

Abstract
Due to the wide presence of carbohydrates in nature and their crucial roles in numerous important biological processes, oligosaccharides have attracted a lot of attention in synthetic organic chemistry community. Many innovative synthetic methods have been developed for oligosaccharide synthesis, among which the pre-activation based glycosylation is particularly noteworthy. Traditionally, glycosylation reactions are carried out when the glycosyl donor and the acceptor are both present when the promoter is added. In comparison, the pre-activation based glycosylation is unique, where the glycosyl donor is activated by the promoter in the absence of the acceptor. Upon complete donor activation, the acceptor is added to the reaction mixture enabling glycosylation. The key step in any oligosaccharide synthesis is the stereoselective formation of the glycosidic bond. As donor activation and acceptor glycosylation are temporally separated, pre-activation based glycosylation can bestow unique stereochemical control. This review systematically discusses factors impacting the stereochemical outcome of a pre-activation based glycosylation reaction including substituents on the glycosyl donor, reaction solvent, and additives. Applications of pre-activation based stereoselective glycosylation in assembly of complex oligosaccharides are also discussed.
Keywords: diastereoselectivity, glycosides, glycosylation, pre-activation, synthesis design
Graphical abstract
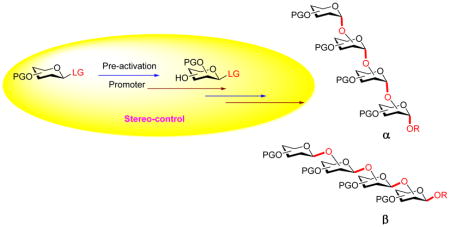
Pre-activation based glycosylation separates donor activation and acceptor glycosylation into two distinct steps. As a result, unique stereo-control can be achieved by judicious choices of protective groups, anomeric groups, additives and solvents. Some highly challenging glycosyl linkages including β-mannosides and deoxy glycosides can now be prepared with high stereoselectivities.
Introduction
Carbohydrates play important roles in many biological events.[1–2] To expedite biological studies of carbohydrates, chemical synthesis is of high current interests. Compared to peptides and nucleic acids, carbohydrate synthesis has a unique stereochemical challenge as the anomeric oxygen atom can be either axial or equatorial relative to the pyranose ring. Since anomeric configuration can significantly impact the structure and function of the glycosides, the ability to stereoselectively form the glycosyl linkage is critical.
The synthesis of the axial linkage typically relies on the anomeric effect, as axial glycosides are generally more stable than the equatorial counterpart (Scheme 1a). To prepare the equatorial glycosides, when the neighboring carbon center bears an equatorial hydroxyl or amine moiety, an acyl type protective group can be installed (Scheme 1b). Through anchimeric assistance of the neighboring protective group, the equatorial product is favored. While many glycosides with the desired stereochemistry have been successfully prepared through these approaches, anomeric mixtures are often formed especially in the absence of neighboring group participation, requiring tedious separation to obtain pure products. Thus, new methodologies enabling stereoselective glycosylation are desired.
Scheme 1.

a) Formation of axial glycoside commonly relies on the anomeric effect due to the higher thermodynamic stability of the axial product. b) Participation by neighboring protective groups on equatorial C-2 of the glycosyl donor can typically result in high selectivity in forming the equatorial glycoside.
The majority of glycosylation reactions are performed by pre-mixing the donor and acceptor together followed by addition of the promoter for donor activation (Scheme 2a). Recently, an alternative strategy has been established, which pre-activates the glycosyl donor first in the absence of the acceptor (Scheme 2b). Upon complete donor activation, the acceptor is added for glycoside formation. As the donor activation and acceptor glycosylation are carried out in two distinct steps using this protocol, unique stereo- and chemo-selectivity can be achieved compared to the more classical pre-mixed approach.[3–4] In this review, we will summarize how pre-activation can aid in the stereoselective formation of glycosides as well as the mechanistic investigation of glycosylation.
Scheme 2.

a) Traditional glycosylation typically employs the pre-mixed approach with both the donor and the acceptor mixed together when the promoter is added; b) The pre-activation based glycosylation strategy activates the glycosyl donor in the absence of the acceptor, which temporally separates the donor activation step from acceptor glycosylation.
The substituents on a glycosyl donor can significantly impact the structures of the reactive intermediates upon pre-activation, thus influencing stereochemical outcome of the glycosylation. With pre-activation, the intermediates can be characterized spectroscopically prior to acceptor addition, which helps to rationalize glycosylation results and guide further reaction design. In addition, additives and reaction solvents can be used to tune the intermediate structures, bestowing the ability to control stereoselectivity through reagents rather than substrates.
In this review, we will first systemically discuss the effects of glycosyl donor substituents on the stereoselectivity of a glycosylation reaction. This will be followed by the discussions of the influence of reaction solvent and additives.
1. The Effect of 4, 6-O-Benzylidene Acetal on Stereoselectivity
The formation of 1,2-cis-β-D-mannopyranosyl linkage had been viewed as one of the most difficult glycosylation reactions due to the steric hindrance posed by the cis-2-O substituent and its lower thermodynamic stability, while the corresponding 1,2-trans-α-D-mannoside has additional electronic stabilization due to the anti-bonding orbital of C2-O linkage with the anti-bonding orbital of C1-O.[5] A breakthrough was achieved when the Crich group discovered that specific 4, 6-benzylidene protected mannosyl sulfoxides could give excellent β-selectivities under the pre-activation condition with a wide range of acceptors.[3, 6] As an example, the mannosyl sulfoxide donor 5 was pre-activated by trifluoromethane sulfonyl anhydride (Tf2O), which was followed by the addition of acceptor 6 giving disaccharide 7 in an excellent 90% yield and good β selectivity (α:β = 1:5) (Scheme 3). Pre-activation was found to be critical, as pre-mixing the donor and acceptor under otherwise identical reaction condition led to much lower β-selectivity (α:β = 8:1).[7] The high β-selectivity from pre-activation was not limited to glycosyl sulfoxide donors, as 4,6-benzylidene protected thiomannoside,[8] 2-(hydroxycarbonyl)benzyl mannoside[9] or mannosyl hemiacetal[10] all gave excellent β-selectivities. The presence of benzylidene on the glycosyl donor was crucial for high β-selectivity as conformationally more labile mannosyl donors with all ether type protective groups gave poor stereoselectivity.[11]
Scheme 3.

Pre-activation based glycosylation of 5 and 6 gave high selectivity of β-linked disaccharide 7 (α:β = 1:5). The traditional pre-mixed approach led to much lower β-selectivity (α:β = 8:1, 80% yield).
Extensive mechanistic studies have been performed to better understand the high β-selectivity obtained with benzylidene protected mannosyl donors (Scheme 4).[6] Upon pre-activation by Tf2O, donor 5 was converted to 8, which could evolve into several species including oxocarbenium ion 9 and α-triflate 10. Low temperature NMR analysis of the reaction mixture following pre-activation indicated the α-glycosyl triflate was the major resting intermediate.[11] This was attributed to the cyclic benzylidene group locking the C5-C6 bond in trans-gauche conformation.[12] Consequently, the C6-O6 bond is kept antiperiplanar to C5-O5 bond, maximizing its electron-withdrawing effect and destabilizing the electron deficient 9.[3, 13] In addition, the torsional strain bestowed by the benzylidene would favor the glycosyl triflate 10 further shifting the equilibrium away from 9.[3, 14] Upon addition of the acceptor, nucleophilic attack of the α-glycosyl triflate through an exploded transition state with significant oxocarbenium ion character would result in highly stereoselective formation of β-mannosides.[15–16] In contrast, direct nucleophilic attack of the oxocarbenium ion 9 by the acceptor should yield the α-mannoside 12 as the major product (Scheme 4).
Scheme 4.

Proposed mechanism for high β-selectivity in pre-activation of mannosyl donor 5.
Protective groups on other positions of glycan rings can impact the stereoselectivity of 4, 6-O-benzylidene directed β-mannosylation. The reaction of the 3-O-TBDMS-4,6-O-benzylidene protected mannosyl donor 13 with acceptor 14 using the pre-activation protocol gave the desired disaccharide 15 in 77% yield with a low anomeric selectivity (α:β = 1.8:1) (Scheme 5).[17–18] This was attributed to the steric buttressing effect due to the presence of the bulky TBDMS substituent on O-3 position. It was hypothesized that the TBDMS group could push the 2-O-benzyl (Bn) towards the anomeric center of the α-glycosyl triflate intermediate thus shielding the β-face from nucleophilic attack and reducing β-selectivity. This was supported by the observation that replacing the 3-TBDMS with 3-OBn led to the β-linked disaccharide 17 as the sole anomer.[18]
Scheme 5.

The 3-O protective group can significantly influence the β selectivity of benzylidene protected mannosyl donors with the bulky 3-O TBDMS group significantly reducing the β selectivity. Reagents and conditions: (a) Tf2O, −78°C, 2,6-di-tert-butyl-4-methylpyridine (DTBMP), CH2Cl2, then 14.
The understanding of the steric impact of protective groups was applied to the synthesis of a mannohexaose, which is the partial structure of cell surface mannan from Rhodotorula glutinis.[19–20] In this synthesis, coupling of disaccharide donor 18 with acceptor 19 led to trisaccharide 20 with no β-selectivity (Scheme 6).[21] This lack of selectivity is most likely due to the bulky glycan on 3-O of the reducing end glycan of the donor resulting in an unfavorable steric buttressing effect. To overcome this hurdle, the less bulky propargyl ether group was utilized as the 2-O protective group. The new disaccharide donor 21 with the 2-O propargyl ether gave a much enhanced β-selectivity (β : α = 5:1).[21]
Scheme 6.

Installation of the less sterically encumbered 2-O propargyl group on the disaccharide donor significantly enhanced the β-selectivity in mannosylation reactions. Reagents and conditions: (a) Tf2O, 1-benzenesulfinyl piperidine (BSP), 2,4,6-tri-tert-butylpyrimidine (TTBP), CH2Cl2, −60 °C, then 19, −60 °C to 0 °C.
In contrast to the excellent β-selectivity from the mannosyl series, the corresponding 4,6-O-benzylidene protected glucosyl donors gave high α-selectivity with a variety of acceptors (Scheme 7).[22] Low-temperature NMR studies following pre-activation identified the α-glucosyl triflate as the dominant intermediate formed.
Scheme 7.

In contrast to β-selective mannosylation reaction with benzylidene protected mannosyl donors, high α-selectivities were obtained using the corresponding benzylidene bearing glucosyl donors. Reagents and conditions: (a) PhSOTf, DTBMP, CH2Cl2, −78 °C, then ROH, −78 °C to 0 °C.
The opposing stereoselectivity to the mannosyl series was rationalized by the influence of O2-C2-C3-O3 interaction during transformation of the glycosyl triflate (Scheme 8).[23] For the mannose series, when the α-triflate 29 converts to the oxocarbenium ion, which should be either in the 4H3 chair 30 or the B2,5 conformer 31 based on computation studies,[13] the O2-C2-C3-O3 torsional angle decreases while O2-C2-C1-O5 torsional angle increases. As a result, the C2-O2 electron-withdrawing effect would be enhanced resulting in higher barrier for conversion for the covalent glycosyl triflate to the oxocarbenium ion. In comparison, the O2-C2-C3-O3 torsional angle increases during the transition of covalent glycosyl triflate to the oxocarbenium ion in the gluco series. Therefore, with the α-mannosyl triflate, the acceptor glycosylation would be more SN2 like leading to higher β-selectivity. The important roles of O2-C2-C3-O3 torsion in influencing β-selectivity were further confirmed with 2-deoxy donor 35, 3-deoxy donor 36, 2-deoxy-2-fluoro and 3-deoxy-3-fluoro donors 38–40. As these donors lack the important O2-C2-C3-O3 interactions, they gave only modest β-selectivity for both mannosyl and glucosyl donors.[23–24]
Scheme 8.

Torsional angle values for reactive intermediates from mannosyl and glucosyl donors respectively.
Inspired by the profound influence of 4,6-O-benzylidene acetal on stereoselectivity, Crich and coworkers tested the effect of 4,6-O-phenylboronate ester on the stereoselectivity in galacto-, gluco- and mannopyranosyl thioglycosides.[25] Similar to the 4,6-O-benzylidene acetal, the 4,6-O-phenylboronate gave high β-selectivity in mannosylation. This enabled the attachment of mannosyl donors to solid phase via the styrylboronate ester 41 (Figure 1). Successful diastereoselective β-mannosylation reactions were performed on solid phase with a range of primary, secondary, and tertiary glycosyl acceptors.
Figure 1.
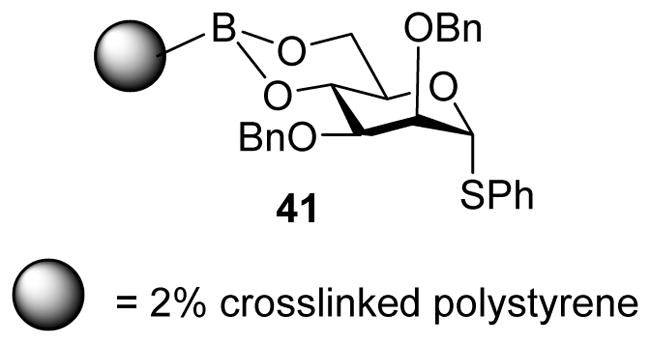
Styrylboronate ester linked mannosyl donor 41 for diastereoselective β-mannosylation on solid phase.
2. The Effects of 3,4-O- and 2,3-O- Cyclic Carbonate
Cyclic carbonate can bestow unique influences on stereoselectivity.[26] When the 2,3-O-carbonate 4,6-O-benzylidene bearing mannosyl donor 42 was pre-activated followed by the addition of acceptor 43, the α-glycoside product 44 was formed in 60% yield with no corresponding β-glycoside observed (Scheme 9). In this case, the α-directing effect of 2, 3-O-carbonate group overrode the β-directing effect of 4,6-O-benzylidene acetal. To explain this stereoselectivity, it was suggested that the α-glycosyl triflate formed after pre-activation of donor 42 would adopt 0H5 conformation as indicated by NMR of similar substrates.[26] This would lower the barrier converting the α-triflate and the half chair shaped reactive oxocarbenium ion, thus shifting the equilibrium toward the oxocarbenium ion, favoring the formation of the more thermodynamic stable α-glycoside. Similar high α selectivity was also observed with 2,3-O-carbonate protected rhamnosyl donors.[27]
Scheme 9.

Stereo-directing effect of 2, 3-O-carbonate protecting group. Reagents and conditions: (a) AgOTf, PhSCl, TTBP, CH2Cl2, −60 °C, then 43, −60 °C to 0 °C.
With 3,4-O-carbonate protected donors, completely different stereoselectivity was obtained from those bearing 2,3-O-carbonate.[27] Coupling of 3,4-O-carbonate donor 45 to glucose acceptor 46 under the pre-activation condition gave disaccharide 47 in 77% favoring the β-isomer and glycosylation of 1-adamantanol 48 led to β-linked 49 exclusively (Scheme 10a). The 3, 4-O-carbonate protecting group may exert its directing effects by conformational and/or electron-withdrawing effect. To decipher the dominant factor, the 3, 4-O-carbonate group in 45 was replaced with 3, 4-O-isopropylidene acetal group (donor 50), which should only bestow the conformational effect. The same pre-activation protocol led to the isolation of disaccharide 51 only in α-form (Scheme 10b). These results suggest that the β-stereoselectivity most likely results from electron-withdrawing effect of the 3,4-O-cyclic carbonate, which can stabilize the α-glycosyl triflate intermediate for SN2 like reaction to give β-isomer. The detailed knowledge on how the position of the carbonate group can influence stereoselectivity can enable judicious choice of suitable protective group patterns to achieve the desired stereochemistry of target molecules.
Scheme 10.

a) β-Directing effect of the 3, 4-carbonate protecting group; b) Reversal of stereoselectivity in the case of 3, 4-isopropylidene acetal group. Reagents and conditions: (a) Tf2O, BSP, CH2Cl2, −60 °C, then 46 or 48, −60 °C to 0 °C.
The impacts of cyclic carbonate have also been examined in glucosyl donors.[28] For 2, 3-O-carbonate protected glucose donor, moderate to good β-selectivity was obtained, in contrast to the high α-selectivity observed in the mannose series (Scheme 11). For 3,4-O-carbonate protected glucose donor, the stereoselectivity was lost. As shown in Scheme 12, coupling of donor 57 to acceptor 55 gave an α/β mixture (1:1) of glycoside products 58. Low-temperature NMR studies showed that glucosides also formed the α-glycosyl triflate intermediate upon activation. Further work is needed to better understand this change of selectivity of glucosyl donors.
Scheme 11.

Stereo-directing effect of the 2, 3-O-carbonate protecting group. Reagents and conditions: (a) Tf2O, BSP, CH2Cl2, −60 °C, then ROH, −60 °C to 0 °C.
Scheme 12.

Loss of stereoselectivity for 3, 4-O-carbonate protected glucosyl donor. Reagents and conditions: (a) Tf2O, BSP, CH2Cl2, −60 °C, then 55, −60 °C to 0 °C.
The stereoselective assembly of 2-deoxyglycosyl linkages can be challenging since 2-deoxyglycosyl donors lack a substituent at the C-2 position that can be used to direct stereochemistry. Utilizing the cyclic carbonate, Ye and coworkers reported a new method for highly α-selective constructions of 2-deoxy and 2, 6-dideoxy glycosides.[29] Under the pre-activation protocol, donor 59 was coupled to various acceptors in high yields and α-stereoselectivities (Scheme 13). Similar results were achieved for 2, 6-dideoxysugars. In contrast, pre-mixing the donor and acceptor followed by addition of the promoter resulted in much lower α-selectivity, highlighting the advantage of pre-activation. In a later study by the same group, per-acetylated thioglycosyl 2-deoxy and 2,6-dideoxy glycosyl donors were found to give good α selectivities,[30] which suggests the high α-selectivity bestowed by the cyclic carbonate in these deoxy sugar series were likely due to the electron withdrawing properties of the substituents rather than through conformation rigidifying effects.
Scheme 13.

Stereoselective glycosylation of 2-deoxygalactose/glucose. Reagents and conditions: (a) Tf2O, BSP, TTBP, CH2Cl2, −72 °C, then ROH, −72 °C to 0 °C.
3. 3, 4-Bisacetal Effect
3, 4-Bisacetal is another type of trans-fused bicyclic protecting group, which can selectively protect 3,4-hydroxyl groups of mannosides and glucosides and affect the stereochemical outcome of glycosylation. For instance, the mannosyl donor 64 bearing 3,4-bisacetal coupled with acceptor 43 leading to disaccharide 65 in a highly α-selective manner (Scheme 14).[26] Similarly, donor 66 also gave high α-selectivity in glycosylating acceptor 43, suggesting the two axial methoxy groups in 67 do not have much influence on stereoselectivity (Scheme 15).
Scheme 14.
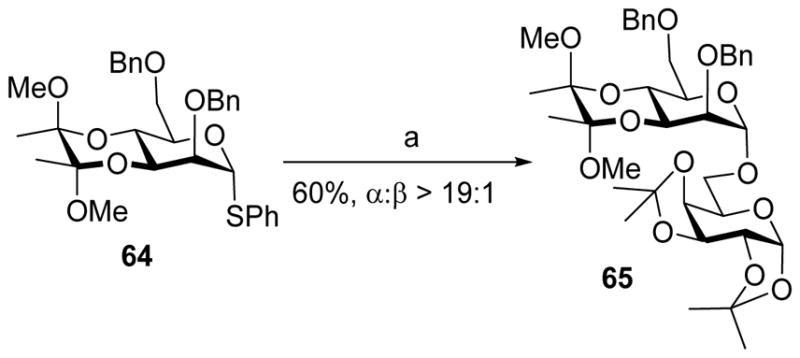
α-Selective glycosylation with 3, 4-bisacetal protected donor 64. Reagents and conditions: (a) PhSOTf, DTBMP, CH2Cl2, −78 °C, then 43, −78 °C to 0 °C.
Scheme 15.
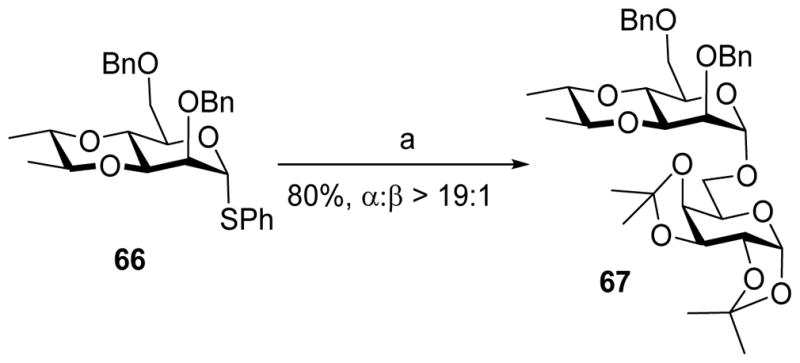
α-Selective glycosylation with 3, 4-bisacetal protected donor 66. Reagents and conditions: (a) PhSOTf, DTBMP, CH2Cl2, −78 °C, then 43, −78 °C to 0 °C.
The dichotomy between glucose-mannose continued for the 3, 4-bisacetal protecting group. Pre-activation based glycosylation of glucosyl donor 68 containing the 3, 4-bisacetal with acceptors gave high β-selectivity (Scheme 16).[31] Interestingly, glucosyl donor 70 without the two axial methoxy groups on its bisacetal group was coupled to acceptor 61 producing a 1:1 α:β mixture of disaccharide products 71 (Scheme 17). These results could be explained as the following: pre-activation of donor 68 would produce the α-glycosyl triflate, which is in equilibrium with the oxocarbenium ion. The preferred conformation of the oxocarbenium ion was 4H3 half-chair, resulting in a steric clash between the axial methoxy group and the 2-OBn.[32–33] This would shift the equilibrium towards the α-glycosyl triflate, thus favoring the SN2 like glycosylation pathway for β-glycoside formation. The removal of the axial methoxy group in donor 70 would reduce this unfavorable steric interaction, shifting the equilibrium towards the oxocarbenium ion and leading to a loss of stereoselectivity.
Scheme 16.

β-Selective glycosylation of donor 68. Reagents and conditions: (a) Tf2O, BSP, TTBP, CH2Cl2, −60 °C, then ROH, −60 °C to 0 °C.
Scheme 17.

Loss of stereoselectivity in the case of glycosylation of glucosyl donor 70 as compared to donor 68. Reagents and conditions: (a) Tf2O, BSP, TTBP, CH2Cl2, −60 °C, then 61, −60 °C to 0 °C.
4. 2,3-Oxazolidinone Effect
Similar to the cyclic carbonate group, oxazolidinone group installed on glycosyl donors can significantly affect stereochemical outcome of glycosylation reactions.[34–45] Pre-activation of 2,3-oxazolidinone protected glucosamine thioglycosyl donor 72 in the presence of a bulky base 2,4,6-tri-tbutyl pyrimidine (TTBP) followed by addition of the acceptor produced β-glycoside in high yields.[46] This protocol was applied to a wide range of acceptors as shown in Scheme 18. Interestingly, in the absence of TTBP, the stereochemistry of the glycoside product was completely reversed giving α-linked glycoside products. The effect of the base on stereochemical outcome was found to be dependent upon the N-substituent of the oxazolidinone and protective group on the glycan ring.[47–48] In addition to TTBP, other additives such as tetrabutylammonium bromide (TBAB), tetrabutylammonium iodide (TBAI) and thiophene had been investigated to modulate stereoselectivity in pre-activation of oxazolidinone-protected amino sugar thioglycoside donors.[49] Excellent α-anomeric selectivities were acquired with thiophene as the additive. For TBAI, the amount of the reagent added controlled stereoselectivity with catalytic amount of TBAI giving α-selectivity while stoichiometric amount favoring β-selectivity. Reagent control is an attractive strategy for stereoselective glycosylation as it enables the desired anomeric configuration without the need to synthesize multiple sets of building blocks. The generality of these stereochemical modulation effects remained to be established beyond the oxazolidinone-protected amino sugar thioglycoside donors.
Scheme 18.

Reagent controlled stereoselective glycosylation of oxazolidinone protected donor 72. Reagents and conditions: (a) Tf2O, BSP, TTBP, CH2Cl2, −73 °C, then ROH, −73 °C to 0 °C.
Sialic acid is a common motif at the non-reducing end of many naturally existing glycans.[50] Formation of the α-sialyl linkage is a challenging task due to the low reactivity of the sialyl donors and the lack of substituents on C-3 of sialic acid for stereochemical control. Oxazolidinone protection of the 4-O and 5-N groups of sialyl donors has been found to significantly enhance both the yield and the stereoselectivity of sialylation.[37–39, 43–44] The Sun group has applied the pre-activation protocol to oxazolidinone protected sialyl donor 81 (Scheme 19).[51] A wide range of thioglycosyl acceptors could be glycosylated in high yields and α-selectivity with donor 81. The resulting thioglycosyl disaccharide could be directly used as donors for further chain elongation without the need for anomeric leaving group adjustment.
Scheme 19.

Pre-activation based glycosylation using sialyl donor 81 gave high α-selectivity. Reagents and conditions: (a) p-TolSCl (1 eq), CH2Cl2/CH3CN, −75 °C, then 82.
5. Effect of Protective Group on C-2 Position
As discussed in the introduction, the most classical method in facilitating 1,2-trans glycosyl linkage formation in glycosylation reaction is the installation of a participating neighboring group such as carboxylic ester on 2-O and amide/imide for 2-amino sugars. This approach has been widely applied in pre-activation based glycosylation method in total synthesis of complex glycans.[52–53]
Besides the desired glycoside, a side product that can form from a donor bearing a C-2 carboxylic ester is the orthoester. This results from nucleophilic attack by the acceptor on the partially positively charged bridging carbon in the dioxolenium ion rather than the anomeric carbon (Scheme 20). Aided by low temperature NMR, Crich and coworkers found that the presence of a base in pre-activation based glycosylation of xyloside donors was the determining factor in orthoester formation.[54] Pre-activation of the xyloside donor 89 produced the dioxalenium ion 91 as observed by low temperature NMR (Scheme 21). Upon addition of an acceptor to the reaction mixture, orthoester 90 was formed when an exogenous base DTBMP was added. In the absence of the base, β-xyloside 92 was exclusively formed presumably due to the conversion of orthoester to glycoside catalyzed by the acid produced during the reaction.
Scheme 20.

Possible pathway for the formation of orthoester.
Scheme 21.

The addition of a base in glycosylation favor orthoester formation. Reagents and conditions: (a) Tf2O, DTBMP, CH2Cl2, −78 °C, then ROH.
In the case of 2-amino sugar donors, oxazoline can form as a major side product (Scheme 22). In pre-activation based synthesis of hyaluronan oligosaccharides using trichloroacetamide (TCA) protected thioglycosyl donor 99,[55] even without a base in the reaction mixture, oxazoline 102 was found to be a major side product (40%) (Scheme 23). Various acids were examined to improve the yield of glycosylation and TMSOTf was found to be the most effective, leading to the desired glycoside 101 in 82% yield. Presumably TMSOTf activates oxazoline 102 in situ for glycosylation, enhancing the yield of the desired glycoside.
Scheme 22.

Formation of oxazoline upon pre-activation.
Scheme 23.

Synthesis of fully protected tetrasaccharide 101. Addition of TMSOTf to the reaction mixture suppressed the amount of oxazoline side product. Reagents and conditions: (a) AgOTf, p-TolSCl, TMSOTf, CH2Cl2, −78 °C, then 100.
To avoid the orthoester formation during glycosylation, Yamago and coworkers reported dialkylphosphates such as 2, 2-dimethyltrimethylene (DMTM) phosphate as the stereodirecting group for 1,2-trans glycoside formation (Scheme 24).[56] Glycosyl phosphate donor 103 was pre-activated by BSP/Tf2O, followed by the addition of cyclohexanol to produce 104 with excellent β-selectivity. The phosphate ester could be deprotected by base treatment. Low temperature NMR analysis showed that the dominant resting intermediate following pre-activation was the α-triflate 106, which underwent “SN2-like” attack by the acceptor to afford glycoside 104 (Scheme 25).[57]
Scheme 24.

The usage of dialkylphosphate DMTM in glycosylation to favor the formation of 1,2-trans glycosides. Reagents and conditions: (a) BSP, Tf2O, CH2Cl2, −60 °C, then cyclohexanol; (b) NaOH, EtOH/H2O, 60 °C, 1 h.
Scheme 25.

Plausible reaction mechanism using dialkylphosphate as the 2-O protective group.
While neighboring group participation has been traditionally utilized to favor 1,2-trans linkage, Boons group reported a novel strategy using chiral auxiliary such as ethyl mandelate[58] and phenyl-2-(phenylsulfanyl)ethyl[59] to protect C-2 hydroxyl group and assist the creation of 1, 2-cis glycosyl linkages. Upon pre-activation by TMSOTf, the (1S)-phenyl-2-(phenylsulfanyl)ethyl bearing donor 108 was converted to a reactive intermediate (Scheme 26).[59] Based on NMR analysis, the intermediate was consistent with the structure of 109 with a trans-decalin system. Addition of the acceptor 110 following pre-activation led to disaccharide 111 in 94% yield with a high 1,2-cis selectivity from SN2 like opening of the trans-decalin like moiety in 109. The configuration of the auxiliary played an important role in determining selectivity. Switching the configuration from S to R resulted in loss of α-selectivity. This was rationalized by the unfavorable steric strain of the axial substituent in the auxiliary bearing R configuration.
Scheme 26.

Chiral auxiliary assisted stereoselective glycosylation via pre-activation. Reagents and conditions: (a) TMSOTf, CH2Cl2, −78°C to − 10°C. (b) 110, DTBMP.
Besides the chiral auxiliary, other substituents on the glycan ring could also affect the stereochemical outcome of glycosylation.[60] When donor 112 bearing phenyl-2-(phenylsulfanyl)ethyl auxiliary and multiple electron donating groups reacted with acceptor 61, disaccharide 113 was isolated in 70% with no stereoselectivity (Scheme 27). Although the β-sulfonium ion 116 was the only detectable resting intermediate by NMR, small amount of oxocarbenium ion 115 could exist upon pre-activation, which may be the reactive intermediate undergoing SN1 like reaction leading to anomeric mixtures upon addition of the acceptor (Scheme 28). In contrast, for donor with electron-withdrawing groups such as 108, oxocarbenium ion 115 was destabilized so that the reaction would presumably go through SN2 like attack on the sulfonium ion 116 to generate α-glycoside. These results highlight the subtlety of building block design and the impact of the nature of remote protective groups on stereoselectivity.
Scheme 27.

Low anomeric selectivity from donor 112. Reagents and conditions: (a) TMSOTf, CH2Cl2, −78 °C to 0 °C; then 61, DTBMP, −78 °C to rt.
Scheme 28.

Protective groups on glycosyl donor 114 can significantly impact stereoselectivity with more electron rich donors giving lower α selectivity than the corresponding electron poor donors. Reagents and conditions: (a) TMSOTf, CH2Cl2, −78 °C to 0 °C; then acceptor R1OH, −78 °C to rt.
The utility of this innovative chiral auxiliary method has been demonstrated in total synthesis, including solid phase supported synthesis. Targets successfully produced include the branched α-glucan 121, which bears 1, 2-cis linkage between each sugar unit (Scheme 29).[61]
Scheme 29.

Stereoselective solid phase supported synthesis of pentasaccharide 121 with all α linkages. Reagents and conditions: (a) 2, TMSOTf, CH2Cl2, MS 4 Å, 15 min, −40 °C then added to 119, DTBMP, CH2Cl2, MS 4 Å, 16 h, −40 °C to room temperature, double coupling.
Inspired by the power of chiral auxiliary to control stereoselectivity, the Boltje group investigate its applicability to β-mannosylation. Carboxybenzyl (CB)[9] donor 122 was designed with a locked 1C4 conformation through a 3,6-lactone bridge (Scheme 30a).[62] It was envisioned that upon pre-activation, the 1C4 conformation of the ring would enable the participation of the C-2 auxiliary to form the trans-decalin bearing intermediate. Subsequent SN2-like attack by the acceptor would lead to the β-mannoside. Indeed, glycosylation of donor 122 with acceptor 110 gave excellent selectivities of β-mannoside products (Scheme 30a). As a control, donor lacking the lactone ring resulted in mainly α-mannosides. Interestingly, glycosylation reactions using the 3,6-lactone bearing donor 124 without the auxiliary turned out to be highly β-selective as well (Scheme 30b). To investigate the reaction mechanism, 124 was pre-activated, which formed 126 and benzyl triflate (Scheme 30c). This result suggested the remote participation of C4-O-benzyl group in the activation step, which would lead to the formation of 1,2-cis-β-mannoside upon nucleophilic attack by the acceptor.
Scheme 30.

3,6-Lactone bridged mannosyl donors can lead to high selectivities in β-mannoside formation.
Another type of C2-protective group to facilitate 1,2-cis glycoside formation was the 2-O-cyanobenzyl ether developed by the Liu group.[63] Pre-activation of thioglycoside 127 containing the 2-cyanobenzyl ether moiety on 2-O followed by addition of an acceptor bearing electron withdrawing groups formed 1,2-cis linked α-glycosides selectively (Scheme 31). Interestingly, when electron rich thus more nucleophilic acceptor was employed, complete reversal of anomeric preference was observed with β-linked glycoside as the major product. From computation analysis and low temperature NMR studies, it was proposed that the more nucleophilic acceptor could directly attack in SN2-like fashion the nitrilium ion b formed through neighboring group participation, leading to β-glycoside (Scheme 32). In contrast, due to the decreased nucleophilicity, the acceptor with electron-withdrawing groups reacts with the oxocarbenium ion instead. The presence of the cyano moiety was found to be critical for 1,2-cis product formation, which was proposed to form hydrogen bonding with the acceptor hydroxyl group for intramolecular delivery favoring the generation of α glycoside. The nucleophilicities of acceptors have also been found by Codée and coworkers to directly influence the stereoselectivities of glycosylations without 2-cyanobenzyl ether moiety especially for benzylidene protected glucose donors.[64]
Scheme 31.

Acceptor nucleophilicity can direct the stereoselectivities of 2-O-cyanobenzyl ether bearing donor 127.
Scheme 32.

Mechanism of acceptor directed stereoselective glycosylation by 2-O-cyanobenzyl ether bearing donor 127.
6. The Impact of Anomeric Groups on Stereoselectivities
The Bennett group developed β-specific glycosylation with 2-deoxy sugars.[65] Hemiacetal donors of 2-deoxy sugars were treated with N-toluene sulfonyl imidazole, which presumably formed α-glycosyl tosylate in situ. The intermediate could glycosylate phenolic acceptors or thiols, forming β-aryloxy glycosides and thioglycosides. However, glycosylation with carbohydrate acceptors under this protocol was unsuccessful. To overcome this obstacle, the more potent sulfonylating agent p-toluenesulfonic anhydride was utilized to favor the formation of α-glycosyl tosylates (Scheme 33), which was detected via low temperature NMR.[66] With this new protocol, both primary and secondary carbohydrate acceptors were glycosylated by 2-deoxy hemiacetal donors with exclusive β-selectivities likely through a SN2 like substitution of the α-glycosyl tosylate.
Scheme 33.

Pre-activation of a 2-deoxy hemiacetal donor to glycosyl tosylate favored the formation of β-linked 2-deoxy glycoside. Reagents and conditions: (a) TTBP, KHMDS, THF, −78°C: (b) Ts2O; (c) Acceptor 132, −78°C to rt.
Instead of converting the anomeric hydroxyl group to tosylate as a leaving group, the Zhu group reversed the polarities of coupling partners by deprotonating deoxy-glycosyl hemiacetal and using the resulting alkoxide ion as a nucleophile to displace the triflate leaving group on the acceptor.[67] As shown in Scheme 34, treatment of disaccharide hemiacetal 134 with NaH followed by addition of triflate 135 gave tetrasaccharide 136 in an excellent 95% yield. The 3-OH of donor 134 needed to be kept free to reduce elimination side product from the donor under the strong basic condition. While the glycosyl donor 134 contained a mixture of anomeric hydroxyl groups, the glycoside product 136 was exclusively β, which was attributed to the rapid equilibration of α- and β-hemiacetal under the reaction condition and the higher nucleophilicity of β-alkoxide formed due to the kinetic anomeric effect.
Scheme 34.

Umpolung approach for the formation of 2-deoxy β-glycosides. Reagents and conditions: a) 3 eq. NaH, 1.5 eq. 15-C-5, 1,4-dioxane, r.t, 24 h.
The umpolung approach was also applied to stereoselective synthesis of both α- and β- S-linked 2-deoxyglycosides.[68] The 2-deoxy phenyl thioglycoside 137 was reductively pre-activated with lithium 4,4′-di-tertbutylbiphenyl (LiDBB), which afforded α-glycosyl lithium 138 at −78 °C (Scheme 35a). This nucleophile can attack asymmetric disulfide 139 producing the desired α-product 140 in excellent stereoselectivity. To obtain β-product 142, the reaction temperature was raised to −20 °C following reductive pre-activation. The led to epimerization of the intermediate, resulting in the thermodynamically more stable equatorial glycosyl lithium species 141, which reacted with disulfide 139 to form the S-linked 2-deoxy-β-glycoside 142. Thus, both α- and β-deoxyglycosides may be conveniently obtained from the same thioglycosyl donor. In addition, this umpolung based S-glycosylation method worked well with thioglycoside acceptor. This opened up a new strategy for stereoselective iterative glycosylation. As an example, reductively activated donor 143 glycosylated phenyl glycoside acceptor 144 producing S-linked disaccharide 145 in 52% yield with excellent β-selectivity (Scheme 35b). Disaccharide 145 then underwent α-selective glycosylation leading to tetrasaccharide 147 containing both α and β linkages en route to S-linked hexasaccharide analog of Landomycin A.[69]
Scheme 35.

Umpolung approach for the stereoselective formation of S-linked 2-deoxy β-glycosides Reagents and conditions: a) LiDBB, THF, −78 °C, 15 min; − 20 °C, 144, 45 min; then −78 °C, overnight; b) LiDBB, THF, −100 °C, 15 min, then 146, −85 °C, overnight.
7. Remote Neighboring Group Participation
Besides C-2 protective groups, substituents on other positions of the glycan ring have often been suggested to participate in glycosylation and contribute to stereoselectivity under the traditional pre-mixed glycosylation condition.[70–80] However, direct evidence for participation has been scarce. Pre-activation approach provides an opportunity to investigate this type of participation since it is possible to trap and characterize the reactive intermediate prior to addition of acceptor.
The Crich group systemically probed remote participation using tert-butoxycarbonyl (Boc) as an acyl type participating group since the loss of a tert-butyl-cation would lead to the formation of a stable cyclic carbonate.[81] Pre-activation of donor 148 with axial 3-O-Boc gave cyclic carbonate 149 (Scheme 36). When an acceptor was added following pre-activation, the desired glycoside with high β-selectivity was isolated, which supported the participation of axial 3-O-Boc. In contrast, when a donor with an equatorial 3-O-Boc was pre-activated, no cyclic carbonate formed. Instead a complex mixture of products retaining 3-O-Boc was found, ruling out the possibility of participation by this group. Similarly, little evidence was observed for 6-O-Boc or equatorial 4-O-Boc group to participate in this glycosylation.
Scheme 36.
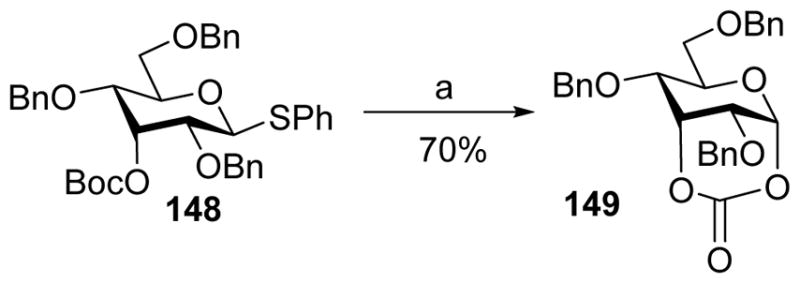
Participation of axial 3-O-Boc group. Reagents and conditions: (a) BSP, Tf2O, CH2Cl2, −60 °C.
For the axial 4-O moiety, 4-O-acyl-galactosyl donors have been shown to give much higher α-selectivity compared to those bearing 4-O-ether protective groups, which hinted the possibility of remote participation by the 4-O-acyl group.[77] To probe this, a new isotopic labeling probe approach was developed (Scheme 37). Donor 150 was pre-activated by BSP/Tf2O. If the 4-benzoyl ester could participate, 152 would be the major intermediate, which upon quenching with 18O labeled water would generate 156 with 18O incorporated to the ester carbonyl on C-4. In the absence of 4-O-Bz participation, 18O incorporation would only be expected at the anomeric center. Further chemical derivatization and mass spectrometry (MS) and nuclear magnetic resonance (NMR) analysis of the reaction product failed to detect any isotopic incorporation to the benzoyl carbonyl group. These results strongly suggest the absence of remote participation by 4-O-Bz under the reaction condition. The reason for the enhancement of α-selectivity observed with 4-O-acyl-galactosyl donor requires further investigation.
Scheme 37.

Isotopic labeling experiment suggests the 4-O acyl moiety on galactosyl donor does not undergo remote participation in glycosylation. Reagents and conditions: (a) BSP, Tf2O, CH2Cl2, −60 °C; (b) Ac2O, pyridine; (c) BF3-Et2O, PhSH.
8. Inductive Effect from Electron Withdrawing Protective Groups to Enhance 1,2-cis Selectivity
As discussed earlier, 4,6-O-benzylidene acetal protection of mannosyl donors could lead to high β-selectivity. However, similar approach is not directly applicable to β-rhamnoside formation as rhamnose lacks a 6-OH group. Schuerch and coworkers found that 2-O-sulfonyl group could stabilize α-mannosyl and α-rhamnosyl glycosyl sulfonate esters for β-selective glycosylation.[82–83] Inspired by this pioneering work, Crich group tested the possibility of direct formation of β-L-rhamnopyranosides by inductively disarming thio-rhamnosyl donor using 2-O-sulfonate ester as a protective group.[84] For example, coupling of donor 158 to 3β-cholestanol under the pre-activation condition gave glycoside 169 with moderate β-selectivity (Scheme 38a). The β-selectivity could be enhanced by installing a second electron-withdrawing group, e.g. a Bz ester onto the donor (Scheme 38b). This was rationalized that upon pre-activation, the α-glycosyl triflate intermediate would be in equilibrium with the oxocarbenium ion. The installation of electron withdrawing groups onto the glycan ring would shift the equilibrium away from the electron deficient oxocarbenium ion and favor the covalent α-glycosyl triflate, thus forming more β-glycosides. However, with secondary carbohydrate acceptor such as 61, the β-selectivity decreased suggesting further improvements are needed (Scheme 38b).
Scheme 38.

a) Stereo-directing effect of 2-O-sulfonate group. b) Installation of a second electron-withdrawing group besides the sulfate ester on rhamnosyl donor 160 led to enhanced β-selectivity. Reagents and conditions: (a) Tf2O, BSP, TTBP, CH2Cl2, −60 °C; then 3β-cholestanol, 46 or 61, −60 °C to 0 °C.
While the 4,6-O-benzylidene protection of mannosyl donor can lead to high β-selectivity, the requirement of the benzylidene can restrict building block design for total synthesis of complex glycans. It would provide a useful alternative if high selectivity can be achieved to form β-mannoside without resorting to benzylidene protection. The influence of electron withdrawing groups on 2-O of mannosyl donors was examined for this purpose. Among various electron-withdrawing groups such as vinylogous esters, phosphates, cyanates and sulfonate ester on donors, the 2-O-sulfonate ester containing donors gave the best β-selectivity (Scheme 39).[85] A 10:1 β:α selectivity was achieved when acceptor 55 was coupled to sulfonate ester protected donor 167, although α-isomer was the major product in the case of acceptor 61 even in the presence of a second electron-withdrawing acetyl group in donor 167.
Scheme 39.

Stereoselectivity for sulfonate ester protected mannoside donor 167. Reagents and conditions: (a) Tf2O, BSP, TTBP, CH2Cl2, −60 °C; then 55 or 61, −60 °C to 0 °C.
1,2-Cis linked N-acetyl-D-mannosaminuronic acid (ManNAcA) is a common motif in many microbial glycans.[86–88] It is challenging to form this linkage as it is thermodynamically less stable than the corresponding 1,2-trans linkage due to the anomeric effect and the steric hindrance posted by the C2 substituent in glycosylation. The van der Marel group developed a series of 2-azido mannosyluronate donors bearing various anomeric leaving groups including thioether, sulfoxide and trifluoroacetimidate (donors 170 – 176).[89] Under the pre-activation protocol, all these glycosyl donors gave good 1,2-cis selectivities. Low temperature NMR studies following pre-activation of the mannuronic ester donors 170 showed the presence of two main conformers of α-anomeric triflates 177 and 178 with the equatorial triflate in 1C4 chair conformation as the main product (Scheme 40). The unexpected observation of equatorial triflate despite the strong anomeric effect of triflate was attributed to stabilization by the electron withdrawing carboxylic ester at C-5. The excellent β-selectivity in glycosylations is postulated to be due to a combination of SN2 like pathway with the glycosyl triflate and stereoselective attack on the 3H4 oxocarbenium ion.
Scheme 40.


Pre-activation based glycosylation of donor 170 with acceptor 46 gave high β-selectivity of disaccharide 179.
9. 2, 3-Anhydrosugar Donor on Stereoselectivity
Arabinofuranoside is an important structural motif in the cell wall of Mycobacterium tuberculosis.[90] The 1,2-cis-β-arabinofuranosyl linkage is challenging to synthesize. The Lowary group developed a synthetic strategy using epoxy thioglycoside or glycosyl sulfoxide 180 as the donor (Scheme 41).[91] Pre-activation of donor 180 followed by addition of the acceptor led to β-arabinofuranoside 181 exclusively. Similarly, pre-activation of donor 182 resulted in α-arabinofuranoside 183 as the sole anomer. A possible mechanistic pathway has been proposed to rationalize the stereochemical outcome of these reactions (Scheme 42). Reaction of 184 with Tf2O led to the formation of intermediate 185, which would evolve to oxocarbenium ion 186. This species would be in equilibrium with α-triflate 187 and β-triflate 188. Once the acceptor was added, it could attack 186 to give α and β mixture in a SN1-like manner or attack 187 or 188 in a SN2-like manner to yield β-arabinofuranoside 189 or α-arabinofuranoside 190 respectively. Computation analysis determined that the α-triflate was lower in energy compared to the β-triflate by several kcal/mol. Low temperature NMR analysis of the reaction mixture right after pre-activation at −78 °C showed a complex mixture of intermediates, which eventually evolved into one major species consistent with the characteristics of α-glycosyl triflate 187 upon warming up to −40 °C. Subsequent addition of an acceptor produced β-anomer glycoside possibly from SN2 like reaction with the α-glycosyl triflate. The major intermediate generated by pre-activating donor 182 was identified as β-glycosyl triflate from computational and low-temperature NMR studies, thus consistent with the α-selectivity observed in glycosylation products from 182. The knowledge on reactive intermediates led to modification of the reaction protocol. The original operation was to add the acceptor 61 to the reaction mixture right after pre-activation of donor 184, which produced disaccharide product 191 in 71% yield with a β:α ratio of 5:1. In the revised protocol, following pre-activation, the reaction was warmed to −40 °C for the intermediates to converge on the α-glycosyl triflate as the resting state. Addition of the acceptor to reaction produced pure β-glycoside 192 in 77% yield (Scheme 43). These results highlighted the importance of mechanistic understanding.
Scheme 41.

Stereoselective synthesis of arabinofuranoside. Reagents and conditions: (a) Tf2O, DTBMP, CH2Cl2, −78 °C, then n-octanol, −60 °C to 0 °C.
Scheme 42.

Proposed mechanism for arabinofuranosylation.
Scheme 43.

Stereoselective synthesis of arabinofuranoside. Reagents and conditions: (a) Tf2O, DTBMP, CH2Cl2, −78 °C, then acceptor 61, −78 °C to rt. (b) Tf2O, DTBMP, CH2Cl2, −78 °C to −40 °C, then acceptor 61, −40 °C to rt.
10. 2-Deoxyglycoside and 2, 6-Dideoxyglycoside Donors on Stereoselectivity
2-Deoxy- and 2, 6-dideoxyglycosides are essential to the functions of many biologically active compounds including antibiotics and they can exist in both α and β linkages.[92–95] Due to the lack of C2 functionality, stereoselective synthesis of 2-deoxy-glycosides is challenging.[96] β-Linked 2-deoxy glycosides could be formed through judicious choice of anomeric group as discussed earlier.[65–67] For stereoselective α-linked 2-deoxyglycosyl bond formation, Wang group applied the pre-activation based glycosylation approach[97] (Scheme 44).[98] They pre-activated donor 131 with p-TolSCl/AgOTf followed by the addition of acceptor 193 leading to α-deoxyglycoside 194 exclusively (Scheme 44). Similarly, pre-activation of 2, 6-dideoxy donor 195 with p-TolSCl/AgOTf followed by addition of the acceptor 193 yielded dideoxyglycoside 196 with high α-stereoselectivity (α/β = 12:1) (Scheme 44). A variety of acceptors including primary and secondary acceptors can be coupled with both 2-deoxy donors and 2, 6-dideoxy donors to yield desired product with high α-stereoselectivity.
Scheme 44.

Stereoselective synthesis of deoxyglycosides. Reagents and conditions: (a) AgOTf, p-TolSCl, −78 °C, 15 min, then 193, −78 °C, 2–3 h.
To better understand the reaction outcome, the structure of the intermediate was analyzed.[98] Rather than glycosyl triflate, glycosyl chloride 197 was identified as the major resting state upon donor activation (Scheme 45). This is presumably due to the de-activation of AgOTf by the presence of Lewis basic molecular sieves in the reaction mixture.[97] As a result, the deoxy thioglycosyl donor 131 was presumably directly activated by p-TolSCl, leading to the formation of glycosyl chloride 197. AgOTf could promote the reaction of 197 with the acceptor, which possibly went through SN1-like reaction with oxocarbenium ion 198. The high α-selectivity of product might be due to the anomeric effect or through equilibria between β-glycosyl triflate, β-contact ion pair and solvent separated ion pair.[16]
Scheme 45.

Proposed reaction mechanism of α-selective 2-deoxy glycosylation mediated through glycosyl chloride intermediate.
11. Effects of Reaction Solvents and Additives on Stereoselectivity
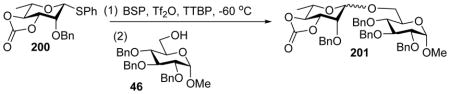
Solvents can significantly impact stereochemical outcome of glycosylation reactions.[99] Nitrile based solvent is well known to favor the formation of equatorial glycosyl linkage.[100–103] This is also the case for pre-activation based glycosylation. As shown in Table 1, addition of a small amount of nitrile (<10%) can help moderately improve the β-stereoselectivity,[104] while reaction ran in pure nitrile solvent gave much reduced yield. Interestingly, when glycosylation of donor 160 with acceptor 46 was performed in a mixed solvent of CH2Cl2/CH3CN (7:3), compound 202 was isolated in 30% yield (Scheme 46). This reaction presumably went through intermediate 203, resulting from nucleophilic addition of CH3CN to the anomeric center. The formation of compound 202 is one of the few direct evidences supporting the participation of CH3CN in glycosylation.
Table 1.
Effect of nitrile solvent ratio on stereoselectivity.
| |||
|---|---|---|---|
|
| |||
| Entry | Solvent (v/v) | Yield (%) of 437 | β:α ratio |
| 1 | EtCN/CH2Cl2 0:100 | 81 | 6:1 |
| 2 | EtCN/CH2Cl2 5:95 | 80 | 8:1 |
| 3 | EtCN/CH2Cl2 100:0 | <10 | >5:1 |
Scheme 46.

Evidence for acetonitrile participation of glycosylation reaction.
Huang and coworkers investigated the effect of a variety of solvents in pre-activation of thioglycoside donors in the absence of neighboring group participation.[105] CH2Cl2 and diethyl ether gave the highest yields, while tetrahydrofuran (THF), toluene, toluene/1, 4-dioxane, and neat acetonitrile did not result in productive coupling. When the reaction of donor 204 and acceptor 82 was carried out in CH2Cl2 using the pre-activation protocol, disaccharide 205 was isolated in 90% yield favoring the β anomer. In contrast, when diethyl ether was used as the solvent, 205 was isolated in 69% yield favoring α product (Scheme 47). This trend was found applicable to a wide range of donor/acceptor pairs.[100, 103–104] Increasing the amount of AgOTf in the reaction led to a mixture of α and β anomers (Scheme 48). The effects of solvent and AgOTf were rationalized based on the α-glycosyl triflate formed as the major intermediate upon pre-activation with p-TolSCl/AgOTf (Scheme 49).[53] When diethyl ether was used as the solvent, it would act as a nucleophile to afford intermediate 207, and subsequent displacement of the ether molecule by the acceptor in SN2-like fashion would lead to α-glycoside as the major product (Pathway a, Scheme 49). When CH2Cl2 is used as the solvent, the reaction would directly go through SN2-like pathway to afford β-glycoside as the major product (Pathway b). In the presence of excess AgOTf, it is possible that AgOTf would coordinate with the oxygen atom of the triflate, leading to its activation to favor the formation of oxocarbenium ion 211 leading to SN1 like pathway and a mixture of anomers as products (Pathway c).
Scheme 47.

Effect of solvent on stereoselective glycosylation.
Scheme 48.

Effect of AgOTf on stereoselective glycosylation.
Scheme 49.

Proposed mechanism for effects of solvents and AgOTf on stereoselectivity.
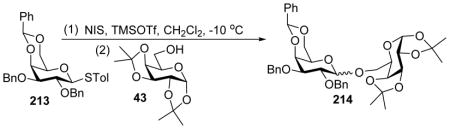
To construct 1, 2-cis linkages, Mong and coworkers developed a novel strategy using N,N-dimethylformamide (DMF) as a modulating molecule.[106] For the reaction of 212 and 43 under the pre-activation condition, as the amount of DMF increased from 1.5 equiv. to 6.0 equiv., the α:β selectivity increased from 1:1 to 19:1 (Table 2). The pre-activation method gave higher α-selectivity than the traditional pre-mixed approach with donor and acceptor mixed together during promoter addition (Entries 2 vs 3). The modulating effect of DMF was found to be general for a wide range of donor/acceptor pairs. The effect is specific to DMF as N,N-dimethylacetamide (DMA) gave lower α-selectivity. However, later this method was shown to be impractically slow for glycosylations of secondary acceptors with 2-azido-2-deoxythioglucosyl donors. To overcome this limitation, a variety of additives were screened and N-formylmorpholine (NFM) was identified as the most effective modulator leading to high yields and α-selectivities.[107]
Table 2.
Modulating effect of DMF on stereoselectivity.
| |||
|---|---|---|---|
|
| |||
| Entry | DMF (equiv.) | Yield (%) of 214 | α:β ratio |
| 1a | 0 | 90 | 1:1 |
| 2a | 1.5 | 82 | 6:1 |
| 3b | 1.5 | 80 | 8:1 |
| 4b | 3.0 | 87 | 15:1 |
| 5b | 6.0 | 87 | 19:1 |
Entries 1 and 2: Non-preactivation;
Entries 3, 4 and 5: Pre-activation.
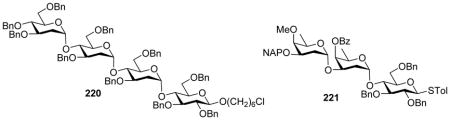
A mechanism has been proposed to explain the DMF effect (Scheme 50). Pre-activation of the thioglycoside donor in the presence of DMF would form the α- and β-glycosyl imidate intermediates 216 and 217 through addition of DMF to the oxocarbenium ion. Similar to the in situ anomerization protocol pioneered by Ray Lemieux, the α- and β-anomers of glycosyl imidates could exist in dynamic equilibrium with the latter being more reactive toward the acceptor to form α-glycoside 219 preferentially. Low temperature NMR analysis of the reaction mixture upon pre-activation suggested the α-glycosyl imidate being the major resting state. Although no β-glycosyl imidate was detected by NMR, it is possible that the acceptor preferentially reacts with more reactive β-glycosyl imidate existing in small amounts. This would shift α/β-glycosyl imidate toward the β-anomer leading to α-glycoside as the major product. The utility of the DMF-modulated glycosylation has been demonstrated in iterative one pot synthesis of multiple oligosaccharides, which include α-selective assembly of 2- and 6-deoxysugar oligosaccharides 220 and 221.[106, 108–110]
Scheme 50.

Possible mechanism for DMF-modulated stereoselective glycosylation.
The Bennett group reported selective synthesis of 1,2-cis-α-linked glycosides with TBAI as the additive for iterative oligosaccharide synthesis (Scheme 51).[111] Pre-activation of thioglycoside donor 204 with Ph2SO/Tf2O followed by TBAI (5 eq) led to the formation of glycosyl iodide intermediate, which underwent glycosylation with thioglycoside acceptor 222 to afford disaccharide 223 in high α-selectivity. Subsequently, compound 223 was directly used as donor to glycosylate acceptor 46 generating trisaccharide 224 in moderate yield and excellent α-selectivity. The mechanism of TBAI facilitating α-glycoside formation was most likely going through the in situ anomerization of α-glycosyl iodide to the more reactive β-glycosyl iodide for SN2 like nucleophilic displacement by the acceptor similar to DMF modulation effect.
Scheme 51.

TBAI mediated α-selective glycosylation. Reagents and conditions: (a) Ph2SO, Tf2O, TTBP (3 eq), N-methylmaleimide, CH2Cl2, −78°C, then TBAI (5 eq), then 222 or 46, 1,4-dioxane, −78°C to rt.
The Taylor group developed a series of innovative borinic acid reagents to effect regio- and stereo-selective glycosylations.[112–113] Pre-activation of the hemiacetal donor 225 with methanesulfonic anhydride (Ms2O) and an amine base 1,2,2,6,6-pentamethylpiperidine presumably formed the glycosyl mesylate 226 in situ.[112] Addition of the partially protected acceptor 227 led to disaccharide 228 with α anomer as the major product (α: β = 2:1) (Scheme 52). Interestingly, addition of the borinic acid catalyst 229 with the acceptor significantly changed the stereoselectivity, favoring the β-linked disaccharide 228 as the major product (α: β = 1:10). The reversal of stereoselectivity was attributed to the formation of borinic acid/acceptor complex 230. The sterically hindered nature of the tetracoordinate borinic ester nucleophile led to the selective acceleration of the 1,2-trans-selective pathway.
Scheme 52.

Addition of a borinic acid catalyst 229 can reverse the stereoselectivity of pre-activation based dehydrative glycosylation of hemiacetal donor 225. Reagents and conditions: a) Ms2O (1.88 equiv.), 1,2,2,6,6-pentamethylpiperidine (4 equiv.), CH2Cl2, 23 °C.
Lewis acids are known to induce the anomerization from 1,2-trans glycosides to 1,2-cis glycosides for pyranosides protected with 2,3-oxazolinidone or 2,3-carbonate groups.[42, 114–116] The isomerization was believed to go through an endocyclic cleavage pathway.[117] Recognizing this, the Ye group investigated Lewis acid as useful additives to mediate α-selective glycoside formation.[118] When the 2,3-O-carbonate-protected donors reacted with acceptors under the pre-activation condition without an additive, only slight α selectivities were obtained (Scheme 53a). However, the addition of a Lewis acid such as 0.2 equiv BF3OEt2 to the reaction significantly enhanced α selectivities (Scheme 53b). This α-selective effect induced by BF3OEt2 could also be extended to 2,3-oxazolidinone protected glucosamine and galactosamine thioglycoside donors.[119]
Scheme 53.

Addition of a Lewis acid to the reaction mixture helped enhance α selectivity. Reagents and conditions: a) Ph2SO, Tf2O, CH2Cl2, − 72 °C; b) BF3.OEt2 (0.2 eq).
Phosphine-oxide has been utilized as an additive to facilitate 1,2-cis-ribofuranoside formation.[120] Pre-activation of donor 233 with tributyl phosphine oxide (Bu3P=O) and Tf2O would presumably generate oxyphosphonium adduct 234 (Scheme 54). Subsequent coupling with acceptors afforded the α-ribofuranosides as major products. A similar strategy was applied to the pyranose hemiacetal donors,[121] glycosyl acetate donors,[122] as well as glycosyl iodides.[123]
Scheme 54.

Tributyl phosphine oxide mediated α-selective glycosylation.
12. Summary and outlook
High stereochemical control is critical for glycosylation reactions. Glycosylations are typically performed by pre-mixing the glycosyl donor and acceptor together followed by donor activation. As the acceptor is present while the reactive intermediate is generated, the reactive intermediate may be quickly consumed by the acceptor. In comparison, the pre-activation based glycosylation temporally separates the donor activation and acceptor glycosylation steps. As a result, the structures of the reactive intermediates can be tuned more easily by the protective group on the glycan ring, additives or solvents present in the reaction mixture. This enables the possibilities to analyze and characterize structures of the intermediates, providing better understanding of glycosylation reactions and factors governing stereochemical control.
Glycosylation reactions commonly generate intermediates with electron deficient anomeric centers, which undergoes nucleophilic substitution reactions with the acceptor. The reaction pathway can range from more SN1 like with oxocarbenium ion intermediates to SN2 like reactions with covalent intermediates such as glycosyl triflate. The protective groups present on the glycosyl donor can tune the intermediate structures by restricting conformational flexibility or through electron withdrawing properties of the substituents. In addition, exogenous additives and solvents can be utilized to favor the formation of intermediates that can lead to the desired stereoisomer product. The possibility of reagent control of stereochemistry is attractive as it reduces the need to synthesize multiple building blocks, thus improving synthetic efficiencies.
During the past two decades, significant progress has been made in pre-activation based stereoselective glycosylation. Some challenging linkages such as β-mannoside can now be synthesized in a straightforward manner through pre-activation. However, glycosylation reactions are intrinsically sensitive to subtle influences of structural features of the building blocks, including structure of the sugar, and conformational rigidity, size and electron withdrawing properties of the protective groups, as well as the presence of exogenous additives and solvents. Further studies are needed to more thoroughly understand the nuances of structural impacts on stereoselectivity, so that automated glycan synthesis can be realized with efficiencies reaching the levels of solid phase supported peptide and nucleic acid synthesis.
Acknowledgments
We are grateful for the financial support from National Science Foundation (CHE 1507226) and the National Institute of General Medical Sciences, NIH (R01GM072667).
List of abbreviations
- Alloc
Allyloxycarbonyl
- Bn
Benzyl
- Boc
tert-Butoxycarbonyl
- BSP
1-Benzenesulfinyl piperidine
- tBu
tert-Butyl
- CB
Carboxybenzyl
- DMA
N,N-Dimethylacetamide
- DMF
N,N-Dimethylformamide
- DMTM
2, 2-Dimethyltrimethylene
- DTBMP
2,6-Di-tert-butyl-4-methylpyridine
- Fmoc
Fluorenylmethyloxycarbonyl
- KHMDS
Potassium bis(trimethylsilyl)amide
- LiDBB
Lithium 4,4′-di-tert-butylbiphenyl
- ManNAcA
N-acetyl-D-mannosaminuronic acid
- MS
Mass spectrometry
- Ms2O
Methanesulfonic anhydride
- NAP
2-Naphthylmethyl
- NFM
N-Formylmorpholine
- NIS
N-Iodosuccinimide
- NMR
Nuclear magnetic resonance
- PMB
p-Methoxybenzyl
- PMP
p-Methoxybenzylidine
- TBAB
Tetrabutylammonium bromide
- TBAI
Tetrabutylammonium iodide
- TBDMS
tert-Butyldimethylsilyl
- Tf
Trifluoromethanesulfonate
- Tf2O
Trifluoromethane sulfonyl anhydride
- THF
Tetrahydrofuran
- TMSOTf
Trimethylsilyl trifluoromethanesulfonate
- TTBP
2,4,6-Tri-tert-butylpyrimidine
Biographies

Bo Yang received his B.Sc. in Chemistry from School of Chemistry and Chemical Engineering, Nanjing University in 2006. He obtained his Ph.D. degree in Organic Chemistry from Michigan State University in 2012 under the supervision of Prof. Xuefei Huang. After one year post-doctoral research work in the same lab, he started his industrial career as a Scientist in Thermo Fisher Scientific in 2013. His research focuses on synthesis of complex oligosaccharides, glycoconjugates, nucleic acids, lipids and proteins.

Weizhun Yang received his Ph.D. degree from East China University of Science and Technology in 2012. During his Ph.D., he worked in Prof. Biao Yu’s lab at Shanghai Institute of Organic Chemistry. Then he moved to Michigan State University and joined the Huang group as a postdoctoral fellow. His research is focused on the synthesis of heparan sulfate glycopeptide and glycoprotein.

Sherif Ramadan obtained a B.Sc. at Faculty of Science, Benha University (Egypt) in 2008, M.Sc. in 2012, and earned his Ph.D. in Organic Chemistry in 2016 from the same School. During his Ph.D., he worked under the supervision of Prof. Xuefei Huang at Michigan State University (USA). He is working as assistant professor at Benha University Chemistry Department since 2016. Currently, he is taking a Postdoctoral Research Position at Michigan State University with Prof Huang. His research interests are focused on developing novel methodologies for assembling biological active oligosaccharide and glycoconjugates.

Xuefei Huang is MSU Foundation Professor of chemistry and biomedical engineering at Michigan State University and a member of the newly formed Institute for Quantitative Health Science and Engineering. His research interests are aimed at studying chemistry and biology of carbohydrates. A major focus is to develop new methodologies for synthesis of complex glycans and glyco-conjugates. In addition, his group is actively investigating novel approaches to boost immune responses against carbohydrate antigens as potential anti-cancer and anti-microbial vaccines. He is also interested in integrating carbohydrate chemistry with nanotechnology for molecular imaging and targeted drug delivery.
References
- 1.Varki A. Glycobiology. 2017;27:3–49. doi: 10.1093/glycob/cww086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dwek RA. Chem Rev. 1996;96:683–720. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 3.Crich D. Acc Chem Res. 2010;43:1144–1153. doi: 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]
- 4.Walvoort MTC, Dinkelaar J, van den Bos LJ, Lodder G, Overkleeft HS, Codee JDC, van der Marel GA. Carbohydr Res. 2010;345:1252–1263. doi: 10.1016/j.carres.2010.02.027. [DOI] [PubMed] [Google Scholar]
- 5.Barresi F, Hindsgaul O. In: Modern Methods in Carbohydrate Synthesis. Khan SH, O’Neil MA, editors. Harwood Academic Publishers; Amsterdam: 1996. pp. 251–276. [Google Scholar]
- 6.Crich D, Sun S. J Org Chem. 1997;62:1198–1199. [Google Scholar]
- 7.Crich D, Sun S. J Org Chem. 1996;61:4506–4507. doi: 10.1021/jo9606517. [DOI] [PubMed] [Google Scholar]
- 8.Crich D, Sun S. J Am Chem Soc. 1998;120:435–436. [Google Scholar]
- 9.Kim KS, Kim JH, Lee YJ, Lee YJ, Park J. J Am Chem Soc. 2001;123:8477–8481. doi: 10.1021/ja015842s. [DOI] [PubMed] [Google Scholar]
- 10.Kim KS, Fulse DB, Baek JY, Lee B-Y, Jeon HB. J Am Chem Soc. 2008;130:8537–8547. doi: 10.1021/ja710935z. [DOI] [PubMed] [Google Scholar]
- 11.Crich D, Sun S. J Am Chem Soc. 1997;119:11217–11223. [Google Scholar]
- 12.Dharuman S, Crich D. Chem Eur J. 2016;22:4535–4542. doi: 10.1002/chem.201505019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jensen HH, Nordstrom LU, Bols M. J Am Chem Soc. 2004;126:9205–9213. doi: 10.1021/ja047578j. [DOI] [PubMed] [Google Scholar]
- 14.Andrews CW, Rodebaugh R, Fraser-Reid B. J Org Chem. 1996;61:5280–5289. doi: 10.1021/jo961115h. [DOI] [PubMed] [Google Scholar]
- 15.Crich D, Chandrasekera NS. Angew Chem Int Ed. 2004;43:5386–5389. doi: 10.1002/anie.200453688. [DOI] [PubMed] [Google Scholar]
- 16.Huang M, Garrett GE, Birlirakis N, Bohé L, Pratt DA, Crich D. Nature Chem. 2012;4:663–667. doi: 10.1038/nchem.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crich D, Jayalath P. Org Lett. 2005;7:2277–2280. doi: 10.1021/ol050680g. [DOI] [PubMed] [Google Scholar]
- 18.Crich D, Dudkin V. Tetrahedron Lett. 2000;41:5643–5646. [Google Scholar]
- 19.Gorin PAJ. Carbohydr Res. 1975;39:3–10. [Google Scholar]
- 20.Gorin PAJ, Horitsu K, Spencer JFT. Can J Chem. 1965;43:950–954. [Google Scholar]
- 21.Crich D, Li W, Li H. J Am Chem Soc. 2004;126:15081–15086. doi: 10.1021/ja0471931. [DOI] [PubMed] [Google Scholar]
- 22.Crich D, Cai W. J Org Chem. 1999;64:4926–4930. doi: 10.1021/jo990243d. [DOI] [PubMed] [Google Scholar]
- 23.Crich D, Vinogradova O. J Org Chem. 2006;71:8473–8480. doi: 10.1021/jo061417b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crich D, Li L. J Org Chem. 2007;72:1681–1690. doi: 10.1021/jo062294y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crich D, Smith M. J Am Chem Soc. 2002;124:8867–8869. doi: 10.1021/ja011406u. [DOI] [PubMed] [Google Scholar]
- 26.Crich D, Cai W, Dai Z. J Org Chem. 2000;65:1291–1297. doi: 10.1021/jo9910482. [DOI] [PubMed] [Google Scholar]
- 27.Crich D, Vinod AU, Picione J. J Org Chem. 2003;68:8453–8458. doi: 10.1021/jo035003j. [DOI] [PubMed] [Google Scholar]
- 28.Crich D, Jayalath P. J Org Chem. 2005;70:7252–7259. doi: 10.1021/jo0508999. [DOI] [PubMed] [Google Scholar]
- 29.Lu Y-S, Li Q, Zhang L-H, Ye X-S. Org Lett. 2008;10:3445–3448. doi: 10.1021/ol801190c. [DOI] [PubMed] [Google Scholar]
- 30.Lu Y-S, Li Q, Wang Y, Ye X-S. Synlett. 2010:1519–1524. [Google Scholar]
- 31.Crich D, Subramanian V, Hutton TK. Tetrahedron. 2007;63:5042–5049. doi: 10.1016/j.tet.2007.03.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nukada T, Berces A, Whitfield DM. Carbohydr Res. 2002;337:765–774. doi: 10.1016/s0008-6215(02)00043-5. [DOI] [PubMed] [Google Scholar]
- 33.Nukada T, Berces A, Wang L, Zgierski MZ, Whitfield DM. Carbohydr Res. 2005;340:841–852. doi: 10.1016/j.carres.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 34.Benakli K, Zha C, Kerns RJ. J Am Chem Soc. 2001;123:9461–9462. doi: 10.1021/ja0162109. [DOI] [PubMed] [Google Scholar]
- 35.Wei P, Kerns RJ. J Org Chem. 2005;70:4195–4198. doi: 10.1021/jo047812o. [DOI] [PubMed] [Google Scholar]
- 36.Manabe S, Ishii K, Ito Y. J Am Chem Soc. 2006;128:10666–10667. doi: 10.1021/ja062531e. [DOI] [PubMed] [Google Scholar]
- 37.Tanaka H, Nishiura Y, Takahashi T. J Am Chem Soc. 2006;128:7124–7125. doi: 10.1021/ja0613613. [DOI] [PubMed] [Google Scholar]
- 38.Farris MD, De Meo C. Tetrahedron Lett. 2007;48:1225–1227. [Google Scholar]
- 39.Crich D, Li W. J Org Chem. 2007;72:2387–2391. doi: 10.1021/jo062431r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crich D, Li W. J Org Chem. 2007;72:7794–7797. doi: 10.1021/jo7012912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka H, Tateno Y, Takahashi T. Org Lett. 2008;10:5597–5600. doi: 10.1021/ol802207e. [DOI] [PubMed] [Google Scholar]
- 42.Olsson JDM, Eriksson L, Lahmann M, Oscarson S. J Org Chem. 2008;73:7181–7188. doi: 10.1021/jo800971s. [DOI] [PubMed] [Google Scholar]
- 43.Hanashima S, Sato K-i, Ito Y, Yamaguchi Y. Eur J Org Chem. 2009:4215–4220. [Google Scholar]
- 44.Hsu C-H, Chu K-C, Lin Y-S, Han J-L, Peng Y-S, Ren C-T, Wu C-Y, Wong C-H. Chem Eur J. 2010;16:1754–1760. doi: 10.1002/chem.200903035. [DOI] [PubMed] [Google Scholar]
- 45.Crich D, Navuluri C. Angew Chem Int Ed. 2010;49:3049–3052. doi: 10.1002/anie.200907178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Geng Y, Zhang L-H, Ye X-S. Chem Commun. 2008:597–599. doi: 10.1039/b712591g. [DOI] [PubMed] [Google Scholar]
- 47.Geng Y, Zhang L-H, Ye X-S. Tetrahedron. 2008;64:4949–4958. [Google Scholar]
- 48.Yang L, Ye X-S. Carbohydr Res. 2010;345:1713–1721. doi: 10.1016/j.carres.2010.05.031. [DOI] [PubMed] [Google Scholar]
- 49.Geng Y, Ye X-S. Synlett. 2015:2506–2512. [Google Scholar]
- 50.Wang B, Brand-Miller J. Eur J Clin Nutr. 2003;57:1351–1369. doi: 10.1038/sj.ejcn.1601704. [DOI] [PubMed] [Google Scholar]
- 51.Sun B, Jiang H. Tetrahedron Lett. 2011;52:6035–6038. [Google Scholar]
- 52.Huang L, Huang X. Chem Eur J. 2007;13:529–540. doi: 10.1002/chem.200601090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zeng Y, Wang Z, Whitfield D, Huang X. J Org Chem. 2008;73:7952–7962. doi: 10.1021/jo801462r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crich D, Dai Z, Gastaldi S. J Org Chem. 1999;64:5224–5229. doi: 10.1021/jo990424f. [DOI] [PubMed] [Google Scholar]
- 55.Lu X, Kamat MN, Huang L, Huang X. J Org Chem. 2009;74:7608–7617. doi: 10.1021/jo9016925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamada T, Takemura K, Yoshida J-I, Yamago S. Angew Chem, Int Ed. 2006;45:7575–7578. doi: 10.1002/anie.200602699. [DOI] [PubMed] [Google Scholar]
- 57.Finley JH, Denney DZ, Denney DB. J Amer Chem Soc. 1969;91:5826–5831. [Google Scholar]
- 58.Kim J-H, Yang H, Boons G-J. Angew Chem, Int Ed. 2005;44:947–949. doi: 10.1002/anie.200461745. [DOI] [PubMed] [Google Scholar]
- 59.Kim J-H, Yang H, Park J, Boons G-J. J Am Chem Soc. 2005;127:12090–12097. doi: 10.1021/ja052548h. [DOI] [PubMed] [Google Scholar]
- 60.Boltje TJ, Kim J-H, Park J, Boons G-J. Org Lett. 2011;13:284–287. doi: 10.1021/ol1027267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boltje TJ, Kim J-H, Park J, Boons G-J. Nat Chem. 2010;2:552–557. doi: 10.1038/nchem.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elferink H, Mensink RA, White PB, Boltje TJ. Angew Chem Int Ed. 2016;55:11217–11220. doi: 10.1002/anie.201604358. [DOI] [PubMed] [Google Scholar]
- 63.Hoang K, Liu X-LMW. Nat Commun. 2014;5:5051. doi: 10.1038/ncomms6051. [DOI] [PubMed] [Google Scholar]
- 64.van der Vorm S, Hansen T, Overkleeft HS, van der Marel GA, Codée JDC. Chem Sci. 2017;8:1867–1875. doi: 10.1039/c6sc04638j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Issa JP, Lloyd D, Steliotes E, Bennett CS. Org Lett. 2013;15:4170–4173. doi: 10.1021/ol4018547. [DOI] [PubMed] [Google Scholar]
- 66.Issa JP, Bennett CS. J Am Chem Soc. 2014;136:5740–5744. doi: 10.1021/ja500410c. [DOI] [PubMed] [Google Scholar]
- 67.Zhu D, Baryal KN, Adhikari S, Zhu J. J Am Chem Soc. 2014;136:3172–3175. doi: 10.1021/ja4116956. [DOI] [PubMed] [Google Scholar]
- 68.Baryal KN, Zhu D, Li X, Zhu J. Angew Chem Int Ed. 2013;52:8012–8016. doi: 10.1002/anie.201301682. [DOI] [PubMed] [Google Scholar]
- 69.Baryal KN, Zhu J. Org Lett. 2015;17:4530–4533. doi: 10.1021/acs.orglett.5b02223. [DOI] [PubMed] [Google Scholar]
- 70.Van Boeckel CAA, Beetz T, Van Aelst SF. Tetrahedron. 1984;40:4097–4107. [Google Scholar]
- 71.Smid P, De Ruiter GA, Van der Marel GA, Rombouts FM, Van Boom JH. J Carbohydr Chem. 1991;10:833–849. [Google Scholar]
- 72.Jin H, Tsai TYR, Wiesner K. Can J Chem. 1983;61:2442–2444. [Google Scholar]
- 73.Chiba S, Kitamura M, Narasaka K. J Am Chem Soc. 2006;128:6931–6937. doi: 10.1021/ja060408h. [DOI] [PubMed] [Google Scholar]
- 74.Ustyuzhanina N, Komarova B, Zlotina N, Krylov V, Gerbst A, Tsvetkov Y, Nifantiev N. Synlett. 2006:921–923. [Google Scholar]
- 75.Dejter-Juszynski M, Flowers HM. Carbohydr Res. 1972;23:41–45. doi: 10.1016/s0008-6215(00)87031-7. [DOI] [PubMed] [Google Scholar]
- 76.Corey EJ, Carpino P. J Am Chem Soc. 1989;111:5472–5474. [Google Scholar]
- 77.Demchenko AV, Rousson E, Boons G-J. Tetrahedron Lett. 1999;40:6523–6526. [Google Scholar]
- 78.Mukaiyama T, Suenaga M, Chiba H, Jona H. Chem Lett. 2002:56–57. [Google Scholar]
- 79.Cheng Y-P, Chen H-T, Lin C-C. Tetrahedron Lett. 2002;43:7721–7723. [Google Scholar]
- 80.De Meo C, Kamat MN, Demchenko AV. Eur J Org Chem. 2005:706–711. [Google Scholar]
- 81.Crich D, Hu T, Cai F. J Org Chem. 2008;73:8942–8953. doi: 10.1021/jo801630m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Awad LF, El Ashry ESH, Schuerch C. Bull Chem Soc Jpn. 1986;59:1587–1592. [Google Scholar]
- 83.Srivastava VK, Schuerch C. J Org Chem. 1981;46:1121–1126. [Google Scholar]
- 84.Crich D, Picione J. Org Lett. 2003;5:781–784. doi: 10.1021/ol0340890. [DOI] [PubMed] [Google Scholar]
- 85.Crich D, Hutton TK, Banerjee A, Jayalath P, Picione J. Tetrahedron: Asymmetry. 2005;16:105–119. [Google Scholar]
- 86.Perkins HR. Biochem J. 1963;86:475–483. doi: 10.1042/bj0860475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nasir ud D, Jeanloz RW. Carbohydr Res. 1976;47:245–260. doi: 10.1016/s0008-6215(00)84190-7. [DOI] [PubMed] [Google Scholar]
- 88.Hase S, Matsushima Y. J Biochem. 1972;72:1117–1128. doi: 10.1093/oxfordjournals.jbchem.a129999. [DOI] [PubMed] [Google Scholar]
- 89.Walvoort MTC, Lodder G, Overkleeft HS, Codee JDC, van der Marel GA. J Org Chem. 2010;75:7990–8002. doi: 10.1021/jo101779v. [DOI] [PubMed] [Google Scholar]
- 90.Gadikota RR, Callam CS, Wagner T, Del Fraino B, Lowary TL. J Am Chem Soc. 2003;125:4155–4165. doi: 10.1021/ja029302m. [DOI] [PubMed] [Google Scholar]
- 91.Callam CS, Gadikota RR, Krein DM, Lowary TL. J Am Chem Soc. 2003;125:13112–13119. doi: 10.1021/ja0349610. [DOI] [PubMed] [Google Scholar]
- 92.He X, Agnihotri G, Liu H-W. Chem Rev. 2000;100:4615–4661. doi: 10.1021/cr9902998. [DOI] [PubMed] [Google Scholar]
- 93.Langenhan JM, Peters NR, Guzei IA, Hoffmann FM, Thorson JS. Proc Natl Acad Sci, USA. 2005;102:12305–12310. doi: 10.1073/pnas.0503270102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.de Lederkremer RM, Marino C. Adv Carbohydr Chem Biochem. 2008;61:143–216. doi: 10.1016/S0065-2318(07)61004-X. [DOI] [PubMed] [Google Scholar]
- 95.Iyer AKV, Zhou M, Azad N, Elbaz H, Wang L, Rogalsky DK, Rojanasakul Y, O’Doherty GA, Langenhan JM. ACS Med Chem Lett. 2010;1:326–330. doi: 10.1021/ml1000933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zeng N, Xu Y, Wang H, Meng L, Wan Q. Sci China Chem. 2017;60:1162–1179. [Google Scholar]
- 97.Huang X, Huang L, Wang H, Ye X-S. Angew Chem, Int Ed. 2004;43:5221–5224. doi: 10.1002/anie.200460176. [DOI] [PubMed] [Google Scholar]
- 98.Verma VP, Wang C-C. Chem Eur J. 2013;19:846–851. doi: 10.1002/chem.201203418. [DOI] [PubMed] [Google Scholar]
- 99.Nigudkar SS, Demchenko AV. Chem Sci. 2015;6:2687–2704. doi: 10.1039/c5sc00280j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Schmidt RR, Behrendt M, Toepfer A. Synlett. 1990:694–696. [Google Scholar]
- 101.Pougny JR, Sinay P. Tetrahedron Lett. 1976;17:4073–4076. [Google Scholar]
- 102.Ratcliffe AJ, Fraser-Reid B. J Chem Soc, Perkin Trans 1. 1990:747–750. [Google Scholar]
- 103.Braccini I, Derouet C, Esnault J, du Penhoat CH, Mallet JM, Michon V, Sinay P. Carbohydr Res. 1993;246:23–41. [Google Scholar]
- 104.Crich D, Patel M. Carbohydr Res. 2006;341:1467–1475. doi: 10.1016/j.carres.2006.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wasonga G, Zeng YL, Huang XF. Sci China Chem. 2011;54:66–73. [Google Scholar]
- 106.Lu S-R, Lai Y-H, Chen J-H, Liu C-Y, Mong K-KT. Angew Chem, Int Ed. 2011;50:7315–7320. doi: 10.1002/anie.201100076. [DOI] [PubMed] [Google Scholar]
- 107.Ingle AB, Chao C-S, Hung W-C, Mong K-KT. Org Lett. 2013;15:5290–5293. doi: 10.1021/ol402519c. [DOI] [PubMed] [Google Scholar]
- 108.Lin YH, Ghosh B, Mong K-KT. Chem Commun. 2012;48:10910–10912. doi: 10.1039/c2cc35032g. [DOI] [PubMed] [Google Scholar]
- 109.liu C-YI, Mulani S, Mong K-KT. Adv Synth Catal. 2012;354:3299–3310. [Google Scholar]
- 110.Mulani SK, Hung W-C, Ingle AB, Shiau K-S, Mong K-KT. Org Biomol Chem. 2014;12:1184–1197. doi: 10.1039/c3ob42129e. [DOI] [PubMed] [Google Scholar]
- 111.Chu A-HA, Nguyen SH, Sisel JA, Minciunescu A, Bennett CS. Org Lett. 2013;15:2566–2569. doi: 10.1021/ol401095k. [DOI] [PubMed] [Google Scholar]
- 112.D’Angelo KA, Taylor MS. J Am Chem Soc. 2016;138:11058–11066. doi: 10.1021/jacs.6b06943. [DOI] [PubMed] [Google Scholar]
- 113.Gouliaras C, Lee D, Chan L, Taylor MS. J Am Chem Soc. 2011;133:13926–13929. doi: 10.1021/ja2062715. [DOI] [PubMed] [Google Scholar]
- 114.Satoh H, Hutter J, Luthi HP, Ishii K, Ito Y. Eur J Org Chem. 2009:1127–1131. [Google Scholar]
- 115.Manabe S, Ishii K, Hashizume D, Koshino H, Ito Y. Chem Eur J. 2009;15:6894–6901. doi: 10.1002/chem.200900064. [DOI] [PubMed] [Google Scholar]
- 116.Manabe S, Ishii K, Satoh H, Ito Y. Tetrahedron. 2011;67:9966–9974. [Google Scholar]
- 117.Satoh H, Manabe S, Ito Y, Luthi HP, Laino T, Hutter J. J Am Chem Soc. 2011;133:5610–5619. doi: 10.1021/ja201024a. [DOI] [PubMed] [Google Scholar]
- 118.Geng Y, Qi Q, Ye X-S. J Org Chem. 2012;77:5255–5270. doi: 10.1021/jo3002084. [DOI] [PubMed] [Google Scholar]
- 119.Qi Q, Xiong D-C, Ye X-S. Carbohydr Res. 2015;403:104–144. doi: 10.1016/j.carres.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 120.Mukaiyama T, Suda S. Chem Lett. 1990;19:1143–1146. [Google Scholar]
- 121.Mossotti M, Panza L. J Org Chem. 2011;76:9122–9126. doi: 10.1021/jo2015856. [DOI] [PubMed] [Google Scholar]
- 122.Kobashi Y, mukaiyama T. Chem Lett. 2004;33:874–875. [Google Scholar]
- 123.Oka N, Kajino R, Takeuchi K, Nagakawa H, Ando K. J Org Chem. 2014;79:7656–7664. doi: 10.1021/jo500632h. [DOI] [PubMed] [Google Scholar]