

Abstract
The realization of the therapeutic potential of targeting the M1 muscarinic acetylcholine receptor (mAChR) for the treatment of cognitive decline in Alzheimer’s disease has prompted the discovery of M1 mAChR ligands showing efficacy in alleviating cognitive dysfunction in both rodents and humans. Among these is GSK1034702 (7-fluoro-5-methyl-3-[1-(oxan-4-yl)piperidin-4-yl]-1H-benzimidazol-2-one), described previously as a potent M1 receptor allosteric agonist, which showed procognitive effects in rodents and improved immediate memory in a clinical nicotine withdrawal test but induced significant side effects. Here we provide evidence using ligand binding, chemical biology and functional assays to establish that rather than the allosteric mechanism claimed, GSK1034702 interacts in a bitopic manner at the M1 mAChR such that it can concomitantly span both the orthosteric and an allosteric binding site. The bitopic nature of GSK1034702, together with the intrinsic agonist activity and a lack of muscarinic receptor subtype selectivity reported here, all likely contribute to the adverse effects of this molecule in clinical trials. Although they impart beneficial effects on learning and memory, we conclude that these properties are undesirable in a clinical candidate due to the likelihood of adverse side effects. Rather, our data support the notion that “pure” positive allosteric modulators showing selectivity for the M1 mAChR with low levels of intrinsic activity would be preferable to provide clinical efficacy with low adverse responses.
Introduction
The M1 muscarinic acetylcholine receptor (mAChR) has emerged as an attractive molecular target to overcome cognitive decline associated with cholinergic degeneration in Alzheimer disease (AD) (Anagnostaras et al., 2003). Activation of M1 mAChRs, which are abundantly expressed in the amygdala, cerebral cortex, striatum, and hippocampus (Buckley et al., 1988; Levey et al., 1995), has been reported to rescue learning and memory deficits associated with neurodegeneration in a number of mouse models (Lange et al., 2015; Puri et al., 2015; Vardigan et al., 2015; Bradley et al., 2017). Translating these promising findings to successful clinical candidates has, however, been challenging due to adverse effects associated with a lack of selectivity of orthosteric M1 mAChR agonists. This is exemplified by the M1/M4-preferring mAChR agonist xanomeline, which significantly improved cognitive function in patients with AD (Bodick et al., 1997) but ultimately failed due to adverse effects attributed to activation of peripheral cholinergic signaling likely through M2 and M3 mAChRs (Langmead et al., 2008).
There is, therefore, an urgent need to develop novel approaches to build selectivity into M1 mAChR ligands. Two related approaches to this problem have been taken: 1) the development of positive allosteric modulators (PAMs) that bind to a site that is topographically distinct from that of the endogenous ligand acetylcholine (ACh) (May et al., 2007) and 2) development of a newer generation of more selective agonists (whose mechanism of action is not always well defined) (Langmead et al., 2008). Allosteric modulators enhance ACh binding and/or signaling, the magnitude of which can vary with different degrees of positive cooperativity (Langmead and Christopoulos, 2006; May et al., 2007; Conn et al., 2009). They can also possess direct allosteric agonist activity (intrinsic activity). M1 mAChR allosteric modulators with high functional selectivity over other muscarinic receptor subtypes (i.e., M2–M5 mAChRs) have been reported to reverse phenotypes associated with neurodegenerative disease (Ma et al., 2009; Puri et al., 2015; Vardigan et al., 2015; Bradley et al., 2017) while showing no adverse side effects in animal models (Bradley et al., 2017).
Less well described in the literature is the development of a new generation of M1 mAChR agonists, which have variously been described as “ectopic” (Spalding et al., 2002), “allosteric” (Langmead and Christopoulos, 2006; Jones et al., 2008; Budzik et al., 2010), “atypical” (Lebon et al., 2009), and “bitopic” (Keov et al., 2011). With but a few exceptions (Keov et al., 2014), this broad nomenclature reflects a paucity in understanding of mechanism(s) of action; it is not clear whether some of these ligands exert true allosteric agonism (i.e., bind solely to an allosteric binding site to activate the receptor), are subtype-selective orthosteric agonists, or represent a hybrid of the two (with a pharmacophore that engages both orthosteric and allosteric binding pockets).
GSK1034702 (7-fluoro-5-methyl-3-[1-(oxan-4-yl)piperidin-4-yl]-1H-benzimidazol-2-one) has been described as an allosteric M1 mAChR agonist; it was identified from a series of benzimidazolones and reversed scopolamine-induced amnesia in rodents (Budzik et al., 2010) and had positive effects on cognitive function in humans. Unfortunately, it also induced gastrointestinal adverse effects, consistent with activation of peripheral mAChRs (Nathan et al., 2013).
Here we conduct a comprehensive pharmacological analysis of GSK1034702 and show that this molecule is not a purely allosteric ligand as previously suggested. Radioligand binding, using [3H]-N-methylscopolamine (NMS) and [3H]-GSK1034702, and functional inositol phosphate (IP) accumulation studies at the muscarinic M1 receptor reveal that GSK1034702 interacts with the orthosteric ACh binding site and likely, concomitantly, with an allosteric binding site. Importantly, this bitopic mode of action is able to mediate beneficial effects on learning and memory but the lower degree of selectivity of GSK1034702 compared with ligands that engage solely with an allosteric binding site, together with the intrinsic agonist activity reported here, may account for the adverse effects observed with this molecule in the clinic.
Materials and Methods
Materials.
GSK1034702 and xanomeline were synthesized by Eli Lilly (Windlesham, Surrey, UK). IP-One and extracellular signal–regulated kinase 1/2 (ERK1/2) phosphorylation assay kits were purchased from Cisbio Assays (Codolet, France). TBPB (1-[1′-(2-methylbenzyl)-1,4′-bipiperidin-4-yl]-1,3-dihydro-2H-benzimidazol-2-one) was obtained from Tocris Bioscience (Bristol, UK). All other chemicals and reagents were purchased from Sigma-Aldrich Company Ltd. (Dorset, UK).
Mouse Maintenance and Diet.
All experiments were performed under a project license from the British Home Office (United Kingdom) under the Animals (Scientific Procedures) Act of 1986. C57Bl/6J mice used in this study were purchased from Charles River (Margate, UK). Mice were fed ad libitum with a standard mouse chow and were maintained within the animal facility at least 1 week prior to experiments.
Fear Conditioning.
C57Bl/6J male mice (aged 8–12 weeks) were acclimatized to the behavioral testing suite at least 2 hours prior to the test. Mice were injected (intraperitoneally) with vehicle (5% glucose) or scopolamine (1.5 mg/kg) alone or in combination with xanomeline, GSK1034702, or TBPB 30 minutes prior to training. Mice were placed in the conditioning chamber (ANY-maze Fear Conditioning System; Stoelting, Dublin, Ireland); after a 2-minute adaptation period, they received three tone/foot shock pairings where the foot shock (unconditioned stimulus; 2 seconds; 0.4 mA) always co-terminated with a tone (conditioned stimulus; 2.8 kH; 85 dB; 30 seconds). The conditioned stimulus–unconditioned stimulus pairings were separated by 1-minute intervals. After the mice completed training, they remained in the conditioning chamber for 1 minute and were then returned to their home cages. The next day, the mice were placed back in the conditioning chamber, and time spent immobile was recorded for 3 minutes to assess context-dependent learning. Data were analyzed using ANY-maze software (Stoelting).
Mouse Pharmacokinetics.
Pharmacokinetic analyses were conducted as previously described (Witkin et al., 2017). Compounds were administered via intraperitoneal injection (in 5% glucose) 30 minutes prior to blood collection. Mice were anesthetized with 3% isoflurane (2 l/min), and blood was collected by cardiac puncture of the left ventricle. Blood was immediately transferred to EDTA tubes and centrifuged at 1000g for 10 minutes at 4°C; the supernatant was collected and frozen. Brains from each mouse were also dissected and snap-frozen on dry ice.
Brain samples were homogenized in three volumes of methanol/water [1:4 (v/v)] by weight. A 25-μl aliquot of each study sample, calibration standard, and control sample was added to a 96-well plate and mixed with 180 μl acetonitrile/methanol [1:1 (v/v)] containing internal standard. The samples were subsequently centrifuged, and the resulting supernatants were diluted 12.5-fold with methanol/water [1:1 (v/v)] prior to analyzing 10-μl aliquots by liquid chromatography–tandem mass spectrometry as previously described (Bradley et al., 2017).
Equilibrium [3H]-NMS Binding.
Chinese hamster ovary (CHO) Flp-In cells expressing the wild-type M1 mAChR (Bmax = 870 fmol/mg) or the M1 designer receptor exclusively activated by designer drug (DREADD) (Bmax = 2400 fmol/mg) were plated at 7500 cells/well in clear 96-well plates and grown to confluence. Prior to the experiment, cells were washed with 100 μl phosphate-buffered saline. Increasing concentrations of test compounds and an approximate equilibrium dissociation constant (KD) concentration of [3H]-NMS (KD (nM) for [3H]-NMS binding to CHO Flp-In cells expressing wild-type M1 mAChR or M1 DREADD were 0.37 ± 0.10 and 18.70 ± 3.49 (n = 3) were incubated with cells overnight at room temperature in a final volume of 100 μl binding buffer of the following composition: 110 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 1 mM MgSO4, 25 mM glucose, 20 mM HEPES, and 58 mM sucrose, pH 7.4. Binding was terminated by rapid aspiration followed by two washes with 200 μl ice-cold 0.9% NaCl. Bound radioactivity was determined by liquid scintillation (Ultima Gold; PerkinElmer, Boston, MA) counting. Nonspecific binding was determined in the presence of 10 μM atropine.
For competition binding experiments at M1, M2, M3, M4, and M5 mAChRs, CHO membranes were purchased from PerkinElmer. All experiments were performed in assay buffer of the following composition: 20 mM HEPES, 100 mM NaCl, and 10 mM MgCl2, pH 7.5, and used 10 µg protein/well in a total assay volume of 1 ml using deep well blocks. CHO cell membranes overexpressing human mAChR M1–M5 subtypes were incubated with a concentration of [3H]-NMS that was close to the calculated KD for each receptor (M1: 200 pM, KD = 196 pM; M2: 700 pM, KD = 769 pM; M3: 700 pM, KD = 642 pM; M4: 200 pM, KD 143 pM; M5: 400 pM, KD = 410 pM), in the presence or absence of 11 different concentrations of compound. Nonspecific binding was determined in the presence of 10 µM atropine. All assay incubations were initiated by the addition of membrane suspensions and deep well blocks were shaken for 5 minutes to ensure complete mixing. Incubation was then carried out for 2 hours at 21°C. Binding reactions were terminated by rapid filtration through GF/A filters (PerkinElmer) presoaked with 0.5% (w/v) polyethylenimine for 1 hour. Filters were then washed three times with 1 ml ice-cold assay buffer. Dried filters were counted with Meltilex A scintillant using a Trilux 1450 scintillation counter (PerkinElmer). The specific bound counts (in disintegrations per minute) were expressed as a percentage of the maximal binding observed in the absence of test compound (total) and nonspecific binding determined in the presence of 10 µM atropine.
Kinetic [3H]-NMS Binding.
For determination of [3H]-NMS dissociation kinetics, membranes (5 μg/tube) expressing the M1 mAChR were preincubated with [3H]-NMS for 1 hour at 37°C in binding buffer containing 100 mM NaCl, 10 mM MgCl2, and 20 mM HEPES, pH 7.4. Dissociation of the bound radioligand was initiated by the addition of atropine (10 μM) alone or atropine (10 μM) plus 100 μM GSK1034702 added in a reverse time course protocol. Reactions were terminated by rapid filtration onto GF/B filter paper (Whatman, Maidstone, UK) and three washes with 3 ml ice-cold 0.9% NaCl using a Brandel harvester (M-24TI; Brandel, Fort Lauderdale, FL). Membrane bound radioactivity was determined by liquid scintillation (Ultima Gold; PerkinElmer) counting.
IP-One Accumulation Assay.
Stimulation of IP accumulation was determined using the Cisbio IP-One Gq assay kit per the manufacturer’s instructions. For agonist concentration-response curves, agonists (2× concentrated) were added to 384-well white ProxiPlates (PerkinElmer) in 7 μl stimulation buffer. CHO Flp-In cells stably expressing the human M1 mAChR were grown to confluence in T75 cell culture flasks at 37°C. Cells were washed with warm phosphate-buffered saline and detached using phosphate-buffered saline with 0.1 M EDTA. Detached cells were centrifuged at 1000g and the cells were resuspended in stimulation buffer. Seven microliters of this cell suspension (1.43 × 106 cells/ml) was added to each well, and cells were stimulated for 45 minutes at 37°C.
For functional interaction studies, CHO Flp-In cells stably expressing the human M1 mAChR were seeded at 5000 cells/well in 384-well white ProxiPlates. Experiments were conducted 48 hours later. Cells were washed once with 50 μl phosphate-buffered saline and then incubated in F12 media containing phenoxybenzamine (where applicable) at 37°C for 30 minutes. Cells were washed with 50 μl phosphate-buffered saline and incubated in stimulation buffer containing agonists in a final volume of 14 μl for 45 minutes at 37°C.
All IP-One stimulations were terminated by the addition of 3 μl/well IP1-d2 solution, followed by 3 μl/well anti–IP1-cryptate solution and incubation for 1 hour at room temperature with shaking. Fluorescence emission at two different wavelengths (665 and 620 nm) was measured with a PHERAstar plate reader (BMG Labtech, Ortenberg, Germany).
ERK1/2 Phosphorylation.
Stimulation of phospho-ERK1/2 (Thr 202/Tyr 204) was determined using the Cisbio Phospho-ERK Cellular Assay Kit. CHO Flp-In cells stably expressing the human M1 receptor were seeded onto transparent 96-well plates at 20,000 cells/well and grown to confluence. Cells were serum starved overnight prior to the experiment. Prior to the stimulations, cells were washed with 100 μl phosphate-buffered saline and then incubated in serum free F12 medium at 37°C. Cells were stimulated with ligands for 5 minutes at 37°C in a final volume of 200 μl. The stimulations were terminated by rapid aspiration and addition of 50 μl lysis buffer supplemented with blocking reagent per the manufacturer’s instructions, followed by gentle agitation at room temperature for 30 minutes. Subsequently, 16 μl of this lysate was transferred to a 384-well white ProxiPlate (PerkinElmer) and incubated with 4 μl premixed antibody solution for 2 hours at room temperature. Fluorescence emission at two different wavelengths (665 and 620 nm) was measured with a PHERAstar plate reader (BMG Labtech).
Native Tissue GTPɣ[35S] Binding Assays.
GTPɣ-[35S] ([35S]ɣ[guanosine-5′-O-(3-[35S]thio)triphosphate] binding in rat membranes was determined in triplicate using an antibody capture technique in 96-well plate format (DeLapp et al., 1999). Native rat membranes were prepared as follows. All procedures were performed at 4°C. Ten to fifteen milliliters of sucrose buffer (10 mM HEPES, 1 mM EGTA, 1 mM dithiothreitol, 10% sucrose, and one tablet/50 ml Complete Protease Inhibitor Cocktail, pH 7.4) was added to each tissue sample and homogenized for 10 strokes using an electric IKA RW20 homogenizer (800 rpm) (IKA, Staufen, Germany) with glass homogenizer. The homogenate was centrifuged at 1000g for 10 minutes at 4°C and the supernatant collected. The pellet was rehomogenized and centrifuged again as above and the supernatant pooled and centrifuged at 11,000g for 20 minutes at 4°C. The resulting pellet was suspended in 40 ml final storage buffer (10 mM HEPES, 1 mM EGTA, 1 mM MgCl2, and 1 mM dithiothreitol, pH 7.4) and centrifuged at 27,000g for 20 minutes at 4°C. The supernatant was removed and the final pellet was suspended in 2 ml final storage buffer. The protein concentration was measured using the Bradford method (Coomassie Plus, Bio-Rad protein assay kit; Bio-Rad, Hercules, CA) with bovine gamma globulin standards. Samples were aliquoted and stored at −80°C. Membrane aliquots (15 µg/well) were then incubated with test compound and GTPɣ[35S] (500 pM/well) for 30 minutes. Labeled membranes were then solubilized with 0.27% Nonidet P-40 plus Gqα antibody (E17; Santa Cruz Biotechnology, Dallas, TX) at a final dilution of 1:200 and 1.25 mg/well anti-rabbit scintillation proximity beads. Plates were left to incubate for 3 hours and then centrifuged for 10 minutes at 2000 rpm. Plates were counted for 1 minute/well using a Wallac MicroBeta Trilux scintillation counter (PerkinElmer). All incubations took place at room temperature in GTP-binding assay buffer of the following composition: 20 mM HEPES, 100 mM NaCl, and 5 mM MgCl2, pH 7.5. Data were converted to the percentage of response compared with oxotremorine-M (100 µM) or the percentage over basal and EC50 values were generated (four-parameter logistic curve) using GraphPad Prism 6 software (GraphPad Software Inc., La Jolla, CA).
[3H]-GSK1034702 Binding.
[3H]-GSK1034702 (specific activity 3.65 TBq/mmol) was synthesized by direct titration on Pd Black (performed by Quotient Bioresearch, Manchester, UK). All experiments were performed in assay buffer of the following composition: 20 mM HEPES, 100 mM NaCl, and 10 mM MgCl2, pH 7.5, and used 50 µg protein/well in a total assay volume of 250 μl.
CHO cell membranes overexpressing human M1 mAChR (PerkinElmer) were incubated with [3H]-GSK1034702 (20 nM) in the presence of 11 concentrations of test compound. All assay incubations were initiated by the addition of membrane suspensions. Incubation was then carried out for 4 hours at 21°C. Binding reactions were terminated by rapid filtration through GF/A filters (PerkinElmer) presoaked with 0.5% (w/v) polyethylenimine for 1 hour. Filters were then washed six times with 1 ml ice-cold assay buffer. Dried filters were counted using Meltilex A scintillant using a Trilux 1450 scintillation counter (PerkinElmer). The specific bound counts (in disintegrations per minute) were expressed as a percentage of the maximal binding observed in the absence of test compound (total) and nonspecific binding determined in the presence of 100 µM nonradiolabeled GSK1034702.
Isolated Rat Atria and Ileum Contraction Experiments.
Adult Wistar rats were humanely euthanized and the left atria and the ileum were placed in an organ bath containing oxygenated Tyrode solution. Measurement of negative inotropic responses in atria or contraction of ileum was performed as described by Lambrecht et al. (1989). For atrial responses, ligands were administered for 5 minutes at 32°C in McEwen’s buffer (pH 7.4) in a bath volume of 10 ml. Agonist responses were measured as negative inotropy relative to 1 μM methacholine, and antagonism was measured as inhibition of methacholine (1 μM)–induced negative ionotropic response. For rat ileum experiments, ligands were administered for 5 minutes at 32°C in Krebs’ buffer (pH 7.4) in a bath volume of 10 ml. Contraction of ileum as a percentage of methacholine-induced contraction was measured, and antagonistic effects were measured by inhibition of methacholine-induced responses.
Data Analysis.
Inhibition binding data were curve-fit using GraphPad Prism 6 to derive the potency (IC50) of the test compound. The equilibrium dissociation constant (KI) of the test compound was then calculated with the Cheng–Prusoff equation: KI = IC50/[1 + ([L]/KD)] using the KD value derived separately from saturation binding studies.
Functional concentration-response curves were fitted according to a four-parameter logistic equation (to determine minimum and maximum asymptotes, LogEC50, and slope; GraphPad Prism 6). For ACh curves in the presence of multiple concentrations of GSK1034702 or TBPB (after phenoxybenzamine treatment), the following form of the Gaddum and Schild equations was applied globally to the datasets:
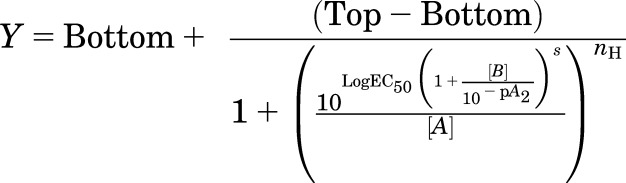 |
where top represents the maximal asymptote of the curves, bottom represents the minimum asymptote of the curves, LogEC50 represents the logarithm of the ACh EC50 in the absence of GSK1034702 or TBPB, [A] represents the concentration of ACh, [B] represents the concentration of GSK1034702 or TBPB, nH represents the Hill slope of the agonist curve, s represents the Schild slope for the antagonist, and pA2 represents the negative logarithm of the concentration of antagonist that shifts the agonist EC50 by a factor of 2. In the absence of antagonist ([B] = 0), this equation becomes the standard four-parameter logistic equation for fitting agonist concentration-response data.
[3H]-NMS binding interaction studies with benzyl quinolone carboxylic acid (BQCA) were fitted to an allosteric ternary complex model (Leach et al., 2010):
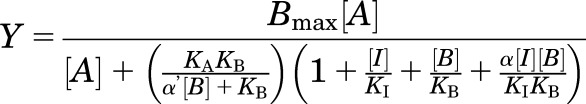 |
where Bmax represents the total number of receptors; [A], [B], and [I] are concentrations of radioligand, allosteric modulator, and orthosteric ligand, respectively; and KA, KB, and KI represent equilibrium dissociation constants of radioligand, allosteric modulator, and orthosteric ligand, respectively. α′ and α are the binding cooperativities between the allosteric modulator and radioligand and the allosteric modulator and the orthosteric ligand, respectively. An α value of >1 denotes positive cooperativity, a value of <1 denotes negative cooperativity, and a value of 1 denotes neutral cooperativity of binding.
To assess agonist bias, the same concentration-response curves were analyzed according to a modified form of the operational model of agonism, recast to directly yield a transduction ratio (Log[τ/KA]; van der Westhuizen et al., 2014):
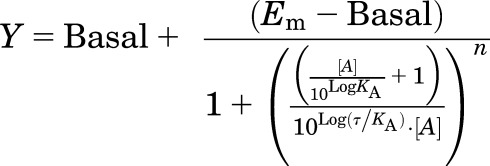 |
where basal represents the response in the absence of agonist, Em represents the maximal response of the assay system, KA represents the equilibrium dissociation constant of the agonist, [A] represents the concentration of agonist, τ is an index of the coupling efficiency (or efficacy) of the agonist, and n is the slope of the transducer function linking agonist occupancy to response. For the analysis, all families of agonist curves at each pathway were globally fitted to the model with the parameters, basal, Em, and n shared between all agonists. For full agonists, the LogKA was constrained to a value of zero, whereas for partial agonists this was directly estimated by the curve fitting procedure; the Log(τ/KA) parameter was estimated as a unique measure of activity for each agonist. Agonist bias factors (10^ΔΔLog[τ/KA]) were calculated as described in van der Westhuizen et al. (2014)).
Results
GSK1034702, TBPB, and Xanomeline Reverse Scopolamine-Induced Deficits in Fear Conditioning.
It is well established that muscarinic receptor agonists and PAMs can reverse deficits in learning and memory induced by the administration of a broad-spectrum muscarinic antagonist such as scopolamine (Young et al., 1995; Ma et al., 2009). Here, doses of scopolamine above 1 mg/kg administered to mice 30 minutes prior to fear conditioning training were sufficient to induce a significant reduction in contextual fear conditioning learning and memory (Supplemental Fig. 1). The effects of muscarinic receptor agonists on this deficit were tested by the coadministration of scopolamine (1.5 mg/kg) with escalating intraperitoneal doses of xanomeline (Fig. 1A), GSK1034702 (Fig. 1B), or TBPB (Fig. 1C). All three agents significantly improved learning and memory compared with vehicle controls (5% glucose solution in double distilled water) (P < 0.05 vs. administration of 1.5 mg/kg scopolamine alone; one-way analysis of variance with the Tukey multiple comparisons test). Free brain concentrations of xanomeline and GSK1034702 determined 30 minutes after administration were seen to increase linearly with escalating doses (Fig. 1, D and E). In contrast, the effects of these compounds on learning and memory were bell shaped, with lower doses improving learning and memory and high doses showing reduced effect (Fig. 1, A and B). This bell-shaped response is characteristic of procognitive agents. Interestingly, brain exposure of TBPB could not be increased beyond that observed at 10 mg/kg, remaining relatively low even after intraperitoneal injection of higher doses (Fig. 1F). This resulted in TBPB effects on learning and memory being similar at both low and high-administered doses with no evidence of a bell-shaped dose response (Fig. 1C).
Fig. 1.

(A–C) Effects of xanomeline (1, 3, 10, and 30 mg/kg) (A), GSK1034702 (0.3, 1, 3, 10, and 30 mg/kg) (B), and TBPB (1, 3, 10, and 30 mg/kg) (C) on scopolamine (1.5 mg/kg)–induced impairments in contextual fear conditioning (inset chemical structures of compounds). Data are expressed as the means ± S.E.M. of eight or more mice per group. Data were analyzed using a one-way analysis of variance with the Tukey multiple comparison test. *P < 0.05; ** P < 0.01; ***P < 0.001 vs. vehicle alone; #P < 0.05 vs. 1.5 mg/kg scopolamine. (D–F) Unbound concentrations (Log M) of xanomeline (D), GSK1034702 (E), and TBPB (F) measured in plasma or brain samples 30 minutes after intraperitoneal injection with increasing concentrations of respective compound. Data are expressed as the means ± S.E.M. of three mice per concentration.
GSK1034702 and TBPB Interact Competitively with [3H]-NMS at M1 mAChRs.
GSK1034702 and TBPB have previously been described as allosteric agonists of the M1 mAChR (Jones et al., 2008; Budzik et al., 2010; Nathan et al., 2013). To test this assertion, [3H]-NMS binding studies were conducted on monolayers of CHO Flp-In cells expressing the human M1 mAChR to determine the nature of their interaction at the receptor. Both GSK1034702 and TBPB fully inhibited binding of [3H]-NMS (0.5 nM) to M1 mAChRs, with estimated pKi values of 6.5 ± 0.2 and 6.8 ± 0.1, respectively, and in a similar manner to the orthosteric agonist, xanomeline (Fig. 2; Table 1). These data suggest that GSK1034702, in contrast to the allosteric mechanism of action previously reported, binds instead in a competitive manner consistent with interaction at the orthosteric site of the M1 mAChR.
Fig. 2.

Displacement of [3H]-NMS binding by ACh, xanomeline, GSK1034702, or TBPB at the human M1 mAChR expressed in CHO Flp-In cell monolayers. Experiments were performed against a KD concentration of [3H]-NMS. Nonspecific binding was determined by the addition of 10 μM atropine. Data are expressed as the means ± S.E.M. of three to five independent experiments performed in duplicate.
TABLE 1.
Affinity estimates for the competition between [3H]-NMS and ACh, xanomeline, GSK1034702, or TBPB at the M1 mAChR
Values stated are the negative logarithms of the equilibrium dissociation constant (pKi). Data are calculated from the means ± S.E.M. of three to five independent experiments performed in duplicate.
| Compound | pKi | n |
|---|---|---|
| ACh | 4.1 ± 0.3 | 5 |
| Xanomeline | 7.0 ± 0.1 | 4 |
| GSK1034702 | 6.5 ± 0.2 | 3 |
| TBPB | 6.8 ± 0.1 | 3 |
GSK1034702 and TBPB Do Not Alter [3H]-NMS Dissociation Kinetics.
As allosteric ligands with high negative cooperativity can still fully inhibit orthosteric ligand binding, kinetic binding experiments were performed to probe any allosteric interactions of GSK1034702 or TBPB with the M1 mAChR. Membranes of CHO Flp-In cells expressing the human M1 mAChR were pre-equilibrated with [3H]-NMS, and bound radioligand dissociated from the receptor with atropine (10 μM) with a rate constant of koff = 0.188 ± 0.009 minute−1. The presence of either GSK1034702 (10 μM) or TBPB (10 μM) had no effect on the [3H]-NMS dissociation rate (Fig. 3). These data further argue against an allosteric mode of action of GSK1034702 at M1 mAChRs as previously reported (Nathan et al., 2013).
Fig. 3.

(A and B) Dissociation of [3H]-NMS with atropine in the absence or presence of GSK1034702 (A) or TBPB (B) in membranes expressing the human M1 receptor. Membranes were incubated with a KD concentration of [3H]-NMS for 60 minutes at 37°C, followed by dissociation of bound radioligand with atropine (10 μM) alone or in the presence of GSK1034702 (10 μM) or TBPB (10 μM). Data shown are the mean of three independent experiments performed in duplicate.
Receptor Alkylation Studies Establish Orthosteric Binding of GSK1034702.
GSK1034702 activity in IP1 accumulation assays was compared with ACh, xanomeline, and TBPB (Fig. 4A; Table 2). GSK1034702 stimulated robust increases in IP1 accumulation, reaching approximately 90% of the maximal response elicited by ACh, with nanomolar potency (pEC50 = 7.1 ± 0.1; Fig. 4A). TBPB behaved as a partial agonist with a modest increase in potency relative to GSK1034702 (pEC50 = 7.6 ± 0.2). In membranes prepared from the rat cortex, GSK1034702 is a partial agonist with respect to Gαq protein coupling, stimulating approximately 37% of the maximum [35S]-GTPγS Gαq binding elicited by the full agonist oxotremorine-M (pEC50 = 6.7 ± 0.1; Fig. 4B).
Fig. 4.

(A) IP accumulation elicited by ACh, xanomeline, GSK1034702, or TBPB via the human M1 receptor expressed in CHO Flp-In cells. Data are expressed as the means ± S.E.M. of 3–10 independent experiments performed in duplicate. (B) [35S]-GTPγS binding to rat frontal cortex membranes. Data are the percentage of the maximal [35S]-GTPγS binding stimulated by oxotremorine-M (mean pEC50 = 6.68 ± 0.13, n = 3). OXO-M, oxotremorine-M.
TABLE 2.
Maximum agonist effect and potency of ACh, xanomeline, GSK1034702, and TBPB at stimulating IP1 accumulation in CHO Flp-In human M1 cells
Data are expressed as the means ± S.E.M. of 3–10 independent experiments performed in duplicate.
| Compound | IP1 |
||
|---|---|---|---|
| Emax | pEC50 | n | |
| ACh | 100 | 7.1 ± 0.1 | 10 |
| Xanomeline | 98.0 ± 1.5 | 8.2 ± 0.1 | 3 |
| GSK1034702 | 90.1 ± 2.9 | 7.1 ± 0.1 | 3 |
| TBPB | 55.2 ± 3.1 | 7.6 ± 0.2 | 4 |
To verify the mechanism of action, we performed receptor alkylation experiments with the orthosteric site covalent binder, phenoxybenzamine, to deplete the level of available and functional muscarinic receptors. Phenoxybenzamine, at a concentration of 3 μM (for 30 minutes), reduced the functional human M1 mAChR population in CHO Flp-In cells by approximately 80%, to an expression level where GSK1034702 had no agonist effect in an IP1 accumulation assay but where ACh still yielded a response (Supplemental Fig. 2, A and B). Under these conditions, establishing whether GSK1034702 and TBPB acted as competitive antagonists with respect to ACh would verify the interaction of these compounds with the orthosteric site.
In phenoxybenzamine-treated cells, GSK1034702 caused a concentration-dependent, parallel rightward shift in ACh-stimulated IP1 accumulation (Fig. 5A) consistent with a competitive antagonist. This effect was similar to that of TBPB (Fig. 5B), which was reported previously to act as a competitive antagonist in a similar preparation (Keov et al., 2013). Analysis of these data using a modified form of the Gaddum and Schild equations yielded Schild slopes approximating to unity and pA2 values of 6.2 ± 0.2 and 7.0 ± 0.1 for GSK1034702 and TBPB antagonism of ACh-stimulated responses, respectively (Table 3).
Fig. 5.

(A and B) GSK1034702 (A) or TBPB (B) antagonism of ACh-stimulated IP accumulation in CHO Flp-In cells expressing the human M1 receptor. Cells were incubated with 3 μM phenoxybenzamine to irreversibly reduce receptor expression prior to the addition of GSK1034702 or TBPB. Data are expressed as the means ± S.E.M. of three independent experiments performed in duplicate.
TABLE 3.
Potency estimates of the antagonism of ACh-stimulated IP1 accumulation by GSK1034702 or TBPB in CHO Flp-In cells expressing the human M1 mAChR
Data are the means ± S.E.M. of three independent experiments performed in duplicate.
| Parameter | GSK1034702 | TBPB | n |
|---|---|---|---|
| pA2 | 6.2 ± 0.2 | 7.0 ± 0.1 | 3 |
| Schild slope | 1.1 ± 0.1 | 1.2 ± 0.1 | 3 |
The Prototypical PAM, BQCA, Potentiates ACh, But Not GSK1034702 Affinity.
Having established that GSK1034702 interacts competitively with ACh at the orthosteric site, radioligand binding experiments were designed to establish whether the mode of GSK1034702 binding at the orthosteric pocket was equivalent to ACh. In these studies, the potentiation of orthosteric agonist binding by a PAM was used to probe the nature of GSK1034702 and TBPB binding. BQCA, a PAM selective for the M1 mAChR, has previously been shown to potentiate ACh affinity by approximately 100-fold (Ma et al., 2009; Butcher et al., 2016). Consistent with these previous studies, we show here that BQCA potentiates the ACh-mediated displacement of [3H]-NMS, thereby demonstrating positive cooperativity for ACh binding of approximately 35-fold, consistent with previous reports of modulation according to a two-state model (Canals et al., 2012; Fig. 6A). Such actions would predict a similar (if less substantial) effect on GSK1034702 or TBPB. However, the displacement of [3H]-NMS by GSK1034702 (Fig. 6B) or TBPB (Fig. 6C) was not modulated by BQCA. These data demonstrate the probe dependency of BQCA and indicate that either: 1) there is neutral cooperativity between BQCA and GSK1034702/TBPB, or 2) that the binding site of GSK1034702 or TBPB simultaneously overlaps with those of both ACh and BQCA.
Fig. 6.

(A–C) Displacement of [3H]-NMS binding by ACh (A), GSK1034702 (B), or TBPB (C) in the presence of increasing concentrations of BQCA at the human M1 mAChR expressed in CHO Flp-In cell monolayers. Experiments were performed against a KD concentration of [3H]-NMS. Nonspecific binding was determined by the addition of 10 μM atropine. Data are expressed as the means ± S.E.M. of three independent experiments performed in duplicate.
[3H]-GSK1034702 Binding Studies Further Confirm Novel Orthosteric Binding Pose.
To further characterize the binding site of GSK1034702 at the M1 mAChR, we generated a radiolabeled version of the GSK1034702 compound ([3H]-GSK1034702) and conducted binding interaction experiments in membranes expressing the human M1 mAChR. [3H]-GSK1034702 bound in a monophasic manner with moderate affinity (KD = 550 nM; Bmax = 2.6 pmol/mg protein) (Supplemental Fig. 3, A–C). Membranes were incubated with 20 nM [3H]-GSK1034702 in the absence and presence of increasing concentrations of unlabeled GSK1034702, TBPB, ACh, and xanomeline (Fig. 7). GSK1034702 and TBPB fully displaced specific [3H]-GSK1034702 binding to the M1 mAChR in a monophasic manner, whereas ACh and xanomeline only partially displaced [3H]-GSK1034702 binding. These data indicate either an allosteric interaction between GSK1034702 and ACh/xanomeline or a bitopic mechanism consistent with GSK1034702 spanning a binding pocket at the M1 mAChR partially shared with that of ACh.
Fig. 7.

Displacement of [3H]-GSK1034702 binding by ACh, xanomeline, GSK1034702, or TBPB at membranes expressing the human M1 mAChR. Experiments were performed against 20 nM [3H]-GSK1034702 and nonspecific binding was determined in the presence of 100 μM nonradiolabeled GSK1034702. Data are expressed as the means ± S.E.M. of three independent experiments performed in duplicate.
DREADD Pharmacology Confirms an Atypical Mechanism of Action of GSK1034702.
By introducing point mutations (Y106C and A196G) into the orthosteric binding pocket of the M1 mAChR, an M1 DREADD mutant is created (Abdul-Ridha et al., 2013) that displays reduced responsiveness to ACh but instead is activated by clozapine-N-oxide, a ligand that shows little activity at the wild-type M1 mAChR. We investigated the ability of GSK1034702 to interact with the M1 DREADD by conducting [3H]-NMS binding, functional IP1 accumulation, and ERK1/2 phosphorylation studies in CHO Flp-In cells expressing the humanized M1 DREADD (Figs. 8 and 9; Supplemental Tables 1 and 2). The affinity of GSK1034702 for the M1 DREADD was not significantly different from the affinity for binding at the wild-type M1 mAChR (pKi = 6.5 ± 0.2 and 6.0 ± 0.2 for the wild type and DREADD, respectively; Fig. 8). In addition, as reported previously (Armbruster et al., 2007; Abdul-Ridha et al., 2013), M1 mAChR orthosteric agonists, ACh and xanomeline, showed a significant reduction in potency at the M1 DREADD (Fig. 9). In contrast, GSK1034702 activated the M1 DREADD with comparable potency and efficacy compared with its activity at the wild-type receptor. Similar results were obtained with TBPB, confirming previous observations for this compound (Abdul-Ridha et al., 2014) (Fig. 9, D and E). Furthermore, we assessed the ability of ACh and GSK1034702 to stimulate phosphorylation of the M1 mAChR at serine 228 using a phosphorylation-specific antibody (Butcher et al., 2016). Both ACh and GSK1034702 stimulated a concentration-dependent increase in pSer228 immunoreactivity at the wild-type receptor (Fig. 10). The potency of GSK1034702 to stimulate phosphorylation at serine 228 was unchanged at the M1 DREADD, whereas ACh failed to stimulate a response. These data support the notion that GSK1034702 has a distinct binding mode at the orthosteric site from that of ACh.
Fig. 8.

(A) Displacement of [3H]-NMS binding by ACh, CNO, xanomeline, GSK1034702, or TBPB at the humanized M1 DREADD expressed in CHO Flp-In cell monolayers. Experiments were performed against a KD concentration of [3H]-NMS. Nonspecific binding was determined by the addition of 10 μM atropine. Data are expressed as the means ± S.E.M. of three to five independent experiments performed in duplicate. (B) Comparison of pKi values for each of the compounds used at the wild-type M1 mAChR or the mutant M1 DREADD. CNO, clozapine-N-oxide; WT, wild type.
Fig. 9.

(A–E) IP accumulation (left) or ERK1/2 phosphorylation (right) elicited by ACh (A), xanomeline (B), CNO (C), GSK1034702 (D), or TBPB (E) via the humanized M1 DREADD expressed in CHO Flp-In cells. The dashed curve represents the response of the ligand at the wild-type M1 mAChR. Data are expressed as the means ± S.E.M. of three to four independent experiments performed in duplicate. CNO, clozapine-N-oxide; PI, phosphoinositide.
Fig. 10.

(A) Phosphorylation at serine 228 elicited by ACh (left) or GSK1034702 (right) in CHO Flp-In cells expressing either M1 WT (top) or M1 DREADD (bottom). (B) Mean densitometric data showing phosphorylation at serine 228 as a percentage of the maximal response. Data were normalized to the total receptor expression, assessed using an HA antibody. Data are expressed as the means ± S.E.M. of three independent experiments. HA, hemagglutinin; WT, wild type.
GSK1034702 and TBPB Are Differentially Biased Agonists at M1 mAChR.
Convergent evidence from radioligand binding and functional studies (vide supra and Supplemental Fig. 4; Supplemental Table 3) suggests that GSK1034702 and TBPB bind to the orthosteric site of the M1 mAChR, but with an orientation or pose that distinguishes them from ACh and xanomeline. To further interrogate their pharmacology, we assessed their ability to engender biased signaling by application of an operational model of agonism to the concentration-response curves of either xanomeline, GSK1034702, or TBPB in both IP1 and ERK1/2 phosphorylation assays (Evans et al., 2011; Keov et al., 2011; Kenakin et al., 2012) (Table 4). These analyses generated transduction coefficient values for each of these agonists at the two different pathways and allowed us to calculate the bias factor between IP1 and ERK1/2 phosphorylation, revealing significant differences between ACh and xanomeline/TBPB (P < 0.001; one-way analysis of variance), with the latter displaying bias toward IP1 responses (bias factor IP1 − ERK1/2 phosphorylation = 1.1 and 1.4 for xanomeline and TBPB, respectively). However, no significant differences were revealed for GSK1034702 and ACh, suggesting that GSK1034702 and TBPB, despite apparently similar binding modes, engender differential signaling from the receptor.
TABLE 4.
Transduction coefficients [Log10(τ/KA)], normalized (reference ligand ACh) transduction coefficients [ΔLog10(τ/KA)], and bias factors [ΔΔLog10(τ/KA)] for IP1 accumulation and phosphorylation of ERK1/2 at the wild-type M1 mAChR
| Compound | IP1 Accumulation |
ERK1/2
Phosphorylation |
Log Bias Factor
IP1 – ERK1/2 |
||
|---|---|---|---|---|---|
| Log10(τ/KA) | ΔLog10(τ/KA) | Log10(τ/KA) | ΔLog10(τ/KA) | ΔΔLog10(τ/KA) | |
| ACh | 7.10 ± 0.1 | 0.0 ± 0.1 | 7.1 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.1 |
| Xanomeline | 8.2 ± 0.1 | 1.1 ± 0.1 | 7.1 ± 0.3 | 0.0 ± 0.3 | 1.1 ± 0.1 |
| GSK1034702 | 7.4 ± 0.2 | 0.3 ± 0.2 | 7.2 ± 0.1 | 0.1 ± 0.1 | 0.2 ± 0.2 |
| TBPB | 8.2 ± 0.2 | 1.1 ± 0.2 | 6.9 ± 0.4 | −0.3 ± 0.4 | 1.4 ± 0.2 |
GSK1034702 Shows a Lack of Selectivity for M1 mAChRs.
We evaluated the ability of GSK1034702 to bind to other mAChR subtypes by conducting equilibrium-binding studies on membranes expressing the M1, M2, M3, M4, or M5 mAChR, and we found that GSK1034702 could inhibit [3H]-NMS binding at all muscarinic receptor subtypes, albeit with much lower affinity for the M3 mAChR (Fig. 11A; Table 5). We further assessed the functional activity of GSK1034702 at the M2, M3, M4, and M5 mAChRs in the ERK1/2 phosphorylation assay. GSK1034702 exhibited partial agonist activity at M2, M4, and M5 mAChRs but was devoid of activity at the M3 mAChR in this assay (Fig. 11B). Finally, we investigated the ability of GSK1034702 to stimulate negative inotropic responses in isolated rat atria (Fig. 12, A and B) or contraction of rat ileum (Fig. 12, C and D), indicative of activity at M2 and M3 mAChRs, respectively. GSK1034702 elicited a robust response in the rat atria, reaching a maximal response equivalent to that of methacholine, with micromolar potency (Fig. 12A). Furthermore, GSK1034702 could inhibit methacholine-induced responses with an IC50 of 8 μM (Fig. 12B). In the rat ileum, GSK1034702 stimulated approximately 50% of the maximal methacholine-induced contraction, with an EC50 of 7 μM (Fig. 12C), and inhibited methacholine-induced contraction with an IC50 of 46 μM (Fig. 12D).
Fig. 11.

(A) Displacement of [3H]-NMS binding by GSK1034702 in CHO membranes expressing M1, M2, M3, M4, or M5 mAChRs. Experiments were performed against a KD concentration of [3H]-NMS. Nonspecific binding was determined by the addition of 10 μM atropine. Data are expressed as the means ± S.E.M. of three independent experiments. (B) ERK1/2 phosphorylation elicited by GSK1034702 at the M1, M2, M3, M4, or M5 mAChR expressed in CHO cells. Data are expressed as a percentage of the maximum response stimulated by ACh and are the means ± S.E.M. of three experiments performed in duplicate.
TABLE 5.
Negative logarithms of the equilibrium dissociation constant (pKi) of GSK1034702 binding to M1–M5 mAChRs
Data are calculated from the means ± S.E.M. of three independent experiments performed in duplicate.
| mAChR subtype | pKi | n |
|---|---|---|
| M1 | 6.0 ± 0.1 | 3 |
| M2 | 5.4 ± 0.1 | 3 |
| M3 | n.d. | 3 |
| M4 | 5.7 ± 0.1 | 3 |
| M5 | 5.2 ± 0.1 | 3 |
n.d., not determined.
Fig. 12.

(A–D) Assessment of the effects of GSK1034703 activity at M2 (A and B) and M3 (C and D) mAChRs in ex vivo tissue preparations. The ability of GSK1034702 to stimulate negative inotropy in rat atria (A) or inhibit methacholine (1 μM)–induced responses (B) was assessed. Activity of GSK1034702 at the M3 mAChR was assessed by measurement of rat ileum contraction relative to methacholine responses (C) or inhibition of methacholine-induced contraction (D). Data shown are a single experiment.
Discussion
The development of the selective M1 mAChR allosteric agonist GSK1034702 provided the opportunity to test the hypothesis that allosteric M1 mAChR drugs might provide a clinical advantage over orthosteric M1 mAChR agents due to increased selectivity while yielding fewer side effects. In the nicotine abstinence model of cognitive dysfunction, GSK1034702 significantly improved immediate memory recall but also induced adverse responses consistent with activation of other muscarinic receptor subtypes (Nathan et al., 2013). At first glance, these data might suggest that allosteric M1 mAChR drugs offer little or no safety benefit compared with previous investigational agents targeting mAChRs. However, here we provide direct pharmacological evidence that GSK1034702 is not a pure allosteric agonist as previously reported, but rather interacts with the orthosteric binding site and broadly mimics the pharmacology of the known bitopic ligand, TBPB. Based on radioligand binding and functional studies, coupled with the structural similarity between GSK1034702 and TBPB, we conclude that GSK1034702 likely interacts concomitantly with both allosteric and orthosteric sites on the M1 mAChR in a bitopic manner.
The conclusion that GSK1034702 interacts with the orthosteric site is primarily based on the full inhibition of [3H]-NMS binding and a lack of cooperative effects on [3H]-NMS, features consistent with an orthosteric rather than prototypical allosteric mechanism. In functional assays after receptor alkylation (to diminish its agonist response), GSK1034702 causes a nonsaturable, concentration-dependent parallel rightward shift in the ACh-mediated IP response, further confirming an orthosteric mode of action.
That GSK1034702 might also interact with a site distinct from the orthosteric site was indicated most clearly in functional assays at the M1 mAChR DREADD and by characterizing the binding of [3H]-GSK1034702 to the wild-type receptor. The M1 mAChR DREADD contains mutations at key residues within the orthosteric binding pocket, which yields a receptor that is poorly responsive to the cognate ligand, ACh, but instead is activated by an otherwise inert chemical ligand, clozapine-n-oxide (Armbruster et al., 2007; Roth, 2016). As predicted from previous reports (Abdul-Ridha et al., 2014), the potencies of orthosteric ligands ACh and xanomeline in IP signaling, ERK1/2 phosphorylation, and M1 receptor phosphorylation were significantly reduced at the M1 DREADD. However, the potency and efficacy of GSK1034702 and TBPB was unaffected by the DREADD mutations, suggesting that GSK1034702, like TBPB, is able to activate the M1 mAChR with a binding mode that is subtly distinct from that of prototypical orthosteric ligands. In support of this conclusion, GSK1034702 and TBPB fully inhibit the binding of [3H]-GSK1034702 to the M1 mAChR, whereas the prototypical orthosteric agonists, ACh and xanomeline, only partially inhibit its binding. This indicates that GSK1034702 can still bind to the M1 mAChR when ACh or xanomeline occupy the orthosteric site, suggesting that it can interact with the receptor via an allosteric binding site. The display of apparently both orthosteric (competitive) and allosteric behaviors depending on test system is typical of bitopic ligands that are able to “flip-flop” between binding poses (Valant et al., 2012).
Greater clarity around the receptor mechanism of action of GSK1034702, revealed here, has implications for drug design aimed at the treatment of AD. We have previously demonstrated that the learning and memory deficit observed in murine prion disease is due to a loss of cholinergic signaling in the hippocampus and as such, this model replicates one of the key pathologic hallmarks associated with human AD (Bradley et al., 2017). In the prion model, we found that both M1 mAChR orthosteric agonists and allosteric modulators completely rescue the learning and memory deficit observed in prion disease. However, we also found that the orthosteric ligand, xanomeline, gave adverse responses consistent with the activation of other muscarinic receptor subtypes, whereas the PAM, BQCA, gave no detectable adverse responses at doses that rescued learning and memory (Bradley et al., 2017). Although these results together with other studies on the cognitive responses of M1 mAChR allosteric modulators (Ma et al., 2009; Lange et al., 2015; Puri et al., 2015; Vardigan et al., 2015) support the potential clinical benefit of this class of ligand, it is also clear that allosteric modulators that show direct agonism in addition to cooperativity in rodent models result in adverse effects (Alt et al., 2016; Davoren et al., 2016, 2017). Hence, we conclude here that to avoid adverse effects, clinical candidates targeting the M1 mAChR in AD would require the following properties: 1) high levels of receptor subtype selectivity as would be seen with an allosteric modulator and 2) low levels of intrinsic agonist activity.
Acknowledgments
The authors acknowledge the Biological Services Unit facilities at the Cancer Research UK Beatson Institute (C596/A17196).
Abbreviations
- ACh
acetylcholine
- AD
Alzheimer disease
- BQCA
benzyl quinolone carboxylic acid
- CHO
Chinese hamster ovary
- DREADD
designer receptor exclusively activated by designer drug
- ERK
extracellular signal–regulated kinase
- GTPɣ[35S], [35S]ɣ [guanosine-5′-O-(3-[35S]thio)triphosphate]; GSK1034702
7-fluoro-5-methyl-3-[1-(oxan-4-yl)piperidin-4-yl]-1H-benzimidazol-2-one
- IP
inositol phosphate
- mAChR
muscarinic acetylcholine receptor
- NMS
N-methylscopolamine
- PAM
positive allosteric modulator
- TBPB
1-[1′-(2-methylbenzyl)-1,4′-bipiperidin-4-yl]-1,3-dihydro-2H-benzimidazol-2-one
Authorship Contributions
Participated in research design: Bradley, Sexton, Felder, Christopoulos, Broad, Tobin, Langmead.
Conducted experiments: Bradley, Molloy, Bundgaard, Mogg, Thompson, Dwomoh, Sanger, Crabtree, Brooke.
Contributed new reagents or analytic tools: Felder, Broad.
Performed data analysis: Bradley, Christopoulos, Tobin, Langmead.
Wrote or contributed to the writing of the manuscript: Bradley, Christopoulos, Broad, Tobin, Langmead.
Footnotes
This work was supported in part by the Wellcome Trust [Collaborative Award 201529/Z/16/Z], the Royal Society [International Exchanges Scheme Award IE131060], the Research Councils UK [Medical Research Council Industry Collaboration Agreement Award MR/P019366/1], the University of Glasgow Lord Kelvin Adam Smith Fellowship, and the Eli Lilly Company.
 This article has supplemental material available
at molpharm.aspetjournals.org.
This article has supplemental material available
at molpharm.aspetjournals.org.
References
- Abdul-Ridha A, Lane JR, Sexton PM, Canals M, Christopoulos A. (2013) Allosteric modulation of a chemogenetically modified G protein-coupled receptor. Mol Pharmacol 83:521–530. [DOI] [PubMed] [Google Scholar]
- Abdul-Ridha A, López L, Keov P, Thal DM, Mistry SN, Sexton PM, Lane JR, Canals M, Christopoulos A. (2014) Molecular determinants of allosteric modulation at the M1 muscarinic acetylcholine receptor. J Biol Chem 289:6067–6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alt A, Pendri A, Bertekap RL, Jr, Li G, Benitex Y, Nophsker M, Rockwell KL, Burford NT, Sum CS, Chen J, et al. (2016) Evidence for classical cholinergic toxicity associated with selective activation of M1 muscarinic receptors. J Pharmacol Exp Ther 356:293–304. [DOI] [PubMed] [Google Scholar]
- Anagnostaras SG, Murphy GG, Hamilton SE, Mitchell SL, Rahnama NP, Nathanson NM, Silva AJ. (2003) Selective cognitive dysfunction in acetylcholine M1 muscarinic receptor mutant mice. Nat Neurosci 6:51–58. [DOI] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. (2007) Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci USA 104:5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodick NC, Offen WW, Levey AI, Cutler NR, Gauthier SG, Satlin A, Shannon HE, Tollefson GD, Rasmussen K, Bymaster FP, et al. (1997) Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch Neurol 54:465–473. [DOI] [PubMed] [Google Scholar]
- Bradley SJ, Bourgognon JM, Sanger HE, Verity N, Mogg AJ, White DJ, Butcher AJ, Moreno JA, Molloy C, Macedo-Hatch T, et al. (2017) M1 muscarinic allosteric modulators slow prion neurodegeneration and restore memory loss. J Clin Invest 127:487–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley NJ, Bonner TI, Brann MR. (1988) Localization of a family of muscarinic receptor mRNAs in rat brain. J Neurosci 8:4646–4652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budzik B, Garzya V, Shi D, Walker G, Woolley-Roberts M, Pardoe J, Lucas A, Tehan B, Rivero RA, Langmead CJ, et al. (2010) Novel N-substituted benzimidazolones as potent, selective, CNS-penetrant, and orally active M1 mAChR agonists. ACS Med Chem Lett 1:244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher AJ, Bradley SJ, Prihandoko R, Brooke SM, Mogg A, Bourgognon JM, Macedo-Hatch T, Edwards JM, Bottrill AR, Challiss RA, et al. (2016) An antibody biosensor establishes the activation of the M1 muscarinic acetylcholine receptor during learning and memory. J Biol Chem 291:8862–8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canals M, Lane JR, Wen A, Scammells PJ, Sexton PM, Christopoulos A. (2012) A Monod-Wyman-Changeux mechanism can explain G protein-coupled receptor (GPCR) allosteric modulation. J Biol Chem 287:650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW. (2009) Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov 8:41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davoren JE, Garnsey M, Pettersen B, Brodney MA, Edgerton JR, Fortin JP, Grimwood S, Harris AR, Jenkinson S, Kenakin T, et al. (2017) Design and synthesis of γ- and δ-lactam M1 positive allosteric modulators (PAMs): convulsion and cholinergic toxicity of an M1-selective PAM with weak agonist activity. J Med Chem 60:6649–6663. [DOI] [PubMed] [Google Scholar]
- Davoren JE, Lee CW, Garnsey M, Brodney MA, Cordes J, Dlugolenski K, Edgerton JR, Harris AR, Helal CJ, Jenkinson S, et al. (2016) Discovery of the potent and selective M1 PAM-agonist N-[(3R,4S)-3-hydroxytetrahydro-2H-pyran-4-yl]-5-methyl-4-[4-(1,3-thiazol-4-yl)benzyl]pyridine-2-carboxamide (PF-06767832): evaluation of efficacy and cholinergic side effects. J Med Chem 59:6313–6328. [DOI] [PubMed] [Google Scholar]
- DeLapp NW, McKinzie JH, Sawyer BD, Vandergriff A, Falcone J, McClure D, Felder CC. (1999) Determination of [35S]guanosine-5'-O-(3-thio)triphosphate binding mediated by cholinergic muscarinic receptors in membranes from Chinese hamster ovary cells and rat striatum using an anti-G protein scintillation proximity assay. J Pharmacol Exp Ther 289:946–955. [PubMed] [Google Scholar]
- Evans BA, Broxton N, Merlin J, Sato M, Hutchinson DS, Christopoulos A, Summers RJ. (2011) Quantification of functional selectivity at the human α(1A)-adrenoceptor. Mol Pharmacol 79:298–307. [DOI] [PubMed] [Google Scholar]
- Jones CK, Brady AE, Davis AA, Xiang Z, Bubser M, Tantawy MN, Kane AS, Bridges TM, Kennedy JP, Bradley SR, et al. (2008) Novel selective allosteric activator of the M1 muscarinic acetylcholine receptor regulates amyloid processing and produces antipsychotic-like activity in rats. J Neurosci 28:10422–10433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S. (2012) A simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci 3:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keov P, López L, Devine SM, Valant C, Lane JR, Scammells PJ, Sexton PM, Christopoulos A. (2014) Molecular mechanisms of bitopic ligand engagement with the M1 muscarinic acetylcholine receptor. J Biol Chem 289:23817–23837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keov P, Sexton PM, Christopoulos A. (2011) Allosteric modulation of G protein-coupled receptors: a pharmacological perspective. Neuropharmacology 60:24–35. [DOI] [PubMed] [Google Scholar]
- Keov P, Valant C, Devine SM, Lane JR, Scammells PJ, Sexton PM, Christopoulos A. (2013) Reverse engineering of the selective agonist TBPB unveils both orthosteric and allosteric modes of action at the M(1) muscarinic acetylcholine receptor. Mol Pharmacol 84:425–437. [DOI] [PubMed] [Google Scholar]
- Lambrecht G, Feifel R, Wagner-Roder M, Strohmann C, Zilch H, Tacke R, Waelbroeck M, Christophe J, Boddeke H, Mutschler E. (1989) Affinity profiles of hexahydro-sila-difenidol analogues at muscarinic receptor subtypes. Eur J Pharmacol 168:71–80. [DOI] [PubMed] [Google Scholar]
- Lange HS, Cannon CE, Drott JT, Kuduk SD, Uslaner JM. (2015) The M1 muscarinic positive allosteric modulator PQCA improves performance on translatable tests of memory and attention in rhesus monkeys. J Pharmacol Exp Ther 355:442–450. [DOI] [PubMed] [Google Scholar]
- Langmead CJ, Christopoulos A. (2006) Allosteric agonists of 7TM receptors: expanding the pharmacological toolbox. Trends Pharmacol Sci 27:475–481. [DOI] [PubMed] [Google Scholar]
- Langmead CJ, Watson J, Reavill C. (2008) Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol Ther 117:232–243. [DOI] [PubMed] [Google Scholar]
- Leach K, Loiacono RE, Felder CC, McKinzie DL, Mogg A, Shaw DB, Sexton PM, Christopoulos A. (2010) Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology 35:855–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G, Langmead CJ, Tehan BG, Hulme EC. (2009) Mutagenic mapping suggests a novel binding mode for selective agonists of M1 muscarinic acetylcholine receptors. Mol Pharmacol 75:331–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey AI, Edmunds SM, Koliatsos V, Wiley RG, Heilman CJ. (1995) Expression of m1-m4 muscarinic acetylcholine receptor proteins in rat hippocampus and regulation by cholinergic innervation. J Neurosci 15:4077–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Seager MA, Wittmann M, Jacobson M, Bickel D, Burno M, Jones K, Graufelds VK, Xu G, Pearson M, et al. (2009) Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation [published correction appears in Proc Natl Acad Sci USA (2009) 106:18040]. Proc Natl Acad Sci USA 106:15950–15955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulos A. (2007) Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol 47:1–51. [DOI] [PubMed] [Google Scholar]
- Nathan PJ, Watson J, Lund J, Davies CH, Peters G, Dodds CM, Swirski B, Lawrence P, Bentley GD, O’Neill BV, et al. (2013) The potent M1 receptor allosteric agonist GSK1034702 improves episodic memory in humans in the nicotine abstinence model of cognitive dysfunction. Int J Neuropsychopharmacol 16:721–731. [DOI] [PubMed] [Google Scholar]
- Puri V, Wang X, Vardigan JD, Kuduk SD, Uslaner JM. (2015) The selective positive allosteric M1 muscarinic receptor modulator PQCA attenuates learning and memory deficits in the Tg2576 Alzheimer’s disease mouse model. Behav Brain Res 287:96–99. [DOI] [PubMed] [Google Scholar]
- Roth BL. (2016) DREADDs for neuroscientists. Neuron 89:683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spalding TA, Trotter C, Skjaerbaek N, Messier TL, Currier EA, Burstein ES, Li D, Hacksell U, Brann MR. (2002) Discovery of an ectopic activation site on the M(1) muscarinic receptor. Mol Pharmacol 61:1297–1302. [DOI] [PubMed] [Google Scholar]
- Valant C, Robert Lane J, Sexton PM, Christopoulos A. (2012) The best of both worlds? Bitopic orthosteric/allosteric ligands of g protein-coupled receptors. Annu Rev Pharmacol Toxicol 52:153–178. [DOI] [PubMed] [Google Scholar]
- van der Westhuizen ET, Breton B, Christopoulos A, Bouvier M. (2014) Quantification of ligand bias for clinically relevant β2-adrenergic receptor ligands: implications for drug taxonomy. Mol Pharmacol 85:492–509. [DOI] [PubMed] [Google Scholar]
- Vardigan JD, Cannon CE, Puri V, Dancho M, Koser A, Wittmann M, Kuduk SD, Renger JJ, Uslaner JM. (2015) Improved cognition without adverse effects: novel M1 muscarinic potentiator compares favorably to donepezil and xanomeline in rhesus monkey. Psychopharmacology (Berl) 232:1859–1866. [DOI] [PubMed] [Google Scholar]
- Witkin JM, Ornstein PL, Mitch CH, Li R, Smith SC, Heinz BA, Wang XS, Xiang C, Carter JH, Anderson WH, et al. (2017) In vitro pharmacological and rat pharmacokinetic characterization of LY3020371, a potent and selective mGlu2/3receptor antagonist. Neuropharmacology 115:100–114. [DOI] [PubMed] [Google Scholar]
- Young SL, Bohenek DL, Fanselow MS. (1995) Scopolamine impairs acquisition and facilitates consolidation of fear conditioning: differential effects for tone vs context conditioning. Neurobiol Learn Mem 63:174–180. [DOI] [PubMed] [Google Scholar]