

Abstract
Engineering the bacteria present in animal microbiomes promises to lead to breakthroughs in medicine and agriculture, but progress is hampered by a dearth of tools for genetically modifying the diverse species that comprise these communities. Here we present a toolkit of genetic parts for the modular construction of broad-host-range plasmids built around the RSF1010 replicon. Golden Gate assembly of parts in this toolkit can be used to rapidly test various antibiotic resistance markers, promoters, fluorescent reporters and other coding sequences in newly isolated bacteria. We demonstrate the utility of this toolkit in multiple species of Proteobacteria that are native to the gut microbiomes of honey bees (Apis mellifera) and bumble bees (Bombus sp.). Expressing fluorescent proteins in Snodgrassella alvi, Gilliamella apicola, Bartonella apis, and Serratia strains enables us to visualize how these bacteria colonize the bee gut. We also demonstrate CRISPRi repression in B. apis and use Cas9-facilitated knockout of an S. alvi adhesion gene to show that it is important for colonization of the gut. Beyond characterizing how the gut microbiome influences the health of these prominent pollinators, this bee microbiome toolkit (BTK) will be useful for engineering bacteria found in other natural microbial communities.
Keywords: host-associated microbiome, probiotics, symbiotic bacteria, colony collapse disorder
Introduction
Symbiotic communities of microorganisms live in close association with many animals and plants. Bacteria in these communities influence the development, metabolism, and health of their hosts.1,2 Genetically engineering bacteria in these microbiomes to manipulate these interactions and add novel functions has enormous practical potential, including such diverse applications as treating human disease,3–5 deploying environmental biosensors,6–9 controlling crop pests,10 and mitigating the spread of disease by insect vectors.11,12 However, these communities are largely composed of bacteria that have not yet been extensively characterized in the laboratory, which presents a major obstacle to achieving these goals.
Decades of study and recent advances in synthetic biology have generated fully featured toolsets for genetically manipulating model microbial species such as Escherichia coli13,14 and Saccharomyces cerevisiae.15 For these organisms, complex synthetic assemblies of heterologous genes can be designed in silico,16 built from sets of well characterized genetic parts, and then tested. One approach for genetically manipulating a microbiome is to add engineered versions of these platform organisms, but model bacteria that function robustly under laboratory conditions often die or fail to proliferate and persist when introduced into natural microbial communities.
A promising alternative for therapeutic applications in animal microbiomes is engineering the bacteria that naturally live in these environments. This is difficult, however, because technologies for genetically manipulating recently isolated bacteria are limited. While some genetic tools exist for human gut-associated bacteria, notably Bacteroides thetaiotaomicron,9,17–19 few bacteria from other gut microbiomes have received such attention. Extensive trial and error is required to validate transformation methods, antibiotic markers, plasmid backbones, promoters, and ribosome binding sites (RBS) in a new bacterial species before it can be genetically engineered.20
One natural community worth engineering is the gut microbiome of bees. Bee pollination of crops is a multibillion dollar industry that is crucial to agriculture, and honey bee colonies are currently suffering high mortality rates for reasons that are not yet fully understood.21 In Western honey bees (Apis mellifera), the hindgut community is dominated by eight core bacterial species that can all be cultivated in the laboratory, and most of these also occur in guts of bumble bees (Bombus sp.).22–24 The bee gut microbiome (BGM) contributes to host nutrition and growth25 and to protection against pathogens.26 Like the human gut microbiome, the BGM is socially acquired and transmitted27 and has a history of antibiotic exposure.22,26,28,29 Major members of the core BGM — Snodgrassella alvi, Gilliamella apicola, and Bartonella apis — have been the subject of detailed genomic analyses.30–32 But aside from random transposon mutagenesis in S. alvi,33 no genetic tools have been reported for these species.
Here, we describe a plasmid toolkit for combining a broad-host-range (BHR) replicon with a set of modular genetic parts and its application to bacteria from the honey bee and bumble bee gut microbiomes. We show that plasmids constructed using this bee microbiome toolkit (BTK) function reliably in multiple species of Proteobacteria found in the BGM. The BTK can be used to express heterologous genes or to repress or disrupt genes in the bacterial chromosome. We generate fluorescent reporter strains of S. alvi, B. apis, G. apicola and a pathogenic Serratia marcescens34 isolate to visualize how they colonize honey bee guts. Finally, we use the BTK to validate the importance of a specific adhesion gene (staA) for S. alvi colonization of the gut. The broad-host-range replicon used in the BTK promises to make it suitable for use in other newly isolated or poorly characterized bacterial species found in animal and plant microbiomes.
Results and Discussion
Design of the bee microbiome toolkit (BTK)
We performed a preliminary screen with a variety of broad-host-range plasmids with different replication origins (RP4, pBBR1, RSF1010) and antibiotic resistance markers (kanamycin, ampicillin, chloramphenicol, spectinomycin) for their ability to be transferred by conjugation and stably maintained in two bacterial species, S. alvi and G. apicola, which are both abundant in the honey bee gut (Supplementary Table 1). Plasmid pMMB67EH, a synthetic plasmid containing an RSF1010 origin,35 was the most versatile: it replicated in both species. Plasmids containing an RSF1010 origin are known to be extremely broad-host-range (BHR) because they encode multiple ORFs that make them less dependent on the presence of specific proteins in a host cell for replication.36,37 Additionally, they contain a promiscuous origin of transfer (oriT) that enables one-way transfer of the plasmid to a recipient cell from a donor cell encoding a conjugation apparatus in trans on the chromosome, such as E. coli MFDpir.38
Because of these characteristics of pMMB67EH, we decided to create a toolkit of genetic parts for hierarchical and combinatorial assembly into its RSF1010-derived backbone for testing in additional species (Figure 1A). These BTK parts are compatible with the Golden Gate cloning scheme used by the Yeast Toolkit(YTK)15, and connector parts from the YTK are required for BTK assembly. BTK parts are classified into eight types defined by the specific flanking overhangs generated by type IIS restriction enzyme cleavage. Entry vectors containing any complete set of parts labeled 1-8 can be combined via one BsaI Golden Gate assembly reaction into a complete plasmid (Figure 1B). This assembly creates a Stage 1 plasmid comprising parts 2-4 flanked by assembly connector parts 1 and 5 with vector backbone components in parts 6-8. Transcriptional units from multiple Stage 1 plasmids that have matching sets of connector parts can be further composed into one vector by Stage 2 assembly using BsmBI (Figure 1C).
Figure 1.
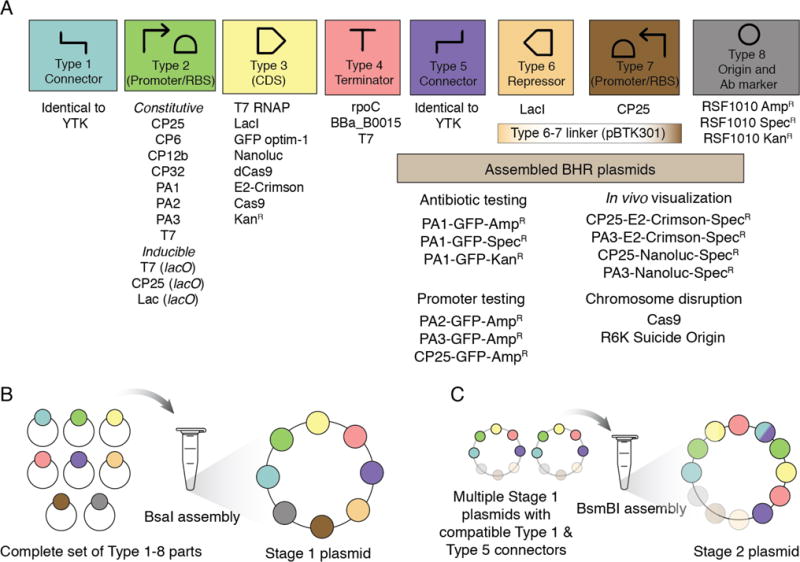
Design of the bee microbiome toolkit (BTK) and schematic assembly. (A) The BTK was designed for Golden Gate assembly according to a scheme with eight part types compatible with the yeast toolkit (YTK).15 Parts of each type generated in this study are shown in the top panel. Type 1-5 and Type 8 parts are defined as in the YTK except that Type 3 open-reading frames include the stop codon. Type 6 and 7 parts are either replaced with a linker part or used to incorporate a reverse reading frame encoding a transcriptional regulator for inducible expression of the main Type 3 open-reading frame and its promoter, respectively, during Stage 1 assembly, so that costly or toxic genes can be repressed while they are assembled into transcriptional units. (B) Schematic of Stage 1 (BsaI) assembly. Plasmid parts are shown, but PCR products with appropriate overhangs can be substituted. (C) Schematic Stage 2 (BsmBI) assembly. Compatible Stage 2 connectors are described in the YTK documentation.
To create BTK vector backbone parts, we replaced the high-copy number bacterial ColE1 origin of YTK part 8 plasmids (AmpR, KanR, SpecR) with the RSF1010 origin from pMMB67EH. These backbones retain the oriT for delivery into recipient cells via conjugation, which is useful for genetically modifying bacterial species and strains lacking established chemical or electrical transformation techniques. In the original YTK, Type 6 and 7 parts encode a yeast marker and a yeast origin, respectively. We repurposed Type 6 part overhangs for flanking a DNA sequence encoding an optional additional CDS (such as a repressor) in the reverse orientation relative to the Type 2 part CDS, and Type 7 part overhangs for incorporating an optional reverse promoter for driving expression of the part 6 CDS. A combined Type 6-7 linker (pBTK301) can be used in lieu of these parts to create constructs lacking this extra reverse gene.
We used these vectors to construct a variety of plasmids containing a single fluorescent protein driven by a broad-host-range promoter. To build more complex assemblies, such as those with T7 RNA polymerase driving inducible expression of GFP, we combined multiple vectors from BsaI Stage 1 assembly in a Stage 2 BsmBI assembly. Notable components of the BTK that expand on the set of genetic parts available in the YTK for compatible assembly include:
3 BHR plasmids with different antibiotic resistance cassettes and oriT as Type 8 origin parts for Stage 1 assembly
2 BHR plasmids ready for Stage 2 assembly (SpecR, KanR)
11 bacterial promoter/RBS combinations as Type 2 parts
6 new CDSs including E2-Crimson39 and Nanoluc40 for in vivo visualization as Type 3 parts
3 bacterial terminators as Type 4 parts
1 transcriptional repressor (LacI) as a Type 6 part
2 R6K-origin plasmid backbones to assemble suicide plasmids for gene disruption or chromosomal modification
Pre-assembled plasmids with BHR promoters for immediate testing in new bacterial strains
Dataset S1 summarizes BTK parts, assembled BTK plasmids, and their validation. In the next sections, we describe and evaluate the functions of plasmids for control of gene expression and disruption of chromosomal genes in non-model bacteria from the honey bee (A. mellifera) and bumble bee (Bombus sp.) gut microbiomes. Component and assembled BTK plasmids have been deposited with Addgene (accession number pending acceptance) for distribution to other researchers.
BTK plasmids function in diverse bacterial species found in the bee gut
We next sought to explore the host range of the RSF1010 origin used as a basis for the BTK in the context of a larger set of bee-associated bacterial strains. Simultaneously, we needed to identify antibiotic resistance genes able to function in each bacterial strain. To do so, we constructed three BTK plasmids, each with a different antibiotic resistance marker and encoding GFP driven by the PA1 promoter: pBTK501 (AmpR), pBTK519 (KanR), pBTK520 (SpecR). We performed biparental matings between E. coli MFDpir donors containing each plasmid and bee gut-associated strains (see Methods). Stable transconjugants were obtained for all of the Gram-negative strains we tested with at least one of these three plasmids, as verified by further passaging on antibiotic-containing media, PCR amplification of plasmid sequences, and GFP expression (Figure 2A). Successfully transformed bacterial species include Alpha-, Beta-, and Gammaproteobacteria and strains isolated from different bee species (A. mellifera, Bombus terrestris, Bombus impatiens, and Bombus pensylvanicus). Several of the bacterial species (S. alvi, G. apicola, B. apis, and Parasaccharibacter apium) are phylogenetically distant from any established model organisms and have no previously reported genetic tools. Transfer of the BTK plasmids was efficient, with >10−3 transconjugants per CFU for four diverse bacterial species (Figure 2B).
Figure 2.
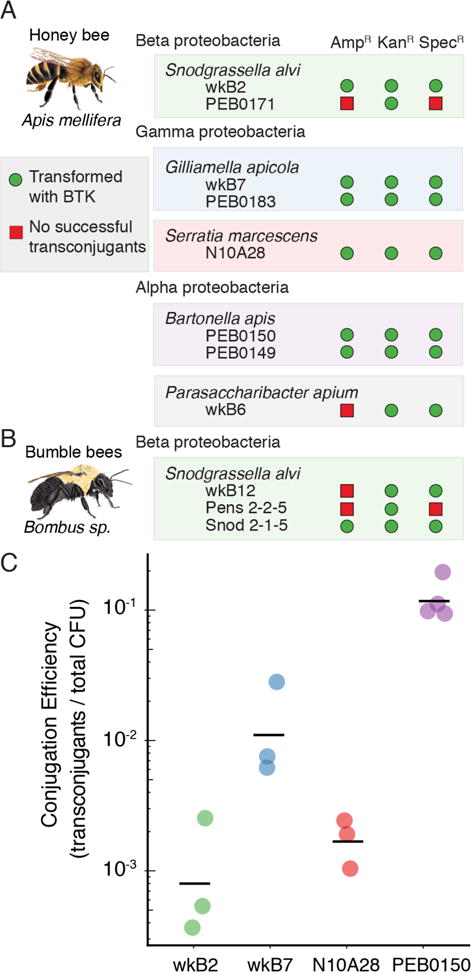
The bee microbiome toolkit (BTK) functions in diverse bee-associated bacteria. (A) Replication of the BTK backbone and the function of three antibiotic resistance cassettes were tested in eight honey bee-associated bacterial strains as described in the Methods. At least one antibiotic resistance cassette functioned in each strain, and the kanamycin cassette functioned in all eight strains. (B) Replication of the BTK backbone and the function of three antibiotic resistance cassettes were tested in three bumble bee-associated bacterial strains. Again, the plasmid with kanamycin resistance was maintained in all three bacteria. (C) Conjugation frequency in four bee gut-associated strains. Black bars are the geometric mean and each point is an independent conjugation. Conjugation in B. apis is the most efficient, with conjugation efficiency approximately 10%.
Identifying functional promoters in BGM species
While some sequence features of transcriptional promoters are conserved across bacterial species, there is no guarantee that promoters designed to function in model organisms will function effectively in new bacterial isolates from a natural community of interest.19 The BTK includes BHR promoters and RBS combinations as Type 2 parts that can be used to build plasmids to identify functional sequences for driving protein expression in new bacterial hosts. We compared the function of the BHR promoters PA1 (pBTK501), PA2 (pBTK509), PA3 (pBTK510), and CP25 (pBTK503) in S. alvi wkB2, G. apicola wkB7, B. apis PEB0150, and S. marcescens N10A28, all isolated from honey bee gut communities. Promoters PA1, PA2, PA3 are strong early promoters from bacteriophage T7.41 The synthetic CP25 promoter was originally designed to function in Lactococcus strains,42 and the BTK includes other promoters from this series.
Using flow cytometry, we characterized fluorescent protein expression from these promoters (Figure 2A). These promoters display significant variability in activity across strains when they are all tested with the same RBS. As expected, the promoter-RBS pairs function most strongly in S. marcescens, which is most closely related to E. coli. In the other BGM strains, expression was weaker, but there was a signal above background for most promoters that were tested with this RBS. Fluorescence is generally lower in S. alvi, G. apicola, and B. apis than it is in E. coli. In E. coli the distributions of fluorescent per cell for the PA2, PA3, and CP25 promoters are noticeably bimodal. This may be an intrinsic property of the promoter or due to the accumulation of “broken” plasmids with mutations that inactivate burdensome GFP expression43. In BGM strains, with the exception of CP25 in G. apicola, these distributions are unimodal, indicating consistent fluorescent expression across single cells. PA3 expression was strong in S. alvi, and we used this observation to design a constitutive E2-Crimson-expressing plasmid (pBTK570) to test expression in vivo, as described in later sections. Validation of additional parts (E2-Crimson, Nanoluc, and other CP-series promoters) is available in Supplemental Figures S1–S3.
Inducible gene expression in BGM species
Induction systems are required for the temporal control of gene expression, and are useful for testing the functional roles of microbes in gut environments.18 We tested two lacI induction systems: one simple system composed of a modified CP25 promoter with lacO sites and a more complex system that uses T7 RNA polymerase (T7 RNAP). We tested IPTG-induction of these systems in E. coli MFDpir, S. alvi wkB2, G. apicola wkB7, B. apis PEB0150, and S. marcescens N10A28. The simple system (pBTK552) showed robust induction of GFP in all strains tested (Figure 3B). Interestingly, G. apicola GFP expression with this system surpassed that of E. coli and S. marcescens.
Figure 3.
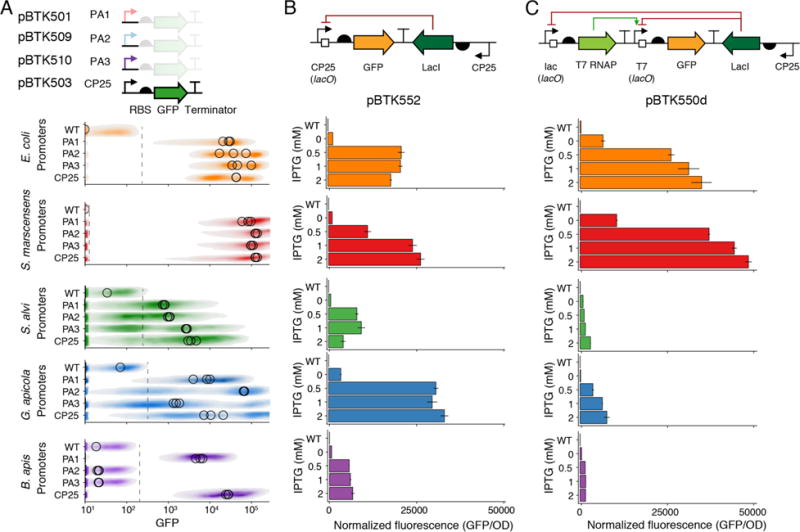
Constitutive and inducible control of in vitro gene expression in bee gut bacteria. (A) Flow cytometry results of GFP fluorescence from four broad-host-range promoters in each of four honey bee-associated bacterial strains and an E. coli control. One representative fluorescence distribution for each promoter is shown, with the medians from three biological replicates plotted as open circles. Spotted grey line indicates maximum detected fluorescence in wild-type cells. Median fluorescent values were calculated from cells more fluorescent than wild-type. (B) GFP fluorescence from a designed CP25 (lacO) promoter at different levels of IPTG-induction, measured in four BGM strains and an E. coli control. All tested species are responsive to IPTG induction, and G. apicola shows the highest expression across all strains. (C) GFP fluorescence from a T7 (lacO) promoter at different levels of IPTG-induction of T7 RNAP expression in the same four BGM strains. Schematics in (A)—(C) show the design of tested constructs using Synthetic Biology Open Language (SBOL) standard glyphs.62 Error bars are standard deviations (n = 3).
For the T7 RNAP system, we built two transcriptional unit plasmids (pBTK549d, pBTK541), one bearing lacI driven by the CP25 promoter and T7 RNAP under control of the inducible lac promoter and the other with GFP expressed from a T7 promoter with lacO sites, and combined them into a composite plasmid (pBTK550d). (Figure 3C). Expression was strong in S. marcescens N10A28 and E. coli MFDpir, with maximal GFP expression after induction surpassing the simpler system in which lacI directly regulates GFP expression. However, in G. apicola wkB7, S. alvi wkB2, and B. apis PEB0150, we saw weaker induction of GFP compared to the simpler system. The cause of this weak expression is unknown. It may be due to poor transcription from the lac promoter driving T7 RNAP or to an intrinsic incompatibility between T7 RNAP and the intracellular environment in the BGM species tested. In all strains, the inducible T7 RNAP construct showed appreciable background expression when not induced.
CRISPRi repression of chromosomal gene expression in Bartonella apis
We next used the BTK to suppress gene activity in a BGM bacterium. Catalytic mutants of Cas9 (dCas9) have been used to reduce transcription of target genes, an approach termed CRISPR interference (CRISPRi), in diverse mammalian and bacterial systems.44 To expand this approach to new non-model bacterial species, we established a modified dCas9 system in which targeting is achieved by a BTK part encoding a small guide RNA (sgRNA) (Figure 4A).45 To test the system, we targeted the sgRNA to a PA1-driven GFP gene in B. apis PEB0150, which we inserted into the chromosome using Tn7-based integration.46 GFP expression was significantly reduced in the presence of a sgRNA targeted to the GFP sequence (Figure 4B). Coupled with the induction system, this ability to repress a target gene enables functional studies of essential genes that cannot be disrupted entirely.
Figure 4.
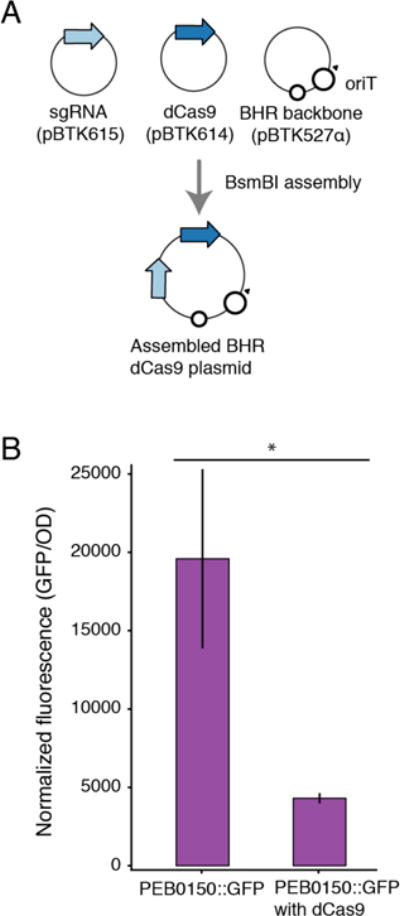
dCas9 gene silencing in Bartonella apis. (A) Schematic assembly of dCas9 plasmids for gene suppression. (B) Fluorescence from chromosomally integrated GFP in PEB0150 in the presence and absence of dCas9 and sgRNA targeting GFP. Background fluorescence of wild-type PEB0150 was subtracted. GFP fluorescence decreased in presence of dCas9 targeting GFP (p = 0.004, Kruskal-Wallis rank sum test). Error bars are 95% confidence intervals (n = 4).
Cas9-assisted gene disruption in the BGM
Gene disruption is an important tool for establishing gene function and for studying interactions between genes. After identifying functional antibiotic cassettes in our earlier plasmid-replication screen, we attempted to use homologous recombination to disrupt chromosomal genes in our BGM strains. To improve the efficacy of targeted gene disruption, we also implemented a two-step approach based on using Cas9 cleavage for chromosomal modifications44 (see Methods). In step one, Cas9 is introduced into a cell on the BTK backbone (pBTK601) without any targeting sgRNA. In step two, a second round of conjugation is used to deliver a suicide plasmid with the replacement cassette (~1000 bp homology flanking a functional antibiotic resistance gene) and the sgRNA targeting the desired chromosomal location. The suicide plasmid is made with Golden Gate assembly using repurposed Type 2-4 overhangs and an R6K origin of replication (Figure 5A–B). The sgRNA can be retargeted using MEGAWHOP cloning47 (see Methods). A detailed description of suicide plasmid assembly and validation of mutants is shown in Supplemental Figure S4. We expected that Cas9 cleavage might facilitate recombination into the chromosome and that it would also select against single-crossover integrations, in which the suicide plasmid backbone is incorporated into the chromosome, because they preserve the cleavage site, whereas double-crossover integrations result in replacement of the targeted gene sequence with just the antibiotic resistance cassette and delete the cleavage site.
Figure 5.
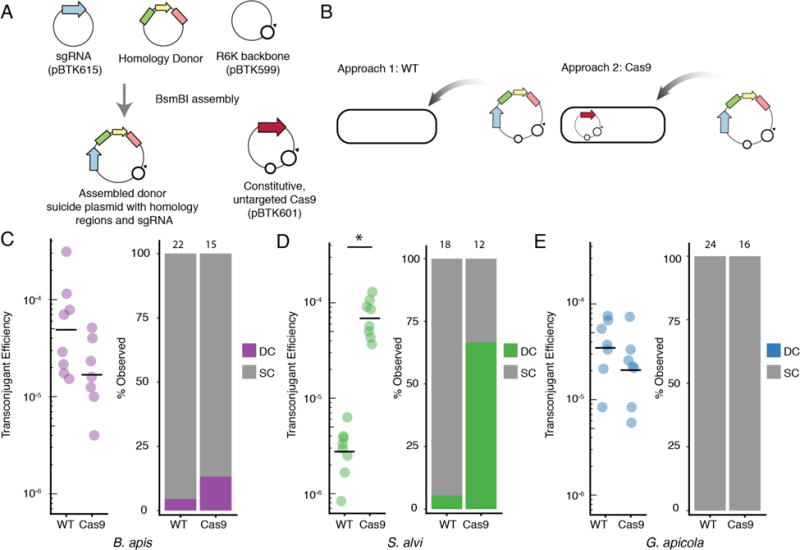
Cas9 assisted gene disruption in species from the bee gut microbiota. (A) Schematic assembly of R6K-based suicide plasmids. Assembly strategy and validation primers are described in Supplementary Figure S4. (B) The two tested approaches for gene disruption. The suicide plasmids were introduced into either wild-type bacteria or bacteria possessing the constitutively active Cas9 (pBTK601). (C) Transconjugation frequency and percent of desired mutants in B. apis, in the presence and absence of Cas9. The Cas9 plasmid did not increase the efficiency of genome modification. Numbers above each bar indicate the number of clones evaluated. (D) Transconjugation frequency and proportion of desired mutants in S. alvi. S. alvi wkB2 showed increased efficiency of genome modification in the presence of the Cas9 plasmid (p = 0.0007, Kruskal-Wallis rank sum test). (E) Transconjugation frequency and proportion of desired mutants in G. apicola. Each point in C-E is from an independent conjugation experiment. Bars in C-E represent the geometric mean of estimated transconjugation efficiencies.
To test the utility of this scheme, we attempted to generate gene disruptions in three BGM species. In S. alvi wkB2 we targeted staA (SALWKB2_RS11470), an adhesion gene previously implicated in a genome-wide screen as important for gut colonization (Supplemental Figure S5).33 In G. apicola wkB7 we targeted acetate kinase ackA (A9G17_RS12535) (Supplemental Figure S6), and in B. apis PEB0150 we targeted nitrate reductase narG (PEB0150_RS00755) (Supplemental Figure S7). We designed our homology regions to be internal to each coding sequence, so that even single-crossover events would disrupt gene function. For S. alvi wkB2, our multi-step system showed higher efficiency compared to basic homologous recombination not using Cas9. In the presence of Cas9, wkB2 mutants were obtained more frequently and were more often double-crossover mutants (Figure 5D). In contrast, B. apis PEB0150 showed relatively high gene disruption efficiency even in the absence of Cas9, and the Cas9 system had little effect on improving the number of double-crossover mutants (Figure 5C). In G. apicola wkB7 the Cas9 was also not helpful, and we obtained no double-crossover mutants (Figure 5E). The G. apicola wkB7 mutants isolated showed irregular PCR amplification at the expected junctions (Supplemental Figure S6), indicating we could not effectively disrupt ackA, perhaps because it is an essential gene in this species. While we validated this general approach to gene disruption in multiple BGM species, it will be necessary to repeat this procedure on more target genes in these species to gain a broader understanding of its efficiency and the effect of Cas9.
Engineered strains colonize bees and can be directly visualized in the ileum
We next tested the ability of engineered BGM strains to colonize newly emerged worker bees removed from the hive before they acquire a normal microbiota. Previously, S. alvi and G. apicola have been visualized in bees using fluorescent in situ hybridization.48 However, this technique can only be used at one time point because it requires sacrificing the bee. In contrast, fluorescent reporter strains can be used to non-destructively estimate bacterial abundance and observe how bacterial community structure changes over time in live bees. Previous studies have examined how S. alvi colonizes the honey bee gut,31,49 but colonization by G. apicola, B. apis, and S. marcescens has not been investigated.26,34
We inoculated newly emerged workers with ~104 CFU per bee of either S. marcescens N10A28 or S. alvi wkB2, each carrying a constitutively expressed E2-Crimson fluorescent protein (pBTK570). After 5 days, we dissected bees from each group and examined their guts (Figure 6). Fluorescent bacteria were successfully imaged directly in guts without preparation or fixation, preserving natural community structure. S. marcescens N10A28 shows robust colonization in all gut compartments, while other species show spatially restricted colonization. As previously reported, S. alvi wkB2 robustly colonizes the ileum, with little colonization in the midgut and rectum.
Figure 6.
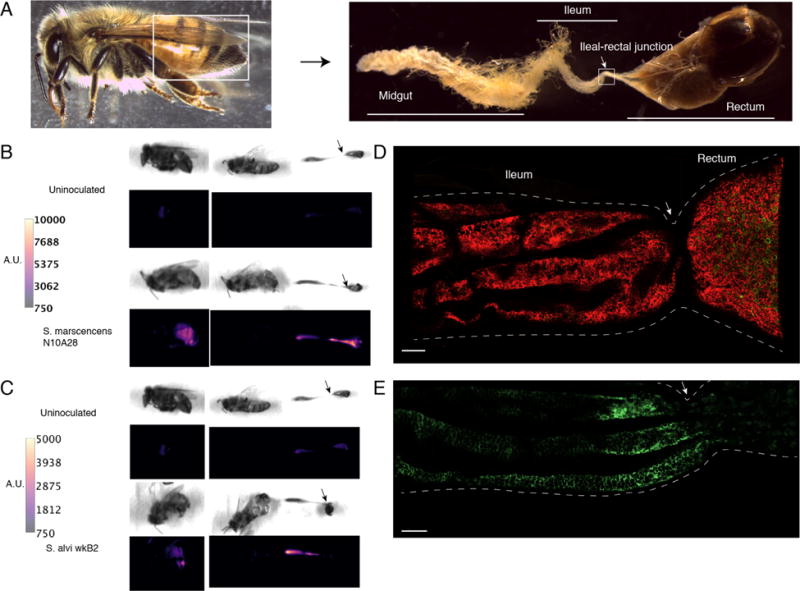
Visualization of engineered bacteria in the honey bee gut. (A) Intact honey bee worker and dissection of honey bee gut showing brightfield microscopy of midgut, ileum, and rectum. (B) Fluorescent imaging of whole bee (left) and dissected bee (right) 5 days after inoculation with S. marcescens N10A28 expressing E2-Crimson (plasmid pBTK570). Control bee is uninoculated. Color corresponds to pixel fluorescence intensity. Engineered S. marcescens N10A28 is present in the midgut, ileum, and rectum. (C) Similar to (B), with S. alvi wkB2 expressing E2-Crimson as inoculum. Control bee is identical to (B), but different fluorescent intensity scales are used for comparison between bees inoculated with S. alvi and S. marcescens. Engineered S. alvi wkB2 is visibly fluorescent in the midgut and ileum. (D) Confocal imaging of partial ileum and rectum in bees inoculated with S. marcescens N10A28 expressing E2-Crimson (red). As in (B), S. marcescens can be seen robustly colonizing throughout the ileum and rectum. (E) Similar to (D), with S. alvi wkB2 expressing E2-Crimson (green). Snodgrassella alvi wkB2 colonizes the ileum, but not the rectum. Scale bars in (D) and (E) are 100 μm. Images are representative of multiple bees inspected (n = 3-5 per condition) for (B–E). White and black arrows correspond to the ileum-rectum junction across images (A–E).
Additionally, we performed co-inoculations with S. alvi wkB2 and either B. apis PEB0150 or G. apicola wkB7 engineered to express GFP (pBTK520). We again dissected the guts of colonized bees and were able to fluorescently image in vivo co-colonization of these defined communities (Figure 7A–B). While both B. apis and G. apicola are found in the ileum co-located with S. alvi, they also colonize the rectum, in contrast to S. alvi.
Figure 7.
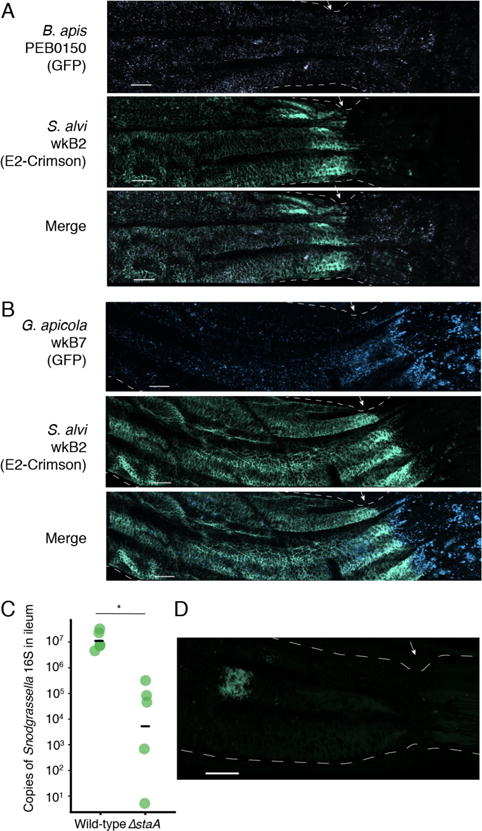
Visible co-inoculation of the bee gut with species from the bee gut microbiota and the role of staA in colonization. (A) The ileum-rectum junction imaged by confocal fluorescence microscopy 5 days after co-inoculating B. apis PEB0150 (purple) and S. alvi wkB2 (green). When co-inoculated, B. apis and S. alvi are co-located in the ileum, but only B. apis colonizes the rectum. (B) Similar to (A), with images taken 5 days after co-inoculation of G. apicola wkB7 (blue) and S. alvi wkB2 (green). As in (A), S. alvi remains restricted to the ileum, while G. apicola is present in both ileum and rectum. Scale bars are 100 μm. Images are representative of multiple bees inspected (n = 3 per condition). (C) The number of S. alvi 16S ribosomal DNA copies 5 days after inoculating newly emerged worker bees with the S. alvi WT or ΔstaA mutant, based on quantitative PCR. Horizontal bars represent means per condition (n = 5). The ΔstaA has a significant colonization defect compared to WT (p = 2.7 × 10−6, Kruskall-Wallis rank sum test). (D) The ileums of bees inoculated with S. alvi wkB2ΔstaA expressing E2-Crimson (pBTK570) were imaged 5 days after colonization. Mutants achieved lower colonization levels than did S. alvi WT (see Fig. 6E). Localized colonization was typical of multiple ileums inspected (n = 3). Scale bar is 100 μm.
Snodgrassella staA contributes to gut colonization in vivo
Finally, we sought to validate the usefulness of the BTK for disrupting specific genes in BGM species in order to investigate their function. StaA belongs to a family of YadA-like adhesion proteins important for colonization and pathogenicity in multiple host-associated species.50 These trimeric autotransporter proteins localize to the bacterial membrane and form “lollipop” structures that allow bacteria to adhere to epithelial cells.51,52 Orthologs of these genes are found in multiple S. alvi genomes, including those from honey bee- and bumble bee-associated strains.31 Our previous work screening a transposon mutant library identified staA (SALWKB2_RS11470) as necessary for the fitness of S. alvi during gut colonization.33 However, we were unable to isolate a mutant from our library with a transposon disrupting staA, and thus we could not fully characterize and validate the role of this gene.
Using the BTK, we generated a ΔstaA mutant in S. alvi wkB2 (as described above). We then labeled the ΔstaA mutant and a wild-type control with a BTK plasmid expressing E2-Crimson (pBTK570) to assess the effects of disrupting this gene in the context of the bee gut. The wkB2 ΔstaA mutant shows reduced colonization efficacy compared to a wild-type control, as measured by qPCR of S. alvi 16S rRNA gene copies (Figure 7C). The colonization pattern of this mutant in terms of its localization within the gut (Figure 7D) is distinct from that of wild-type S. alvi (Figure 6E). After 6 days, the mutant does not form the contiguous, robust colonization of the ileum wall seen for the wild type strain. Instead, colonization is apparently restricted to small patches, while the majority of the ileum remains uncolonized.
Summary
We built the BTK, the first Golden Gate toolkit designed specifically for the combinatorial assembly of broad-host-range plasmids, with the aim of expanding synthetic biology into diverse bacteria native to non-laboratory environments. In this study, we applied the BTK to modify bacteria found in the honey bee gut microbiome. These species are typical of many other bacteria in natural microbial communities of interest: they have only been cultured recently, are phylogenetically diverse, and have few or no established genetic tools. We validated fundamental BTK components needed for genetic modification, including antibiotic selection markers, conjugation procedures, and promoters to express proteins under constitutive or inducible control. BGM strains engineered with the BTK colonize the guts of newly emerged bees, and fluorescent in vivo imaging revealed a characteristic spatial distribution of each species in the gut.
The species we engineered are within the Proteobacteria, a diverse Gram-negative phylum that is a common component of animal- and plant-associated communities. Although we have not yet tested it more broadly, BTK components should also be useful for genetically modifying other bacteria native to other natural communities. The core of the BTK is the RSF1010 plasmid origin, which is known to replicate in diverse bacterial lineages including Cyanobacteria, Agrobacterium, and others.36,37,53,54 The BTK also includes promoters that have previously been shown to function in both Gram-negative and Gram-positive bacteria.42 The E2-Crimson reporter gene fluoresces at far-red excitation wavelengths, which is ideal for in vivo imaging of bacteria through tissue in host-associated systems.39 While broad-host-range plasmids have already been extensively used to study newly isolated bacteria in the past,54 the combinatorial nature of this new toolkit makes it possible to test multiple antibiotic resistance markers and promoters, which are more difficult to replace in plasmids that rely on classical cloning approaches.
Standard part definitions enable researchers to customize a toolkit by adding new capabilities for their own applications, as we did with re-using parts from the yeast toolkit (YTK).15 Several aspects of the BTK could be improved and fleshed out in future work. Separating antibiotic resistance cassettes and replication origins into different subparts and adding to the library of choices available for each function would allow more combinations of antibiotics and origins to be tested when first working with a new species. Gram-positive origins, such as pAMβ1,54 would be especially useful for the manipulation of other common host-associated phyla such as Firmicutes and Actinobacteria.55 Further validation of some BTK parts, such as the dCas9 and Cas9 systems, is needed to conclude that they will function reliably across diverse species. Other established broad-host-range tools—such as Tn7-transposon integration46, Group II intron-based gene disruption57, and emerging CRISPR methods for targeted mutagenesis58—could also be incorporated into the BTK-compatible Golden Gate framework in the future.
Application of the BTK to engineering bee gut bacteria enables new approaches to microbiome research in these insect species that are important pollinators and model systems for studying social behavior and learning. For example, gene disruption combined with fluorescent visualization of bacterial cells in living bees can be used to improve our understanding of the molecular basis of host-microbe and microbe-microbe interactions and their relevance to host health. The BTK can also be used to implement and test biotechnological approaches for mitigating threats to bee health. For example, it could be used to engineer commensal gut bacteria to degrade pesticides or suppress pathogen populations (i.e., paratransgenesis)59. These efforts could one day profoundly affect the health of the bee colonies that sustain modern agriculture.
Methods
Bacterial Culture
A complete list of bacterial strains used in this work and their sources is available as Table S2. Unless otherwise specified, bacterial strains S. alvi wkB2, G. apicola wkB7, Parasaccharibacter apium wkB6, B. apis PEB0150, G. apicola PEB0183, B. apis PEB0149, and S. alvi PEB0171, S. marcescens N10A28 were grown on Columbia agar supplemented with 5% sterile sheep’s blood (B-COL) and incubated at 35°C in a 5% CO2 atmosphere as static cultures. E. coli were cultured at 37°C with orbital shaking at 225 rpm over a 1-inch diameter. E. coli MFDpir was grown in LB supplemented with 0.3 mM diaminopimelic acid (DAP). E. coli EC100D and E. coli DH5α were grown in LB.
For antibiotic selection, the following concentrations were used: ampicillin (100 μg/mL E. coli, 30 μg/mL S. alvi, 30 μg/mL G. apicola, 30 μg/mL B. apis, 300 μg/mL S. marcescens), kanamycin (50 μg/mL E. coli, 20 μg/mL S. alvi, 20 μg/mL G. apicola, 20 μg/mL B. apis), spectinomycin (60 μg/mL E. coli, 30 μg/mL for S. alvi, 30 μg/mL G. apicola, 30 μg/mL B. apis, 30 μg/mL P. apium, 180 μg/mL S. marcescens).
Biparental Conjugation
MFDpir with mobilizable plasmid (“donor strain”) was grown overnight, shaking in LB with appropriate selective antibiotics and DAP (0.3 mM) supplementation. Recipient strains (wkB2, wkB7, PEB0150, PEB0183, PEB0171, N10A28, wkB6, wkB12, Snod 2-15, Pens 2-2-5) were grown overnight on solid media. Recipient and donor strains were washed in 1 mL PBS, spun down (1006 × g for 5 minutes), and resuspended with 1 mL of PBS. These two suspensions were mixed in a 9:1 OD ratio of recipient:donor and spotted (without filter) onto a B-COL plate supplemented with 0.3 mM DAP. Conjugations proceeded overnight (~12-14 hours) and were scraped from the plate into PBS the next morning. Conjugation mixtures were again gently spun down (1006 × g) and washed twice in PBS to remove residual DAP. Approximately 100 μL of this mixture (and 1:10, 1:100 dilutions) was plated onto selective antibiotic plates and incubated 2-3 days to obtain transconjugant colonies. Transconjugants were passaged again on selective media and confirmed by PCR amplification of a plasmid sequence and visible fluorescence, when appropriate. For the initial broad-host-range plasmid screen, transconjugants were further verified by plasmid re-isolation and electroporation into E. coli DH5α cells. To determine conjugation efficiency, mating mixtures were serially diluted and plated on selective and non-selective plates. Conjugation frequency was calculated as the number of fluorescent transconjugant CFUs on selective plates per total CFUs on non-selective plates.
BTK construction
Construction of the BTK backbone was carried out with Gibson assembly60 following established protocols. New part plasmids were constructed using a previously published BsmBI assembly protocol for the yeast toolkit (YTK)15 with inserts synthesized as double-stranded DNA gBlocks (IDT). New parts were cloned into the pYTK001 entry vector. The BTK kit uses the entry vector plasmid, connector parts (Type 1 and Type 5), and part sequence overhangs of the YTK. In contrast to the YTK, Type 3 parts of the BTK include a stop codon, as the Type 4 terminators do not include a stop codon. The entire list of BTK parts is available in Dataset S1. A complete list of non-BTK plasmids used to generate data for this work is available in Table S3.
Measuring BGM GFP in vitro
To measure fluorescence, 50 μL of ~0.2 OD bacterial cultures were pooled on B-COL agar plates and incubated for 48 hours. Cells were scraped into PBS and then loaded into wells of a 96 well plate to measure fluorescent excitation using a Tecan Spark 10M multimode microplate reader at excitation/emission wavelengths of 485/535. Fluorescent readings were corrected with blank values, and then normalized by OD. Gain was set manually and consistent throughout experiments.
Flow cytometry analysis of GFP expression
As with plate reader measurements, we pooled 50 μL of ~0.2 OD bacterial culture onto B-COL agar plates and incubated for 48 hours. Bacteria were scraped into PBS, washed, and then gently spun down (1006 × g for 5 minutes). Cells were resuspended vigorously to disrupt any biofilm, and then diluted to ~0.1 OD in HPLC-grade water. We counterstained cells with SYTO 17 red nucleic acid stain (Thermo-Fisher), and then ran samples on a BD LSRFortessa SORP Flow Cytometer at the UT Austin flow cytometry core. Data were acquired with FACSDiva v6.1.3, and then analyzed with FlowJo v10.4.2. All samples were run under identical conditions. GFP-A voltage was consistent throughout experiments. Non-fluorescent controls were used to determine forward-scatter, side-scatter, and APC-A (counterstain) gates that were then set individually for each species.
Tn7-transposition in B. apis
For the chromosomal insertion of gfp into B. apis, a tri-parental mating was performed with B. apis, E. coli MFDpir with pTNS2, and E. coli MFDpir with pTN7-PA1-gfp-kan in an 8:1:1 ratio. Conjugation proceeded for 12 hours, and transformed B. apis was selected with kanamycin as in biparental conjugation.
CRISPRi gene repression
Broad-host-range dCas9 plasmids are created by BsmBI assembly of 3 parts plasmids containing: (1) the sgRNA transcriptional unit (pBTK615), (2) the dCas9 transcriptional unit (pBTK614), and (3) the broad-host-range backbone with ConLE and ConRE connector sequences (pBTK527a). To repress gfp expression in B. apis, we targeted the gfp non-template strand by using the N20 sequence: 5′–CGTCTAATTCCACGAGGATT. The sgRNA plasmid can be retargeted using MEGAWHOP cloning47. Briefly, in MEGAWHOP cloning a double-stranded PCR product containing the sequence change to be introduced, but otherwise identical to a portion of the plasmid, is used as a “megaprimer” to re-amplify the whole plasmid in a second PCR reaction. Because the sgRNA targeting sequence is short, it is possible to include a new target sequence flanked by 20 bp of homology to the plasmid on either side in one of the primers used in the initial PCR reaction to generate the megaprimer. The fully assembled CRISPRi plasmid (pBTK618) was conjugated into B. apis with chromosomally integrated PA1-gfp, and GFP fluorescence was measured as above.
Chromosomal disruption using Cas9 and homologous recombination
Plasmid pBTK601 contains Cas9 driven by the kanamycin resistance gene promoter on the broad-host-range backbone. This plasmid was conjugated into S. alvi wkB2, G. apicola wkB7, and B. apis PEB0150 and maintained with spectinomycin. The CP25-driven sgRNA is on plasmid pBTK615 and can be retargeted using MEGAWHOP cloning.47 A full description of homology donor plasmid assembly is available in Supplemental Figure S4. Briefly, a genomic homology segment upstream of the gene of interest to disrupt or replace is amplified with Type 2 part overhangs, and a downstream genomic homology segment is amplified as a Type 4 part. Upstream homology, antibiotic resistance cassette (Type 3), and downstream homology are combined in a single BsaI reaction with ConLE and ConRE to form a Stage 1 assembly of the replacement cassette. The final BsmBI assembly includes: (1) the sgRNA plasmid, (2) the replacement cassette plasmid, and (3) pBTK599 (R6K suicide plasmid backbone). This final assembly must be transformed into pir+ strains, such as EC100D or MFDpir.
Efficiency of chromosomal disruption with and without Cas9
Recipient BGM strains (wkB2, wkB2∷pBTK601, wkB7, wkB7∷pBTK601, PEB0150, PEB0150∷pBTK601) were grown on B-COL plates for 48 hours prior to conjugation. Donor E. coli strains were grown in liquid culture overnight prior to conjugation. Donor and recipients were washed in PBS and mixed in a 1:9 ratio (by OD), and 100 μL was plated on B-COL + 0.3 mM DAP media for overnight conjugation. After 14 hours, the entire conjugation mixtures were scraped into PBS and washed twice to remove residual DAP, and dilutions were plated on selective agar plates (B-COL + Kanamycin 20 μg/mL) and non-selective agar plates (B-COL). Efficiency of gene disruption was calculated as (# of transconjugant cells)/(# of total cells). To identify single-crossover and double-crossover mutants, a series of PCR reactions were conducted as described in Supplemental figure S4. Briefly, transconjugants were screened for the appropriate upstream and downstream junctions with colony PCR. Potential double-crossover mutants were then further screened for the size of the disrupted region, and loss of the suicide plasmid backbone.
Laboratory care of honey bees
Microbiota-free bees were obtained and raised using methods described previously.31 Briefly, pupae were pulled under sterile conditions from brood combs obtained from outdoor hives. These pupae emerged in a sterile incubator (becoming newly emerged adult workers) and were then sorted into individual cup cages for further development in the laboratory. Prior to inoculation, newly emerged workers were allowed to feed on sterile irradiated pollen (Betterbee) and 50% sucrose solution ad libitum. For any individual experiment, all pupae were obtained from the same hive. When raised in this manner, Apis mellifera workers remain uncolonized by core BGM bacteria species and show very low levels of environmental bacteria in their guts.27 It is critical to pull the pupae from frames at an early stage, before the mouthparts have hardened, as later pupal stages will begin to ingest hive material and may be colonized.
Mono- and Co-inoculation of engineered BGM into honey bees
After obtaining newly emerged workers, bees were chilled at 4°C for 30 minutes and then coated in sugar syrup containing resuspended bacterial inoculum, transferred to cup cages, and allowed to groom each other. The inoculum generally contained 200 μL of OD ~0.1 bacterial suspension combined with 800 μL of 1:1 sucrose:water solution. Approximately 30 μL of this solution per bee was used for inoculations (corresponding to 104 bacteria per bee to ensure robust inoculation). Plate counts of the inoculum were used to confirm concentrations.
In vivo imaging of bacterial burden using E2-Crimson
To visualize in vivo expression of E2-Crimson in living bees, we used a Syngene G:Box Chemi XX6 gel doc system at the UT ICMB Microscopy Core. Bees were chilled on ice for 30 min to minimize movement, then imaged using manufacturers recommended instructions for far-red fluorescent probe visualization: “Red LED” light source and “Filter 705M” emission filter. All bees were imaged under identical conditions: 5 minutes exposure time for whole bee and 30 seconds for bees with dissected guts. Images were saved as TIFF files for further analysis in FIJI.61 In FIJI, fluorescence intensity was mapped to the “mpl-magma” scale. A representative bee for each condition is shown. No further image manipulation was performed. Different scales are used for comparing fluorescent S. marcescens and fluorescent BGM species due to the increased fluorescent protein production and titer of S. marcescens.
Confocal fluorescence microscopy
Fluorescent images were obtained at the UT ICMB Microscopy core on a Zeiss 710 Laser Scanning Confocal microscope. Bees were chilled and then dissected to expose rectum, ileum, and midgut. Without puncturing the gut, the entire gut compartment was transferred to an Ibidi μ-Dish 35 mm (CAT #81156) and then placed on the microscope. Images were taken with a 20× objective and tiled using Zeiss software. Z-stack 2-channel fluorescent images were taken and combined using Imaris software. Intensity on individual channels was false colored to correspond to species-specific coloring. Display intensity of individual channels was scaled linearly to aid in visualization of different species, but no further transformations or background reduction was used.
qPCR to assess colonization of staA mutant
Absolute quantification of 16S rRNA gene copies specific to S. alvi was performed as described previously33. Cohorts of newly emerged bees were hand-fed with equal amounts (~104 CFU/bee) of either wild-type S. alvi or the staA mutant. Control bees were maintained identically but remained uninoculated. After five days, five bees from each group were dissected and DNA was isolated from individual bee guts using the cetyltrimethylammonium bromide (CTAB) extraction method outlined previously27. After extraction, S. alvi-specific primers were used for quantitative PCR and absolute quantification based on 10-fold dilution of the target sequence in a pGEM-T plasmid vector. Reactions were run in triplicate.
Quantification and Statistical Analysis
All data processing and statistical analyses were done in R. Kruskal-Wallis rank sum tests were used to assess significance in the dCas9 gene repression experiment and the Cas9-assisted genome modification experiments.
Supplementary Material
Supplementary Table S1. Broad-host-range plasmid screen in bee gut bacteria
Supplementary Table S2. Bacterial strains
Supplementary Table S3. Non-BTK plasmids
Supplementary Table S4. Oligonucleotides
Supplementary Table S5. Assembled BTK plasmids
Supplementary Figure S1. Expression of Nanoluc in BGM strains
Supplementary Figure S2. Expression of E2-Crimson in BGM strains
Supplementary Figure S3. Validation of additional BTK promoters in E. coli
Supplementary Figure S4. Construction of replacement cassette plasmids
Supplementary Figure S5. Schematic and validation of staA disruption in S. alvi
Supplementary Figure S6. Schematic and validation of ackA disruption in G. apicola
Supplementary Figure S7. Schematic and validation of narG disruption in B. apis
Supplementary Dataset S1. BTK parts
Supplementary Dataset S2. Complete sequences for BTK plasmids
Acknowledgments
We thank members of the Moran, Barrick, Davies, and Ellington laboratories for discussion and ideas. Special thanks to Kim Hammond for care and maintenance of bee hives and laboratory equipment. Thanks to Dan Deatherage for assistance with the Flow Cytometry experiment. Thanks to Vidya Pandarinath and Kate Elston for help organizing BTK parts for submission to Addgene. Thanks to Waldan Kwong, Phillip Engel and Hauke Koch for bacterial strains used in this study. Thanks to Julie Hayes and Anna Webb of the UT Microscopy and Imaging Facility for confocal microscopy assistance and instruction.
Funding
This work was supported by DARPA (HR0011-15-C0095 to NAM, ADE, BWD, JEB) and the NIH (1R01GM108477-01 to NAM).
Abbreviations
- BTK
Bee microbiome toolkit
- BHR
Broad-host-range
- BGM
Bee gut microbiome
Footnotes
Competing Interests
N.A.M, J.E.B., and S.P.L. have filed a provisional patent application (62/529,754) for engineering bee gut bacteria with BTK components to improve bee health.
Author Contributions
All authors conceived the study. JP designed the BTK assembly scheme. SPL, JP, JEP, MB, SK, PG, DR conducted experiments. SPL, JEB, and NAM wrote the manuscript. SPL and JEP performed data analysis and imaging. All authors participated in manuscript revision.
References
- 1.Sommer F, Bäckhed F. The gut microbiota — masters of host development and physiology. Nat Rev Microbiol. 2013;11:227–238. doi: 10.1038/nrmicro2974. [DOI] [PubMed] [Google Scholar]
- 2.Shin SC, Kim SH, You H, Kim B, Kim AC, Lee KA, Yoon JH, Ryu JH, Lee WJ. Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science. 2011;334:670–674. doi: 10.1126/science.1212782. [DOI] [PubMed] [Google Scholar]
- 3.Shen TCD, Albenberg L, Bittinger K, Chehoud C, Chen YY, Judge CA, Chau L, Ni J, Sheng M, Lin A, Wilkins BJ, Buza EL, Lewis JD, Daikhin Y, Nissim I, Yudkoff M, Bushman FD, Wu GD. Engineering the gut microbiota to treat hyperammonemia. J Clin Invest. 2015;125:2841–2850. doi: 10.1172/JCI79214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palmer JD, Piattelli E, McCormick BA, Silby MW, Brigham CJ, Bucci V. Engineered probiotic for the inhibition of Salmonella via tetrathionate-Induced production of microcin H47. ACS Infect Dis. 2017;4:39–45. doi: 10.1021/acsinfecdis.7b00114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mimee M, Citorik RJ, Lu TK. Microbiome therapeutics — advances and challenges. Adv Drug Delivery Rev. 2016;105:44–54. doi: 10.1016/j.addr.2016.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daeffler KN-M, Galley JD, Sheth RU, Ortiz-Velez LC, Bibb CO, Shroyer NF, Britton RA, Tabor JJ. Engineering bacterial thiosulfate and tetrathionate sensors for detecting gut inflammation. Mol Syst Biol. 2017;13:923. doi: 10.15252/msb.20167416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riglar DT, Giessen TW, Baym M, Kerns SJ, Niederhuber MJ, Bronson RT, Kotula JW, Gerber GK, Way JC, Silver PA. Engineered bacteria can function in the mammalian gut long-term as live diagnostics of inflammation. Nat Biotechnol. 2017;105:1–8. doi: 10.1038/nbt.3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kotula JW, Kerns SJ, Shaket LA, Siraj L, Collins JJ, Way JC, Silver PA. Programmable bacteria detect and record an environmental signal in the mammalian gut. Proc Natl Acad Sci U S A. 2014;111:4838–4843. doi: 10.1073/pnas.1321321111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mimee M, Tucker AC, Voigt CA, Lu TK. Programming a human commensal bacterium, Bacteroides thetaiotaomicron, to sense and respond to stimuli in the murine gut microbiota. Cell Syst. 2015;1:62–71. doi: 10.1016/j.cels.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Abrieux A, Chiu JC. Oral delivery of dsRNA by microbes: Beyond pest control. Commun Integr Biol. 2016;9:e1236163. doi: 10.1080/19420889.2016.1236163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang S, Dos-Santos ALA, Huang W, Liu KC, Oshaghi MA, Wei G, Agre P, Jacobs-Lorena M. Driving mosquito refractoriness to Plasmodium falciparum with engineered symbiotic bacteria. Science. 2017;357:1399–1402. doi: 10.1126/science.aan5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang S, Ghosh AK, Bongio N, Stebbings KA, Lampe DJ, Jacobs-Lorena M. Fighting malaria with engineered symbiotic bacteria from vector mosquitoes. Proc Natl Acad Sci U S A. 2012;109:12734–12739. doi: 10.1073/pnas.1204158109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iverson SV, Haddock TL, Beal J, Densmore DM. CIDAR MoClo: improved MoClo assembly standard and new E. coli part library enable rapid combinatorial design for synthetic and traditional biology. ACS Synth Biol. 2015;5:99–103. doi: 10.1021/acssynbio.5b00124. [DOI] [PubMed] [Google Scholar]
- 14.Moore SJ, Lai HE, Kelwick RJR, Chee SM, Bell DJ, Polizzi KM, Freemont PS. EcoFlex: a multifunctional MoClo kit for E. coli synthetic biology. ACS Synth Biol. 2016;10:1059–1069. doi: 10.1021/acssynbio.6b00031. [DOI] [PubMed] [Google Scholar]
- 15.Lee ME, DeLoache WC, Cervantes B, Dueber JE. A highly characterized yeast toolkit for modular, multipart assembly. ACS Synth Biol. 2015;4:975–986. doi: 10.1021/sb500366v. [DOI] [PubMed] [Google Scholar]
- 16.Nielsen AAK, Der BS, Shin J, Vaidyanathan P, Paralanov V, Strychalski EA, Ross D, Densmore D, Voigt CA. Genetic circuit design automation. Science. 2016;352:aac7341–aac7341. doi: 10.1126/science.aac7341. [DOI] [PubMed] [Google Scholar]
- 17.Salyers AA, Shoemaker NB, Guthrie EP. Recent advances in Bacteroides genetics. Crit Rev Microbiol. 1987;14:49–71. doi: 10.3109/10408418709104435. [DOI] [PubMed] [Google Scholar]
- 18.Lim B, Zimmermann M, Barry NA, Goodman AL. Engineered regulatory systems modulate gene expression of human commensals in the gut. Cell. 2017;169:547–558. doi: 10.1016/j.cell.2017.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whitaker WR, Shepherd ES, Sonnenburg JL. Tunable expression tools enable single-cell strain distinction in the gut microbiome. Cell. 2017;169:538–538. doi: 10.1016/j.cell.2017.03.041. e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salyers AA, Bonheyo G, Shoemaker NB. Starting a new genetic system: lessons from Bacteroides. Methods. 2000;20:35–46. doi: 10.1006/meth.1999.0903. [DOI] [PubMed] [Google Scholar]
- 21.Cox-Foster DL, Conlan S, Holmes EC, Palacios G, Evans JD, Moran NA, Quan PL, Briese T, Hornig M, Geiser DM, Martinson V, vanEngelsdorp D, Kalkstein AL, Drysdale A, Hui J, Zhai J, Cui L, Hutchison SK, Simons JF, Egholm M, Pettis JS, Lipkin WI. A metagenomic survey of microbes in honey bee colony collapse disorder. Science. 2007;318:283–287. doi: 10.1126/science.1146498. [DOI] [PubMed] [Google Scholar]
- 22.Kwong WK, Moran NA. Gut microbial communities of social bees. Nat Rev Microbiol. 2016;14:374–384. doi: 10.1038/nrmicro.2016.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kwong WK, Moran NA. Cultivation and characterization of the gut symbionts of honey bees and bumble bees: description of Snodgrassella alvi gen. nov., sp. nov., a member of the family Neisseriaceae of the Betaproteobacteria, and Gilliamella apicola gen. nov., sp. nov., a member of Orbaceae fam. nov., Orbales ord. nov., a sister taxon to the order “Enterobacteriales” of the Gammaproteobacteria. Int J Syst Evol Microbiol. 2013;63:2008–2018. doi: 10.1099/ijs.0.044875-0. [DOI] [PubMed] [Google Scholar]
- 24.Moran NA, Hansen AK, Powell JE, Sabree ZL. Distinctive gut microbiota of honey bees assessed using deep sampling from individual worker bees. PLoS One. 2012;7:e36393. doi: 10.1371/journal.pone.0036393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng H, Powell JE, Steele MI, Dietrich C, Moran NA. Honeybee gut microbiota promotes host weight gain via bacterial metabolism and hormonal signaling. Proc Natl Acad Sci U S A. 2017;114:4775–4780. doi: 10.1073/pnas.1701819114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raymann K, Shaffer Z, Moran NA. Antibiotic exposure perturbs the gut microbiota and elevates mortality in honeybees. PLoS Biol. 2017;15:e2001861. doi: 10.1371/journal.pbio.2001861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Powell JE, Martinson VG, Urban-Mead K, Moran NA. Routes of acquisition of the gut microbiota of the honey bee Apis mellifera. Appl Environ Microbiol. 2014;80:7378–7387. doi: 10.1128/AEM.01861-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ludvigsen J, Porcellato D, L’Abée-Lund TM, Amdam GV, Rudi K. Geographically widespread honeybee-gut symbiont subgroups show locally distinct antibiotic-resistant patterns. Mol Ecol. 2017;26:6590–6607. doi: 10.1111/mec.14392. [DOI] [PubMed] [Google Scholar]
- 29.Tian B, Fadhil NH, Powell JE, Kwong WK, Moran NA. Long-term exposure to antibiotics has caused accumulation of resistance determinants in the gut microbiota of honeybees. mBio. 2012;3:e00377–12. doi: 10.1128/mBio.00377-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng H, Nishida A, Kwong WK, Koch H, Engel P, Steele MI, Moran NA. Metabolism of toxic sugars by strains of the bee gut symbiont Gilliamella apicola. mBio. 2016;7:e01326–16. doi: 10.1128/mBio.01326-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwong WK, Engel P, Koch H, Moran NA. Genomics and host specialization of honey bee and bumble bee gut symbionts. Proc Natl Acad Sci U S A. 2014;111:11509–11514. doi: 10.1073/pnas.1405838111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Segers FH, Kešnerová L, Kosoy M, Engel P. Genomic changes associated with the evolutionary transition of an insect gut symbiont into a blood-borne pathogen. ISME J. 2017;60:810. doi: 10.1038/ismej.2016.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Powell JE, Leonard SP, Kwong WK, Engel P, Moran NA. Genome-wide screen identifies host colonization determinants in a bacterial gut symbiont. Proc Natl Acad Sci U S A. 2016;113:13887–13892. doi: 10.1073/pnas.1610856113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burritt NL, Foss NJ, Neeno-Eckwall EC, Church JO. Sepsis and hemocyte loss in honey bees (Apis mellifera) infected with Serratia marcescens strain Sicaria. PLoS One. 2016;11:e0167752. doi: 10.1371/journal.pone.0167752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fürste JP, Pansegrau W, Frank R, Blöcker H, Scholz P, Bagdasarian M, Lanka E. Molecular cloning of the plasmid RP4 primase region in a multi-host-range tacP expression vector. Gene. 1986;48:119–131. doi: 10.1016/0378-1119(86)90358-6. [DOI] [PubMed] [Google Scholar]
- 36.Jain A, Srivastava P. Broad host range plasmids. FEMS Microbiol Lett. 2013;348:87–96. doi: 10.1111/1574-6968.12241. [DOI] [PubMed] [Google Scholar]
- 37.Meyer R. Replication and conjugative mobilization of broad host-range IncQ plasmids. Plasmid. 2009;62:57–70. doi: 10.1016/j.plasmid.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferrières L, Hémery G, Nham T, Guérout AM, Mazel D, Beloin C, Ghigo JM. Silent mischief: bacteriophage Mu insertions contaminate products of Escherichia coli random mutagenesis performed using suicidal transposon delivery plasmids mobilized by broad-host-range RP4 conjugative machinery. J Bacteriol. 2010;192:6418–6427. doi: 10.1128/JB.00621-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strack RL, Hein B, Bhattacharyya D, Hell SW, Keenan RJ, Glick BS. A rapidly maturing far-red derivative of dsRed-Express2 for whole-cell labeling. Biochemistry. 2009;48:8279–8281. doi: 10.1021/bi900870u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hall MP, Unch J, Binkowski BF, Valley MP, Butler BL, Wood MG, Otto P, Zimmerman K, Vidugiris G, Machleidt T, Robers MB, Benink HA, Eggers CT, Slater MR, Meisenheimer PL, Klaubert DH, Fan F, Encell LP, Wood KV. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol. 2012;7:1848–1857. doi: 10.1021/cb3002478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siebenlist U. Nucleotide sequence of the three major early promoters of bacteriophage T7. Nucleic Acids Res. 1979;6:1895–1907. doi: 10.1093/nar/6.5.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jensen PR, Hammer K. The sequence of spacers between the consensus sequences modulates the strength of prokaryotic promoters. Appl Environ Microbiol. 1998;64:82–87. doi: 10.1128/aem.64.1.82-87.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sleight SC, Bartley BA, Lieviant JA, Sauro HM. Designing and engineering evolutionary robust genetic circuits. J Biol Eng. 2010;4:12. doi: 10.1186/1754-1611-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barrangou R, Horvath P. A decade of discovery: CRISPR functions and applications. Nat Microbiol. 2017;2:1–9. doi: 10.1038/nmicrobiol.2017.92. [DOI] [PubMed] [Google Scholar]
- 45.Bikard D, Jiang W, Samai P, Hochschild A, Zhang F, Marraffini LA. Programmable repression and activation of bacterial gene expression using an engineered CRISPR-Cas system. Nucleic Acids Res. 2013;41:7429–7437. doi: 10.1093/nar/gkt520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi KH, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP. A Tn7-based broad-range bacterial cloning and expression system. Nat Meth. 2005;2:443–448. doi: 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
- 47.Miyazaki K. MEGAWHOP cloning: a method of creating random mutagenesis libraries via megaprimer PCR of whole plasmids. Meth Enz. 2011;498:399–406. doi: 10.1016/B978-0-12-385120-8.00017-6. [DOI] [PubMed] [Google Scholar]
- 48.Martinson VG, Moy J, Moran NA. Establishment of characteristic gut bacteria during development of the honeybee worker. Appl Environ Microbiol. 2012;78:2830–2840. doi: 10.1128/AEM.07810-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Engel P, James RR, Koga R, Kwong WK, McFrederick QS, Moran NA. Standard methods for research on Apis mellifera gut symbionts. J of Api Res. 2013;52:1–24. [Google Scholar]
- 50.Linke D, Riess T, Autenrieth IB, Lupas A, Kempf VAJ. Trimeric autotransporter adhesins: variable structure, common function. Trends Microbiol. 2006;14:264–270. doi: 10.1016/j.tim.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 51.Ribet D, Cossart P. How bacterial pathogens colonize their hosts and invade deeper tissues. Microbes Infect. 2015;17:173–183. doi: 10.1016/j.micinf.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 52.Tahir El Y, Skurnik M. YadA, the multifaceted Yersinia adhesin. Int J Med Microbiol. 2001;291:209–218. doi: 10.1078/1438-4221-00119. [DOI] [PubMed] [Google Scholar]
- 53.Taton A, Unglaub F, Wright NE, Zeng WY, Paz-Yepez J, Brahamsha B, Palenik B, Peterson TC, Haerizadeh F, Golden SS, Golden JW. Broad-host-range vector system for synthetic biology and biotechnology in cyanobacteria. Nucleic Acids Res. 2014;42:gku673–e136. doi: 10.1093/nar/gku673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.pAMB1 cite. Clewell DB, Yagi Y, Dunny GM, Schultz SK. Characterization of three plasmid deoxyribonucleic acid molecules in a strain of Streptococcus faecaelis: identification of a plasmid determining erythromycin resistance. J Bacteriol. 1974;117:283–289. doi: 10.1128/jb.117.1.283-289.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robinson CJ, Bohannan JM, Young VB. From structure to function: the ecology of host-associated microbial communities. Microbiol Mol Biol Rev. 2010;74:453–476. doi: 10.1128/MMBR.00014-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martínez-García E, Aparicio T, Goñi-Moreno A, Fraile S, de Lorenzo V. SEVA 2.0: an update of the Standard European Vector Architecture for de-/re-construction of bacterial functionalities. Nucleic Acids Res. 2015;43:D1183–D1189. doi: 10.1093/nar/gku1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Enyeart PJ, Chirieleison SM, Dao MN, Perutka J, Quandt EM, Yao J, Whitt JT, Keatinge-Clay AT, Lambowitz AM, Ellington AD. Generalized bacterial genome editing using mobile group II introns and Cre-lox. Mol Syst Biol. 2013;9:685–685. doi: 10.1038/msb.2013.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Banno S, Nishida K, Arazoe T, Mitsunobu H, Kondo A. Deaminase-mediated multiplex genome editing in Escherichia coli. Nat Microbiol. 2018;339:819. doi: 10.1038/s41564-017-0102-6. [DOI] [PubMed] [Google Scholar]
- 59.Rangberg A, Diep DB, Rudi K, Amdam GV. Paratransgenesis: an approach to improve colony health and molecular insight in honey bees (Apis mellifera)? Integr Comp Biol. 2012;52:89–99. doi: 10.1093/icb/ics089. [DOI] [PubMed] [Google Scholar]
- 60.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Meth. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 61.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Meth. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Galdzicki M, Clancy KP, Oberortner E, Pocock M, Quinn JY, Rodriguez CA, Roehner N, Wilson ML, Adam L, Anderson JC, Bartley BA, Beal J, Chandran D, Chen J, Densmore D, Endy D, Grünberg R, Hallinan J, Hillson NJ, Johnson JD, Kuchinsky A, Lux M, Misirli G, Peccoud J, Plahar HA, Sirin E, Stan GB, Villalobos A, Wipat A, Gennari JH, Myers CJ, Sauro HM. The Synthetic Biology Open Language (SBOL) provides a community standard for communicating designs in synthetic biology. Nat Biotechnol. 2014;32:545–550. doi: 10.1038/nbt.2891. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Broad-host-range plasmid screen in bee gut bacteria
Supplementary Table S2. Bacterial strains
Supplementary Table S3. Non-BTK plasmids
Supplementary Table S4. Oligonucleotides
Supplementary Table S5. Assembled BTK plasmids
Supplementary Figure S1. Expression of Nanoluc in BGM strains
Supplementary Figure S2. Expression of E2-Crimson in BGM strains
Supplementary Figure S3. Validation of additional BTK promoters in E. coli
Supplementary Figure S4. Construction of replacement cassette plasmids
Supplementary Figure S5. Schematic and validation of staA disruption in S. alvi
Supplementary Figure S6. Schematic and validation of ackA disruption in G. apicola
Supplementary Figure S7. Schematic and validation of narG disruption in B. apis
Supplementary Dataset S1. BTK parts
Supplementary Dataset S2. Complete sequences for BTK plasmids