

Abstract
Chronic elevation of interferon (IFN)-response genes (IRG) in a subset of patients with systemic immune-dysregulatory diseases, including the Mendelian Type-I IFN-mediated autoinflammatory diseases and some autoimmune diseases suggest a causative role of excessive IFN signaling in the disease pathogenesis and as target for treatment. We developed a 28-IFN response gene scoring system to calculate either a standardized or geomean score by customizing a NanoString assay to quantify the expression of putative IRGs. The gene targets were selected in patients with the IFN-mediated disease chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) and an adult patient with chronic hepatitis C who received the first dose of pegylated interferon alpha-2a. The putative target genes were validated in patients with STING-associated vasculopathy with onset in infancy (SAVI), a monogenic autoinflammatory disease caused by gain-of-function mutations in TMEM173 that encodes the viral sensor stimulator of IFN genes (STING), and had low expression in clinically active patients with the monogenic IL-1-mediated autoinflammatory disease, neonatal-onset multisystem inflammatory disease (NOMID) and in healthy controls. The score calculation on the NanoString assay is rapid and showed high reproducibility and low intra-, and interassay variability. The utility of this 28-gene IFN score may be explored in the diagnosis of patients with presumed interferonopathies and as a biomarker to assess disease activity, long-term outcome, and treatment responses.
Keywords: : IFN score, NanoString assay, blood IFN signature, assay validation, IFN-mediated autoinflammatory diseases, interferonopathies
Introduction
Interferons or IFNs (Type I, II, and III) control and coordinate numerous homeostatic and pathological processes in infections, cancer, autoimmunity, and innate inflammatory and metabolic disorders through upregulation of “IFN regulated genes” (compiled in a database: www.interferome.org) (Rusinova and others 2013) that regulate cell proliferation, survival, migration, and other specialized functions. Elevated IFN levels have been detected in noninfectious diseases, such as systemic lupus erythematosus (SLE) and Sjögren's syndrome and other autoimmune diseases since the 1970s (Hooks and others 1979; Ytterberg and Schnitzer 1982; Ronnblom and Alm 2001). Chronic elevation of IRG expressions were identified in gene expression studies and are reported in a growing number of conditions that includes their description in lupus erythematosus (Baechler and others 2003; Bennett and others 2003) and in juvenile dermatomyositis (JDM) (Baechler and others 2007). The IFN signature is referred to in the literature as “IFN-regulated gene signature” (IRS) or IFN-stimulated genes (ISGs) and includes viral response genes (Der and others 1998).
In 2006, the discovery of loss-of-function (LOF) mutations in the exonuclease TREX1 as genetic cause of a subset of noninfectious encephalopathies that present with high IFN alpha levels in the cerebrospinal fluid, called Aicardi–Goutières syndrome (AGS) spearheaded discoveries of LOF mutations in other enzymes that regulate nucleic acid metabolism (Crow and others 2006 and reviewed in Rodero and Crow 2016). These findings unleashed basic research that uncovered elevated cytoplasmic levels of self-nucleic acids that trigger viral nucleic acid sensors as molecular mechanisms that lead to a chronically elevated IFN-regulated gene signature in AGS, and forged the concept of a group of conditions with presumed IFN-mediated pathology termed “Type I interferonopathies,” (Crow 2011; Rodero and Crow 2016).
The IFN signature had historically been described in patients with autoimmune diseases, where the organ damage had been attributed to predominantly adaptive immune cell dysregulation. The discovery that mutations in innate immune pathways and homeostatic pathways that regulate nucleic acid sensing cause AGS, followed by gain-of-function (GOF) mutations in the viral sensor adaptor, TMEM173, that encodes the stimulator of IFN genes (STING) protein cause the Mendelian autoinflammatory disease, SAVI (Liu and others 2014), and by LOF mutations in PSMB8 and PSMB4, and other proteasome components that regulate protein homeostasis cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) (Liu and others 2012; Brehm and others 2015), respectively, suggests a role of type I IFN in amplifying innate immune dysregulation as a major driver of the disease pathogenesis (Kim and others 2016).
Although multiple assays to detect the IRS have been described, they are typically limited to 5–6 target genes using quantitative polymerase chain reaction (qPCR) assays (Feng and others 2006; Yao and others 2009; Higgs and others 2011; Rice and others 2013, 2017), which can be difficult to standardize, particularly between centers (Fryer and others 2008; Bustin and others 2009). Furthermore, a systematic validation of the specific IRS target genes in known IL-1-mediated diseases has not been done.
We aimed to define an IRS score in CANDLE and SAVI patients that would allow us to rapidly distinguish patients with presumed IFN-mediated disease from those with presumed IL-1-mediated autoinflammatory diseases, particularly the prototypic IL-1β-mediated disease, neonatal-onset multisystem inflammatory disease (NOMID), which is caused by GOF mutations in NLRP3, which leads to constitutive activation of the NLRP3 inflammasome and IL-1β production (Hoffman and Broderick 2016). Additionally, we wanted to develop an assay that could quantify the IFN signature rapidly and reproducibly, thus allowing for comparison between batches of samples and longitudinally. In search of a reliable technology, we found NanoString Technologies (Seattle, WA), which uses molecular “barcodes” and an optical scanner to detect and simultaneously count up to several hundred unique target gene sequences in a single hybridization reaction without requiring amplification (Geiss and others 2008).
The rapid turnaround and the reliability in producing selected gene expression signatures allows the development of a clinical test to assess and quantify IFN response gene expression in patients with presumed interferonopathies and potential use as a biomarker for longitudinal monitoring of disease activity or treatment responses.
Materials and Methods
Patients and healthy controls
The initial target gene selection was based on a peripheral blood microarray expression profile from 1 CANDLE patient with active disease before Janus kinase (JAK) inhibitor treatment and from 1 adult male patient with chronic hepatitis C obtained before starting and 6 h after the first dose of pegylated interferon alpha-2a (Pegasys) 180 μg subcutaneously (Rotman and others 2014).
For validation, we utilized blood samples from 26 healthy controls (HCs), 11 patients with genetically defined CANDLE before JAK inhibitor therapy, 7 patients with genetically defined SAVI before JAK inhibitor therapy, and 18 patients with active NOMID, of which 17 have heterozygous germline or mosaic mutations in NLRP3. Samples from 16 NOMID patients were obtained before initiation of IL-1 blocking therapies (Table 1) (1 NOMID patient was on a suboptimal dose of anakinra at the time of blood draw). SAVI is used as a positive IFN control as it is caused by GOF mutations in the viral sensor STING, which lead to constitutive STAT1 phosphorylation IFNβ expression and high expression of IRGs on gene expression data (Liu and others 2014). NOMID patients have IL-1-mediated inflammation and serve as negative controls (Goldbach-Mansky and others 2006). For later characterization of IFN score in HCs, the 7 additional HCs who had no known illnesses or chronic inflammatory illnesses were used (total n = 33).
Table 1.
Demographics
| Diagnosis | % Female | Age (median, min–max years old) | Ethnicity |
|---|---|---|---|
| CANDLEa (n = 11) | 45.5 | 6.1 (1.7–19.5) | 4 white |
| 4 Hispanic | |||
| SAVIb (n = 7) | 42.9 | 16.9 (0.7–24.0) | 7 white |
| NOMIDc (n = 18) | 38.9 | 8.5 (0.8–42.2) | 12 white |
| 3 Hispanic | |||
| HCd (n = 26) | 65.4 | 25.9 (0.6–53.6) | 15 white |
| 8 Hispanic |
Other ethnicities: 2 black, 1 unknown. All with clinically active disease before Janus kinase inhibitor therapy, 7 with elevated C-reactive protein (CRP), 4 with rash.
All clinically active, 6 with elevated CRP, 1 with elevated erythrocyte sedimentation rate.
Other ethnicities: 1 black, 1 Asian, 1 multiple race. All clinically active; 16 before IL-1 blockade with elevated CRP, 2 on early IL-1 blockade with persistent cerebrospinal fluid pleocytosis.
Other ethnicities: 1 black, 1 multiple race, 1 unknown.
CANDLE, chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature; HC, healthy control; NOMID, neonatal-onset multisystem inflammatory disease; SAVI, STING-associated vasculopathy with onset in infancy; STING, stimulator of IFN genes.
The hepatitis patient and all other patients were recruited to IRB-approved studies, a clinical study (NCT00718172), and natural history protocol (NCT02974595) respectively; all patients, their parents, or the HCs had signed informed consent. The inclusion of increasing the number of CANDLE and SAVI patients over time in the validation experiments reflects on the addition of newly referred patients with CANDLE and SAVI to our cohort.
Materials
PAXgene tube (Qiagen, Germantown, MD) samples were collected and total RNA was extracted as recommended by the manufacturer (Qiagen). RNA integrity was analyzed with an Agilent 2100 Bioanalyzer. Initial gene selection was based on prior microarray data with Affymetrix HU-133 Plus 2.0 gene chips based on differential expression and pathway analysis as previously described (Liu and others 2012).
Gene expression measurement by NanoString and data processing
Gene expression of various target genes that are listed in the respective tables (Supplementary Fig. S1A, B; Supplementary Data are available online at www.liebertpub.com/jir) was measured initially using the NanoString custom-designed “CodeSets” (Supplementary Fig. S1A), and later for stable longitudinal use, the nCounter Elements Technology System (Supplementary Fig. S1B) was used, both according to the manufacturer's recommendations (NanoString Technologies, Seattle, WA) (www.nanostring.com).
Briefly, NanoString technology uses complementary DNA synthetic oligos to directly bind, capture, and tag mRNA molecules for counting with minimal sample preparation and without reverse transcription or amplification. The cartridges provided by the manufacturer allow 12 samples to be run at once and 200 ng of total RNA are loaded for each sample. The quality of the RNA extracted from total blood collected in PAXgene tubes is usually high, with an RNA integrity number (RIN) above 8. We have obtained results with RNA that had lower RIN numbers and with lower RNA concentrations, but we have not systematically tested the reliability of the assay at these low RNA concentrations with poor quality.
For the Elements system, Capture (probe A) and tag (probe B) probe DNA oligos were designed by NanoString and synthesized by Integrated DNA Technologies (IDT, Coralville, IA). Probe A and B each contain the gene-specific sequences; probe A also has a gene-specific sequence that binds the reporter tag, whereas probe B binds to a universal capture tag. Nucleotide sequences for probes A and B are listed in Supplementary Table S1A and B, respectively. After hybridization at 67°C for 24 h, samples can be stored at 4°C for up to 24 h. Both nCounter “Standard” and nCounter “Elements” sample runs were then processed using the “High Sensitivity” protocol option on the nCounter Prep Station and counted on nCounter Digital Analyzer using maximal data resolution (1,155 fields of view).
Data were processed with nSolver software (NanoString Technologies, Seattle, WA), which included assessment of quality of the runs. Data were combined, normalized, and analyzed in Excel (Microsoft Corporation, Seattle, WA). JMP Version 13 was used for further statistical analysis and plotting (SAS Corporation, Cary, NC).
As per the manufacturer's recommendation, two normalization processes were performed to generate normalized counts: (1) Six internal positive control sequences that are not native to any known organism, are run with each cartridge. The geometric mean is calculated for these positive control sequences at each sample run, and a scaling (normalization) factor is calculated for each sample to equilibrate each count for the respective gene to the geometric means of the positive controls. (2) A second normalization (content normalization) is performed by using the same process by applying a scaling factor that “normalizes” the geometric mean of the housekeeping genes (ALAS1, HPRT1, TBP, and TUBB) for each sample to 1,000. These normalized counts are used to calculate the scores.
During target gene selection process and validation, we combined data from different runs (generated with different CodeSets and different reagent lots) that were generated from sample runs on the “Standard” or “Elements” system. As the detection efficiency for each gene can vary between the different methods, manufacturer-recommended procedures were followed to scale the data generated so that it could be combined. Briefly, to combine data from different target gene lists and from runs performed on the “Standard” or “Elements” system, at least 5 identical samples were run on each run with data to be combined. For each gene, the geometric mean of the sample was calculated and the ratio of the geometric mean was used to scale the data for each gene separately to combine data generated with different target gene lists and in runs with different systems (“Standard” and “Elements”). Technical replicates that were included in the different runs were plotted on an XY scatter plot. Linear regression that yielded an R2 of less than 0.95 were eliminated or rerun. Remaining technical replicates were averaged for statistical analysis.
Generation of calibration standards
Synthetic DNA oligonucleotides of each of the 28 IRG target genes included in the score and the 4 housekeeping genes were designed by NanoString and synthesized by IDT (Coralville, IA) (Supplementary Table S1C). Dilutions for each gene, which approximate the respective counts expected in an active CANDLE or SAVI patient, were produced. These synthetic DNA oligonucleotides are used as a calibration standard to check run and reagent lot consistency. To compare different lots within the Elements system, multiple calibration standard results were averaged and linear regression was performed between each individual standard run against the average. An R2 of greater than 0.95 and a slope between 0.9 and 1.1 was considered consistent. For all runs assessed, the correlation was within the parameters considered consistent and no adjustment or scaling has been required.
Calculation of an IFN score by 2 methods: a z-score-based standardized score and a geomean score
Z-score-based standardized IFN score calculation
Z-scores for each of the 28 genes were calculated using the mean and standard deviation of a cohort of HCs (n = 18) using the following equation for each gene:
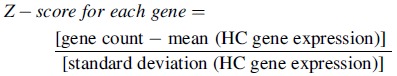 |
The 28 IRG (or 25 IRG) standardized (STD) score was calculated by summing the 28 (or 25) z-scores for each sample. The calculation for each z-score is relative to the mean and standard deviation of HCs, therefore, each z-score and the summary z-score can become negative if gene expression is below the mean of the HCs.
Geomean score calculation
Alternatively, the geometric mean or geomean of the counts of each of the 28 genes for each sample was calculated to generate the 28 IRS score using the equation below:
 |
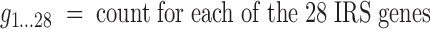 |
The geometric mean characterizes the central tendency of a group of values that is robust against outliers. Furthermore, the score can be calculated without an HC reference. The geomean was calculated for baseline CANDLE, SAVI, and NOMID patients and for HCs. For the score reporting, the geomean was divided by 10, which scales the ranges of z-score-based (standardized) IFN scores and of the geomean-based (geomean) score to be similar.
Validation and comparison of ISGs by analysis in the type I IFN-mediated diseases CANDLE, SAVI, and the IL-1-mediated disease NOMID, and HCs
To validate the genes that were highly expressed both in CANDLE we selected samples from patients with another type-I IFN-mediated disease SAVI (validation cohort) and from patients with the active IL-1-mediated disease NOMID as we expected the IFN response genes to be negative in NOMID similarly to HCs (negative control groups). We compared log-transformed gene-specific expression using uncorrected t-tests. Individuals with more than 1 assessment had values averaged for statistical comparison. Genes that did not follow this pattern were eliminated. We also assessed overlapping expression ranges between CANDLE and SAVI versus NOMID and HC and eliminated 4 genes (CD274, PLSCR1, IFNA2, and SAMD9) due to overlap.
The geomean and standardized scores were compared stratified by diagnosis and HC groups using uncorrected t-tests. Individual standardized scores and geomeans for the individuals in the 4 baseline groups were compared by linear regression.
Characterization of distribution of the IFN score in HCs
We assessed the distribution of the 28-gene standardized score and the geomean score in the 33 healthy adult and pediatric controls described above. For the assessment of gender and age differences, standardized scores and geomeans were compared stratified by gender and age, the latter based on a cutoff of < and ≥18 years of age as well as < and ≥10 years of age using Mann–Whitney U-nonparametric test. In 2 HCs, 5 blood samples were collected over an 8-h period to assess for diurnal variability. The lowest and highest scores or values were compared by a paired t-test and coefficient of variation (CV) was calculated for each individual. We compared the 25-IRS gene standardized score with the 28-gene standardized score and the 28-IRS gene standardized score with the geomean. We determined the 95th percentile of the 33 HCs for the 28-IRS gene standardized score, 25-IRS gene standardized score, and the 28-IRS geomean, as well as geomean score, and suggest this value as a preliminary cutoff for normal and elevated scores.
Assessment of technical variability
Patient samples and/or calibration standards were run using different gene lists and runs or different operators or different lots of reagents. For all correlations, these samples were analyzed by linear regression and R2 of greater than 0.95 and a slope between 0.9 and 1.1 was considered concordant.
Results
Selection of the IRG targets
An initial set of candidate genes of IRG was selected by choosing 2-fold up-, or downregulated genes from whole blood microarray gene expression data generated from 2 samples from a patient with chronic hepatitis C drawn just before, and 6 h after initial pegylated interferon alpha-2a (Pegasys), then analyzed by comparing posttreatment versus pretreatment samples, and 1 CANDLE patient assessed at 3 time points before the use of a JAK inhibitor compared with samples from 3 healthy pediatric controls. Up and downregulated genes that were among the “interferon pathway genes” in Ingenuity Pathway Analysis (IPA, www.ingenuity.com) (Liu and others 2012) were selected (Table 2). IFN alpha, beta, and gamma genes (IFNA1, IFNA2, IFNA5, IFNA6, IFNA7, IFNA8, IFNA10, IFNA16, IFNA21, IFNB1, and IFNG) are low expressing and were too low to reliably detect on microarray; they were, however, added as possible candidate genes, as NanoString is more sensitive in assaying low expressing genes than microarray (Geiss and others 2008). IFNA4, IFNA13, IFNA14, and IFNA17 could not be uniquely targeted and were not included. Furthermore, IFN receptor genes (IFNAR1, IFNAR2, IFNGR1, and IFNGR2) and 6 possible housekeeping genes (ALAS1, G6PD, HPRT1, POLR2A, TBP, and TUBB) were added.
Table 2.
List of Initial Set of Genes Analyzed on NanoString
| Entrez gene symbol | Entrez gene name | IFN probe set ID | Fold change | CANDLE probe set ID | Fold change | Genes removed |
|---|---|---|---|---|---|---|
| Upregulated genesa | ||||||
| >2 × increased in CANDLE | ||||||
| AIM2 | Absent in melanoma 2 | 206513_at | 5.77 | x | ||
| APOL1 | Apolipoprotein L, 1 | 209546_s_at | 2.26 | x | ||
| APOL6 | Apolipoprotein L, 6 | 219716_at | 3.44 | 241869_at | 5.02 | |
| BCL2L1 | BCL2-like 1 | 206665_s_at | 2.25 | 215037_s_at | 3.80 | |
| BIRC5 | Baculoviral IAP repeat-containing 5 | 202094_at | −2.66 | 202095_s_at | 3.70 | x |
| C1QB | Complement component 1, q subcomponent, B chain | 202953_at | 2.16 | x | ||
| CASP1 | Caspase 1, apoptosis-related cysteine peptidase (interleukin 1, beta, convertase) | 1552703_s_at | 2.96 | x | ||
| CCNA1 | Cyclin A1 | 205899_at | 7.71 | x | ||
| CCNA2 | Cyclin A2 | 203418_at | 2.85 | 203418_at | 2.19 | x |
| CCRL2 | Chemokine (C-C motif) receptor-like 2 | 211434_s_at | 2.36 | x | ||
| CD274 | CD274 molecule | 223834_at | 6.70 | 227458_at | 3.81 | |
| CD80 | CD80 molecule | 1554519_at | 2.05 | x | ||
| CLTCL1 | Clathrin, heavy chain-like 1 | 205944_s_at | 2.80 | x | ||
| CXCL10 | Chemokine (C-X-C motif) ligand 10 | 204533_at | 12.92 | 204533_at | 4.03 | |
| DDX58 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 58 | 218943_s_at | 4.65 | 242961_x_at | 6.53 | |
| DDX60 | DEAD (Asp-Glu-Ala-Asp) box polypeptide 60 | 218986_s_at | 2.00 | 218986_s_at | 4.60 | |
| EIF2AK2 | Eukaryotic translation initiation factor 2-alpha kinase 2 | 204211_x_at | 2.53 | 204211_x_at | 2.72 | |
| FCGR1A | Fc fragment of IgG, high-affinity Ia, receptor (CD64) | 216951_at | 2.13 | 216951_at | 5.22 | |
| FCGR1B | Fc fragment of IgG, high-affinity Ib, receptor (CD64) | 214511_x_at | 4.34 | x | ||
| GBP1 | Guanylate-binding protein 1, interferon-inducible | 202269_x_at | 4.85 | 231578_at | 4.45 | |
| GBP2 | Guanylate-binding protein 2, interferon-inducible | 242907_at | 2.01 | x | ||
| H6PD | Hexose-6-phosphate dehydrogenase (glucose 1-dehydrogenase) | 226160_at | 2.28 | x | ||
| HERC5 | Hect domain and RLD 5 | 219863_at | 5.56 | 219863_at | 6.50 | |
| HERC6 | Hect domain and RLD 6 | 219352_at | 2.23 | 219352_at | 4.63 | |
| IFI27 | Interferon alpha-inducible protein 27 | 202411_at | 2.28 | 202411_at | 72.76 | |
| IFI35 | Interferon-induced protein 35 | 209417_s_at | 3.51 | 209417_s_at | 2.80 | |
| IFI44 | Interferon-induced protein 44 | 214059_at | 4.01 | 214059_at | 9.41 | |
| IFI44L | Interferon-induced protein 44-like | 204439_at | 2.94 | 204439_at | 13.36 | |
| IFI6 | Interferon alpha-inducible protein 6 | 204415_at | 6.22 | 204415_at | 3.94 | |
| IFIH1 | Interferon induced with helicase C domain 1 | 216020_at | 4.41 | 1555464_at | 3.52 | |
| IFIT1 | Interferon-induced protein with tetratricopeptide repeats 1 | 203153_at | 5.85 | 203153_at | 8.68 | |
| IFIT2 | Interferon-induced protein with tetratricopeptide repeats 2 | 217502_at | 31.77 | 217502_at | 2.52 | |
| IFIT3 | Interferon-induced protein with tetratricopeptide repeats 3 | 204747_at | 11.11 | 204747_at | 7.16 | |
| IFIT5 | Interferon-induced protein with tetratricopeptide repeats 5 | 203595_s_at | 2.96 | 203596_s_at | 3.63 | |
| IGLL1/IGLL5 | Immunoglobulin lambda-like polypeptide 1 | 217235_x_at | 2.15 | x | ||
| IL10 | Interleukin 10 | 207433_at | 2.18 | x | ||
| IL15 | Interleukin 15 | 205992_s_at | 2.52 | x | ||
| IL18R1 | Interleukin 18 receptor 1 | 206618_at | 2.67 | x | ||
| IL1RN | Interleukin 1 receptor antagonist | 212657_s_at | 2.45 | 216243_s_at | 2.43 | |
| IRF7 | Interferon regulatory factor 7 | 208436_s_at | 2.19 | 208436_s_at | 3.09 | |
| ISG15 | ISG15 ubiquitin-like modifier | 205483_s_at | 7.20 | 205483_s_at | 7.05 | |
| LAMP3 | Lysosomal-associated membrane protein 3 | 205569_at | 4.99 | 205569_at | 4.24 | |
| LGALS3BP | Lectin, galactoside-binding, soluble, 3-binding protein | 200923_at | 5.39 | x | ||
| LY6E | Lymphocyte antigen 6 complex, locus E | 202145_at | 3.08 | 202145_at | 6.19 | |
| MMP1 | Matrix metallopeptidase 1 (interstitial collagenase) | 204475_at | 4.25 | x | ||
| MMP9 | Matrix metallopeptidase 9 (gelatinase B, 92 kDa gelatinase, 92 kDa type IV collagenase) | 203936_s_at | 3.27 | 203936_s_at | 3.53 | x |
| MTMR3 | Myotubularin-related protein 3 | 202198_s_at | 2.33 | x | ||
| MUC1 | Mucin 1, cell surface-associated | 213693_s_at | 2.84 | x | ||
| MX1 | Myxovirus (influenza virus) resistance 1, interferon-inducible protein p78 (mouse) | 202086_at | 4.19 | 202086_at | 5.53 | |
| NF1 | Neurofibromin 1 | 204323_x_at | 2.31 | 216115_at | 3.00 | |
| OAS1 | 2′-5′-Oligoadenylate synthetase 1, 40/46 kDa | 202869_at | 2.12 | 205552_s_at | 8.06 | |
| OAS2 | 2′-5′-Oligoadenylate synthetase 2, 69/71 kDa | 204972_at | 2.22 | 228607_at | 4.26 | |
| OAS3 | 2′-5′-Oligoadenylate synthetase 3, 100 kDa | 218400_at | 2.66 | 218400_at | 9.20 | |
| OASL | 2′-5′-Oligoadenylate synthetase-like | 205660_at | 6.72 | 210797_s_at | 5.44 | |
| PLSCR1 | Phospholipid scramblase 1 | 202430_s_at | 2.51 | 202446_s_at | 3.65 | |
| PML | Promyelocytic leukemia | 206503_x_at | 2.57 | 211012_s_at | 3.07 | |
| PRDX2 | Peroxiredoxin 2 | 211658_at | 2.05 | x | ||
| RBX1 | Ring-box 1, E3 ubiquitin protein ligase | 218117_at | 2.64 | x | ||
| RSAD2 | Radical S-adenosyl methionine domain-containing 2 | 213797_at | 4.39 | 213797_at | 12.91 | |
| RTP4 | Receptor (chemosensory) transporter protein 4 | 219684_at | 3.20 | 219684_at | 5.05 | |
| SAMD9 | Sterile alpha motif domain-containing 9 | 219691_at | 3.00 | 219691_at | 3.00 | |
| SEPT4 | Septin 4 | 210657_s_at | 7.52 | x | ||
| SOCS1 | Suppressor of cytokine signaling 1 | 210001_s_at | 5.65 | 210001_s_at | 2.13 | |
| STAT1 | Signal transducer and activator of transcription 1, 91 kDa | AFFX-HUMISGF3A/M97935_5_at | 2.07 | AFFX-HUMISGF3A/M97935_MA_at | 3.25 | |
| STAT2 | Signal transducer and activator of transcription 2, 113 kDa | 205170_at | 2.45 | 205170_at | 2.18 | |
| TAP2 | Transporter 2, ATP-binding cassette, subfamily B (MDR/TAP) | 208428_at | 3.89 | x | ||
| TDRD7 | Tudor domain-containing 7 | 213361_at | 2.73 | 213361_at | 2.05 | |
| USP18 | Ubiquitin-specific peptidase 18 | 219211_at | 12.11 | 219211_at | 13.86 | |
| USP6NL | USP6N terminal-like | 238164_at | 4.61 | x | ||
| ZC3HAV1 | Zinc finger CCCH-type, antiviral 1 | 213051_at | 2.14 | 220104_at | 2.33 | |
| >2 × increased in IFN probe set, but not increased in CANDLE | ||||||
| ATF3 | Activating transcription factor 3 | 202672_s_at | 9.33 | |||
| BST2 | Bone marrow stromal cell antigen 2 | 201641_at | 2.25 | |||
| CCL2 | Chemokine (C-C motif) ligand 2 | 216598_s_at | 45.74 | x | ||
| DEFB1 | Defensin beta 1 | 210397_at | 3.98 | x | ||
| FAS | Fas (TNF receptor superfamily, member 6) | 215719_x_at | 2.75 | |||
| HSH2D | Hematopoietic SH2 domain-containing | 1552623_at | 3.68 | x | ||
| IDO1 | Indoleamine 2,3-dioxygenase 1 | 210029_at | 2.39 | |||
| IL1B | Interleukin 1, beta | 205067_at | 3.58 | |||
| IRF1 | Interferon regulatory factor 1 | 202531_at | 2.16 | |||
| IRF2 | Interferon regulatory factor 2 | 203275_at | 2.25 | x | ||
| ITGB3 | Integrin, beta 3 (platelet glycoprotein IIIa, antigen CD61) | 204626_s_at | 2.03 | |||
| KLF6 | Kruppel-like factor 6 | 208960_s_at | 2.72 | |||
| MDM2 | Mdm2 p53-binding protein homolog (mouse) | 211832_s_at | 2.28 | |||
| MX2 | Myxovirus (influenza virus) resistance 2 (mouse) | 204994_at | 2.13 | |||
| PELI1 | Pellino E3 ubiquitin protein ligase 1 | 218319_at | 2.21 | |||
| PRKCD | Protein kinase C delta | 202545_at | 2.16 | x | ||
| PRKD2 | Protein kinase D2 | 209282_at | 2.28 | |||
| PTAFR | Platelet-activating factor receptor | 206278_at | 3.22 | |||
| PTGES | Prostaglandin E synthase | 210367_s_at | 2.58 | |||
| SOD2 | Superoxide dismutase 2, mitochondrial | 215078_at | 2.41 | |||
| SP100 | SP100 nuclear antigen | 202864_s_at | 2.22 | |||
| SP110 | SP110 nuclear body protein | 208392_x_at | 3.47 | |||
| SPI1 | Spi-1 proto-oncogene | 205312_at | 2.54 | |||
| TLR4 | Toll-like receptor 4 | 221060_s_at | 2.10 | |||
| TNFRSF1A | TNF receptor superfamily member 1A | 207643_s_at | 2.05 | |||
| TNFSF10 | TNF superfamily member 10 | 202687_s_at | 2.39 | |||
| TNFSF13B | TNF superfamily member 13b | 223502_s_at | 2.47 | |||
| TREX1 | Three prime repair exonuclease 1 | 205875_s_at | 3.56 | |||
| TRIM14 | Tripartite motif-containing 14 | 203147_s_at | 2.45 | |||
| TYMP | Thymidine phosphorylase | 217497_at | 4.08 | |||
| Downregulated genesb | ||||||
| >2 × decreased in CANDLE | ||||||
| ADORA2B | Adenosine A2b receptor | 205891_at | −2.55 | x | ||
| ALOX15 | Arachidonate 15-lipoxygenase | 207328_at | −4.15 | |||
| AUTS2 | Autism susceptibility candidate 2 | 212599_at | −3.65 | x | ||
| CAMK4 | Calcium/calmodulin-dependent protein kinase IV | 241871_at | −3.27 | x | ||
| CCL5 | Chemokine (C-C motif) ligand 5 | 204655_at | −2.11 | |||
| CCR5 | Chemokine (C-C motif) receptor 5 | 206991_s_at | −2.39 | x | ||
| CD2 | CD2 molecule | 205831_at | −2.74 | x | ||
| CD40LG | CD40 ligand | 207892_at | −4.33 | x | ||
| CIITA | Class II, major histocompatibility complex, transactivator | 205101_at | −2.26 | |||
| CXCL1 | Chemokine (C-X-C motif) ligand 1 (melanoma growth-stimulating activity, alpha) | 204470_at | −2.40 | |||
| CXCR3 | Chemokine (C-X-C motif) receptor 3 | 207681_at | −2.03 | |||
| DPP4 | Dipeptidyl-peptidase 4 | 203717_at | −2.28 | x | ||
| EEF1A1 | Eukaryotic translation elongation factor 1 alpha 1 | 227708_at | −2.27 | x | ||
| GATA3 | GATA-binding protein 3 | 209604_s_at | −3.17 | x | ||
| HLA-DQB1 | Major histocompatibility complex, class II, DQ beta 1 | 212999_x_at | −3.53 | x | ||
| IL23A | Interleukin 23, alpha subunit p19 | 240102_at | −3.08 | x | ||
| IL24 | Interleukin 24 | 206569_at | −2.13 | |||
| IL32 | Interleukin 32 | 203828_s_at | −2.31 | |||
| IL8 | Interleukin 8 | 202859_x_at | −2.40 | 202859_x_at | −5.24 | |
| KLRK1 | Killer cell lectin-like receptor subfamily K, member 1 | 1555691_a_at | −2.61 | x | ||
| MARCKSL1 | MARCKS-like 1 | 200644_at | −2.58 | x | ||
| NCR3 | Natural cytotoxicity-triggering receptor 3 | 211583_x_at | −3.11 | x | ||
| PIGR | Polymeric immunoglobulin receptor | 226147_s_at | −2.03 | x | ||
| PTGS2 | Prostaglandin-endoperoxide synthase 2 (prostaglandin G/H synthase and cyclooxygenase) | 1554997_a_at | 2.82 | 1554997_a_at | −4.94 | x |
| TBX21 | T-box 21 | 220684_at | −2.14 | x | ||
| >2 × decreased in IFN probe set, but not increased in CANDLE | ||||||
| CCDC75 | Coiled-coil domain-containing 75 | 228495_at | −2.30 | |||
| CXCL2 | Chemokine (C-X-C motif) ligand 2 | 209774_x_at | −2.88 | |||
| ELAVL1 | ELAV (embryonic lethal, abnormal vision, Drosophila)-like 1 (Hu antigen R) | 244660_at | −5.24 | x | ||
| ICOSLG | Inducible T cell costimulator ligand | 213450_s_at | −2.36 | x | ||
| ITGAV | Integrin, alpha V (vitronectin receptor, alpha polypeptide, antigen CD51) | 202351_at | −2.17 | x | ||
| PARVG | Parvin, gamma | 244229_at | −2.20 | |||
| TLR3 | Toll-like receptor 3 | 206271_at | −2.04 | |||
| Added later for initial NanoString IRG listc | ||||||
| C1QC | Complement component 1, q subcomponent, C chain | x | ||||
| CEACAM1 | Carcinoembryonic antigen-related cell adhesion molecule 1 (biliary glycoprotein) | x | ||||
| CYSLTR2 | Cysteinyl leukotriene receptor 2 | x | ||||
| IGFBP4 | Insulin-like growth factor-binding protein 4 | x | ||||
| IRF3 | Interferon regulatory factor 3 | x | ||||
| IFN and IFNR genesd | ||||||
| IFNA1 | Interferon, alpha 1 | 208375_at | x | |||
| IFNA10 | Interferon alpha 10 | 208261_x_at | x | |||
| IFNA16 | Interferon alpha 16 | 208448_x_at | x | |||
| IFNA2 | Interferon alpha 2 | 211338_at | ||||
| IFNA21 | Interferon, alpha 21 | 211145_x_at | x | |||
| IFNA5 | Interferon alpha 5 | 214569_at | x | |||
| IFNA6 | Interferon alpha 6 | 208548_at | x | |||
| IFNA7 | Interferon, alpha 7 | 208259_x_at | x | |||
| IFNA8 | Interferon alpha 8 | 207932_at | x | |||
| IFNAR1 | Interferon alpha and beta receptor subunit 1 | 204191_at | ||||
| IFNAR2 | Interferon (alpha, beta, and omega) receptor 2 | 204785_x_at | ||||
| IFNB1 | Interferon, beta 1, fibroblast | 208173_at | ||||
| IFNG | Interferon, gamma | 210354_at | ||||
| IFNGR1 | Interferon gamma receptor 1 | 202727_s_at | ||||
| IFNGR2 | Interferon gamma receptor 2 | 201642_at | ||||
| Housekeeping genese | ||||||
| ALAS1 | Delta-aminolevulinate synthase 1 | |||||
| G6PD | Glucose-6-phosphate dehydrogenase | x | ||||
| HPRT1 | Hypoxanthine phosphoribosyltransferase 1 | |||||
| POLR2A | RNA polymerase II subunit A | x | ||||
| TBP | TATA box-binding protein | |||||
| TUBB | Tubulin beta class I | |||||
Increased expression versus HCs potential IFN target genes.
Decreased expression versus HCs IFN target genes.
Added afterwards.
IFN or IFNR gene added.
Housekeeping gene.
This list of genes is based on previous microarray data from an IFN-alpha-treated hepatitis patient and 1 CANDLE patient at multiple time points with microarray probe set ID and fold change comprising the initial 152 candidate gene list and 6 housekeeping genes. Genes have been grouped alphabetically within each category as described. In the right-most column, x indicates genes removed from initial candidate list to get to target gene list 2, which has 92 IRS genes and 4 housekeeping genes.
IFN, interferon; IRG, IFN response gene; IRS, IFN-regulated gene signature.
Of the initial set of 152 IRG candidate genes and 6 housekeeping genes, 19 genes that were only upregulated in the CANDLE patient and not in the IFN alpha-2a-treated hepatitis patient, and 6 genes that are not specific for IFN signaling (CCNA2, MMP9, CCL2, DEFB1, HSH2D, and PRKCD) and IRF2, which was not elevated in CANDLE, were removed. Five genes that were only up in the IFN alpha-2a-treated patient, but not in CANDLE were also removed, along with IRF1. Among the downregulated genes, 1 gene was only down in the hepatitis patient (ICOSLG), 1 gene was only down in CANDLE, but up in hepatitis patient (PTGS2), and 7 other genes down in CANDLE, but not in the hepatitis patient (ADORA2B, CCR5, EEF1A1, HLA-DQB1, IL-23A, PIGR, and TBX21) were also removed from the candidate list.
The remaining 118 target genes and 6 potential housekeeping genes (124 genes included in target IRG gene list 1) were evaluated in 4 additional clinically active CANDLE patients (5 total CANDLE patients, all with elevated inflammatory markers) using the NanoString custom gene expression assay, which showed a gene expression profile similar to the gene expression profile on microarray observed in the first CANDLE patient. Most target genes were elevated in the additional CANDLE patients, except for 8 low-expressing interferon alpha genes, IRF2, IRF3, 9 downregulated genes in CANDLE, 6 upregulated genes in CANDLE that were not more than 2-fold upregulated in the hepatitis patient, and 5 genes that are not IFN specific (C1QC, CEAVAM1, CYSLTR2, IGFBP4, and HSH2D) (Table 2). We also excluded 2 of the potential housekeeping genes (G6PD and POLR2A), which were upregulated in the patients compared with HCs.
The reduced set of 88 potential IFN-regulated genes and 4 potential housekeeping gene probes (92 genes included in target IRG gene list 2) was assessed in 8 CANDLE patients, in 3 patients with a suspected but genetically undefined interferonopathy (all with active but variable disease activity), and in 4 HCs. Maximum z-scores were calculated for these 92 genes from all samples run with target IRG gene list 1 and 2 and z-scores were ranked (Supplementary Table S2). Genes with high z-scores of 46 or greater seemed more specifically IFN regulated compared with lower ranking genes that were also upregulated in non-IFN-regulated inflammatory pathways, such as IL8, STAT1, ATF3, IFNGR1, PTGES, IL1RN, TNFSF10, IL24, TLR3, APOL6, CXCL2, MDM2, TRIM14, TLR4, CIITA, SP110, ALOX15, IFNAR1, FAS, IL32, and IL1B (Supplementary Table S2). The eliminated genes were later assessed in a gene expression data set from peripheral blood samples from CANDLE, SAVI, NOMID, and HCs and were also elevated in patients with NOMID, an IL-1-mediated disease (data not shown). We subsequently compared our gene list with lists previously published in autoimmune diseases (Bennett and others 2003; Yao and others 2009) and added PLSCR1 back, which was included in one of the IRG gene lists and had a borderline z-score of 42.5 in our dataset. After the exclusions, we had generated a list of 31 target genes and 4 housekeeping genes listed in target IRG gene list 3 (Supplementary Table S2).
Validation of score in patients with SAVI (validation group) and patients with the IL-1-mediated disease NOMID (negative control group) resulted in identification of 5 genes that did not differentiate between disease groups
We assessed the remaining 31 target genes (target IRG gene list 3) in 5 patients with SAVI (positive IFN validation control), and reassessed them in 10 active patients with CANDLE (before JAK inhibitor therapy), 2 CANDLE patients had not previously been assessed. We also assessed 18 NOMID patients (negative IFN control group as described above), and 16 HCs (see Table 1 for details).
Most genes were highly expressed in CANDLE and SAVI patients, but not in HCs and active NOMID patients; however, there was overlap between some genes in active CANDLE and either SAVI or CANDLE. To identify genes that were also elevated in NOMID, we compared gene expression for each candidate gene in SAVI and CANDLE patients versus both HCs and NOMID. For most target genes, disease comparisons between CANDLE and SAVI each compared with NOMID and HC were statistically significant (P < 0.05) and both CANDLE and SAVI patients had little or no overlap with NOMID and HC (Fig. 1). However, 3 genes did not meet these criteria and were therefore removed from the final target list: BCL2L1 and IDO1 were not significantly different in SAVI patients compared with HC, and PLSCR1 had significant overlap between CANDLE and SAVI patients, and NOMID patients and HCs (Supplementary Fig. S2A and Supplementary Table S3).
FIG. 1.

Comparison of IFN response genes included in the IFN score in CANDLE, SAVI, NOMID, and HCs. The figure panels depict the expression patterns of the 28 genes that were kept in the final score. For each example, a representative graph is shown with a list of the other genes with similar patterns. Box and whisker plots of log2 normalized counts of each gene are shown, with individual dots for each patient stratified by diagnosis (CANDLE, SAVI, and NOMID) and for healthy control subjects (HC). The middle line indicates the median. Uncorrected t-tests were performed to compare between diagnosis and control groups. At the right of each panel, each circle represents a diagnosis group, with the center at the level of the mean of the respective group. See Supplementary Fig. S2 legend for details. (A) Thirteen genes are highly expressed, significantly higher in CANDLE and SAVI than in NOMID and HC, expression in NOMID is significantly lower than in HCs. (P value range for CANDLE and NOMID were between 3.01 × 10−30 and 7.24 × 10−18, for SAVI and HC between 8.70 × 10−27 and 1.33 × 10−14), and SAVI and NOMID (P value range, 1.01 × 10−28 and 1.40 × 10−16). There is no statistically significant difference between CANDLE and SAVI, but expression in NOMID patients is significantly lower than in HC (P value range, 7.46 × 10−6 to 0.0496). (B) Nine genes are highly expressed in CANDLE and SAVI with similar low expression in NOMID and HC. P values are significant for CANDLE versus NOMID (P value range, 4.34 × 10−25 to 1.27 × 10−8), SAVI versus HC (P value range, 1.94 × 10−22 to 1.17 × 10−4), and SAVI versus NOMID (P value range, 8.61 × 10−23 to 7.47 × 10−6). There is no significant difference between CANDLE and SAVI or NOMID and HC. (C) Three genes are significantly highly expressed in CANDLE than SAVI (P value range, 1.16 × 10−5 to 0.042), further expression in CANDLE patients is significantly higher than in NOMID patients (P value range, 1.08 × 10−25 to 4.19 × 10−16), SAVI expression is significantly higher than in NOMID (P value range, 1.43 × 10−19 to 4.97 × 10−8) and in HC (P value range, 3.67 × 10−16 to 3.25 × 10−5). Gene expression in NOMID was significantly higher than in HC (P value range, 1.67 × 10−4 to 8.86 × 10−4) for SPATS2L and LAMP3, but the P value was not significant for GBP1. (D) One gene is highly expressed in SAVI than CANDLE (P value of 0.01), although both high, and NOMID lower than HC (P value of 0.0002), although both low. P values are as follows: CANDLE versus NOMID (2.05 × 10−26), SAVI versus HC (3.38 × 10−25), and SAVI versus NOMID (6.57 × 10−27). (E) Two genes have NOMID higher than HC (P values 0.013 and 0.025), although both are lower than CANDLE and SAVI. P value are as follows: CANDLE versus NOMID (2.25 × 10−17 to 4.32 × 10−14), SAVI versus HC (6.25 × 10−19 to 4.10 × 10−8), and SAVI versus NOMID (7.09 × 10−5 and 1.21 × 10−15). For SOCS1, the gene expression is significantly higher in NOMID than in HC (02.47 × 10−4), but expression of IFIT5 is not significantly different. (F) Standardized 28-IFN gene scores in CANDLE and SAVI patients were each significantly higher than in NOMID and HC (P values <2 × 10−9), scores in CANDLE and SAVI patients are not significantly different and scores in NOMID patients and HC are not significantly different. (G) The geomean scores in patients with CANDLE and SAVI are significantly higher than in patients with NOMID and in HC (P values <2 × 10−13). CANDLE, chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature; IFN, interferon; NOMID, neonatal-onset multisystem inflammatory disease; SAVI, STING-associated vasculopathy with onset in infancy; STING, stimulator of IFN genes.
Based on literature review, 3 genes that were not included in the initial candidate list (SIGLEC1, SPATS2L/DNAPTP6, and EPSTI1) (Yao and others 2009) were added to target IRG gene list 4. These genes were not on the IPA list for IFN response genes and had therefore not been on the initial candidate gene list. They were, however, validated in CANDLE, SAVI, NOMID, and HCs (Supplementary Fig. S2B and Supplementary Table S3).
The final set of genes (30 genes, including the 3 added genes) was reassessed in 11 CANDLE (1 newly added since previous assessment), 7 SAVI (2 newly added from previous assessment), and 18 active NOMID patients, and 26 HCs (8 additional controls from previous assessment). IFNA2 and SAMD9 had significant overlap of either CANDLE or SAVI with either NOMID (SAMD9) or HC (IFNA2) (Supplementary Fig. S2A and Supplementary Table S3) and were removed resulting in the final list of 28 genes. The housekeeping genes are not significantly different among the 4 conditions (Supplementary Fig. S2C).
We plotted the gene expression pattern for each gene for CANDLE, SAVI, NOMID, and HC groups and noted differences that could be grouped in 5 expression patterns (Fig. 1). All genes included in the final 28 gene score were highly expressed in CANDLE and SAVI, with significantly lower expression in NOMID and HC with P values below 0.01 (Supplementary Table S3).
Most genes (13) had high expression in CANDLE and SAVI with NOMID significantly lower than HC with P values <0.05 (Pattern 1, Fig. 1A), 9 genes had no significant difference between CANDLE and SAVI (both high) or NOMID and HC (both low) (Pattern 2, Fig. 1B). Three genes had significantly higher expression in CANDLE than in SAVI patients with P values <0.05 (Pattern 3, Fig. 1C). One gene had significantly higher expression in SAVI patients than in CANDLE patients with a P value of 0.01 (Pattern 4, Fig. 1D); 2 genes had higher expression in NOMID than in HC with P values <0.03 (Pattern 5, Fig. 1E).
The standardized 28-gene score and geomean significantly differed between CANDLE and SAVI compared with NOMID and HC with P values <2 × 10−9 (Fig. 1F, G). As we used a 25-gene set (without SIGLEC1, SPATS2L/DNAPTP6, and EPSTI1) in the validation of the IFN score in a clinical study (data not shown), we correlated the 25-gene and 28-gene IRS scores for the CANDLE, SAVI, NOMID, and HCs, which, as expected, had a high correlation by linear regression with an R2 of 1.000 (Supplementary Fig. S3A). Similarly, when comparing the standardized score versus the geomean, we found a reasonable correlation with an R2 of 0.753 (Supplementary Fig. S3C). All the outliers noted have a relatively high contribution of the IFI27 gene z-score to the 28-gene standardized z-score (Supplementary Table S2). After removal of the top 4 outliers, the correlation increased to an R2 of 0.979 (Supplementary Fig. S3D) confirming that a very high IFI27 expression can skew the standardized 28-gene score to overall higher scores.
Assessment of reproducibility, interassay, and interoperator variance
To assess reproducibility, 11 samples that were run twice by the same operator on the NanoString Elements system, were compared; and counts for each gene highly correlated (R2 of 0.999) (Fig. 2A). To assess operator variability, we compared 4 samples that had been run by 2 different operators, and standardized counts for each gene highly correlated (R2 of 0.991) (Fig. 2B). To assess interassay variability on NanoString systems, we assessed the consistency between the Standard and Elements NanoString system based on 5 patient samples that were run on both systems. Linear regression showed high consistency (R2 of 0.936) (Supplementary Fig. S3B).
FIG. 2.

Assessment of interassay and interoperator variability. (A) 11 samples were assayed at 2 different time points by the same operator. The counts for each gene were plotted. (B) Interoperator variability was compared in 4 samples that were assayed by 2 operators each.
Determination of variance (age, gender, diurnal variation) and normal range in HCs
We investigated the impact of age, gender on the standardized and geomean score in 33 HCs, and the diurnal variation of the scores in 2 HCs. Gender and age stratification, using a cutoff of 10 and 18 years, showed no significant differences (Supplementary Fig. S4A, B and Fig. S5A, B). To assess diurnal variation, 2 HCs had 5 IFN scores each assessed at 9 am, 10:30 am, 11:45 am, 3 pm, and 5 pm. Paired t-tests between the minimum and maximum standardized score per person were not significantly different (P values of 0.16 and 0.06, respectively) and the coefficient of variance for the standardized 28-IRS score was 35.1% for HC1 and 19.6% for HC2, and for the geomean 3.9% for HC1 and 8.5% for HC2, respectively (Supplementary Fig. S4C, C). To determine a cutoff that may distinguish HCs from patients with elevated scores, we calculated the 95th percentile among 33 HCs for the 28-gene standardized score and the geomean score (geomean divided by 10), which is 48.9 and 178.1 respectively. The scale was skewed by one individual who had no clinical indication that would justify exclusion as an HC, but who had a high IFN score. When excluded from the calculations, the 95th percentile dropped to 25.2 and 132.7 for the 28-IRS gene standardized score and geomean score, respectively. (Supplementary Figs. S4D and S5D).
Discussion
In summary, we have customized a NanoString assay that allows detection and quantification of an IFN response gene (IRS) signature in peripheral blood in real time by assessing 28-IFN-target genes. This assay may be valuable as a diagnostic tool for identifying patients with IFN-mediated diseases and in the assessment of the IFN score as biomarker in treatment studies or in pathogenesis studies. We have validated the 28 genes in patients with the IL-1-mediated disease, NOMID, who do not have significant IRG expression and together with HCs serve as negative disease controls, and in patients with SAVI, a disease caused by constitutive IFNβ transcription that served as a positive control.
As many of the genes that are IFN stimulated can also be upregulated by other proinflammatory cytokines, we used the Mendelian diseases, CANDLE and SAVI, which present with systemic inflammation (high erythrocyte sedimentation rate and high C-reactive protein), a strong persistent IFN response gene signature on gene expression studies, and with mutations that affect IFN production and signaling, as positive controls. In contrast, the systemic inflammatory disease NOMID, which is treated with targeted biologics that block a single cytokine, IL-1, has no strong IFN response gene signature, although some genes that are considered to be IRGs are also upregulated in untreated patients (Liu and others 2012, 2014; de Jesus and others 2015).
We therefore attempted to develop an IRG score that discriminates between IFN-regulated genes that distinguish IL-1-mediated autoinflammatory diseases from those with presumed IFN-mediated pathogenesis. The final gene set which we selected includes genes that are elevated in CANDLE and SAVI patients, but low in active NOMID patients and HCs. The list of 28 genes is more extensive than other qPCR assays that have been used in different diseases, including SLE, JDM, and systemic sclerosis (Feng and others 2006; Yao and others 2009; Higgs and others 2011; Rice and others 2013, 2017) (Table 3).
Table 3.
Comparison of Selected Interferon-Regulated Gene Lists
| 28 IFN final | Feng and others (2006)a | Yao and others (2009)a | Higgs and others (2011)a | Rice and others (2013)a, Rice and others (2017)a,c |
|---|---|---|---|---|
| CANDLE, SAVIb | SLEb | SLEb | DM, SLE, SSc, RAb | AGSbSAVI, SPENCD, JSLE, JDMc |
| CXCL10 | ||||
| DDX60 | ||||
| EPSTI1 | EPSTI1 | |||
| GBP1 | ||||
| HERC5 | HERC5 | |||
| HERC6 | ||||
| IFI27 | IFI27 | IFIT27 | IFIT27 | |
| IFI44 | IFI44 | IFI44 | ||
| IFI44L | IFI44L | IFI44L | IFI44L | |
| IFI6 | IFI6 | IFI6 | ||
| IFIT1 | IFIT1 | IFIT1 | ||
| IFIT2 | ||||
| IFIT3 | IFIT3 | |||
| IFIT5 | ||||
| ISG15 | ISG15 | ISG15 | ISG15 | |
| LAMP3 | LAMP3 | |||
| LY6E | LY6E | LY6E | ||
| MX1 | MX1 | MX1 | ||
| OAS1 | OAS1 | OAS1 | ||
| OAS2 | OAS2 | |||
| OAS3 | OAS3 | |||
| OASL | OASL | |||
| PLSCR1d | ||||
| RSAD2 | RSAD2 | RSAD2 | RSAD2 | |
| RTP4 | RTP4 | |||
| SIGLEC1 | SIGLEC1 | SIGLEC1 | ||
| SOCS1 | ||||
| SPATS2L | SPATS2L | |||
| USP18 | USP18 |
Seven genes in bold are only in this 28 IRG list (CXCL10, DDX60, GBP1, HERC6, IFIT2, IFIT5, and SOCS1) and 1 gene of the other listed gene lists is not in the 28 IRG list (PLSCR1).
References for IRG as listed also in main references.
The respective genes below are part of the interferon-regulated gene list developed in this/these condition(s) with respective reference above with this 28 IRG score list on the left.
The 6 gene ISG signature from Rice and others (2013) developed in AGS was assessed to be significantly higher in the 4 listed conditions (SAVI, SPENCD, JSLE, JDM) with individuals from at least 5 different families versus HCs from this publication (Rice and others, 2017)
This gene is not in the 28 gene ISG score.
SLE, systemic lupus erythematosus; DM, dermatomyositis; SSc, systemic scleroderma; RA, rheumatoid arthritis; SPENCD, spondyloenchondrodysplasia; JSLE, juvenile systemic lupus erythematosus; JDM, juvenile dermatomyositis; AGS, Aicardi–Goutières syndrome; ISG, IFN-stimulated gene.
Interestingly, the list of genes we selected in CANDLE and SAVI patients is similar to the list that Medimmune developed to assess the IFN response gene signature in SLE patients that was generated before the discovery of most of the monogenic interferonopathies (Yao and others 2009). Our data therefore confirm the “universality of these IFN IRGs” in autoimmune and autoinflammatory diseases. It remains to be investigated whether there is benefit of a more extensive list of genes compared with a shorter list. With the NanoString platform, a set of 28 genes can simultaneously be assayed and adding more target genes does not extend the time to assay the samples and allows this gene set to be further customized. The gene set is not specific for type I IFN responses, and are reported to be upregulated by IFN-γ (www.interferome.com). As no gene amplification is required, there is little source of handling variability that could influence the determination of the gene score.
As many biomarker assays have high interassay variability, which limits their use in patient diagnostics and in assessing longitudinal trends, the low interassay variability and excellent reproducibility of the assay makes it valuable in the diagnosis and in assessing the value of the IFN score as a potential biomarker in predicting responses to treatment with immunosuppressive and targeted therapies. Furthermore, most qPCR-based assays calculate a z-score for each gene that is summed up (Feng and others 2006) or a median fold change versus HCs (Yao and others 2009; Higgs and others 2011; Rice and others 2013, 2017), both requiring an HC cohort for reference. The high reproducibility of the standardized counts in different assays and assay systems (“Standard” and “Elements”), and by different operators, allowed us to calculate geomeans of the gene expression of all 28 genes without the need for an HC reference cohort. Validation of the geomean by comparing HC and patients showed that the geomean performs similarly to the z-score-based score and may allow for direct comparison of scores that are generated at different centers with different machines, which needs to be assessed.
As our ability to assess and quantify an IRG signature expands, more patients and cohorts with IRG signatures are being identified and an increasing number of intracellular pathways that lead to IFN production are being described. For example, type I IFN transcription/production can be triggered by signaling through the intracellular viral or self-RNA/DNA sensors, cGAS/STING or RIG-I, or MDA-5/MAVS as is the case for patients with AGS (Rodero and Crow 2016), through endosomal sensors, including TLR3 or TLR9 as in patients with spondyloenchondrodysplasia (Shinohara and others 2006; Briggs and others 2011), through constitutive activation of STING as in SAVI (Liu and others 2014), and through the TNF/NFkB pathway (Baccala and others 2007) and more, but we currently have no clinical tests that allow us to determine the intracellular signaling pathways that lead to the IRG signature. Whether the IRG signature identified in these diseases is an epiphenomenon or disease causing, and thus a target for therapy, needs to be further evaluated.
The NanoString assay we customized is based on the selection of a set of genes that can discriminate IFN- and IL-1-mediated inflammatory diseases and can be scored and compared longitudinally. The assay lends itself to reliable and reproducible results and is well suited as a potential standardized clinical assay that allows for the comparison of scores between different diseases and different centers. Although the assay was customized with blood samples, the assay could be used to assess the IFN score in RNA samples obtained from biopsies and other samples where RNA extraction is possible (NanoString Technologies Inc; nCounter Elements General Purpose Reagent Package Insert; version 3; 2013). Similar to an FDA-approved NanoString-based assay, PAM50 gene signature Prosigna Breast Cancer Prognostic Gene Signature Assay that is used in the diagnosis of breast cancer in postmenopausal women with hormone receptor-positive breast cancers to estimate the risk of distant recurrence of disease (Nielsen and others 2014), the current assay may be used for diagnostic purposes in the future. The included exact probe sequences and calculations to calibrate the assay allow for a setup that allows the measurement of the IRG signature in other patients.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases (NIAID), the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), and the Clinical Center (CC), all of the National Institutes of Health (NIH). The authors would like to thank Ms. Nicole Plass (RN), Dawn Chapelle (NP), Michelle O'Brien (RN), and Samantha Dill (NP) for their help with clinical sample collection.
Funding: The Intramural Research Program of the NIH, NIAID, the NIAMS, NIDDK, and the CC (ClinicalTrials.gov No. NCT02974595 and NCT00718172).
Author Disclosure Statement
The authors have no relevant disclosures. R.G.M. has received grant support from SOBI, Novartis, Regeneron, and Eli Lilly for clinical studies.
References
- Baccala R, Hoebe K, Kono DH, Beutler B, Theofilopoulos AN. 2007. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med 13(5):543–551 [DOI] [PubMed] [Google Scholar]
- Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, Gregersen PK, Behrens TW. 2003. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A 100(5):2610–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baechler EC, Bauer JW, Slattery CA, Ortmann WA, Espe KJ, Novitzke J, Ytterberg SR, Gregersen PK, Behrens TW, Reed AM. 2007. An interferon signature in the peripheral blood of dermatomyositis patients is associated with disease activity. Mol Med 13(1–2):59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, Pascual V. 2003. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 197(6):711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, Montealegre G, Biancotto A, Reinhardt A, Almeida de Jesus A, Pelletier M, Tsai WL, Remmers EF, Kardava L, Hill S, Kim H, Lachmann HJ, Megarbane A, Chae JJ, Brady J, Castillo RD, Brown D, Casano AV, Gao L, Chapelle D, Huang Y, Stone D, Chen Y, Sotzny F, Lee CC, Kastner DL, Torrelo A, Zlotogorski A, Moir S, Gadina M, McCoy P, Wesley R, Rother K, Hildebrand PW, Brogan P, Kruger E, Aksentijevich I, Goldbach-Mansky R. 2015. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest 125(11):4196–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs TA, Rice GI, Daly S, Urquhart J, Gornall H, Bader-Meunier B, Baskar K, Baskar S, Baudouin V, Beresford MW, Black GC, Dearman RJ, de Zegher F, Foster ES, Frances C, Hayman AR, Hilton E, Job-Deslandre C, Kulkarni ML, Le Merrer M, Linglart A, Lovell SC, Maurer K, Musset L, Navarro V, Picard C, Puel A, Rieux-Laucat F, Roifman CM, Scholl-Burgi S, Smith N, Szynkiewicz M, Wiedeman A, Wouters C, Zeef LA, Casanova JL, Elkon KB, Janckila A, Lebon P, Crow YJ. 2011. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat Genet 43(2):127–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. 2009. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55(4):611–622 [DOI] [PubMed] [Google Scholar]
- Crow YJ. 2011. Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci 1238:91–98 [DOI] [PubMed] [Google Scholar]
- Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, Lindahl T. 2006. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet 38(8):917–920 [DOI] [PubMed] [Google Scholar]
- de Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R. 2015. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol 33:823–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der SD, Zhou A, Williams BR, Silverman RH. 1998. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A 95(26):15623–15628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X, Wu H, Grossman JM, Hanvivadhanakul P, FitzGerald JD, Park GS, Dong X, Chen W, Kim MH, Weng HH, Furst DE, Gorn A, McMahon M, Taylor M, Brahn E, Hahn BH, Tsao BP. 2006. Association of increased interferon-inducible gene expression with disease activity and lupus nephritis in patients with systemic lupus erythematosus. Arthritis Rheum 54(9):2951–2962 [DOI] [PubMed] [Google Scholar]
- Fryer JF, Baylis SA, Gottlieb AL, Ferguson M, Vincini GA, Bevan VM, Carman WF, Minor PD. 2008. Development of working reference materials for clinical virology. J Clin Virol 43(4):367–371 [DOI] [PubMed] [Google Scholar]
- Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, Fell HP, Ferree S, George RD, Grogan T, James JJ, Maysuria M, Mitton JD, Oliveri P, Osborn JL, Peng T, Ratcliffe AL, Webster PJ, Davidson EH, Hood L, Dimitrov K. 2008. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol 26(3):317–325 [DOI] [PubMed] [Google Scholar]
- Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, Kim HJ, Brewer C, Zalewski C, Wiggs E, Hill S, Turner ML, Karp BI, Aksentijevich I, Pucino F, Penzak SR, Haverkamp MH, Stein L, Adams BS, Moore TL, Fuhlbrigge RC, Shaham B, Jarvis JN, O'Neil K, Vehe RK, Beitz LO, Gardner G, Hannan WP, Warren RW, Horn W, Cole JL, Paul SM, Hawkins PN, Pham TH, Snyder C, Wesley RA, Hoffmann SC, Holland SM, Butman JA, Kastner DL. 2006. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med 355(6):581–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs BW, Liu Z, White B, Zhu W, White WI, Morehouse C, Brohawn P, Kiener PA, Richman L, Fiorentino D, Greenberg SA, Jallal B, Yao Y. 2011. Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann Rheum Dis 70(11):2029–2036 [DOI] [PubMed] [Google Scholar]
- Hoffman HM, Broderick L. 2016. The role of the inflammasome in patients with autoinflammatory diseases. J Allergy Clin Immunol 138(1):3–14 [DOI] [PubMed] [Google Scholar]
- Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, Notkins AL. 1979. Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med 301(1):5–8 [DOI] [PubMed] [Google Scholar]
- Kim H, Sanchez GA, Goldbach-Mansky R. 2016. Insights from mendelian interferonopathies: comparison of CANDLE, SAVI with AGS, monogenic lupus. J Mol Med (Berl) 94(10):1111–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, Lee CC, DiMattia MA, Cowen EW, Gonzalez B, Palmer I, DiGiovanna JJ, Biancotto A, Kim H, Tsai WL, Trier AM, Huang Y, Stone DL, Hill S, Kim HJ, St Hilaire C, Gurprasad S, Plass N, Chapelle D, Horkayne-Szakaly I, Foell D, Barysenka A, Candotti F, Holland SM, Hughes JD, Mehmet H, Issekutz AC, Raffeld M, McElwee J, Fontana JR, Minniti CP, Moir S, Kastner DL, Gadina M, Steven AC, Wingfield PT, Brooks SR, Rosenzweig SD, Fleisher TA, Deng Z, Boehm M, Paller AS, Goldbach-Mansky R. 2014. Activated STING in a vascular and pulmonary syndrome. N Engl J Med 371(6):507–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S, Kim PW, Sheikh A, Lee CC, Chen Y, Vera A, Zhang X, Goldbach-Mansky R, Zlotogorski A. 2012. Mutations in proteasome subunit beta type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum 64(3):895–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen T, Wallden B, Schaper C, Ferree S, Liu S, Gao D, Barry G, Dowidar N, Maysuria M, Storhoff J. 2014. Analytical validation of the PAM50-based Prosigna Breast Cancer Prognostic Gene Signature Assay and nCounter Analysis System using formalin-fixed paraffin-embedded breast tumor specimens. BMC Cancer 14:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, Ackroyd S, Allcock R, Bailey KM, Balottin U, Barnerias C, Bernard G, Bodemer C, Botella MP, Cereda C, Chandler KE, Dabydeen L, Dale RC, De Laet C, De Goede CG, Del Toro M, Effat L, Enamorado NN, Fazzi E, Gener B, Haldre M, Lin JP, Livingston JH, Lourenco CM, Marques W, Jr., Oades P, Peterson P, Rasmussen M, Roubertie A, Schmidt JL, Shalev SA, Simon R, Spiegel R, Swoboda KJ, Temtamy SA, Vassallo G, Vilain CN, Vogt J, Wermenbol V, Whitehouse WP, Soler D, Olivieri I, Orcesi S, Aglan MS, Zaki MS, Abdel-Salam GM, Vanderver A, Kisand K, Rozenberg F, Lebon P, Crow YJ. 2013. Assessment of interferon-related biomarkers in Aicardi-Goutieres syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol 12(12):1159–1169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Melki I, Fremond ML, Briggs TA, Rodero MP, Kitabayashi N, Oojageer A, Bader-Meunier B, Belot A, Bodemer C, Quartier P, Crow YJ. 2017. Assessment of type I interferon signaling in pediatric inflammatory disease. J Clin Immunol 37(2):123–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodero MP, Crow YJ. 2016. Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J Exp Med 213(12):2527–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnblom L, Alm GV. 2001. An etiopathogenic role for the type I IFN system in SLE. Trends Immunol 22(8):427–431 [DOI] [PubMed] [Google Scholar]
- Rotman Y, Noureddin M, Feld JJ, Guedj J, Witthaus M, Han H, Park YJ, Park SH, Heller T, Ghany MG, Doo E, Koh C, Abdalla A, Gara N, Sarkar S, Thomas E, Ahlenstiel G, Edlich B, Titerence R, Hogdal L, Rehermann B, Dahari H, Perelson AS, Hoofnagle JH, Liang TJ. 2014. Effect of ribavirin on viral kinetics and liver gene expression in chronic hepatitis C. Gut 63(1):161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, Hertzog PJ. 2013. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res 41(Database issue):D1040–D1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara ML, Lu L, Bu J, Werneck MB, Kobayashi KS, Glimcher LH, Cantor H. 2006. Osteopontin expression is essential for interferon-alpha production by plasmacytoid dendritic cells. Nat Immunol 7(5):498–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Higgs BW, Morehouse C, de Los Reyes M, Trigona W, Brohawn P, White W, Zhang J, White B, Coyle AJ, Kiener PA, Jallal B. 2009. Development of potential pharmacodynamic and diagnostic markers for anti-IFN-alpha monoclonal antibody trials in systemic lupus erythematosus. Hum Genomics Proteomics 2009; DOI: 10.4061/2009/374312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ytterberg SR, Schnitzer TJ. 1982. Serum interferon levels in patients with systemic lupus erythematosus. Arthritis Rheum 25(4):401–406 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.