H), 6.48 (d, J = 8.8 Hz, 2H), 4.82 (s, 1H), 3.73 (s, 3H), 3.22 (dtt, J = 11.1, 7.4, 3.4 Hz, 1H), 3.13–3.05 (m, 1H), 2.40 (s, 3H), 2.09 (d, J = 12.3 Hz, 1H), 1.96 (d, J = 13.3 Hz, 1H), 1.86–1.79 (m, 1H), 1.75 (dt, J = 14.0, 3.9 Hz, 1H), 1.33–1.20 (m, 1H), 1.17–1.07 (m, 1H), 0.98 (td, J = 12.4, 11.8, 8.7 Hz, 2H); 13C NMR (151 MHz, CDCl3): δ 152.7, 143.4, 140.5, 138.4, 129.8, 127.0, 115.5, 115.0, 55.9, 52.2, 51.8, 40.3, 33.7, 32.4, 22.4, 21.7; HRMS (ESI-QToF) m/z: [M+H]+ Calcd for C20H27N2O3S 375.1742; Found 375.1736; IR: (cm−1, neat): 3372, 3292, 2962, 1511, 1423, 1319, 1290, 1233, 1155, 109, 1038, 904, 812, 669; Mp: 130–134 °C; TLC: Rf (2:1 Hexanes/EtOAc) 0.16, visualized with UV light; HPLC: Chiralpak AD-H column, 1.0 mL/min, 20% EtOH/Hex, 254 nm, peaks observed at 51.0 and 84.7 min. See supporting information for crystal structures of (1R,3S)-cis-7d and (±)-cis-7d.
Methods for Measurement of Conversion by LC/MS
The following methods utilized a Waters CORTECS UPLC C18 (1.6 μm, 3.0 × 50 mm) column at 40 °C with a 0.8 mL/min flow rate.
4a/7a
Gradient conditions with H2O (0.01% formic acid) and MeCN (0.01% formic acid) were utilized as follows: Held at 10% MeCN for 2 min, ramped to 90% MeCN for 12.5 min, ramped to 10% MeCN for 2.5 min, held at 10% MeCN for 2 min. Peaks observed: 4a = 3.53 min; cis-7a = 3.90 min; trans-7a = 3.98 min.
4b/7b
Gradient conditions with H2O (0.01% formic acid) and MeCN (0.01% formic acid) were utilized as follows: Held at 10% MeCN for 2 min, ramped to 90% MeCN for 12.5 min, ramped to 10% MeCN for 2.5 min, held at 10% MeCN for 2 min. Peaks observed: 4b = 3.18 min; cis-7b = 3.77 min; trans-7b = 3.93 min.
4c/7c
Gradient conditions with H2O (0.01% formic acid) and MeCN (0.01% formic acid) were utilized as follows: Held at 10% MeCN for 0.2 min, ramped to 98% MeCN for 3.8 min, held at 98% MeCN for 0.25 min, ramped to 10% MeCN for 0.45 min, held at 10% MeCN for 3.3 min. Peaks observed: 4c = 1.88 min; cis-7c = 1.62 min; trans-7c = 1.65 min.
4d/7d
Gradient conditions with H2O (0.01% formic acid) and MeCN (0.01% formic acid) were utilized as follows: Held at 10% MeCN for 2 min, ramped to 90% MeCN for 12.5 min, ramped to 10% MeCN for 2.5 min, held at 10% MeCN for 2 min. Peaks observed: 4d = 4.80 min; 7d = 4.48 min.
Calculation of Relative Response Factors (RRF) of 7 to 4
To correct for different response factors between ketone 4 and diamine 7 on the LC/MS in conversion measurements, external standard calibration on the compounds’ relative response factors were calculated. The LC/MS was set to monitor from 220 nm to 400 nm, the HPLC was set to monitor 254 nm.
Example Method for LC/MS
To a vial containing 4 or 7 was 10 mL MeCN. From this solution was taken 0.75 mL, which was transferred into a separate LC/MS vial and diluted with 0.25 mL 0.1% aqueous formic acid. The sample was subjected to LC/MS analysis, with the response factor measured.
RRF for cis-7a/4a: 2.48
RRF for trans-7a/4a: 2.60
Average RRF for 7a/4a (used for some screening): 2.54
RRF for cis-7b/4b: 1.25
RRF for trans-7b/4b: 1.08
RRF for cis-7c/4c: 2.90
RRF for trans-7c/4c: 3.87
RRF for cis-7d/4d: 1.66
RRF for trans-7d/4d: 1.69
Average RRF for 7d/4d: 1.67
Example Method HPLC
(This was required for 4d/7d, as cis-7d and trans-7d co-eluted on LC/MS) To a vial containing 7d was added 3.0 mL EtOH. From this solution was taken 0.40 mL, which was transferred into a separate LC/MS vial and diluted with 0.20 mL Hex. The sample was subjected to HPLC analysis, with the response factor measured.
RRF for cis-7d/trans-7d: 1.59
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIH R01-GM096403). We graciously thank Dr. Brandon Q. Mercado for acquiring the X-ray crystallographic data. C.R.S. thanks the National Science Foundation Graduate Research Fellowship Program for funding (DGE1122492). A.L. would like to acknowledge funding from the Yale College Dean’s Office in support of the Science, Technology, and Research Scholars (STARS) summer research program.
Footnotes
Notes
The authors declare no competing financial interests.
The Supporting Information is available free of charge on the ACS Publications website.
Schemes for peptide and substrate synthesis (Schemes S1–S13); 1H and 13C NMR spectra for all compounds; 31P NMR spectra for all peptide catalysts; 1H–1H COSY, 1H–13C HSQC, and 1H–1H NOESY NMR spectra for peptide P25; HPLC traces for compounds 4 and 7; DFT and M06 calculations for 4a, 8a, and 10.
X-ray crystallographic data for compound (±)-trans-7d.
X-ray crystallographic data for compound (1R,3S)-cis-7d.
X-ray crystallographic data for compound (±)-cis-7d.
References
- 1.Yoon TP, Jacobsen EN. Science. 2003;299:1691–1693. doi: 10.1126/science.1083622. [DOI] [PubMed] [Google Scholar]
- 2.(a) Parmar D, Sugiono E, Raja S, Rueping M. Chem Rev. 2014;114:9047–9153. doi: 10.1021/cr5001496. [DOI] [PubMed] [Google Scholar]; (b) Parmar D, Sugiono E, Raja S, Rueping M. Chem Rev. 2017;117:10608–10620. doi: 10.1021/acs.chemrev.7b00197. [DOI] [PubMed] [Google Scholar]
- 3.Terada M. Synthesis. 2010;12:1929–1982. [Google Scholar]
- 4.Akiyama T. Chem Rev. 2007;107:5744–5758. doi: 10.1021/cr068374j. [DOI] [PubMed] [Google Scholar]
- 5.Reid JP, Goodman JM. Chem Eur J. 2017;23:14248–14260. doi: 10.1002/chem.201702019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anantharaman V, Aravind L, Koonin EV. Curr Opin Chem Biol. 2003;7:12–20. doi: 10.1016/s1367-5931(02)00018-2. [DOI] [PubMed] [Google Scholar]
- 7.Tang J, Wong RNS. J Chem Biol. 1987;33:53–63. [Google Scholar]
- 8.Lairson LL, Henrissat B, Davies GJ, Withers SG. Annu Rev Biochem. 2008;77:521–555. doi: 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]
- 9.Werck-Reichhart D, Feyereisen R. Genome Biol. 2000;1:3003–3009. doi: 10.1186/gb-2000-1-6-reviews3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McDonald AG, Tipton KF. FEBS J. 2014;281:583–592. doi: 10.1111/febs.12530. [DOI] [PubMed] [Google Scholar]
- 11.Dixon M, Webb EC. Enzymes. Longmans Green; London: Academic Press; New York: [Google Scholar]
- 12.Colby Davie E, Mennen SM, Xu Y, Miller SJ. Chem Rev. 2007;107:5759–5812. doi: 10.1021/cr068377w. [DOI] [PubMed] [Google Scholar]
- 13.Metrano AJ, Abascal NC, Mercado BQ, Paulson EK, Hurtley AE, Miller SJ. J Am Chem Soc. 2017;139:492–516. doi: 10.1021/jacs.6b11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hunter T. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 15.Yaffe MB, Elia AEH. Curr Opin Cell Biol. 2001;13:131–138. doi: 10.1016/s0955-0674(00)00189-7. [DOI] [PubMed] [Google Scholar]
- 16.Mahajan A, Yuan C, Lee H, Chen ESW, Wu PY, Tsai MD. Sci Signal. 2008;1:151re12. doi: 10.1126/scisignal.151re12. [DOI] [PubMed] [Google Scholar]
- 17.Wilker E, Yaffe MB. J Mol Cell Cardiol. 2004;37:633–642. doi: 10.1016/j.yjmcc.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 18.Barford D, Das AK, Egloff MP. Annu Rev Biomol Struct. 1998;27:133–164. doi: 10.1146/annurev.biophys.27.1.133. [DOI] [PubMed] [Google Scholar]
- 19.El-Faham A, Albericio F. Chem Rev. 2011;111:6557–6602. doi: 10.1021/cr100048w. [DOI] [PubMed] [Google Scholar]
- 20.Rueping M, Sugiono E, Azap C, Theissmann T, Bolte M. Org Lett. 2005;7:3781–3783. doi: 10.1021/ol0515964. [DOI] [PubMed] [Google Scholar]
- 21.Hoffmann S, Seayad AM, List B. Angew Chem Int Ed. 2005;44:7424–7427. doi: 10.1002/anie.200503062. [DOI] [PubMed] [Google Scholar]
- 22.Li G, Liang Y, Antilla JC. J Am Chem Soc. 2007;129:5830–5831. doi: 10.1021/ja070519w. [DOI] [PubMed] [Google Scholar]
- 23.Nguyen TB, Bousserouel H, Wang Q, Guéritte F. Org Lett. 2010;12:4705–4707. doi: 10.1021/ol102043x. [DOI] [PubMed] [Google Scholar]
- 24.Chen MW, Chen QA, Duan Y, Ye ZS, Zhou YG. Chem Commun. 2012;48:1698–1700. doi: 10.1039/c2cc16832d. [DOI] [PubMed] [Google Scholar]
- 25.Li G, Antilla JC. Org Lett. 2009;11:1075–1078. doi: 10.1021/ol802860u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rueping M, Antonchick AP, Theissmann T. Angew Chem Int Ed. 2006;45:3683–3686. doi: 10.1002/anie.200600191. [DOI] [PubMed] [Google Scholar]
- 27.Guo QS, Du DM, Xu J. Angew Chem Int Ed. 2008;47:759–762. doi: 10.1002/anie.200703925. [DOI] [PubMed] [Google Scholar]
- 28.Aillerie A, Lemau de Talancé V, Moncomble A, Bousquet T, Pélinski L. Org Lett. 2014;16:2982–2985. doi: 10.1021/ol5011196. [DOI] [PubMed] [Google Scholar]
- 29.Cai XF, Guo RN, Feng GS, Wu B, Zhou YG. Org Lett. 2014;16:2680–2683. doi: 10.1021/ol500954j. [DOI] [PubMed] [Google Scholar]
- 30.Hantzsch A, Liebigs Justus. Ann Chem. 1882;215:1. [Google Scholar]
- 31.Singh S, Batra UK. Indian J Chem, Sect B. 1989;28:1. [Google Scholar]
- 32.Storer RI, Carrera DE, Ni Y, MacMillan DWC. J Am Chem Soc. 2006;128:84–86. doi: 10.1021/ja057222n. [DOI] [PubMed] [Google Scholar]
- 33.Faisca Phillips AM, Pombeiro AJL. Org Biomol Chem. 2017;15:2307–2340. doi: 10.1039/c7ob00113d. [DOI] [PubMed] [Google Scholar]
- 34.Garrett RH, Grisham CM. Biochemistry. 5. Cengage: United States of America; 2013. [Google Scholar]
- 35.Pollak N, Dolle C, Ziëgler M. Biochem J. 2007;402:205–218. doi: 10.1042/BJ20061638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shugrue CR, Miller SJ. Angew Chem Int Ed. 2015;54:11173–11176. doi: 10.1002/anie.201505898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shugrue CR, Miller SJ. Chem Rev. 2017;117:11894–11951. doi: 10.1021/acs.chemrev.7b00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sorensen EJ, Nicolaou KC. Classics in Total Synthesis: Targets, Strategies, Methods. 5. Wiley VCH; New York: 2008. [Google Scholar]
- 39.Hoffmann S, Nicoletti M, List B. J Am Chem Soc. 2006;128:13074–13075. doi: 10.1021/ja065404r. [DOI] [PubMed] [Google Scholar]
- 40.Khan IA, Saxena AK. J Org Chem Soc. 2013;78:11656–11669. doi: 10.1021/jo4012249. [DOI] [PubMed] [Google Scholar]
- 41.Romney DK, Colvin SM, Miller SJ. J Am Chem Soc. 2014;136:14019–14022. doi: 10.1021/ja508757g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilmot CM, Thornton JM. J Mol Biol. 1988;203:221. doi: 10.1016/0022-2836(88)90103-9. [DOI] [PubMed] [Google Scholar]
- 43.See supporting information for synthetic details.
- 44.Additional peptides in Table 1 were screened and followed the same pattern as P32 and P34, with minute changes to er with marginal or no increase in dr favoring cis-4a.
- 45.Vedejs E, Chen X. J Am Chem Soc. 1997;119:2584–2585. [Google Scholar]
- 46.Dehli JR, Gotor V. Chem Soc Rev. 2002;31:365–370. doi: 10.1039/b205280f. [DOI] [PubMed] [Google Scholar]
- 47.See Supporting Information for computational details.
- 48.Zhao Y, Truhlar DG. Theor Chem Acc. 2008;120:215–241. [Google Scholar]
- 49.For recent energy difference calculations performed on chair conformations using M06-2X functional, see: Jahn MK, Dewald D, Val-lejo-Lopez M, Cocinero EJ, Lesarri A, Grabow J-U. J Phys Chem A. 2013;117:13673–13679. doi: 10.1021/jp407671m.
- 50.For a review on solvation models, see: Tomasi J, Mennucci B, Cammi R. Chem Rev. 2005;105:2999–3093. doi: 10.1021/cr9904009.
- 51.For recent combinations of the M06-2× functional with the IEF-PCM solvation model, see: Simon A, Lam Y-h, Houk KN. J Am Chem Soc. 2016;138:503–506. doi: 10.1021/jacs.5b12097.
- 52.Alford JS, Abascal NC, Shugrue CR, Colvin SM, Romney DK, Miller SJ. ACS Cent Sci. 2016;2:733–739. doi: 10.1021/acscentsci.6b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fulmer GR, Miller AJM, Sherden NH, Gottlieb HE, Nudelman A, Stoltz BM, Bercaw JE, Goldberg KI. Organometallics. 2010;29:2176–2179. [Google Scholar]
- 54.Petrillo DE, Mowrey DR, Allwein SP, Bakale RP. Org Lett. 2012;14:1206–1209. doi: 10.1021/ol203332j. [DOI] [PubMed] [Google Scholar]
- 55.Hollenstein M. Chem Eur J. 2012;18:13320–13330. doi: 10.1002/chem.201201662. [DOI] [PubMed] [Google Scholar]
- 56.Gangamani BP, Kumar VA, Ganesh KN. Tetrahedron. 1996;52:15017–15030. [Google Scholar]
- 57.Sakashita H, Akahoshi F, Toshida T, Kitajima H, Hayashi Y, Ishii S, Takashina Y, Tsutsumiuchi R, Ono S. Bioorg Med Chem. 2007;15:641–655. doi: 10.1016/j.bmc.2006.10.059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.