

Abstract
T cells expressing second-generation chimeric antigen receptors (CARs) specific for CD5, a T-cell surface marker present on normal and malignant T cells, can selectively kill tumor cells. We aimed to improve this killing by substituting the CD28 costimulatory endodomain (28.z) with 4-1BB (BB.z), as 28.z CD5 CAR T cells rapidly differentiated into short-lived effector cells. In contrast, 4-1BB costimulation is known to promote development of the central memory subpopulation. Here, we found BB.z CD5 CAR T cells had impaired growth compared with 28.z CD5.CAR T cells, due to increased T-cell–T-cell fratricide. We demonstrate that TRAF signaling from the 4-1BB endodomain upregulated the intercellular adhesion molecule 1, which stabilized the fratricidal immunologic synapse between CD5 CAR T cells. As the surviving BB.z CD5 CAR T cells retained the desired central memory phenotype, we aimed to circumvent the 4-1BB–mediated toxicity using a regulated expression system that reversibly inhibits CAR expression. This system minimized CAR signaling and T-cell fratricide during in vitro expansion in the presence of a small-molecule inhibitor, and restored CAR expression and antitumor function of transduced T cells in vivo. These studies reveal a mechanism by which 4-1BB costimulation impairs expansion of CD5 CAR T cells and offer a solution to mitigate this toxicity.
Introduction
Incorporation of one or more costimulatory endodomains in chimeric antigen receptors (CARs) increases the expansion and persistence of CAR T cells after tumor challenge (1–3). Clinically used endodomains are commonly derived from either the Ig superfamily gene CD28 or TNFR superfamily members 4-1BB (CD137) and OX40 (CD134; ref. 4), coupled to CD3ζ. T cells expressing CD19 CARs harboring either CD28 or 4-1BB endodomains have induced complete remissions in patients with advanced B-cell malignancies, though each endodomain may produce distinct biological effects (5–8). Incorporation of CD28.z in CD19 CAR T cells results in their rapid expansion and immediate antitumor activity, whereas 4-1BB.z CD19 CAR T cells may persist longer and establish immune control (5–9). These differences could be attributed, at least in part, to the role of CD28-derived PI3K signaling in enhancing T-cell activation and spurring differentiation to cytotoxic but short-lived effector cells (10–12). In contrast, 4-1BB signaling induces more limited T-cell differentiation, favoring the development of central memory T cells with longer-term persistence (13). Despite these differences in biological activity, whether coupling CD19 CARs to CD28 or to 4-1BB signaling domains produces superior clinical outcomes remains unknown. Thus, continued exploration of both classes of signaling endodomains is necessary for future studies of CAR T-cell activity in humans.
Some potentially targetable tumor-associated antigens, particularly those associated with lymphoid malignancies, are also expressed by T cells (14–17). Expression of a CAR specific for these antigens in T cells may promote their fratricide and therefore compromise expansion. This problem is particularly relevant for the development of CAR T-cell–based therapies for T-lineage malignancies. The pan–T-cell marker CD5 is being investigated for the treatment of T-cell malignancies such as T-cell acute lymphoblastic leukemia (T-ALL) and peripheral lymphoma (17). This approach is feasible because CD5 is downregulated from the T-cell surface upon ligation with its ligand or antibody, allowing CD5+ T-cell tumors to be targeted even by CD5 CAR T cells that are themselves CD5+ (17). Thus, expression of a CD28.z CD5 CAR in activated T cells resulted in transient and limited fratricide followed by robust expansion of CD5 CAR T cells. CD28.z CD5 CAR T cells had high cytotoxicity against malignant T cells but limited in vivo persistence. Although the short life span of effector-enriched 28.z CD5 CAR T cells may reduce the extent and duration of potential off-tumor toxicities in patients (e.g., T-cell aplasia), it may also limit the durability of antitumor responses. Therefore, we hypothesized that replacing CD28 with the 4-1BB costimulatory endodomain in CD5 CARs would restrain effector differentiation of CD5 CAR T cells and increase their in vivo persistence.
We found that incorporation of 4-1BB in the CD5 CAR indeed augmented the formation of central memory T cells. We observed that 4-1BB costimulation also enhanced fratricide of CD5 CAR T cells and impaired their expansion, an adverse effect also produced by other TNFR superfamily–derived CAR endodomains. Nonetheless, by developing a CAR expression system that reversibly disrupts this deleterious CAR signaling and prevents CAR T-cell fratricide in vitro, we were able to explore the potential benefits of 4-1BB signaling without promoting fratricide.
Materials and Methods
CAR constructs and vectors
The second-generation CD5 CAR containing a CH3 IgG Fc spacer and CD28-derived transmembrane region and endodomain (28.z) was described previously (17). Other CD5 CAR constructs used in this study were created by replacing the CD28 endodomain with cytoplasmic regions from ICOS, 4-1BB, CD27, CD30, OX40, or HVEM genes. OX40 and 4-1BB endodomains were amplified from GD2- and CD19-specific CARs that are being evaluated clinically at Baylor College of Medicine (BCM). ICOS, CD27, CD30, and HVEM genes were subcloned from a cDNA library of genes expressed in activated T cells using standard molecular cloning techniques. In ICOS.z and HVEM.z CD5 CARs, the CH3 spacer was replaced with the CD8a stalk due to unstable expression in the original configuration. All CAR constructs contained the transmembrane region from CD28 except ICOS.z where the transmembrane region from ICOS was used. TRAF-binding sites in the 4-1BB endodomain were created using site-directed mutagenesis with the InFusion cloning Kit (Clontech). CAR constructs were subcloned into SFG gammaretroviral vector and sequence-verified by Sanger DNA sequencing. Gammaretroviral transduction of T cells was performed as previously described (17). Transduced T cells were expanded in the presence of IL7 (10 ng/mL) and IL15 (10 ng/mL) in RPMI-1640/Click's (CTL) media supplemented with 10% FBS and 1% l-glutamine. Primary human peripheral blood mononuclear cells were isolated from healthy donors after informed consent using the protocol approved by the BCM Institutional Review Board and in accordance with the Declaration of Helsinki.
Flow cytometry
Antibodies were purchased from Beckman Coulter [CD3 (UCHT1), CD4 (SK3), CD5 (UCHT2), CD8 (SK1), CD45RA (2H4)], BD Biosciences [CCR7 (150503)], or Biolegend [intercellular adhesion molecule 1 (ICAM-1; HA-58)]. Annexin V and/or 7-AAD (BD Biosciences) was used to detect apoptotic cells. Goat anti-mouse Fc-specific or Fab-specific polyclonal antibodies (Jackson Immunoresearch) were used to detect CAR expression on the cell surface. Samples were acquired on BD Gallios and analyzed with FlowJo v.9.
Cell culture
Jurkat and CCRF T-ALL cell lines modified to express GFP-FFluc were described previously (17). CD5 CAR T cells were plated with tumor cells at a 1:4 initial effector-to-target ratio and cultured for 72 hours in the absence of exogenous cytokines. Cells were acquired by flow cytometry and quantified at the end of coculture in the presence of 7-AAD and Countbright counting beads (Invitrogen).
For ICAM-1 blockade, CD5 CAR T cells were cultured in the presence of 2 μg/mL anti-ICAM-1-blocking antibodies (84H10; Bio-Rad) or vehicle control for 72 hours. Cell death and counts were measured by flow cytometry as indicated above.
Western blot analysis
Western blot analysis was performed as previously described (18). Briefly, cells were dissociated using lysis buffer with phosphatase and protease inhibitors (Thermo Scientific). Protein concentrations were determined using a Bio-Rad protein assay (BioRad). Samples were denatured in Laemmli buffer containing 2-Mercaptoethanol and boiled at 95°C for 10 minutes. Cell lysates were run on a premade 10% SDS polyacrylamide gel (BioRad) and transferred to nitrocellulose membranes (BioRad). Membranes were then blocked with 5% BSA or milk and probed with primary antibodies followed by horseradish peroxidase-conjugated secondary antibodies. Blots were developed using SuperSignal West Dura Extended Duration Substrate (Thermo Scientific) and exposed to GeneMate Blue Basic Autoradiography Film (BioExpress). Antibodies used in the study are as follows: anti-CD3.ζ (sc-1239; Santa Cruz Biotechnology, Inc.), anti-CD247 (pY142) (558402; BDPharmigen), pIKKα/β and IKKα (#9936; Cell Signaling Technology), and GAPDH (sc-47724; Santa Cruz Biotechnology, Inc.). Secondary antibodies used in the study are as follows: goat anti-mouse (sc-2005; Santa Cruz Biotechnology, Inc.) and goat anti-rabbit (111-035-003; Jackson ImmunoResearch Laboratories, Inc.). ImageJ (National Institutes of Health, Bethesda, MD) was used for Western blot quantification. Protein levels were normalized to loading controls (GAPDH).
Immunofluorescence analysis
For confocal microscopy, conjugates between CAR T cells and target cells were incubated for 30 minutes at 37°C and then fixed and stained for F-actin (Phalloidin 568), Pericentrin (rabbit anti-Pericentrin; Abcam), and ICAM-1 (mouse anti-ICAM-1; BD Biosciences). Pericentrin was detected using Alexa Fluor Pacific Blue secondary antibody to rabbit IgG, and ICAM-1 was detected using 488 Alexa Fluor secondary antibody to mouse IgG. Conjugates were imaged as Z stacks of 0.2 μm thickness to cover the entire volume of the immune synapse (IS), determined individually for each conjugate on a Leica SP8 confocal microscope using a 100× objective. Density of F-actin and ICAM-1 accumulation at the immune synapse of CAR T cells was calculated using the formula [volume × mean fluorescence intensity (MFI)] for an equal number of 1 μm3 boxes selected to cover the IS (19). Microtubule-organizing center (MTOC) to synapse distance was measured as described before (20).
Tet-OFF expression system
Gammaretroviral backbone from pRetroX-Tight-Pur (Clontech) was digested with EcoRI-EcoRV. An expression cassette comprised of the EF1a/LTR hybrid promoter (pCDH-CMV-MCS-EF1-copGFP; System Biosciences), and tTA-Advanced (pRetroX-TetOFF; Clontech) was assembled and inserted into the gammaretroviral backbone using the InFusion Cloning Kit (Clontech). CD5 CAR constructs were subcloned downstream of the Tet-responsive elements and upstream of the EF1a/LTR promoter. Gammaretroviral transduction of T cells was performed as previously described (17). To suppress transgene expression, transduced T cells were cultured in the presence of 10 to 50 ng/mL doxycycline (Sigma Aldrich). Doxycycline was replaced every 48 to 72 hours.
Mouse xenograft model
Eight- to 10-week-old male or female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (The Jackson Laboratory) were inoculated intravenously with 1 × 106 Jurkat-FFluc cells. Mice were then ear-tagged and randomly assigned to experimental groups. Mice received a single intravenous injection of 1.0 × 106 CAR T cells 3 or 7 days after tumor engraftment. Tumor burden was monitored by in vivo imaging with an IVIS Imaging system (Caliper Life Sciences) after injecting D-Luciferin (150 μg/kg i.p.). Mice were euthanized after the tumor burden reached a luminescence level of 108 photons/sec or after displaying signs of distress associated with graft-versus-host disease (GVHD) or high tumor burden. Peripheral blood was collected by tail vein bleeding. All animal experiments were conducted in compliance with the Institutional Animal Care and Use Committee of BCM.
Statistical analysis
Unpaired two-tailed Student t test was used to determine statistical significance for 2-sample comparison, and one-way ANOVA with Bonferroni posttest correction was used for multiple comparisons. P values below 0.05 were considered statistically significant. All statistical analyses were performed in GraphPad Prism 6.
Results
4-1BB costimulation abrogates the expansion of CD5 CAR T cells
We previously reported that T cells expressing a second-generation CD5 CAR with the CD28 costimulatory endodomain (28.z) have antitumor activity (17). To examine the role of 4-1BB costimulation in CD5 CARs, we substituted 28.z with the 4-1BB endodomain (BB.z), leaving the rest of the CAR backbone intact (Fig. 1A). Both 28.z and BB.z CD5 CARs were expressed on the cell surface of transduced T cells, and their expression correlated with the downregulation of CD5 (Fig. 1A), reflecting the rapid internalization of CD5 upon binding to the CAR. Expression of the BB.z CD5 CAR resulted in enrichment for CCR7+ CD45RA− central memory T cells (Fig. 1B); however, the BB.z CD5 CAR T cells failed to expand compared with control or 28.z CD5 CAR T cells (Fig. 1C). The impaired growth of BB.z CD5 CAR T cells correlated with significantly enhanced apoptosis (Fig. 1D), indicating that the expression of BB.z CD5 CAR augmented T-cell death. The increased numbers of 28.z CD5 CAR T cells could not be attributed to an associated functional exhaustion and loss of cytotoxicity or fratricide as these cells retained high cytotoxic activity even 21 days after transduction (Supplementary Fig. S1). To determine whether the increased fratricide was a result of an elevated expression of BB.z CD5 CAR in T cells (Fig. 1A), we increased the expression of 28.z CD5 CAR by replacing the CH3 Fc spacer with a short IgG Fc-derived hinge and evaluated T-cell expansion (Supplementary Fig. S2A and S2B). Elevated 28.z CD5 CAR expression did not abrogate T-cell growth (Supplementary Fig. S2C), indicating that the inability of BB.z CAR T cells to expand is not due to increased CAR expression.
Figure 1.
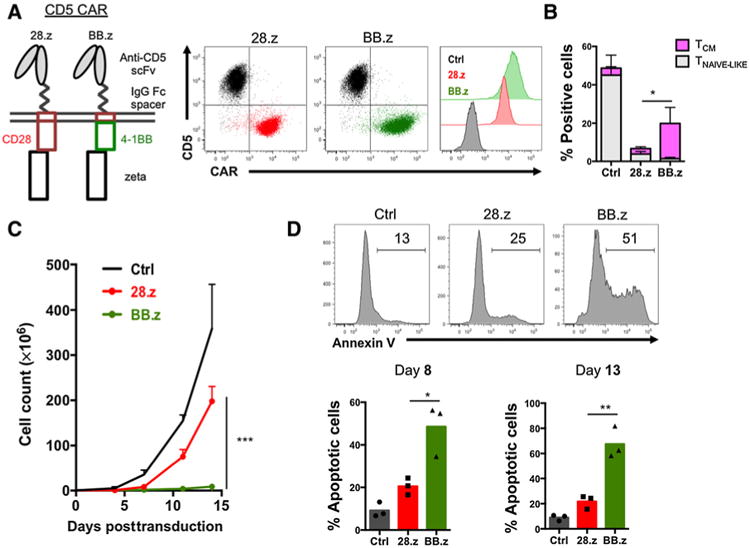
Expression of BB.z CD5 CAR abrogates T-cell expansion. A, Schematic representation of CD5 CAR constructs and their expression in T cells 4 days after transduction. B, Frequency of CCR7+ CD45RA+ (naïve-like) and CCR7+ CD45RA− (central memory) cells among T cells 13 days after transduction with 28.z or BB.z CD5 CAR, compared with nontransduced control T cells. The rest of the cells were comprised by terminally differentiated effector and effector memory T cells. Data are shown as mean ± SD (P = 0.0331 by unpaired Student t test, n = 3). C, Expansion of T cells transduced with 28.z or BB.z CD5 CAR and mock-transduced cells (Ctrl). Data are shown as mean ± SD (n = 3). D, Representative histograms showing Annexin V staining of CD5 CAR T cells. Bar graphs show summarized data from days 8 and 13 after transduction (P = 0.019 and 0.0044 by unpaired Student t test, respectively). Data represent four to six independent experiments.
TNFR superfamily costimulatory endodomains impair expansion of CD5 CAR T cells
To investigate whether only 4-1BB impaired expansion of CD5 CAR T cells, or whether this was a more general phenomenon observed with other TNFR superfamily genes (OX40, CD27, CD30, and HVEM), we investigated the role of each gene in regulating CD5 CAR T-cell expansion. We incorporated these costimulatory endodomains in a panel of second-generation CD5 CARs and then measured T-cell expansion following transduction. We found that T cells expressing CD5 CAR with immunoglobulin superfamily endodomains CD28 or ICOS (28.z and ICOS.z) expanded similarly to those expressing the first-generation CD5 CAR (ζ), whereas CD5 CAR T cells incorporating the TNFR superfamily costimulatory domains OX40, CD27, CD30, and HVEM showed reduced expansion, resembling the defect observed in BB.z CD5 CAR T cells (Fig. 2A). Although all CD5 CARs were expressed normally on T cells 3 days after transduction (Fig. 2B), we observed complete or partial downregulation of CD5 CARs with TNFR costimulatory endodomains in T cells a week later (Fig. 2B). We detected higher frequencies of the central memory subpopulation in all T cells expressing TNFR CD5 CARs compared with CD5 CARs with IgSF superfamily endodomains or no costimulation (Supplementary Fig. S3), indicating all TNFR endodomains can promote central memory formation in CAR-transduced T cells. The poor expansion of all TNFR domain-harboring CD5 CAR T-cell populations was not associated with increased expression of CD5 on the cell surface, as cell-surface expression of CD5 remained equally low in all CD5 CAR T cells (Fig. 2C). Thus, signaling from other TNFR endodomains pro-duced the same effects on phenotype and toxicity as 4-1BB in these CD5 CAR T cells. T cells expressing a third-generation 28.BB.z CD5 CAR behaved like BB.z CAR T cells, and they failed to expand (Fig. 2D). Because the phenotype of BB.z CD5 CAR T cells cannot be reversed by additional CD28 costimulation, 4-1BB signaling may play a dominant role in impairing expansion and survival of CD5 CAR T cells.
Figure 2.
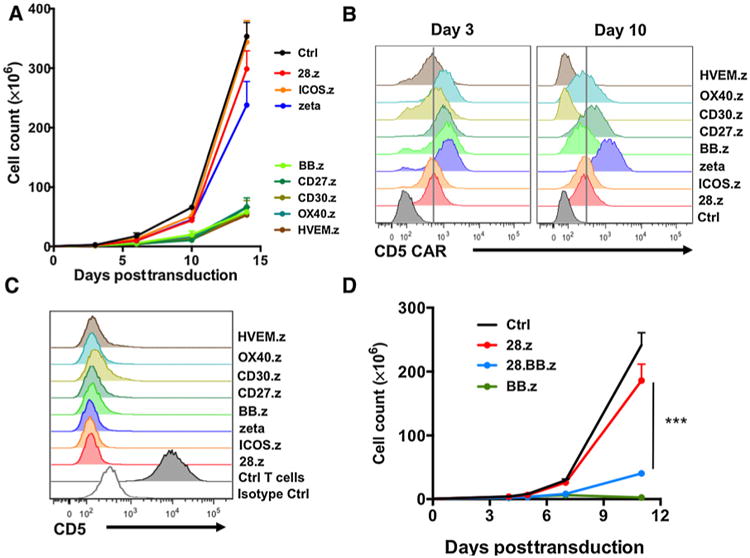
CD5 CARs containing TNFR superfamily endodomains preclude T-cell expansion. A, Expansion of T cells transduced with CD5 CARs containing the endodomains indicated. Nontransduced cells were used as control. Data are shown as mean ± SD (n = 3). B, Representative histograms show expression of CD5 CARs on T cells at the indicated time points after transduction using anti-mouse F(ab)2-specific antibodies. Vertical line denotes mean 28.z CD5 CAR expression. C, Expression of CD5 in CD5 CAR T cells 7 days after transduction. D, Expansion of T cells expressing second- (28.z) or third-generation (28.BB.z) CD5 CARs. Data are shown as mean ± SD (P = 0.0006 by unpaired Student t test, n = 3). Data represent two to three independent experiments.
TRAF2 signaling from the 4-1BB endodomain enhanced apoptosis in CD5 CAR T cells
Although CD5 CAR T cells downregulate surface expression of CD5, CD5 gene transcription remains intact (17). This observation suggests that CD5 CAR can constantly interact with residual ligand and therefore produce tonic CAR signaling, an interpretation supported by the presence of comparable levels of spontaneous phosphorylation of the CAR-derived ζ chain in both 28.z and BB.z CD5 CAR T cells (Fig. 3A). Hence, the selective toxicity of BB.z CD5 CAR is not associated with the differences in the magnitude of tonic signaling. However, unlike CD28, which directly activates PI3K signaling, 4-1BB signals through TRAF adapter proteins leading to the activation of the IkB kinase complex (IKKα/β), which subsequently disinhibits the transcription factor NF-kB prompting its nuclear translocation and transcriptional activity (21–23). We detected robust steady-state phosphorylation of IKKα/β in BB.z CAR T cells compared with the 28.z controls (Fig. 3B). Because 4-1BB and CD28 costimulation elicits different signaling pathways, we determined whether the toxicity of BB.z CD5 CAR in T cells is driven by 4-1BB–derived TRAF signaling. We disrupted the N-terminal (mut1QEED–QAAA) and the C-terminal (mut2PEEEE→PEAAA) motifs in the 4-1BB endo-domain to prevent binding of TRAF2 (mut1) or TRAF1, TRAF2, and TRAF3 (mut2; ref. 21). Alterations in TRAF-binding motifs did not compromise cell surface expression of CD5 CAR (Fig. 3C) but reduced the rate of apoptosis in BB.z CAR T cells (Fig. 3D) and decreased IKKα/β phosphorylation (Supplementary Fig. S4), resulting in normal expansion of T cells expressing either of the mutant BB.z CARs (Fig. 3E). Despite expanding normally, BB.z CD5 CAR T cells with mutated TRAF-binding motifs were unable to sequentially kill CD5+ tumor cells (CCRF); their killing capacity was similar to first-generation CD5 CAR T cells lacking costimulation (Fig. 3F). Moreover, disruption of TRAF2 signaling reduced the frequency of central memory T cells (Supplementary Fig. S5). These results indicate that although tonic 4-1BB–derived TRAF signaling promotes apoptosis in BB.z CAR T cells, acute antigen-driven TRAF2 signaling is essential for the costimulatory properties of the 4-1BB endodomain.
Figure 3.
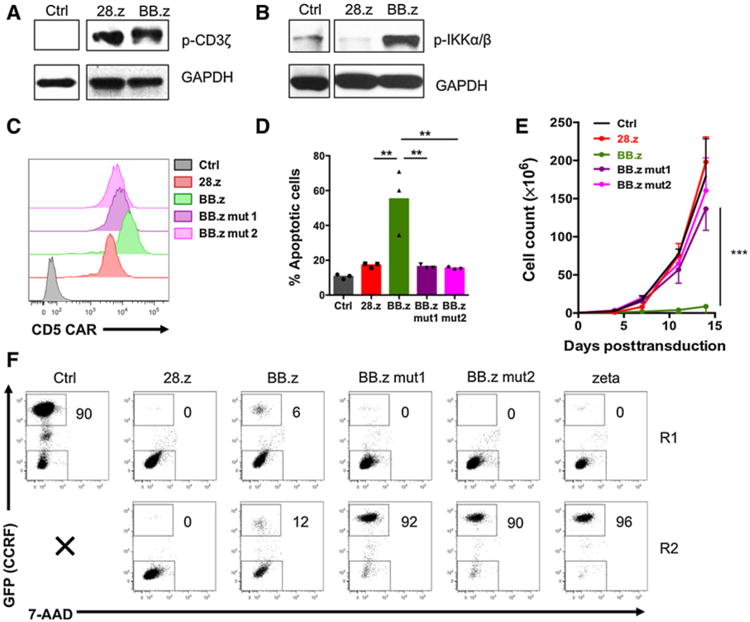
Disrupting TRAF-binding motifs in the 4-1BB endodomain restores T-cell expansion but ablates costimulation. A, Western blot analysis showing phosphorylation of the CAR-embedded ζ chain in CD5 CAR T cells 7 days after transduction. Nontransduced T cells were used as controls. B, Phosphorylation of IKKα/IKKβ in CD5 CAR T cells 7 days after transduction. C, Expression of CD5 CARs with mutated TRAF-binding sites in T cells 7 days after transduction. D, Frequency of apoptotic T cells expressing normal or mutated CD5 CARs 7 days after transduction. P = 0.002 calculated by one-way ANOVA with Bonferroni posttest correction (n = 3). E, Expansion of T cells transduced with normal or mutated CD5 CARs. P = 0.0015 calculated at day 14 by one-way ANOVA with Bonferroni posttest correction (n = 3). F, CD5 CAR T cells were cocultured with GFPþ CCRF cells at an effector-to-target ratio of 1:4 for two 3-day rounds. Fresh CCRF cells were added to CD5 CAR T cells at the end of round 1 (R1) to restore the initial effector-to-target ratio. Representative dot plots show frequency of tumor cells at the end of each round. Data represent two to four independent experiments.
BB.z CD5 CAR T cells form fratricidal immunologic synapses
Because CD5 expression in 28.z CD5 CAR T cells produces transient and limited fratricide, we investigated whether constant TRAF signaling in BB.z CD5 CAR T cells enhanced their fratricide. Consistent with this hypothesis, we observed formation of immunologic synapses between BB.z CD5 CAR T cells characterized by polymerization of actin (refs. 24, 25; Fig. 4A and B). A similar effect was observed in T cells expressing a third-generation 28.BB.z CD5 CAR (Fig. 4C). We also detected increased recruitment of the MTOC to the immunologic synapse in BB.z but not 28.z or mutated BB.z CD5 CAR T cells (Fig. 4D and E), suggesting self-directed cytotoxicity of BB.z CAR T cells (24, 25).
Figure 4.
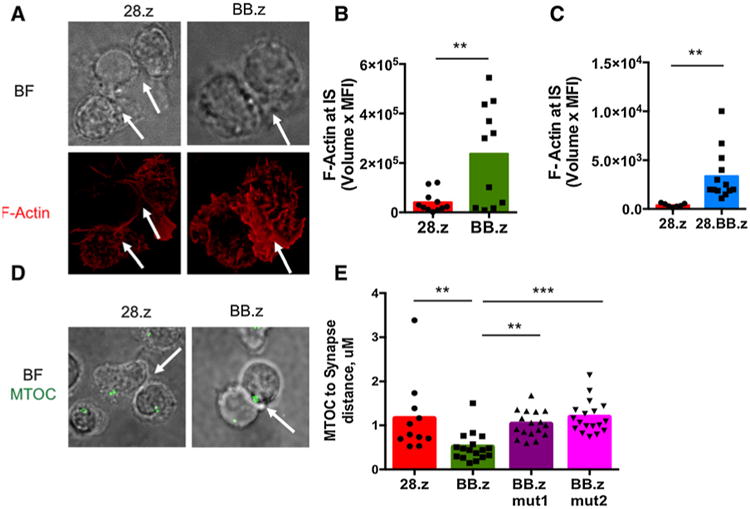
4-1BB endodomain in CD5 CAR enhances formation of immunologic synapses between T cells. A, Confocal representative images (original magnification, ×100) of 28.z and BB.z CD5 CAR T cells. Shown are bright field (BF) and single-color anti-F-actin (red) images. Arrows indicate cell-cell contacts. B and C, CD5 CAR T cells were assessed for accumulation of F-actin at synapses formed with each other. P < 0.01 by unpaired two-tailed Student t test. D, Representative images showing recruitment of MTOC to the immunological synapse between 28.z and BB.z CD5 CAR T cells. E, Distance between MTOC and immunological synapse formed between two CD5 CAR T cells (P = 0.0002 by one-way ANOVA with Bonferroni posttest correction). Cells were imaged as a z stack on a Leica SP8 confocal microscope. Data are representative of 3 independent experiments with n = 3 donors in each.
Engagement of adhesion molecules such as leukocyte function-associated antigen-1 (LFA-1) on T cells and its ICAM ligands on target cells is central to the formation of a stable immunologic synapse (25–27). Because NF-kB can directly activate ICAM-1 gene transcription (28, 29), we investigated whether chronic TRAF signaling can similarly enhance ICAM-1 expression in BB.z CD5 CAR T cells. In fact, surface ICAM-1 expression was significantly elevated in BB.z CD5 CAR T cells compared with 28.z CD5 CAR T cells (Fig. 5A), and blunting TRAF signaling as above in BB.z CD5 CAR prevented this upregulation (Fig. 5A). We also detected significant ICAM-1 upregulation in T cells expressing 28.BB.z CD5 CAR (Fig. 5B) or CD5 CARs with other TNFR endodomains (Fig. 5C), which correlated with the formation of large cell clusters during in vitro expansion (Supplementary Fig. S6). ICAM-1 was recruited to the synapse interface (Fig. 5D and E), potentially strengthening the fratricidal synapse. Blocking ICAM-1 with monoclonal antibodies in BB.z CD5 CAR T cells for 48 hours significantly reduced apoptotic cells (Fig. 5F) and enhanced T-cell expansion (Fig. 5G). These results indicate that constant TRAF2 signaling promoted BB.z CD5 CAR T-cell apoptosis by enhancing ICAM-1 expression and stabilizing the fratricidal immunologic synapse.
Figure 5.
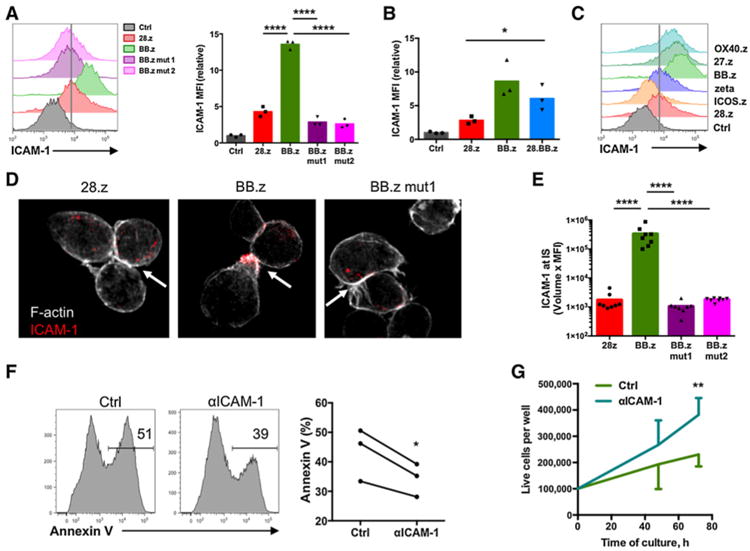
Upregulation of ICAM-1 in T cells expressing CD5 CARs with TNFR costimulatory endodomains promotes fratricide. A, Expression of surface ICAM-1 in CD5 CAR T cells 7 days after transduction. Total relative MFI of ICAM-1 is shown in the bar graph (P < 0.0001 by one-way ANOVA with Bonferroni posttest correction, n = 3). B, Upregulation of ICAM-1 in 28.BB.z CD5 CAR T cells on day 7 after transduction (P = 0.03 by unpaired two-tailed Student t test). C, ICAM-1 expression in CD5 CAR T cells. Vertical line denotes average expression level in 28.z CD5 CAR T cells. D, Confocal representative images (original magnification, ×100) of CD5 CAR T cells showing F-actin (silver) and ICAM-1 (red). Arrow indicates cell-cell contact area. E, CD5 CAR T cells were assessed for accumulation of ICAM-1 at synapses formed with each other. P < 0.0001 by one-way ANOVA with Bonferroni correction. F, BB.z CD5 CAR T cells were cultured in the presence of ICAM-1 blocking antibodies for 72 hours, and apoptosis was measured by Annexin V staining as shown on the representative histograms (P = 0.043 calculated by paired two-tailed Student t test). G, Increased expansion of BB.z CD5 CAR T cells incubated in the presence of ICAM-1 blocking antibodies for 72 hours (P = 0.0098 by paired two-tailed Student t test). Data are representative of two to three independent experiments.
Conditional CAR expression system eliminates fratricide of BB.z CD5 CAR T cells
Because enhanced apoptosis in T cells was a direct consequence of tonic BB.z CD5 CAR activation, we determined whether reducing CAR expression could reverse the toxicity. We attenuated toxic tonic signaling by expressing the BB.z CD5 CAR from a self-inactivating lentiviral vector, which reduced surface expression of the CAR (30). This improved cell viability and decreased tonic NF-kB activation, as reflected by normal ICAM-1 expression (Supplementary Fig. S7). Unfortunately, reduced CAR expression also significantly impaired the cytotoxicity of BB.z CD5 CAR T cells (Supplementary Fig. S7), indicating that the T-cell fratricide and the antitumor activity of BB.z CD5 CAR expression cannot readily be separated. Because such permanent reduction of CAR expression would compromise the antitumor activity of T cells, we evaluated an alternative approach that minimized fratricide during expansion while enabling subsequent antitumor function. We designed a conditional retroviral Tet-OFF expression system in which CAR transcription is driven by a transactivator (rTA) encoded downstream of the CAR under the control of a constitutive synthetic promoter (Fig. 6A). Addition of the small-molecule doxycycline (Dox) during in vitro culture prevented rTA from binding the CAR promoter, resulting in minimal CAR expression on the cell surface (Fig. 6B). We observed normal expansion of BB.z CD5 CAR T cells cultured in the presence of Dox (Fig. 6C), indicating that inhibiting CAR expression during culture prevents CAR T-cell fratricide. The residual CD5 CAR expression observed in Tet-OFF–transduced T cells did not trigger cytotoxicity by CD5 CAR T cells against CD5+ target cells (Supplementary Fig. S8). The phenotype of BB.z Tet-OFF cells was different from the phenotype of 28.z CD5 CAR T cells and mirrored that of the nontransduced T cells (Fig. 6D). Removing Dox from the media resulted in restoration of CAR expression over 4 to 5 days (Fig. 6E) and concomitant downregulation of CD5 (Supplementary Fig. S9). Conversely, adding Dox to CD5 CAR T cells minimized CAR expression within 24 hours (Fig. 6F). After Dox withdrawal, T cells with freshly acquired CAR expression were able to control tumor growth in vitro upon coculture with CD5+ cell lines Jurkat and CCRF-CEM (Fig. 6G) with similar effectiveness as the 28.z control. As in T cells with constitutively expressed BB.z CARs, we observed enriched central memory in Tet-OFF BB.z CD5 CAR T cells 7 days after Dox withdrawal (Fig. 6H). We observed similar increases in the expansion and frequency of undifferentiated T-cell subsets and restoration of cytotoxicity with a Tet-OFF 28.z CD5 CAR (Supplementary Fig. S10). These results indicate that the Tet-OFF CAR expression system produces functional CD5 CAR T cells in the absence of Dox.
Figure 6.
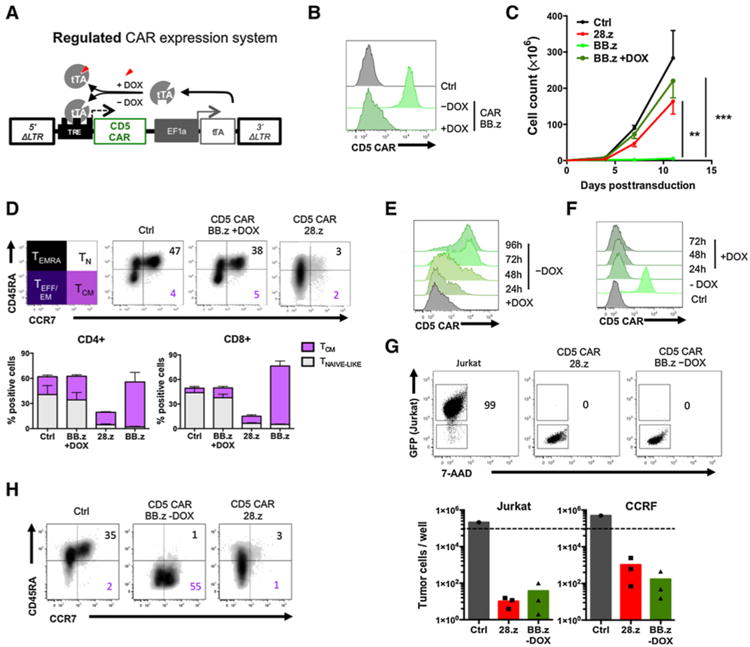
DOX-regulated expression system enables controllable CAR expression and prevents fratricide of BB.z CD5 CAR T cells during expansion. A, Schematic representation of the Tet-OFF expression system. Both 5′ and 3′ LTR elements are mutated to eliminate endogenous promoter activity, and CD5 CAR is encoded downstream of the PTight promoter. Tetracycline transactivator (tTA) is expressed under the constitutive hybrid EF1a/LTR promoter and, in the absence of DOX, activates the PTight promoter and drives CD5 CAR expression. Addition of DOX prevents tTA from activating the promoter and minimizes CD5 CAR expression. B, Activated T cells were transduced with Tet-OFF BB.z CD5 CAR and cultured with or without DOX. Representative histograms show CAR expression in T cells 4 days after transduction. C, Expansion of T cells transduced with Tet-OFF BB.z CD5 CARs with or without DOX, compared with 28.z CD5 CAR T cells or control nontransduced cells. Data are shown as mean ± SD (P = 0.0006 by one-way ANOVA with Bonferroni posttest correction, n = 3). D, Surface phenotype of T cells transduced with 28.z, BB.z, or Tet-OFF BB.z CD5 CAR and expanded in the presence of DOX for 11 days, compared with nontransduced control T cells. Numbers denote the frequency of naïve-like (TN) and central memory (TCM) T cells, and bar graphs show summarized data for CD4+ and CD8+ T cells (mean ± SD, n = 3). E, Tet-OFF BB.z CD5 CAR T cells expanded in the presence of DOX were washed and transferred into cell culture media without DOX. Histogram shows the kinetics of CAR re-expression in T cells after 96 hours of culture. F, Addition of DOX to Tet-OFF BB.z CD5 CAR T cells represses CAR expression after 24 hours of culture. G, Tet-OFF BB.z CD5 CAR T cells were expanded in the presence of DOX for 11 days followed by DOX withdrawal and CAR re-expression. CD5 CAR T cells were then cocultured with CD5+ tumor cell lines Jurkat and CCRF at an effector-to-target ratio of 1:4 for 3 days. Dot plots show representative frequency of GFP+ tumor cells at the end of coculture. Bar graphs show absolute counts of viable tumor cells at the end of coculture (n = 3). Dashed line denotes initial number of tumor cells upon plating. H, Surface phenotype of Tet-OFF BB.z CD5 CAR T cells 7 days after DOX withdrawal, compared with nontransduced and 28. z CAR T cells. Numbers denote the frequency of naïve-like (TN) and central memory (TCM) T cells, similar to D. Data represent three independent experiments.
BB.z Tet-OFF CD5 CAR T cells expand in vivo and exhibit superior antitumor activity
We tested the ability of the BB.z Tet-OFF CAR T cells to protect against systemic T-cell leukemia progression in a mouse xenograft model where NSG mice received a single dose of CAR T cells 3 days after engraftment with Jurkat-FFluc cells (Fig. 7A). We removed Dox from BB.z Tet-OFF CD5 CAR T cells immediately prior to intravenous injection, resulting in minimal CAR expression at the time of injection (Fig. 7B). Despite delayed expression of the BB.z CAR in the Tet-OFF system, these T cells produced superior mouse survival (Fig. 7C) and suppression of leukemia (Fig. 7D) compared with control 28.z CD5 CAR T cells. Restoration of BB.z CAR expression in T cells prior to in vivo administration significantly reduced the ability of CD5 CAR T cells to control the tumor (Supplementary Fig. S11), indicating the need to delay CAR expression following in vivo administration. We observed a significant expansion of BB.z CAR T cells in peripheral blood of tumor-bearing mice (Fig. 7E and F), which correlated with the upregulation of CAR and concomitant downregulation of CD5 (Fig. 7G). These data demonstrate the ability of the Tet-OFF expression system to overcome fratricide and generate functional BB.z CAR T cells in vitro and in vivo.
Figure 7.
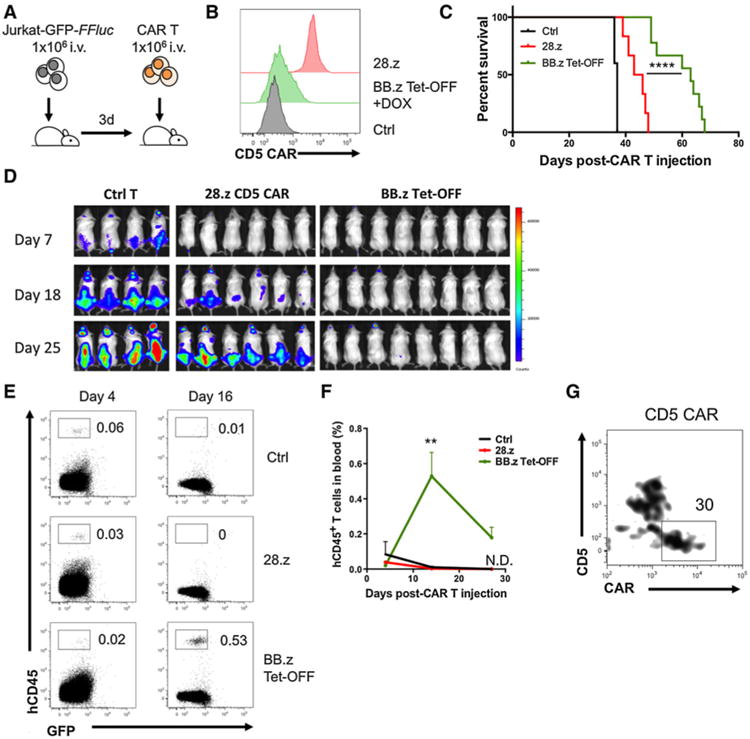
Tet-OFF BB.z CD5 CAR T cells expanded in vivo and protected mice against systemic leukemia. A, Ffluc-expressing GFP+ Jurkat cells were systemically engrafted into NSG mice 3 days before a single intravenous injection of 28.z or Tet-OFF BB.z CD5 CAR T cells. B, Expression of CD5 CAR on T cells before injection. C, Overall survival of animals receiving 28.z or BB.z Tet-OFF CD5 CAR T cells (28.z vs. BB.z, P < 0.0001 by Mantel–Cox test, n = 6–9). D, Development of systemic leukemia in control and CD5 CAR–treated animals. E, T cells transduced with 28.z or BB.z Tet-OFF CD5 CAR were injected into mice 7 days after Jurkat engraftment. Dot plots show expansion of adoptively transferred T cells in peripheral blood at indicated time points after CAR T-cell injection. Numbers indicate relative frequency of human T cells. F, Overall expansion of T cells in peripheral blood of tumor-bearing mice at the indicated time points (P = 0.0046 by one-way ANOVA with Bonferroni posttest correction, n = 3). N.D., not detectable. G, Representative plot showing expression of CD5 and CD5 CAR in Tet-OFF BB.z CD5 CAR T cells 27 days after injection. Cells with upregulated CD5 CAR and downregulated CD5 are gated. Data represent three independent experiments with 3 to 9 animals in each individual experiment.
Discussion
4-1BB costimulation can enhance the in vivo survival and persistence of CAR T cells and may contribute to their antitumor function. In this study, however, we show that when a T-cell malignancy is targeted, the incorporation of 4-1BB or other TNFR-related endodomains into a self-reactive CD5 CAR can also stabilize fratricidal immunologic synapses in a TRAF2- and ICAM-1–dependent manner, decreasing survival of CAR-expressing T cells. We found that a Tet-OFF–inducible retroviral expression system overcomes fratricide by controllably repressing BB.z CAR expression in the presence of doxycycline in vitro while allowing CAR expression and antitumor activity to be restored once transduced T cells are introduced in vivo.
Although both CD28 and 4-1BB endodomains have been used to deliver costimulation in CAR T cells, they have different roles in T-cell antitumor activity and effects on T-cell phenotype. CD28 is expressed by both naïve and effector/memory T cells and is active during the initial priming of naïve T cells (31, 32). CD28 signaling directly activates PI3K signaling, which augments activation of T cells and promotes their differentiation to short-lived cytotoxic effector cells (11, 12). As a result, 28.z CD5 CAR T cells are highly cytotoxic but have limited persistence in mouse xenograft models (17). In contrast to CD28, 4-1BB is upregulated in T cells after initial stimulation, and therefore physiological 4-1BB ligation delivers late costimulatory signals to the activated T cells (31–33). 4-1BB signaling protects T cells against activation-induced cell death (34) and facilitates fatty acid oxidation and increased respiration, enhancing development of central memory T cells (13). We found, however, that constant CAR activation upregulates ICAM-1, which in turn sustains cell contact between CAR T cells and enhances fratricide, thus decreasing expansion of CD5 CAR T cells.
The interaction between ICAM-1 and its receptor LFA-1 promotes T-cell adhesion and intercellular contact. LFA-1 is an integrin expressed by all T cells, and TCR stimulation triggers a conformational change in LFA-1, promoting ligand binding and providing additional costimulation for T cells (35, 36). In contrast, ICAM-1 is mainly present on other cells: expression of ICAM-1 on antigen-presenting cells is critical for efficient T-cell priming and memory differentiation (37–39), whereas upregulation of ICAM-1 on the endothelium facilitates T-cell migration (26, 40). ICAM-1 levels in T cells are usually low, although IL2, IL12, and IFNγ can upregulate its expression (41, 42). Expression of ICAM-1 promotes homotypic T-cell–T-cell interactions, leading to the formation of T-cell clusters that may facilitate paracrine IL2 signaling and regulate T-cell effector functions (42). In our study, we found that expression of BB.z CD5 CARs similarly upregulated ICAM-1 in T cells, which correlated with increased clustering. Although CD5 is rapidly internalized upon ligation (43, 44), our results indicate that the increased intercellular adhesion and continuous CAR signaling promoted formation of immunologic synapses between CD5 CAR T cells manifested in polymerization of actin and recruitment of MTOC. Blocking ICAM-1 with a monoclonal antibody decreased fratricide and augmented survival of BB.z CD5 CAR T cells, indicating increased intercellular adhesion promoted self-elimination.
In addition to 4-1BB, other TNFR superfamily members including OX40 and CD27 have been explored as costimulatory endodomains for CARs. Although 4-1BB costimulates both CD4+ and CD8+ T cells, ligation of OX40 primarily boosts the proliferation and function of CD4+ T cells (33). Integration of both CD28 and OX40 in a GD2-specific CAR enhanced effector function and antitumor activity of GD2 CAR T cells in vitro (45). Similarly, incorporation of the CD27-derived costimulatory endodomain into an FR-specific CAR increased CAR T-cell persistence and antitumor function compared with control T cells expressing 28.z or ζ-only CAR (46). The potential costimulatory function of HVEM and CD30 genes for CARs has not yet been explored. All these TNFR superfamily genes can activate both canonical and noncanonical NF-kB pathways via TRAF2-mediated signaling, albeit by recruiting distinct and partially nonoverlapping sets of TRAF proteins (32, 33). We found that in addition to 4-1BB, other TNFR endodomains also enriched the central memory subpopulation of CD5 CAR T cells, but that incorporation of any of these domains into the CD5 CAR led to similar defects in T-cell expansion associated with upregulation of ICAM-1, highlighting a general mechanism mediated by TRAF2 signaling. These results indicate that 4-1BB and other TNFR superfamily endodomains can be detrimental in CARs specific for antigens that may be expressed in T cells even at a low level. Therefore, the optimal costimulatory endodomain(s) in a CAR may need to be determined both by which population of CAR T cells should be expanded, and the expression pattern of the target antigen.
Several studies have demonstrated the tonic CAR signaling significantly affects T-cell differentiation and antitumor function. Tonic ligand-independent signaling in 28.z GD2 CAR resulted in accelerated terminal differentiation of T cells in vitro and acquisition of an exhausted phenotype (47). In a mouse model, continuous activation through the CD28.z CD19 CAR impaired the ability of T cells to adequately respond to simultaneous TCR stimulation leading to functional deletion of dual-reactive T cells in vivo (48). Although 28.z CD5 CAR T cells also demonstrated enhanced effector differentiation, their cytotoxicity was sustained for at least 3 weeks after stimulation, indicating those cells were not functionally exhausted by the continuous CAR signaling. Therefore, the differential survival of 28.z and BB.z CD5 CAR T cells after CAR transduction was not a result of impaired cytotoxicity of the former cells but rather reflected enhanced fratricide by the latter.
One way to attenuate tonic CAR signaling is to decrease its expression on T cells. We found that a reduction in BB.z CAR expression indeed decreased fratricide of transduced T cells but also compromised their tumor-directed cytotoxicity. Therefore, we created the single-vector gammaretroviral Tet-OFF system to temporally control CAR expression and thus limit both CAR T-cell fratricide and antigen-driven exhaustion. Using this system, CAR expression is suppressed in the presence of the small-molecule doxycycline in vitro but recovers upon introduction of the cells into a doxycycline-free in vivo environment. The Tet-OFF system restores normal expression and function of BB.z CD5 CAR after doxycycline withdrawal, allowing CAR-mediated cytotoxicity and central memory differentiation to be restored to the transduced T cells. Injection of these cells in leukemia-bearing mice protected against systemic leukemia progression resulting in greater T-cell expansion and prolonged suppression of leukemia compared with 28.z CD5 CAR T cells. Re-expression of the BB.z CD5 CAR in vivo did not result in immediate fratricide of CAR T cells, likely due to the excessive dilution of these cells in mouse tissues and competition with malignant blasts for target cells. In addition to enabling normal expansion of CD5 CAR-transduced T cells, this system has several advantages over “always-on” expression systems. Unlike Tet-OFF systems that require cotransduction with two separate vectors (one plasmid containing the transgene and another encoding the transactivator), ours utilizes only one gammaretroviral vector, thus simplifying the manufacturing process and increasing transduction efficiency. Although expression of the synthetic transactivator may promote an immune response against Tet-OFF T cells in humans, it remains to be seen whether the magnitude of this response would limit the expansion or long-term persistence of transgenic T cells.
Taken together, these studies show that TNFR costimulation enhances fratricide of CAR T cells specific for T-cell malignancies. We identified the mechanism of toxicity and described a solution to circumvent this limitation. Our results have direct implications on the design of CARs targeting T-cell antigens for optimal clinical performance.
Supplementary Material
Acknowledgments
This study was supported by a grant from the NIH, NCI (P50 CA126752), by the ASH Scholar Award (to M. Mamonkin), and by a Stand Up To Cancer – St. Baldrick's Pediatric Dream Team Translational Research Grant (SU2C-AACR-DT1113; to M.K. Brenner). Stand Up To Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C. The authors also appreciate the support of shared resources in the Cancer Center (support grant NCIP30CA125123). The imaging work was supported by R01AI067946 (to J.S. Orange).
The authors thank Catherine Gillespie for editing the article.
Footnotes
Authors' Contributions: Conception and design: M. Mamonkin, J.S. Orange, M.K. Brenner
Development of methodology: M. Mamonkin, M. Mukherjee, M. Srinivasan, D. Gomes-Silva, J.S. Orange
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): M. Mamonkin, M. Srinivasan, S. Sharma, F. Mo
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): M. Mamonkin, M. Mukherjee, M. Srinivasan, G. Krenciute, J.S. Orange
Writing, review, and/or revision of the manuscript: M. Mamonkin, M.K. Brenner
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): M. Mamonkin, G. Krenciute, M.K. Brenner
Study supervision: M. Mamonkin, M.K. Brenner
Other (acquisition and analysis of high-resolution microscopic images): M. Mukherjee
Note: Supplementary data for this article are available at Cancer Immunology Research Online (http://cancerimmunolres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest were disclosed.
References
- 1.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor–modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–6. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112:2261–71. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16:1245–56. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev. 2014;257:107–26. doi: 10.1111/imr.12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR–T cells of defined CD4+:CD8+composition in adult B cell ALL patients. J Clin Invest. 2016;126:2123–38. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–28. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor– modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Macintyre AN, Finlay D, Preston G, Sinclair LV, Waugh CM, Tamas P, et al. Protein kinase B controls transcriptional programs that direct cytotoxic T cell fate but is dispensable for T cell metabolism. Immunity. 2011;34:224–36. doi: 10.1016/j.immuni.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. 2012;12:325–38. doi: 10.1038/nri3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boomer JS, Green JM. An enigmatic tail of CD28 signaling. Cold Spring Harb Perspect Biol. 2010;2:a002436. doi: 10.1101/cshperspect.a002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44:380–90. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 14.Hombach A, Heuser C, Sircar R, Tillmann T, Diehl V, Pohl C, et al. An anti-CD30 chimeric receptor that mediates CD3-ζ-independent T-cell activation against Hodgkin's lymphoma cells in the presence of soluble CD30. Cancer Res. 1998;58:1116–9. [PubMed] [Google Scholar]
- 15.Savoldo B, Rooney CM, Stasi AD, Abken H, Hombach A, Foster AE, et al. Epstein Barr virus–specific cytotoxic T lymphocytes expressing the anti-CD30z artificial chimeric T-cell receptor for immunotherapy of Hodgkin disease. Blood. 2007;110:2620–30. doi: 10.1182/blood-2006-11-059139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chu J, Deng Y, Benson DM, He S, Hughes T, Zhang J, et al. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia. 2014;28:917–27. doi: 10.1038/leu.2013.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mamonkin M, Rouce RH, Tashiro H, Brenner MK. A T-cell–directed chimeric antigen receptor for the selective treatment of T-cell malignancies. Blood. 2015;126:983–92. doi: 10.1182/blood-2015-02-629527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krenciute G, Krebs S, Torres D, Wu MF, Liu H, Dotti G, et al. Characterization and functional analysis of scFv-based chimeric antigen receptors to redirect T cells to IL13Rα2-positive glioma. Mol Ther. 2016;24:354–63. doi: 10.1038/mt.2015.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banerjee PP, Orange JS. Quantitative measurement of F-actin accumulation at the NK cell immunological synapse. J Immunol Methods. 2010;355:1–13. doi: 10.1016/j.jim.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mentlik AN, Sanborn KB, Holzbaur EL, Orange JS. Rapid lytic granule convergence to the MTOC in natural killer cells is dependent on dynein but not cytolytic commitment. Mol Biol Cell. 2010;21:2241–56. doi: 10.1091/mbc.E09-11-0930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jang IK, Lee ZH, Kim YJ, Kim SH, Kwon BS. Human 4-1BB (CD137) signals are mediated by TRAF2 and activate nuclear factor-κB. Biochem Biophys Res Commun. 1998;242:613–20. doi: 10.1006/bbrc.1997.8016. [DOI] [PubMed] [Google Scholar]
- 22.Pomerantz JL, Baltimore D. NF-kappaB activation by a signaling complex containing TRAF2, TANK and TBK1, a novel IKK-related kinase. EMBO J. 1999;18:6694–704. doi: 10.1093/emboj/18.23.6694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.So T, Soroosh P, Eun SY, Altman A, Croft M. Antigen-independent signalosome of CARMA1, PKCθ, and TNF receptor-associated factor 2 (TRAF2) determines NF-κB signaling in T cells. Proc Natl Acad Sci USA. 2011;108:2903–8. doi: 10.1073/pnas.1008765108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dustin ML, Cooper JA. The immunological synapse and the actin cytoskeleton: molecular hardware for T cell signaling. Nat Immunol. 2000;1:23–9. doi: 10.1038/76877. [DOI] [PubMed] [Google Scholar]
- 25.Billadeau DD, Nolz JC, Gomez TS. Regulation of T-cell activation by the cytoskeleton. Nat Rev Immunol. 2007;7:131–43. doi: 10.1038/nri2021. [DOI] [PubMed] [Google Scholar]
- 26.Springer TA. Adhesion receptors of the immune system. Nature. 1990;346:425–34. doi: 10.1038/346425a0. [DOI] [PubMed] [Google Scholar]
- 27.Schmits R, Kündig TM, Baker DM, Shumaker G, Simard JJ, Duncan G, et al. LFA-1-deficient mice show normal CTL responses to virus but fail to reject immunogenic tumor. J Exp Med. 1996;183:1415–26. doi: 10.1084/jem.183.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roebuck KA, Finnegan A. Regulation of intercellular adhesion molecule-1 (CD54) gene expression. J Leukoc Biol. 1999;66:876–88. doi: 10.1002/jlb.66.6.876. [DOI] [PubMed] [Google Scholar]
- 29.Melotti P, Nicolis E, Tamanini A, Rolfini R, Pavirani A, Cabrini G. Activation of NF-kB mediates ICAM-1 induction in respiratory cells exposed to an adenovirus-derived vector. Gene Ther. 2001;8:1436–42. doi: 10.1038/sj.gt.3301533. [DOI] [PubMed] [Google Scholar]
- 30.Gomes-Silva D, Mukherjee M, Srinivasan M, Krenciute G, Dakhova O, Zheng Y, et al. Tonic 4-1BB costimulation in chimeric antigen receptors impedes T cell survival and is vector-dependent. Cell Rep. 2017;21:17–26. doi: 10.1016/j.celrep.2017.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharpe AH. Mechanisms of costimulation. Immunol Rev. 2009;229:5–11. doi: 10.1111/j.1600-065X.2009.00784.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–42. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watts TH. Tnf/Tnfr family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 34.Hernandez-Chacon JA, Li Y, Wu RC, Bernatchez C, Wang Y, Weber J, et al. Co-stimulation through the CD137/4-1BB pathway protects human melanoma tumor-infiltrating lymphocytes from activation-induced cell death and enhances anti-tumor effector function. J Immunother. 2011;34:236–50. doi: 10.1097/CJI.0b013e318209e7ec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kinashi T. Intracellular signalling controlling integrin activation in lymphocytes. Nat Rev Immunol. 2005;5:546–59. doi: 10.1038/nri1646. [DOI] [PubMed] [Google Scholar]
- 36.Dustin ML, Springer TA. T-cell receptor cross-linking transiently stimulates adhesiveness through LFA-1. Nature. 1989;341:619–24. doi: 10.1038/341619a0. [DOI] [PubMed] [Google Scholar]
- 37.Parameswaran N, Suresh R, Bal V, Rath S, George A. Lack of ICAM-1 on APCs during T cell priming leads to poor generation of central memory cells. J Immunol. 2005;175:2201–11. doi: 10.4049/jimmunol.175.4.2201. [DOI] [PubMed] [Google Scholar]
- 38.Cox MA, Barnum SR, Bullard DC, Zajac AJ. ICAM-1-dependent tuning of memory CD8 T-cell responses following acute infection. Proc Natl Acad Sci. 2013;110:1416–21. doi: 10.1073/pnas.1213480110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Camacho SA, Heath WR, Carbone FR, Sarvetnick N, LeBon A, Karlsson L, et al. A key role for ICAM-1 in generating effector cells mediating inflammatory responses. Nat Immunol. 2001;2:523–9. doi: 10.1038/88720. [DOI] [PubMed] [Google Scholar]
- 40.Dustin ML, Rothlein R, Bhan AK, Dinarello CA, Springer TA. Induction by IL 1 and interferon-gamma: tissue distribution, biochemistry, and function of a natural adherence molecule (ICAM-1) J Immunol. 1986;137:245–54. [PubMed] [Google Scholar]
- 41.Buckle AM, Hogg N. Human memory T cells express intercellular adhesion molecule-1 which can be increased by interleukin 2 and interferon-gamma. Eur J Immunol. 1990;20:337–41. doi: 10.1002/eji.1830200216. [DOI] [PubMed] [Google Scholar]
- 42.Zumwalde NA, Domae E, Mescher MF, Shimizu Y. ICAM-1-dependent homotypic aggregates regulate CD8 T cell effector function and differentiation during T cell activation. J Immunol. 2013;191:3681–93. doi: 10.4049/jimmunol.1201954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shawler DL, Johnson DE, McCallister TJ, Bartholomew RM, Dillman RO. Mechanisms of human CD5 modulation and capping induced by murine monoclonal antibody T101. Clin Immunol Immunopathol. 1988;47:219–29. doi: 10.1016/0090-1229(88)90074-8. [DOI] [PubMed] [Google Scholar]
- 44.Lu X, Axtell RC, Collawn JF, Gibson A, Justement LB, Raman C. AP2 Adaptor complex-dependent internalization of CD5: differential regulation in T and B cells. J Immunol. 2002;168:5612–20. doi: 10.4049/jimmunol.168.11.5612. [DOI] [PubMed] [Google Scholar]
- 45.Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12:933–41. doi: 10.1016/j.ymthe.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 46.Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood. 2012;119:696–706. doi: 10.1182/blood-2011-03-344275. [DOI] [PubMed] [Google Scholar]
- 47.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21:581–90. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ghosh A, Smith M, James SE, Davila ML, Velardi E, Argyropoulos KV, et al. Donor CD19 CAR T cells exert potent graft-versus lymphoma activity with diminished graft-versus-host activity. Nat Med. 2017;23:242–9. doi: 10.1038/nm.4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.