

Abstract
A longstanding research goal has been to understand the nature and role of copper–oxygen intermediates within copper-containing enzymes and abiological catalysts. Synthetic chemistry has played a pivotal role in highlighting the viability of proposed intermediates and expanding the library of known copper–oxygen cores. In addition to the number of new complexes that have been synthesized since the previous reviews on this topic in this journal (Mirica, L. M.; Ottenwaelder, X.; Stack, T. D. P. Chem. Rev. 2004, 104, 1013–1046 and Lewis, E. A.; Tolman, W. B. Chem. Rev. 2004, 104, 1047–1076), the field has seen significant expansion in the (1) range of cores synthesized and characterized, (2) amount of mechanistic work performed, particularly in the area of organic substrate oxidation, and (3) use of computational methods for both the corroboration and prediction of proposed intermediates. The scope of this review has been limited to well-characterized examples of copper–oxygen species but seeks to provide a thorough picture of the spectroscopic characteristics and reactivity trends of the copper–oxygen cores discussed.
Graphical Abstract
1. INTRODUCTION
Understanding how oxygenations and oxidations of organic molecules operate and developing new selective, green, and efficient methods to perform these transformations are central goals in chemical research.1–4 Such reactions are critically important in myriad processes, including metabolism, synthesis of useful organic compounds, and energy-related conversions. Metal ions play a privileged role as oxygenation and oxidation reagents and catalysts, largely through their ability to activate O2 and to generate structurally intriguing metal–oxygen species that can have the ability to attack even the strongest C–H bonds. Copper ions are particularly prevalent in enzymes,5,6 heterogeneous catalysts,7,8 and soluble reagents9–11 that oxidize organic molecules, and they are also involved in four-electron processes that interconvert O2 and H2O.12–14 A rich variety of mechanisms have been postulated for these systems, which may contain one or more copper ions that generate structurally diverse intermediates. Longstanding goals have been to comprehend these mechanisms, determine the geometries and electronic structures of the key intermediates, and unravel structure/function relationships for the catalytic centers, ultimately to enable the design of new and more selective and/or reactive oxidation catalysts.
A particularly valuable strategy for reaching these goals involves the synthesis, characterization, and detailed evaluation of the reactivity and mechanisms of reactions of discrete molecules that contain copper–oxygen moieties. In this review, we survey recent studies that use this strategy and that have provided unique and fundamental insights into possible structures, properties, and reactivities of copper–oxygen intermediates involved in oxygenation and oxidation reactions in both biological and abiological systems. As two previous comprehensive reviews on the subject were published in this journal in 2004,15,16 we focus on work that has appeared since then, and through August 2016. The reader also is pointed to a number of more narrowly targeted but useful reviews or accounts on this subject that have appeared since 2004.4,9,12,14,17–39
In this section, we set the stage for discussion of the synthetic work by briefly surveying various proposals for copper–oxygen intermediates in biology and in abiological catalysts. The subsequent discussion is organized by the copper ion nuclearity of the synthetic compounds (sections 2–4). The supporting ligands and their abbreviations discussed in all the sections are provided in Charts 1, 2, 3, 4, and 5, organized according to the number and type of donors they contain.
Chart 1.

Ligands Containing Two Nitrogen Donors
Chart 2.

Ligands Containing Three Nitrogen Donors
Chart 3.

Ligands Containing Four Nitrogen Donors
Chart 4.

Ligands Containing Five or More Nitrogen Donors
Chart 5.

Ligands Containing a Mixture of Nitrogen, Sulfur, and Oxygen Donors
1.1. Proposed Copper–Oxygen Intermediates in Biology
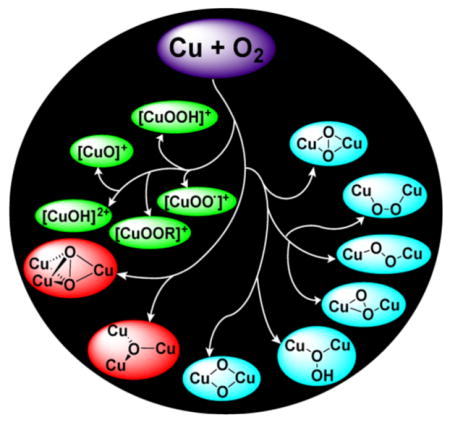
Much of the research on synthetic copper–oxygen compounds is inspired by postulates for active site intermediates and mechanisms in enzymes. A recent comprehensive review describes these enzymes and their copper-containing active sites in detail,5 so here we only briefly summarize some of the proposed copper–oxygen motifs and key issues that have guided synthetic modeling work (Figure 1).
Figure 1.

Proposed copper–oxygen intermediates involved in the reactions of the indicated enzymes. (a) and (b) Superoxo, hydroperoxo, and oxyl intermediates proposed for the monocopper sites in the indicated enzymes, R = H or Me. (c) Possible equilibrium between putative substrate-bound intermediates, either of which could undergo electrophilic attack at the substrate to yield a catecholate species in the monooxygenase reaction of coupled binuclear polyphenol oxidases such as tyrosinase (N indicates nitrogen donor atom of histidine imidazoles). (d) Selected copper–oxygen intermediates speculated to be responsible for C–H bond attack of substrate by particulate methane monooxygenase. (e) Selected tricopper intermediates proposed for reduction of O2 to H2O by the multicopper oxidases.40 The proximate type 1 Cu electron transfer center is not shown. (f) Two key intermediates proposed for reduction of O2 to H2O by the Fe–Cu core of cytochrome c oxidase.
Monocopper species have been proposed as intermediates in hydroxylations catalyzed by dopamine and tyramine β-monooxygenases (DβM and TβM),41–44 peptidylglycine α-hydroxylating monooxogyenase (PHM),45,46 and the more recently characterized lytic polysaccharide monooxygenase (LPMO) (Figure 1a and b).47–51 In DβM, TβM, and PHM, the copper coordination sphere includes two histidine imidazolyls and a methionine thioether, whereas in LPMO a histidine imidazolyl and a “histidine brace” comprising a histidine imidazolyl and the amine terminus of the peptide chain are bound to the active site metal ion. This same “histidine brace” has also been identified in particulate methane monooxygenase (pMMO).52 For all of the monocopper systems, reaction of a Cu(I) form with O2 is proposed to yield a copper(II)-superoxo adduct (X = OO•−).53 Such an adduct has been characterized by X-ray crystallography in an oxygenated precatalytic PHM enzyme complex54 and has been proposed to attack the C–H bond of substrate, primarily on the basis of kinetic data obtained for DβM and PHM.41 The presumed product is a copper(II)-hydroperoxide (X = OOH; also written as [CuOOH]+).55,56 This latter species could also be formed from the superoxo complex by addition of a proton from the medium and an electron from a redox site. Alternatively, a copper(II)-hydroperoxide might also be capable of attacking the substrate, either directly or after O–O bond scission to yield a copper(II)-oxyl (X = O•; also written as [CuO]+). Computational studies aimed at evaluating the feasibility of these intermediates and their ability to attack a substrate C–H bond have indicated that the [CuO]+ unit, best described as having a triplet ground state with a Cu(II) ion weakly bonded to an O-centered radical,17,57 is the least stable species and is the most potent oxidant.58–61 These various ideas concerning the mechanism of substrate attack by the monocopper enzyme sites and the structures of the putative intermediates have inspired numerous attempts to synthesize complexes with the Cu–X (X = OO•−, OOH, O•) cores, and related species, and to understand their properties and reactivities (section 2).
In the coupled binuclear polyphenol oxidases (CB–PPOs, of which tyrosinase and catechol oxidase are the most studied), it is proposed that the substrate binds to the oxy form of the enzyme to generate the “peroxo” intermediate shown in Figure 1c. The μ-η2:η2-peroxo binding mode shown in this intermediate has been conclusively identified by X-ray crystallography in the oxy forms of the O2 binding protein hemocyanin62 and in tyrosinase63 and catechol oxidase,64 as well as by spectroscopy in other enzymes.65 Attack at the substrate by the (μ-η2:η2-peroxo)dicopper intermediate in tyrosinase is a mechanistic paradigm.5,32,66,67 Yet, the elucidation of a facile equilibrium between (μ-η2:η2-peroxo)dicopper and bis(μ-oxo)dicopper cores in synthetic complexes68,69 provides precedence for the postulate of a similar equilibrium in the CB-PPOs. Even though a bis(μ-oxo) species has not been observed in any enzyme, it may still be formed as a transient reactive intermediate, which raises a key question: which core is responsible for the electrophilic attack at the coordinated phenol substrate, in particular to result in hydroxylation of the aromatic ring? This and related questions have stimulated extensive research aimed at understanding the reactivities of complexes that contain the μ-η2:η2-peroxo and bis(μ-oxo) cores (section 3).
This research has also been driven by hypotheses about the involvement of the (μ-η2:η2-peroxo)- and bis(μ-oxo)dicopper cores in particulate methane monooxygenase (pMMO).70–76 Other dicopper species have also been suggested (Figure 1d), in large part stimulated by the identification by X-ray crystallography and EXAFS of a dicopper site in the enzyme.29,77–80 These species include triplet75 or mixed-valent Cu(II)Cu(III)72 variants of the bis(μ-oxo)dicopper core, a (μ-oxo)dicopper(II) unit akin to what has been proposed in Cu-doped zeolite catalysts (see section 1.2),81–83 and dicopper units that incorporate a copper(II)-oxyl moiety.77 Alternative hypotheses of mono-84 and tricopper catalytic sites in pMMO have also been advanced, and proposals of additional tricopper reactive intermediates such as that shown in Figure 1d have been made.85,86 In view of the tentative understanding of the nature of the pMMO active site and the mechanism(s) by which the strong C–H bond of methane is attacked, along with the significance of the reaction it catalyzes, much effort continues to be expended to develop models of the various proposed pMMO di- and tricopper active site intermediates and to evaluate their reactivity (sections 3 and 4).
Tricopper intermediates are involved in the complete 4-electron reduction of O2 to H2O catalyzed by the large and biologically important class of multicopper oxidases, which include laccase, ascorbate oxidase, ceruloplasmin, bilirubin oxidase, cuprous oxidase, and others.5,40 Extensive spectroscopic and computational studies of these enzymes have led to the postulate of two key “peroxo” and “native” intermediates along the 4-electron dioxygen reduction pathway (Figure 1e).5,40 The importance of the oxygen reduction reaction (ORR) (cf. for fuel cell applications)87 and the novel structures proposed for the various enzyme intermediates have inspired efforts to construct multicopper model complexes, as described in section 4.
The ORR is also catalyzed by cytochrome c oxidase (CcO, a member of a broader class of heme copper oxidases), which is the terminal mitochondrial component of the respiratory chain that uses the energy supplied by the ORR to pump protons across the cellular membrane and fuel adenosine triphosphate (ATP) synthesis.5,88,89 The binding and reduction of O2 to H2O by CcO, with avoidance of H2O2 production, occurs at a heterobimetallic active site comprising a heme adjacent to a copper center bound to three histidyl imidazoles, one of which is linked via a post-translational modification to a tyrosine residue. Key proposed intermediates include a peroxo species potentially coordinated to both iron and copper, as well as “PM”, in which the O–O bond is broken and the Fe, Cu, and tyrosine moieties are oxidized (Figure 1f). Approaches toward understanding the detailed mechanism of the ORR by CcO and how partial reduction to yield H2O2 are avoided include targeting reactive heme-copper oxygen species for synthesis and characterization as well as using electrochemical methods to evaluate catalysis in model complexes. The results of such approaches have been reviewed extensively elsewhere, so will not be described herein.12,21,24,90–95
In addition to the multitude of fascinating copper–oxygen motifs proposed as intermediates in enzymes, copper–oxygen species have also been hypothesized to be involved in the generation of “reactive oxygen species” (ROS) by copper complexes targeted as metallodrugs and nucleases.96,97 In most cases, copper–oxygen intermediates have not been identified as distinct intermediates in ROS generation, but data in support of the “intermediacy of a ROS that is intimately bound to the copper center”98 has been presented for copper bound to the amino terminal Cu(II)- and Ni(II)-binding (ATCUN) peptide motif.99–101 The nature of such “intimately bound” ROS/copper species is not known. Copper-promoted generation of ROS has also been implicated in many neurodegenerative diseases,102–105 but we are unaware of experimental evidence for specific copper–oxygen intermediates in these processes. Nonetheless, information gleaned from studies of synthetic copper–oxygen complexes may inform understanding of ROS generation mechanisms by a variety of copper species in a biological context.
1.2. Proposed Copper–Oxygen Intermediates in Abiological Catalysis
Copper–oxygen intermediates akin to those postulated for enzymes may also be involved in oxidations of organic substrates by synthetic catalysts.9,10 In most cases, however, evidence for such intermediates in oxidations is sparse or nonexistent, or pathways involving aerial oxidation of Cu(I) to Cu(II) species are invoked that do not specify the nature of any copper– oxygen species involved.106,107 We note here just a few key examples where experimental support for copper–oxygen intermediates during an oxidation reaction has been provided and/or particularly provocative hypotheses for copper–oxygen intermediates are proposed on the basis of theory.
Particular attention has been focused on the mechanism of the selective oxidation of methane to methanol by copper sites in zeolites.81,82,108–110 An early proposal111 invoking a bis(μ-oxo)dicopper species as being responsible for attacking the strong C–H bond of methane has been supplanted on the basis of extensive spectroscopic data by the hypothesis of a dicopper(II) species with a single oxo bridge82,112–114 that is derived from a μ-η2:η2-peroxo precursor.115 DFT calculations support the notion that the (μ-oxo)dicopper species abstracts a hydrogen atom from substrate.112 A driving force is the formation of a strong O–H bond (calculated to be 90 kcal/mol) to yield the [Cu2(μ-OH)]2+ product, although the reaction step was found to be endothermic by 13.8 kcal/mol. It was further proposed that approach of methane to the oxo-bridged dicopper(II) moiety along the reaction coordinate is accompanied by changes in low-lying singly occupied molecular orbitals, essentially inducing formation of a novel mixed valent oxyl radical species with significant p orbital character on the bridging O atom oriented to facilitate hydrogen atom abstraction from the substrate (Figure 2a). The role of water in methane oxidation by Cu in zeolites has been evaluated by experiment and theory,114,116,117 and suggested to play multiple roles, including to change the nature of the active site structure. More recent theoretical work led to the proposal of a pathway invoking peroxo and terminal hydroxo and oxyl intermediates (Figure 2b).83 An alternative [Cu3(μ-O)3]2+ core has been proposed in mordenite (Figure 2c).118 While formally a mixed valent species (Cu(III)2Cu(II)), the cluster was described as having all Cu(II) ions with radical character on the O atoms on the basis of DFT calculations. In contrast, a monocopper [CuOH]+ species has been suggested to be the oxidant in so-called 8-membered ring zeolites.119 Clearly, the mechanism(s) of O2 activation and methane hydroxylation are controversial, providing much impetus for investigation of putative dicopper species through synthetic modeling approaches.
Figure 2.
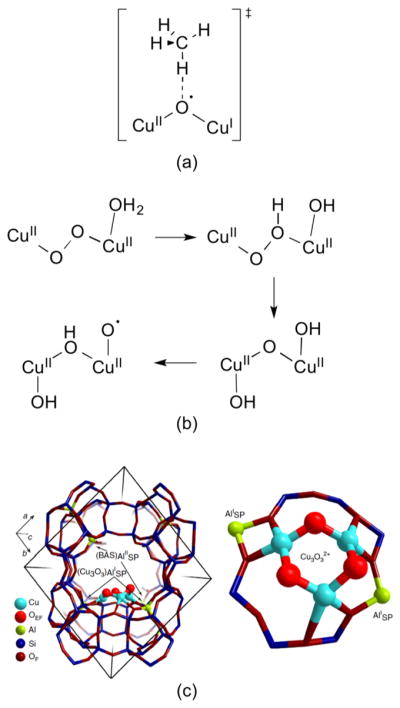
(a) Proposed transition state for hydrogen atom abstraction from methane by the (μ-oxo)dicopper core of Cu-ZSM-5 (ref 112). (b) Alternative O2 activation pathway calculated by DFT for Cu-ZSM-5 (ref 115). (c) Structure and location of [Cu3(μ-O)3]2+ core in mordenite. Reprinted with permission from ref 118. Copyright 2015 Nature Publishing Group.
Another illustrative example of a copper-catalyzed oxidation reaction for which intriguing intermediates are proposed is the hydroxylation of benzoate derivatives (Figure 3).120,121 DFT calculations employed to analyze this process suggested that homolytic scission of the N–O bond in a copper(II) complex of trimethylamine-N-oxide (TMAO) yielded a copper(II)-oxyl intermediate.121 A concerted pathway for hydroxylation of the aromatic ring by this intermediate was found to be favored relative to a stepwise hydrogen atom abstraction/rebound process. Copper(II)-oxyl species have also been proposed in other catalytic reactions. For example, on the basis of DFT calculations such a unit has been suggested to be the active oxidant in the oxidation of alkanes by H2O2 catalyzed by tris(pyrazolyl)hydroborate-based copper complexes.122 These and other examples of copper-catalyzed oxidations for which copper–oxygen species are postulated serve as yet more impetus for studies aimed at understanding the properties of copper–oxygen complexes.
Figure 3.
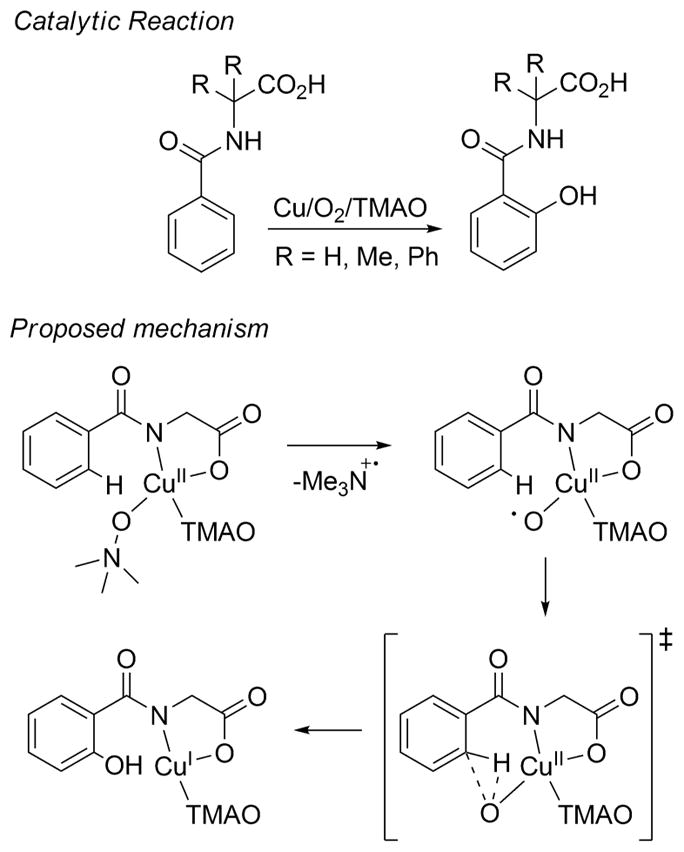
Overall catalytic reaction and proposed mechanism for the hydroxylation of benzoate derivatives (TMAO is trimethylamine-N-oxide) (ref 121).
2. MONOCOPPER COMPOUNDS
In this section, we focus on work reported since 2004 on preparing, characterizing, and understanding the reactivity of mononuclear copper–oxygen complexes. The discussion is divided into three parts: 1:1 Cu:O2 complexes, copper(II) alkyl/hydroperoxide complexes, and high valent [CuO]+/[CuOH]2+ species.
2.1. 1:1 Cu:O2 Complexes
Complexes comprising a copper ion bound to an O2n− unit (n = 1 or 2) model the initial adduct formed upon reaction of Cu(I) biosites with O2 (Figure 4). Such complexes have been prepared by exposure of solutions of Cu(I) complexes to dioxygen or by reaction of a superoxide salt with a Cu(II) precursor, with both types of procedures typically performed at low temperatures in organic solvent. The complexes vary with respect to the way in which the O2n− unit binds (end-on, η1, versus side-on, η2) and the degree of electron transfer from the copper ion to the O2 moiety, with (superoxo)copper(II) and (peroxo)copper(III) representing the two extreme formulations. In many cases, the 1:1 Cu:O2 complexes are observed only as transient intermediates that convert to or interconvert rapidly with dicopper species (section 3). Key research goals have been to elucidate how supporting ligands influence the structural attributes of the 1:1 Cu:O2 adducts and to understand structure/reactivity correlations (See Note Added in Proof).123,124
Figure 4.
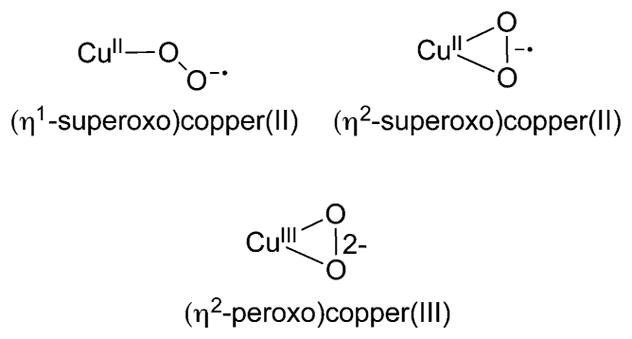
1:1 Cu:O2 core structures.
2.1.1. Structures and Properties
Prior to 2004, only three examples of isolable 1:1 Cu:O2 complexes had been described, with two having been characterized by X-ray crystallography (1a and 3b, Figure 5). Compounds 1, 3, and 4 exhibit side-on (η2) binding of the O2n− fragment. Subsequently, the first X-ray crystal structure of an end-on (η1) (superoxo)copper(II) complex was reported (2),125,126 and a number of other 1:1 Cu:O2 complexes have been described.54,61,123,126–143 The properties of the 1:1 Cu:O2 adducts that have been isolated to date are summarized in Tables 1 and 2 (with several reported earlier than 2004 included for purposes of comparison and discussion).144–151
Figure 5.
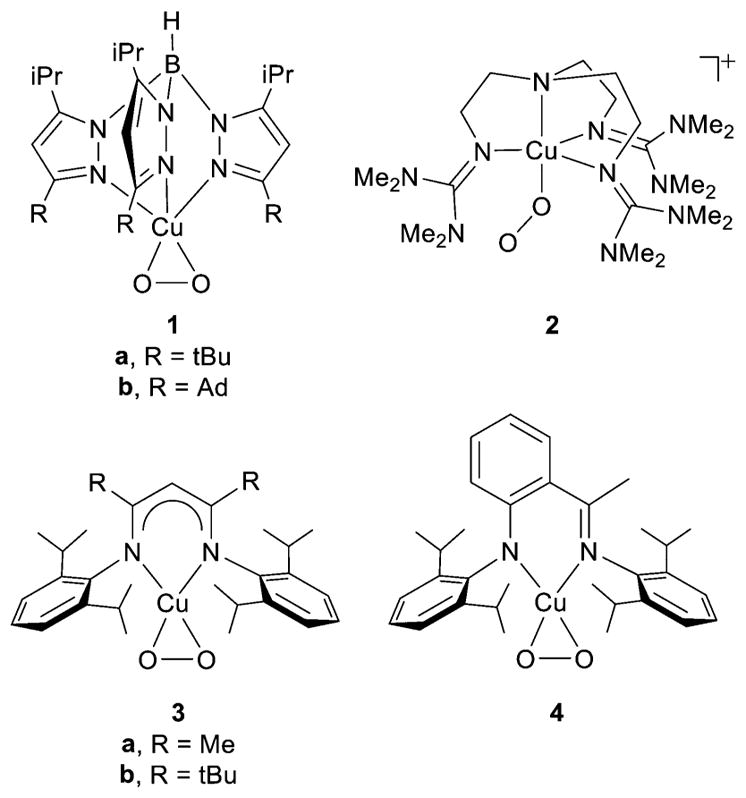
1:1 Cu:O2 adducts defined by X-ray crystallography, supported by ligands L39a,b (1a,b), L44 (2), L2d,e (3a,b), or L3b (4). Reprinted from ref 123. Copyright 2007 American Chemical Society.
Table 1.
Selected Spectroscopic Properties of 1:1 Cu:O2 Complexes
| O2n− hapticity
|
ligand
|
UV–vis
|
Raman (exp, cm−1)
|
Raman (calc, cm−1)
|
ref
|
|
|---|---|---|---|---|---|---|
| λmax (nm) (ε, M−1 cm−1) | ν(O–O) (Δ18O) | ν(Cu–O) (Δ18O) | ν(O–O) (Δ18O) | |||
| η1 | L42a | 412 (480) | 1122 | – | – | 144 |
| η1 | L44 | 444 (3500) | 1117 (28) | 435 (20) | 1218 (32) | 126, 132, 133 |
| η1 | L44 + CF3CO2H | 382 (2600) | 1149 (65) | – | – | 123, 134 |
| η1 | L42b | 434 (3850) | 1096 (67) | 459 (17) | – | 129, 135 |
| η1 | L42d | 416 (5400) | 1120 (61) | 474 (20) | – | 145 |
| η1 | L41b | 418 (4300) | 1121 (63) | 472 (20) | – | 136 |
| η1 | L41c | 409 (4250) | 1121 (63) | 474 (18) | – | 127 |
| η1 | L41d | 410 (3700) | 1130 (63) | 482 (20) | – | 130 |
| η1 | L68 | 418 | 1117 (61) | 460 (20) | – | 137 |
| η1 | L41a | – | – | – | 1251 (33) | 132 |
| η1 | L45 | – | – | – | 1279 (29) | 132 |
| η1 | L28a | 627 (1700) | 1104 (60) | – | 1182 (66) | 128 |
| η1 | L33c | 397 (4200) | 1033 (65) | 457 (15) | – | 131 |
| η1 | L75 | 423 (1800) | 964 (55) | – | – | 146 |
| η2 | L39a | 352 (2330) | 1112 (26) | – | 1124 (31) | 132, 147 |
| η2 | L39b | – | 1043 (59) | 554 (20) | 1040 | 138, 148 |
| η2 | L2d | 385 (2400) | 968 (51) | – | – | 149 |
| η2 | L2e | – | 961 (49) | – | 1013 (28) | 132, 150 |
| η2 | L2g | 415 (1780) | 977 (49) | – | – | 139 |
| η2 | L3b | 390 (7600) | 974 (66) | – | 1041 | 123, 140 |
| η2 | L74a | ~395 | 970(45)/992(67) | 489 (14) | – | 141 |
| η2 | L74b | ~395 | 970(45)/992(67) | 494 (14) | – | 141 |
Table 2.
Selected Interatomic Distances in 1:1 Cu:O2 Complexes
| O2n− hapticity
|
ligand
|
distances (exp, Å)
|
distances (calc, Å)
|
ref
|
||
|---|---|---|---|---|---|---|
| Cu–O | O–O | Cu–O | O–O | |||
| η1 | PHM enzyme | 2.11 | 1.23 | – | – | 54 |
| η1 | LPMO enzyme | – | – | 1.98 | – | 142 |
| η1 | L44 | 1.927(2) | 1.280(3) | - | 1.29 | 126, 132 |
| η1 | L41a | – | – | – | 1.28 | 132 |
| η1 | L45 | – | – | – | 1.27 | 132 |
| η2 | L39a | 1.84(1) | 1.22(3) | – | 1.33 | 132, 147 |
| η2 | L39b | – | – | 1.88 | 1.36 | 138 |
| η2 | L4b | – | – | 1.890 | 1.366 | 143 |
| η2 | L2e | 1.821(5) | 1.392(12) | 1.86 | 1.38 | 132, 138, 151 |
| η2 | L3b | 1.826(2) | 1.392(3) | – | – | 140 |
With few exceptions, the adducts share an intense UV–vis feature ~400 nm (ε ~103 M−1cm−1), the irradiation into which results in enhancement of ν(O–O) and ν(Cu–O) in Raman spectra. Thus, it is assigned as an O2n− → Cu ligand to metal charge transfer (LMCT) transition. The ν(O–O) and ν(Cu– O) fall in the range of 950–1200 cm−1 and 430–560 cm−1, respectively. In general, the complexes assigned as having endon (η1) coordination exhibit ν(O–O) > ~1100 cm−1 commonly associated with superoxide, which also holds for the side-on (η2) complexes supported by the L39 ligands (R = tBu or Ad). The low values <1000 cm−1 for the other η2 complexes implicate a significantly reduced O–O bond order, but these values are higher than typically observed for metal-peroxides (~800–850 cm−1).123 The available measured (X-ray crystallography) and calculated O–O bond distances (Table 2) are consistent with the ν(O–O) differences (higher ν(O–O) = shorter O–O distance).
These and other findings suggest that the degree of electron transfer upon binding of O2 varies, which can be understood within the context of two extreme resonance structure formulations, Cu(II)-O2−• versus Cu(III)-O22−. Evaluation of the electronic structures of several of the adducts (particularly the structurally defined complexes 1–4) has incorporated results from application of Badger’s rule (ν(O–O)/O–O distance relationship), spectroscopy, the oxygen equilibrium isotope effect for O2 binding, and theory.123,138,152,153 From these studies, a bonding picture has evolved of a continuum between the extreme resonance structures with the position on the continuum being determined by the electron-donating power and denticity of the supporting ligands. For example, Badger’s rule plots of O–O distance versus 1/ν2/3 showed good correlations for experimental and calculated data for compounds with a variety of metals and O2n− binding modes, with the only exceptions being a few cases where librational motion led to underestimation of the O–O bond distance determined by X-ray crystallography (including for 1).123,154 The spread of data across O–O between ~1.28–1.39 Å is consistent with O2n− assignments having both integer and noninteger values of n between ~1–2 (i.e., continuum of values).
Complex 2 represents a paradigm for compounds formulated as η1-Cu(II)-O2−• species. NMR132 and variable-temperature variable-field MCD data133 indicated that 2 has a triplet (S = 1) ground state, as determined similarly for the η1-Cu(II)-O2−• species supported by the tren ligand L42b.135 The data for 2 were analyzed and interpreted using DFT calculations, leading to a description involving two singly occupied orthogonal orbitals, one nonbonding orbital localized on the O2n− moiety (π*v) and the other an antibonding orbital with similar Cu and O character (dz2, Figure 6).133 In accordance with TD-DFT calculations, the LMCT band corresponds to the transition from the highest occupied π*σ to the dz2 orbital. More accurate quantum chemical calculations using completely renormalized coupled-cluster theory or multiconfigurational methods led to further understanding of the biradical and multideterminental nature of the η1-Cu(II)-O2−• moiety and a somewhat different orbital description.143 An 18O equilibrium isotope effect of 1.0148 was measured and noted to be larger than those reported for other η1-O2n− adducts in hemes and cobalt compounds (1.0041–1.0066).155,156 The results were interpreted to be consistent with weak covalency in the Cu(II)-O2−• interaction and increased ionic character in the valence bond description.132
Figure 6.
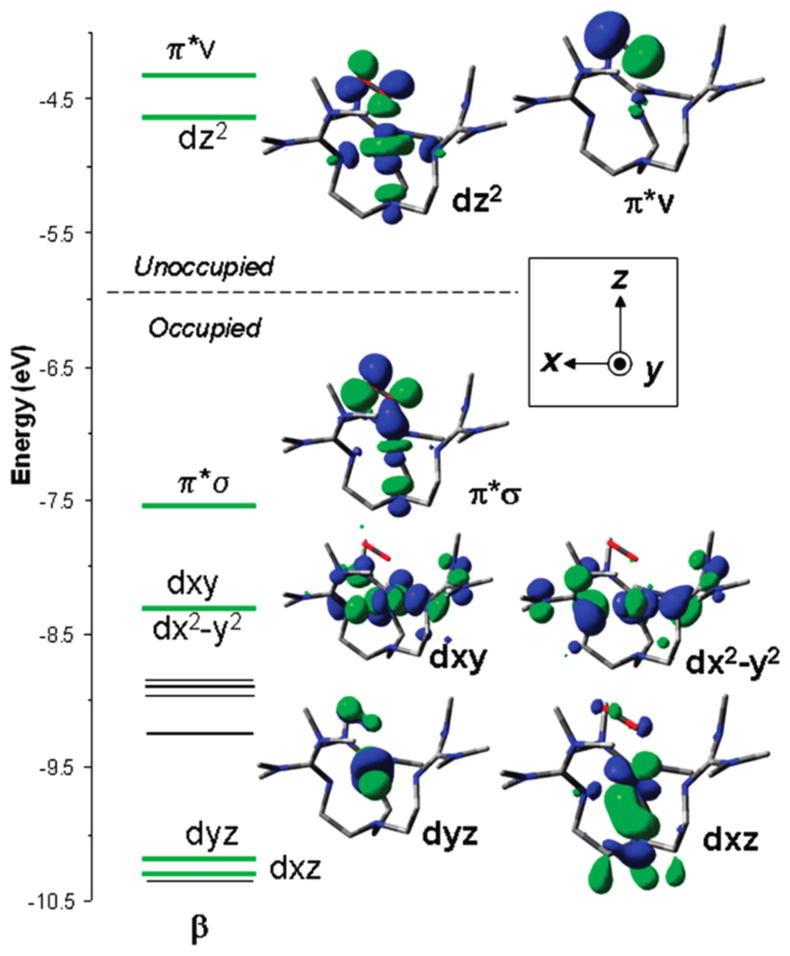
DFT calculated spin down (β) MO diagram of 2. Reprinted from ref 133. Copyright 2010 American Chemical Society.
Intriguing perturbations to the properties of 2, as well as its reactivity (section 2.1.2), were found upon reaction with CF3CO2H.134 The formation of a 1:1 adduct 2·CF3CO2H was reflected by a 62 nm (3655 cm−1) blue shift of the LMCT transition that was reversed by addition of base. The adduct exhibits a ν(O–O) ~30 cm−1 higher than 2, which was unchanged when CF3CO2D was used. NMR and XAS data indicated similar triplet ground states and coordination geometries in 2 and 2·CF3CO2H. Together, the experimental data and accompanying DFT calculations supported the structure for the adduct shown in Figure 7. To rationalize the finding from DFT calculations that H-bonding to the distal oxygen in this model lengthens the O–O bond and lowers ν(O–O) (opposite of experiment), it was proposed that the observed properties of the adduct arose from “the electrostatic interaction with the dipole of CF3CO2H and not a change in orbital covalency imparted by the hydrogen bond.”134
Figure 7.
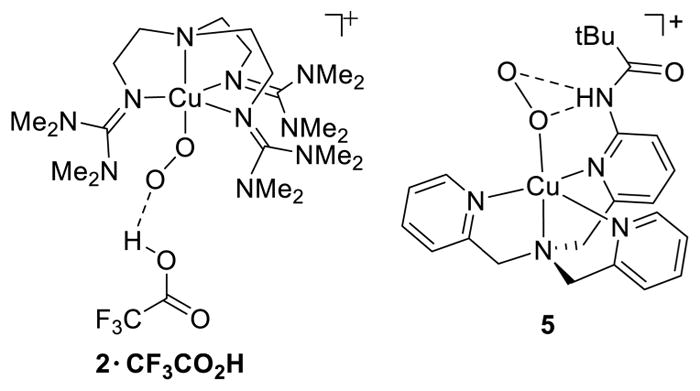
Proposed hydrogen bonding interactions in 2·CF3CO2H and 5 (refs 134 and 130) supported by ligands L44 and L41d, respectively.
The influences of hydrogen bonding on the properties of the η1-Cu(II)-O2−• unit have also been explored in complexes comprising the tris(pyridylmethyl)amine (L41a) ligand frame. While an earlier reported X-ray structure157 purporting to identify intramolecular hydrogen bonding to the [CuO2]+ unit in a complex of L41l was found to be in error,158 complex 5 (Figure 7) was conclusively identified on the basis of UV–vis and resonance Raman spectroscopy.130 Values of 1130 and 482 cm−1 for ν(O–O) and ν(Cu–O), respectively, that are greater than observed in other complexes of L41a derivatives were interpreted using DFT calculations to indicate hydrogen bonding to both the proximal and distal oxygen atoms of the bound superoxide ligand. Importantly, these interactions stabilize the complex sufficiently to enable spectroscopic characterization and reactivity studies (section 2.1.2).
Another η1-Cu(II)-O2−• species with atypical properties is [K(18-crown-6)][(L28a)CuO2] (6).128 While exhibiting a ν(O–O) of 1104 cm−1 consistent with other η1 superoxides, the LMCT absorption feature (assigned by TD-DFT calculations) was at 627 nm, a significantly longer wavelength than all other examples (Table 1). It is likely that the anionic nature of the complex that is reflected in nucleophilic, rather than the typical electrophilic, reactivity of the superoxide moiety (section 2.1.2) underlies the low energy of its LMCT band.
Low ν(O–O) values of 964 and 1033 cm−1 were observed for [CuII(L75)(O2−•) (NEt3)]146 and the adduct supported by L33,131 respectively, both of which were postulated to feature η1 binding of their superoxide ligands. Reasons for these disparities from the norm are unclear, although the similarity of ν(O–O) of [CuII(L75)(O2−•) (NEt3)] to those associated with some η2 complexes could indicate that its assignment as an η1 complex may be incorrect.
Turning next to the smaller set of complexes that exhibit η2-coordination of the O2n− unit, it is here that ligand structural differences have been shown to most significantly influence the degree of charge transfer from the copper ion to the bound O2n− unit. These effects have been most clearly defined in comparisons between 1 versus 3 and 4.123,138,151 All three have singlet ground states, but clear differences in their ν(O–O) values (Table 1) and Cu K- and L-edge XAS data support a Cu(II)-O2−• formulation for 1 but significant Cu(III)-O22− character for 3 and 4 along with a high degree of covalency in the metal–ligand bonding.138 These data and accompanying theoretical calculations show that the more strongly electron-donating L2 and L3 ligands in 3 and 4 play a key role in stabilizing the higher metal oxidation state. Indeed, decreasing the electron donation of L2d by replacement of the backbone methyl groups with CF3 units (L2g) prevents formation of a 1:1 Cu:O2 adduct.139 Other theoretical studies have examined in detail the continuum on which 1, 3, and 4 reside and confirm that the more strongly electron-donating ligands stabilize the singlet with Cu(III)-O22− character.143,153
A unique example of a complex proposed to contain η2-Cu(II)-O2−• with a triplet ground state was recently reported using the supporting ligand L71.159 The assignment was based on UV–vis spectroscopy, the observation of paramagnetically broadened resonances in NMR spectra, and DFT calculations. In the absence of more definitive structural data from additional experiments (i.e., resonance Raman, X-ray crystallography, and EXAFS), however, the formulation of this complex must be regarded as tentative.
2.1.2. Reactivity
We focus on two aspects of reactivity: the process by which 1:1 Cu:O2 adducts form and their subsequent reactions. The kinetics and thermodynamics of the oxygenation of Cu(I) complexes supported by N-donor ligands described extensively in the previous review have been augmented by more recent work132,144,160–164 (Tables 3 and 4, which include previously published data for the systems supported by L41a and L42a).
Table 3.
Selected Kinetic Parameters for the Formation of 1:1 Cu:O2 Adducts
| ligand (solvent) | kon (M−1 s−1) | ΔH‡on (kcal mol−1) | ΔS‡on (cal mol−1 K−1) | koff (M−1 s−1) | ΔH‡off (kcal mol−1) | ΔS‡off (Cal mol−1 K−1) | ref |
|---|---|---|---|---|---|---|---|
| L2d (THF) | (1.560 ± 0.019) × 103a | 4.3 ± 0.5 | −23.9 ± 2.4 | – | – | – | 151 |
| L23a (acetone) | 4.1 × 107b | −1.6 ± 0.2 | −31 ± 1 | 2.4 × 10−2b | 10.5 ± 0.5 | –11 ± 2 | 164 |
| L42a (EtCN) | (9.5 ± 0.4) × 104b | 4.1 ± 0.1 | −12.4 ± 0.7 | (7.0 ± 0.3) × 10−2b | 14.8 ± 0.1 | 18.2 ± 0.7 | 144 |
| L44 (MeTHF) | (2.1 ± 1.0) × 106c | 2 ± 1 | −17 ± 6 | (5.2 ± 2.0) × 102c | 11 ±2 | 10 ± 8 | 160 |
| L41a (THF) | (1.5 ± 0.02) × 108c | 1.82 | −10.8 | 240 ± 6c | 13.9 | 25.1 | 163 |
| L41d (MeTHF) | (6.6 ± 3.5) × 105c | 2.2 ± 0.2 | −23 ± 2 | – | – | – | 160 |
| L43c (THF) | (6.9 ± 0.02) × 107c | 7.67 | 19.1 | 470 ± 0.02c | 15.9 | 37.5 | 161 |
| L40a (THF) | (1.8 ± 0.03) × 108c | 5.59 | 8.39 | 1600 ± 0.05c | 15.4 | 35.9 | 161 |
| L36 (THF) | (7.6 ± 0.2) × 10−1b | 5.83 ± 0.31 | −26.3 ± 1.7 | (1.1 ± 0.1) × 10−3b | 8.10 ± 0.26 | −3 ± 1 | 162 |
At 223 K.
At 183 K.
At 193 K.
Table 4.
Selected Thermodynamic Parameters for the Formation of 1:1 Cu:O2 Adducts
| ligand (solvent) | Keq (M−1) | ΔH° (kcal mol−1) | ΔS° (cal mol−1 K−1) | ref |
|---|---|---|---|---|
| L42a (EtCN) | (1.35 ± 0.04) × 106a | −10.73 ± 0.05 | −30.6 ± 0.2 | 144 |
| L44 (MeTHF) | (6.3 ± 1.9) × 103b | −9.6 ± 0.5 | −32.0 ± 2.6 | 160 |
| L41a (THF) | (6.5 ± 0.02) × 105b | −11.6 | −33.5 | 163 |
| L43c (THF) | (1.5 ± 0.06) × 105b | −8.22 | −18.5 | 161 |
| L40a (THF) | (1.1 ± 0.03) × 105b | −9.82 | −27.2 | 161 |
| L36 (THF) | (7.0 ± 0.1) × 102a | −2.27 ± 0.07 | 0.614 ± 0.382 | 162 |
At 183 K.
At 193 K.
Intriguing variations in kinetic and thermodynamic parameters for oxygenation reactions point to differences in reaction mechanisms for formation of 1:1 Cu:O2 adducts. In a detailed comparison using a “flash and trap” method (irradiation of Cu(I)-CO complexes in the presence of O2) of the systems supported by L40a, L41a, and L43c that feature identical bis(pyridylmethyl)amine units linked to variable fourth donors, positive ΔS‡on values for L40a and L43c contrasted with a negative ΔS‡on value for L41a (all in the same solvent, THF).161 These data were interpreted to indicate divergent dissociative interchange or associative mechanisms, respectively, but with the difference not being due to the order of solvent or O2 binding or loss. Instead, it was hypothesized that O2 binding occurs initially in both cases but with differences in whether electron transfer from Cu(I) to O2 (to yield Cu(II)-O2−• species) occurs before or after solvent dissociation. An interesting parallel was drawn between this notion and the postulated formation of a pre-equilibrium 1:1 Cu:O2 adduct prior to O2 release upon reaction of O2−• with Cu(II) complexes of L41a and L45 examined by stopped-flow kinetics and competitive 18O isotope effects.165
The kinetics and thermodynamics of O2 binding to the Cu(I) complexes of L41a, L41d, and L44 were compared using a direct photolysis method (photoejection of O2 from 1:1 Cu:O2 adducts followed by monitoring of rebinding).160 The L41d and L44 systems exhibited similar ΔH‡on values, but the ΔS‡on value for the former is more negative. These findings were interpreted to indicate that the Ocarbonyl interaction is weak in the Cu(I) complex of L41d, with a more ordered transition state for this system due to simultaneous Ocarbonyl and O2 coordination. A large negative ΔS‡on value was also measured for the system supported by L36, which was suggested to indicate an associative mechanism involving a highly ordered/restricted transition state.162
The mechanism by which O2 reacts with Cu(I) complexes supported by β-diketiminate derivatives (L2d and L2e) was elucidated through a combination of theory and low temperature stopped flow kinetics experiments.151 A dual pathway mechanism was proposed for the reaction that yields complex 3a (Figure 8) on the basis of the results of low-temperature stopped-flow kinetics experiments (in THF solvent) and DFT calculations. The observation of a two-term rate law (eq 1) was interpreted to indicate operation of both pathways A and B, wherein A involves direct rate-determining reaction of O2 with the Cu(I) complex and B is a dissociative route, involving rate-determining solvolysis prior to rapid reaction with O2. Pathway B is rendered effectively inoperative in the presence of excess nitrile, and the presence of bound nitrile in pathway A was confirmed by observation of decreases in rate as a function of para-substituent when para-X-benzonitriles (X = CH3O, CH3, H, F, Cl, and CN) were used (Hammett ρ = −0.34). Both routes operate in the absence of added nitrile, as indicated from plots of kobs versus [O2] that were linear but with nonzero intercepts (kobs = kA[O2] + kB). DFT calculations corroborated this dual pathway model and provided details of the reaction trajectories and structures of transition states and intermediates.
Figure 8.
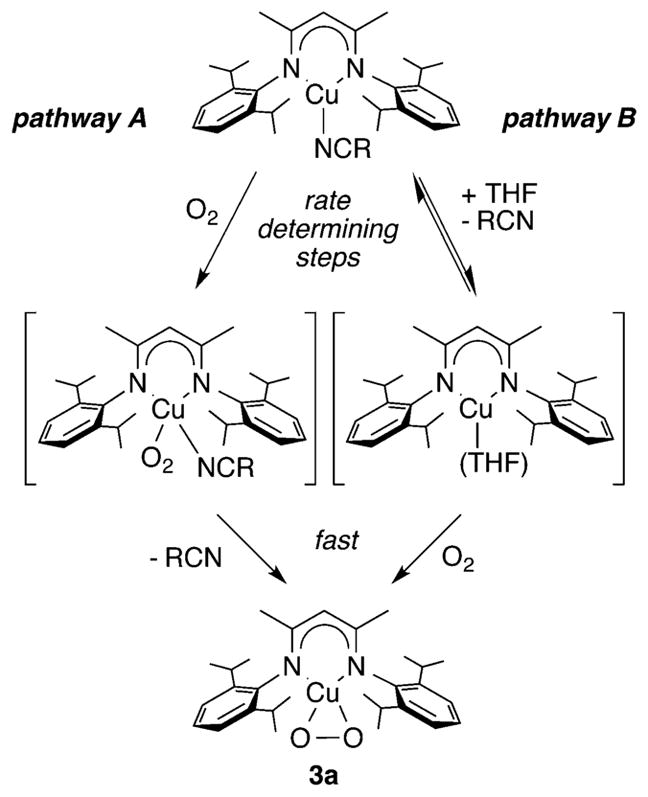
Proposed dual pathway for the oxygenation reaction resulting in formation of complex 3a. Adapted from ref 151.
| (1) |
Finally, with respect to the overall thermodynamics of O2 binding (Table 4), the order of binding strength is tris(2-(dimethylamino)ethyl)amine (L42a) > tris(2-pyridylmethyl)-amine derivatives (L40a ~ L41a ~ L43c) > tris-((tetramethylguanidino)(2-aminoethyl))amine (L44) > 1-isopropyl-5-(2-(2-pyridyl)ethyl)-1,5-diazacyclooctane (L36). The experimental ΔH° and ΔS° values were negative for all complexes, with the exception of ΔS° for the complex supported by L36, as expected for a spontaneous O2 binding reaction where Keq > 1 for all complexes.
Commonly, 1:1 Cu:O2 adducts can react with an additional equivalent of Cu(I) to generate a 2:1 Cu:O2 species (section 3). Indeed, prevention of this process has been key for the isolation and full characterization of 1:1 Cu:O2 adducts such as 1–4, with ligand steric encumbrance being a critical controlling factor. For example, the isolation of 3 and 4 stands in contrast to the formation of bis(μ-oxo)dicopper complexes when Cu(I) complexes of less hindered L2 ligands were used, with both ortho-aryl substituents and backbone groups being impactful (cf. L2f, L74).149,166 The tendency to react with an additional Cu(I) species was used purposefully to help characterize the 1:1 Cu:O2 adduct 6 (Figure 9).128 Treatment of 6, prepared by reaction of a Cu(II) precursor with KO2, with [(L41a)Cu(I)]OTf cleanly yielded the (trans-1,2-peroxo)dicopper complex 7, which was readily identified by its diagnostic UV–vis and resonance Raman features (section 3.2).
Figure 9.
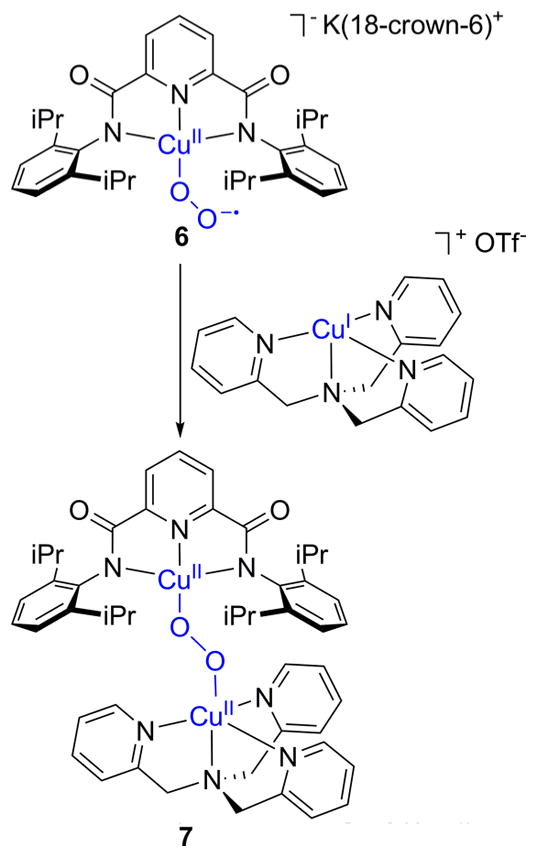
Reaction of a 1:1 Cu:O2 adduct (6, supported by ligand L28a) with a Cu(I) complex to yield a (trans-1,2-peroxo)dicopper complex (7; ref 128).
A hemilabile thioether ligand group enabled controlled isolation of a 1:1 Cu:O2 adduct and subsequent conversion to a bis(μ-oxo)dicopper complex.141 Oxygenation of the Cu(I) complex of L74 (X = Me, Ph) yielded a side-on η2 adduct (8), the properties of which were consistent with minimal interaction with the thioether group and significant Cu(III)-O22− character, just like 3 and 4 (Figure 10). Unlike 3 and 4, however, the binding of O2 was reversible, and upon vigorous purging with Ar, a bis(μ-oxo)dicopper complex (9) formed. It was concluded on the basis of the observations, as well as DFT calculations, that the O2 binding equilibrium involves slow dissociation of O2 (koff) and a large equilibrium constant (Keq).141 The trapping of η2 metal-peroxo complexes was also used to prepare heterobimetallic bis(μ-oxo) complexes comprising CuNi and CuPd pairs,167 and an analogous bis(μ-oxo) complex with a CuGe pair was prepared by oxygenation of a Cu(I)–Ge(II) complex (section 3.1.4).168
Figure 10.
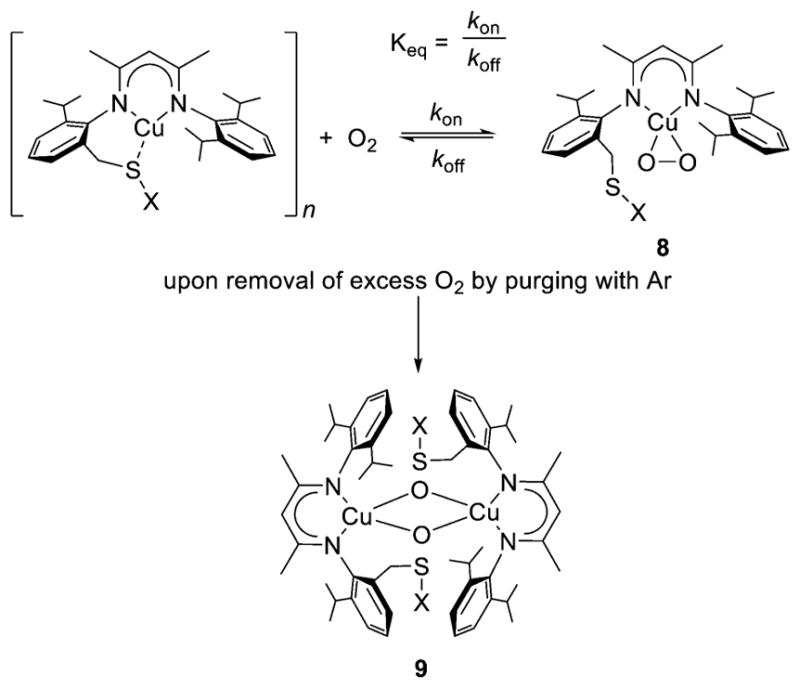
Reversible O2 binding to yield 1:1 Cu:O2 adduct 8 (supported by L74, X = Me or Ph) and its conversion to a bis(μ-oxo)dicopper complex 9. Adapted from ref 141.
In general, the η2 complexes with Cu(III)-O22− character epitomized by complexes 3 and 4 are poor oxidants and do not react with H atom donors like phenols or O atom acceptors like PPh3 (which simply displaces O2 from 3a to yield a Cu(I)-PPh3 complex). Computational studies show that the poor oxidizing ability of these complexes may be traced to the strong electrondonating character of their supporting ligands that render reduction and protonation difficult.123,169 Still, reaction of 3a with [Cu(CH3CN)4]OTf in the presence of 3,5-diphenylpyrazole (pz) resulted in an unusual hydroxylation/oxidation of a ligand aryl ring (Figure 11).170 The product was formulated on the basis of X-ray crystallography as a Cu(II)-semiquinone complex, arising from attack of some copper–oxygen intermediate (unidentified) at a ligand aryl ring and an NIH shift of one of the isopropyl groups.171 The hydroxylation resembles one reported previously upon oxygenation of a fluorinated β-diketiminate Cu(I) complex, for which the nature of copper–oxygen intermediates was not determined.172
Figure 11.
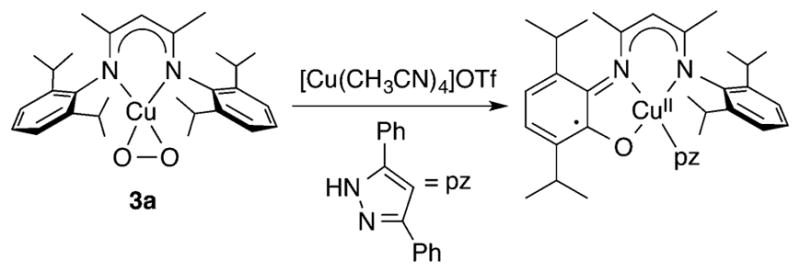
Intramolecular aryl ring hydroxylation/oxidation reaction of 3a (ref 170).
In view of the proposals that a Cu(II)-O2−• species is responsible for attacking a substrate C–H bond in the enzymes PHM, DβM,58 and LPMO,47 relevant reactivity of complexes with this unit have come under scrutiny. The putative η2-Cu(II)-O2−• complex supported by L71 converts 9,10-dihydroanthracene to anthracene, ultimately yielding a bis(μ-hydroxo)-dicopper(II) product via the presumed intermediacy of a [CuOOH]+ complex.159 Several η1-Cu(II)-O2−• complexes exhibited promising reactions with C– H bonds.130,131,137,162,173 Although unreactive with typical substrates with weak C–H bonds like 9,10-dihydroanthracene, xanthene, or 10-methyl-9,10-dihydroacridine, the η1-Cu(II)-O2−• complex 5 (Figure 7) was shown to oxidize BNAH (1-benzyl-1,4-dihydronicotinamide) or BzImH (1,3-dimethyl-2,3-dihydrobenzimidazole) at −125 °C in MeTHF, yielding BNA+ or BzIm as well as a (1,2-trans-peroxo)dicopper complex (Figure 12).130 In addition, kinetic data revealed a significant KIE (12.1) when BNAD was used, with the overall reaction occurring twice as fast with BNAH than with BzImH. These data were interpreted to indicate that the reactions involve initial HAT (homolytic C–H bond cleavage) given that BNAH is a better hydrogen atom donor than BzImH.130
Figure 12.
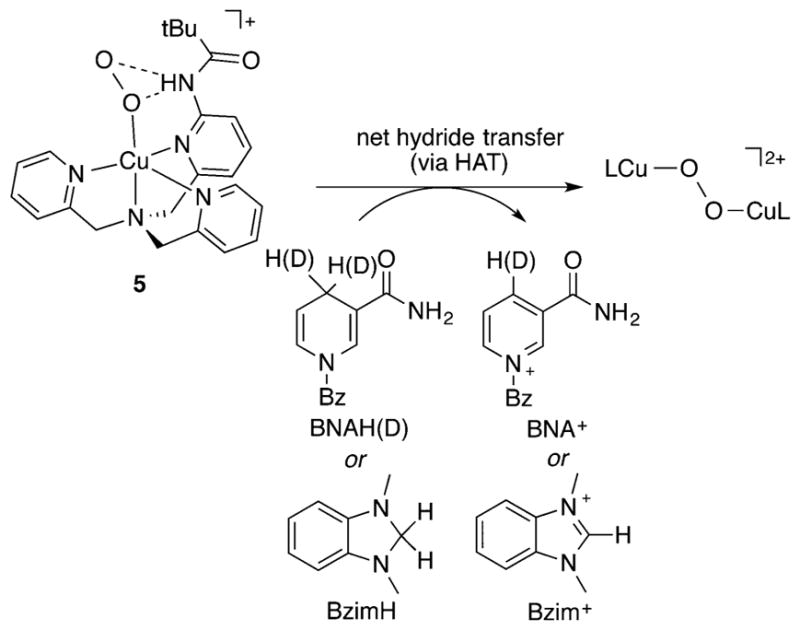
Reactivity of 5 with hydride donors, proposed to involve initial HAT (ref 130).
The η1-Cu(II)-O2−• complex 10 supported by L33c decomposes to yield a Cu(II)-alkoxide resulting from intramolecular hydroxylation of a benzylic C–H bond (Figure 13).131,162 The reaction followed first-order kinetics with a KIE of 4.1 at −65 °C, activation parameters consistent with an intramolecular process (ΔH‡ = 4.54 ± 0.02 kcal mol−1, ΔS‡ = −53 ± 0.1 cal K−1 mol−1), and a Hammett ρ of −0.63 were interpreted to support HAT. On the basis of results from DFT calculations, a pathway involving HAT to yield a [CuOOH]+ intermediate that then “rebounds” its proximal O atom via transition state 11 was favored relative to an alternative distal oxygen transfer.162 In further studies of the reactivity of 10,174 monitoring its decay in the presence of 1-electron reductants enabled estimation of its oxidation potential to be 0.19 ± 0.07 V versus SCE (acetone, 25 °C). In addition, HAT from TEMPOH was observed, but reactions with phenols yielded Cu(II)-phenolate complexes via proton transfer. With para-substituted triaryl phosphines [P(ArY)3; Y = OCH3, H, F, Cl], O atom transfer was observed with a large Hammett ρ of −4.3 indicative of attack by a strong electrophile (either the superoxide in 10 or a derived [CuO]+ species, for which no evidence was available).174
Figure 13.
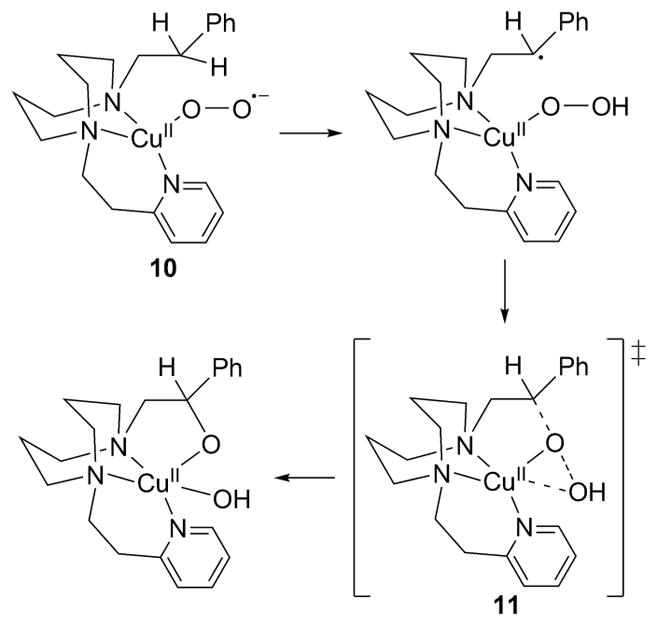
Proposed mechanism for intramolecular hydroxylation by 10 (supported by L33c, refs 131 and 162).
In a comparison of the reactivity of the η1-Cu(II)-O2−• complexes supported by L41b and the mixed N/thioether S donor ligand L68, respectively, reaction of the latter at −135 °C in 4:1 MeTHF:CF3CH2OH with N-methyl-9,10-dihydroacridine or 2,6-di-tert-butyl-4-methoxyphenol yielded 10-methyl-9-acridone or 2,6-di-tert-butyl-1,4-benzoquinone, respectively. These products were not observed with the complex supported by L41b.137 It was concluded that the thioether ligation in the complex of L68, which models that found in the enzymes PHM and DβM, enhances the oxidizing power of the coordinated superoxide ligand, supporting a similar role for the methionine ligand in the biological systems.
Augmenting the examples noted above of η1-Cu(II)-O2−• complexes performing HAT from weak O–H bonds are a number of other explorations of similar reactions. Complexes supported by electron-donating TMPA derivatives, L41b and L41c, and L44 react rapidly with phenols and mechanistic studies have provided key insights.127,136,175 The Cu(II)-O2−• complexes supported by L41b and L44 convert para-MeO-2,6-di-tert-butylphenol to a mixture of the corresponding quinone, hydroperoxide, and radical (in boxes, Figure 14); only quinones are formed from 2,6-di-tert-butylphenol and 2,4,6-tri-tertbutylphenol. 136,175 For the case of L44, an alkoxide complex arising from intramolecular hydroxylation of a ligand methyl group is observed, which was proposed to result from reaction of the [CuOOH]+ species derived from initial HAT from the weak phenol (or TEMPOH) O–H bond (this reaction is discussed in section 2.2).175 In a detailed study of the L41c system with a range of phenols,127 two pathways were identified, a 2-electron oxidation of para-X-2,6-di-tert-butylphenols to the quinone and a 4-electron oxidation of 2,4,6-trialkyl-substituted phenols to the quinone, presumably via loss of alkene. On the basis of kinetic data, a common mechanism involving initial HAT to yield a phenoxyl radical was proposed, with an additional reaction of the radical with another equivalent of the Cu(II)-O2−• complex yielding intermediate 12 at low temperature (Figure 14). For X = alkoxy (illustrated for methoxy), subsequent hydrolysis yields the product quinone, whereas for X = alkyl (illustrated for tert-butyl), alkene loss is the major route toward the quinone product, both of which occur upon warming/workup.127
Figure 14.

Proposed pathways for the generation of oxidized products (in boxes) from the reaction of Cu(II)-O2−• complexes supported by L (L41b and L41c; similar products formed for L44) (refs 127, 136, and 175).
In contrast to the above examples, the reactions of η1-Cu(II)-O2−• complexes supported by L28a and L42b do not readily abstract H atoms from phenols.129,128 The low observed reactivity of the L42b complex with hydroxylamine and phenols (in acetone at −90 °C) was ascribed to poor access of substrate due to the hydrophobic steric encumbrance of the supporting ligand.129 For the complex supported by L28a, reaction with alkyl-substituted phenols was not observed, while deprotonation of nitrophenol was observed, consistent with the nucleophilic/basic character of the anionic complex.128
Finally, we note that 1:1 Cu:O2 adducts have been proposed as intermediates in catalytic reductions of O2 to H2O2 or H2O.176–178 For example, in a study of the influence of added cations on 2-versus 4-electron reductions of O2, the η1-Cu(II)-O2−• complexes supported by L41a or L35a were postulated to be reduced by Fc* or Me2Fc, respectively, in the presence of Sc3+ to yield a Cu(II) intermediate and ScO2+, thus driving the reaction to yield peroxide instead of water.176
2.2. [CuOOR]+ Complexes
The [CuOOR]+ unit has been suggested as a key intermediate in catalytic oxidations by O2 or ROOH (R = H, alkyl, or acyl). In the following subsections, we discuss the syntheses and mechanisms of the formation of [CuOOR]+ species, their properties, and their reactivity.
2.2.1. Syntheses and Mechanism(s) of Formation
The (hydroperoxo)copper(II) unit proposed to be an active oxidant in enzymes may be accessed by the routes outlined in Figure 15. One path involves a 1:1 Cu:O2 adduct reacting with a proton and an electron, either via separate steps or through PCET or hydrogen atom transfer from substrate.175 This route directly models the way the [CuOOH]+ moiety is thought to be generated in biology. Alternative syntheses to [CuOOR]+ (R = H, alkyl, or acyl) involve treatment of copper(I) or copper(II) precursors with H2O2 or ROOH either in the presence or absence of base.179–194 The following examples are illustrative and include the few cases where mechanisms have been examined experimentally.
Figure 15.
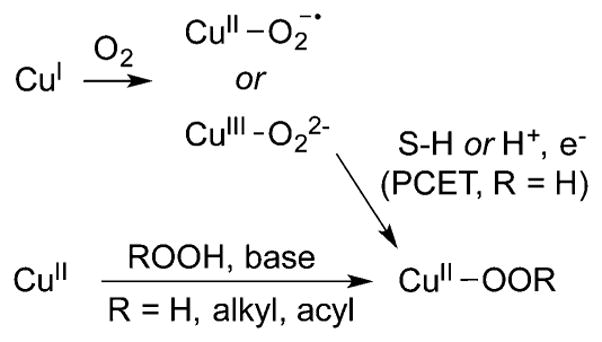
Routes by which [CuOOR]+ (R = H, alkyl, or acyl) complexes may be generated. S–H = substrate C–H or O–H bond. Supporting ligands not shown.
The formation of a [CuOOH]+ intermediate via the PCET pathway shown in Figure 15 was implicated in mechanistic studies of the 2-electron reduction of O2 to H2O2 by ferrocene (Fc) or 1,1′-dimethylferrocene (Me2Fc) by [(L45)Cu]2+ in the presence of HClO4 in acetone.195 In this study encompassing detailed kinetic experiments, the rate of formation of the intermediate [(L45)CuOOH]+ was found to be temperature-independent, which was rationalized by postulating that the negative ΔH for the binding of O2 to [(L45)Cu]+ (formed rapidly by reduction of the Cu(II) precursor by Fc or Me2Fc) is approximately the same as ΔH‡ for the rate-determining PCET reaction of the 1:1 Cu:O2 adduct; this equivalence explains the observed activationless conversion. It is noteworthy that a closely related system, [(L41a)Cu]2+, with one less –CH2– in the ligand backbone, exhibits quite different behavior, such that 1-electron reduction to the Cu(I) form is rate-determining, binuclear 2:1 Cu:O2 intermediates are involved (section 3), and O2 undergoes 4-electron reduction to H2O.178
In another study, kinetics experiments and DFT calculations were used to monitor the reaction of H2O2 in the presence of NEt3 with Cu(II)-solvato (S) complexes of the tridentate ligands L35b and L35c.179 Saturation kinetics were observed and interpreted to indicate rapid equilibrium formation of HOO−Et3NH+ (K), which then formed an initial [CuOOH]+ complex (k1, Figure 16). Conversion of this initial complex to a second [CuOOH]+ species with the hydroperoxide now in the equatorial position was proposed. An alternative hypothesis also consistent with the kinetic data involves loss of a proton and conversion of the –OOH ligand to a η2-peroxide. However, DFT calculations do not support this alternative hypothesis. This work complements a previous study using less sterically encumbered L38a in which analogous saturation kinetics were observed and similarly interpreted, but characterization of the [CuOOH]+ product(s) was hindered by subsequent formation of (μ-η2:η2-peroxo)dicopper(II) species.196
Figure 16.
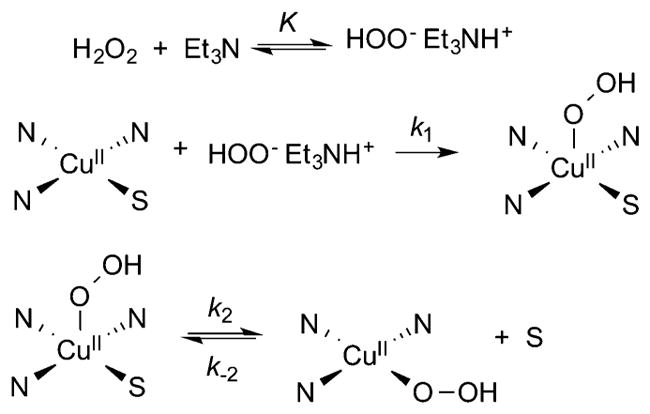
Proposed mechanism for generation of [CuOOH]+ complexes supported by ligand L35b and L35c. Only the donor N atoms of the supporting ligand are shown; S = solvent molecule (ref 179).
Another unusual route to a [CuOOH]+ complex was proposed that involves reaction of a Cu(I) complex with H2O2 in the absence of added base.188 Specifically, reaction of a Cu(I) complex supported by the ligand L41h with 1.5 equiv of H2O2 at −90 °C in acetone yielded 1 equiv. H2O and [(L41h)CuOOH]+, which is stabilized by intramolecular hydrogen bonding. To rationalize this result, and in particular the observed stoichiometry, a Fenton-like reaction to yield a copper-oxyl, [CuO]+ (section 2.3), was proposed (Figure 17). It was suggested that this species is then trapped by the Cu(I) precursor to yield a (μ-oxo)dicopper(II) complex, which reacts with H2O2 to yield the [CuOOH]+ product. An alternative pathway was also considered, whereby reaction of the Cu(I) complex with H2O2 yields hydroxyl radical and a (hydroxo)-copper(II) complex, which then affords the peroxo product upon reaction with H2O2.
Figure 17.
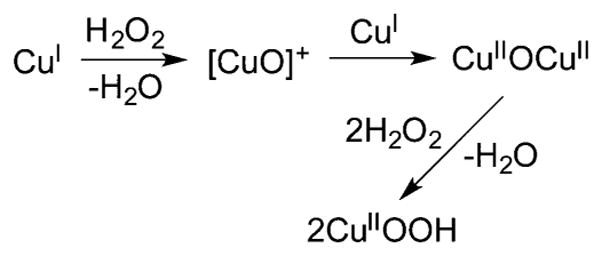
Proposed mechanism for generation of [CuOOH]+ complexes supported by ligand L41h (ligand not shown; ref 188).
A unique route to an alkylperoxide complex was reported involving reaction of copper(II) complexes of ligands L18 with H2O2 in acetone (Figure 18).180,184 An acetone molecule is functionalized to yield the novel species 13, the characterization of which is described below (section 2.2.2). The 2-hydroxy-2-peroxypropane ligand was formed in an analogous way upon reaction of an iron(II) complex with H2O2 in acetone.197 When the reactions of the copper(II) complexes of L18 with H2O2 or cumene hydroperoxide185 were performed in nitrile solvents, simple [CuOOR]+ (R = H or cumyl) complexes formed instead, highlighting a drastic solvent effect on the course of the synthesis.
Figure 18.
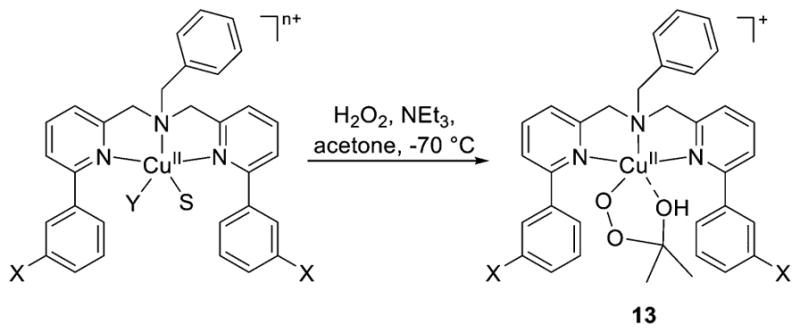
Generation of alkylperoxide complexes supported by L18 via functionalization of acetone solvent. Y = ClO4− or H2O; S = MeCN or H2O; X = NO2, Cl, H, Me, OMe; n = 1 or 2, depending on Y (refs 180, 184, and 185).
The reaction of cumene hydroperoxide with a Cu(I) precursor supported by the highly sterically hindered ligand L42c results in the generation of a complex (18) with a [CuOOR]+ moiety and an anilino radical ligand (Figure 19).194 A mechanism for formation of this unusual product was proposed involving initial generation of a [CuIOOR] complex featuring a protonated aniline arm (15) and hydrogen bonding from an N–H to the bound peroxide. Heterolytic O–O bond scission and release of ROH would generate the copper(II)-hydroxide (17), either stepwise via a copper-oxyl intermediate (16) that then undergoes H atom tautomerization or in concerted fashion. Substitution of the hydroxide in 17 by cumene hydroperoxide would yield the final product (18).
Figure 19.
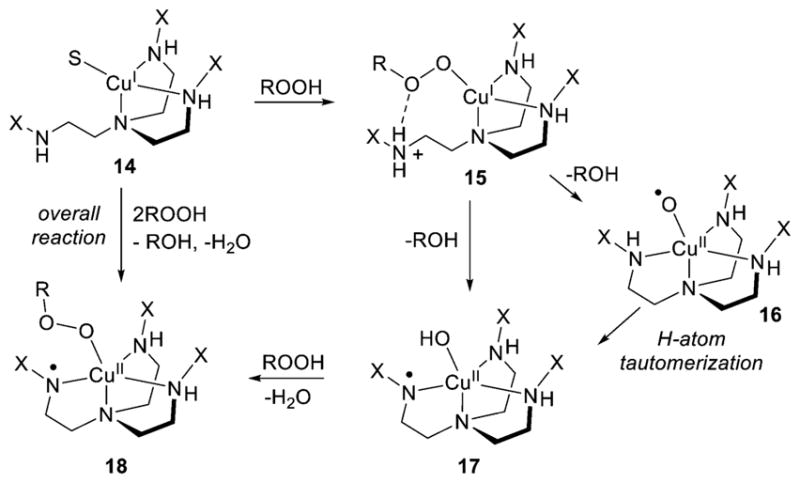
Proposed mechanism for the generation of 18 from reaction of cumene hydroperoxide with the Cu(I) complex (14) of L42c (X = TIPT), R = dimethylbenzyl (cumyl), S = CH3CN (ref 195).
2.2.2. Structures and Properties
Only two complexes with the [CuOOR]+ unit have been characterized by X-ray crystallography; their structures are drawn in Figure 20.198,199 The X-ray structures shown in Figure 20 show similar η1 coordination of the hydro- and alkylperoxo ligands, respectively, and identical O–O distances of 1.460(6) Å consistent with a peroxide formulation.37,199 A key difference is the presence of two hydrogen-bonding interactions in the L41e complex (19) from the amide substituent N–H groups to the proximal oxygen of the peroxide. As noted below (section 2.2.3), these interactions influence the properties and reactivity of the [CuOOH]+ unit.
Figure 20.

X-ray structures of the [CuOOH]+ complex of L41e (19), also drawn in Figure 21), and the [CuOOCm]+ (Cm = cumyl) complex of L39c (20). Selected interatomic distances (Å): (19) Cu–O, 1.888(4); O–O, 1.460(6) (20) Cu–O, 1.816(4); O–O, 1.460(6). (19) Reprinted from ref 37. Copyright 2005 Elsevier. (20) Reprinted from ref 199. Copyright 1993 American Chemical Society.
Other [CuOOR]+ complexes have been identified and characterized via a multitude of spectroscopic techniques (Table 5).175,179–195,200–210 Notably, these complexes show a diagnostic UV–vis feature at ~350 nm assigned as a peroxide→ Cu(II) ligand-to-metal charge transfer (LMCT) transition. In general, this absorption is observed at higher energy and intensity for R = H than for R = alkyl. Excitation into the LMCT band with resonance Raman spectroscopy allows for observation of O-isotope sensitive Cu–O and O–O vibrations. Typically, values of ν(Cu–O) ~550 cm−1 and ν(O–O) ~850 cm−1 are observed, with additional vibrational modes observed for [CuOOR]+ (R = alkyl), including C–C–C and O–C–C stretches. These complexes typically exhibit EPR signals characteristic for Cu(II) sites (data not shown).
Table 5.
Selected Spectroscopic Data for [CuOOH]+ and [CuOOR]+ Complexes
| copper core
|
ligand
|
UV–vis
|
Raman (exp, cm−1)a
|
Raman (calc, cm−1)
|
ref
|
|||
|---|---|---|---|---|---|---|---|---|
| λmax (nm) (ε, M−1 cm−1) | ν(O–O) | ν(Cu–O) | ν(C–O)/ν(C–C) | ν(O–C–C)/ν(C–C–C) | ν(O–O) | |||
| [CuOOH]+ | L83 | 325 (6414) | 822 (41), 836 (45) | – | – | – | – | 203 |
| L38e | 350 (3400) | 834 (42) | – | – | – | – | 204 | |
| L41e | 375 (700) | 860 (45) | – | – | – | – | 205 | |
| L70b | 374 (2589) | 880 (11) | – | – | – | 896 | 193 | |
| L40b | ~380 | 851 (46), 835 (46) | – | – | – | – | 206 | |
| L43a | 381 (1000) | 853 (46) | – | – | – | – | 207 | |
| L43b | 372 (1000) | 848 (45) | – | – | – | – | 207 | |
| L39a | 604 (1180) | 843 (26) | 624 (17) | – | – | – | 208 | |
| L66 | 357 (4300) | 881 (49) | – | – | – | – | 209 | |
| L45 | 332 (4240) | 851 (56) | – | – | – | – | 196 | |
| L33c | 375 (1650) | 831 (43) | – | – | – | – | 174 | |
| L35a | 345 (5000) | 900 (50) | 580 (25) | – | – | 953 | 179 | |
| L42a | 375 (1250) | 846 (48) | 509 (25) | – | – | 854 | 190 | |
| ε-His-Gly-His | 366 (2600) | – | – | – | – | – | 202 | |
| L85 | 380 (2000) | – | – | – | – | – | 187 | |
| [CuOOR]+b | L39c | 572 (3815) | 844 (26) | 652 (19) | 802 (26)/755 (6) | 555 (10)/540 (4) | – | 208 |
| L39a | 603 (5410) | 843 (26) | 645 (16) | 809 (28)/756 (4) | 551 (8)/536 (7) | – | 208 | |
| L39ac | 610 (5000) | 884 (24) | 640 (27) | 834 (37)/754 (15) | 471 (6) | – | 208 | |
| L18bd | 420 (1350) | 855 (30) | 545 (20) | 823 (20)/792 | (7) – | 901 | 180 | |
| L18d | 465 (1100) | 885 (30) | 608 (11) | 841 (33) | 529 (5)/485 (9) | – | 185 | |
| L42a | 440 (280) | 887 (82) | – | 839 (34) | – | 845 | 190 | |
| L69 | 465 | 887 (89) | 610 (7) | 827 (33) | – | – | 192 | |
| L42c | 396 (5400) | 831 (43) | 604 (15) | – | 569 (11)/541 (10) | 833 | 194 | |
| L41ae | 332 (950) | – | – | 1740 (C=O) | – | – | 210 | |
| L39ce | – | – | – | 1640 (C=O) | – | – | 210 | |
D18O indicated in parentheses.
R = Cm, except where indicated.
R = tBu.
2-Hydroxy-2-peroxypropane complex 13.
R = acyl.
An illustrative example is the identification of complex 18 as a [CuOOR]+ species with a bound anilino radical that is based on (a) UV–vis and resonance Raman data typical for the [CuOOR]+ moiety and (b) the observation of ligand vibrations associated with the anilino radical in resonance Raman spectra.194 These assignments were confirmed through comparison to spectra obtained using ligand deuteration on the anilino rings and DFT calculations. The complex is EPR silent, consistent with antiferromagnetic coupling between the radical and the Cu(II) ion.
2.2.3. Reactivity
Variability in the reactivity of [CuOOR]+ complexes has been observed, with some being stable only at low temperature and prone to decomposition upon warming and/or reactions with exogenous substrates and others being quite robust and unreactive. In addition, the reaction pathways are sensitive to the nature of the supporting ligand and the solvent.
Examples of stable, relatively unreactive [CuOOR]+ complexes include those supported by the ligands L35b–c,179 L42a (R = H or Cm),190 L19 (R = H),183 and L41e (R = H).198 DFT calculations aimed at evaluating the reactivity of [(L19)-CuOOH]+ for epoxidation of ethylene revealed a high reaction barrier for O–O bond homolysis consistent with experimental observations (i.e., 40.2 kcal/mol for O–O bond homolysis).183 The stability of the L41e complex 19 (Figure 20 and Figure 21) was attributed to a combination of hydrogen bonds from the amido NH groups to the proximal O atom of the bound hydroperoxo ligand and steric shielding by the tert-butyl substituents.198,211 From a comparative survey of the properties of [CuOOH]+ complexes supported by a series of L41 derivatives with differing hydrogen bonding capabilities and steric influences, it was concluded that hydrogen bonding to the proximal oxygen is correlated with a lower energy peroxo → Cu(II) LMCT transition, higher ν(O–O), lower ν(Cu–O), and slower rates of decomposition. These results are consistent with the hydrogen bond interaction causing a weakening of the Cu– O bond and a strengthening of the O–O bond that is broken in the decomposition process.37 Conversely, a [CuOOH]+ complex supported by L43a (21, Figure 21) was proposed to feature hydrogen bonding to the distal O atom, and it was found to decompose faster than an analog supported by L43b (22) that lacked this distal interaction (Figure 21).37,207
Figure 21.
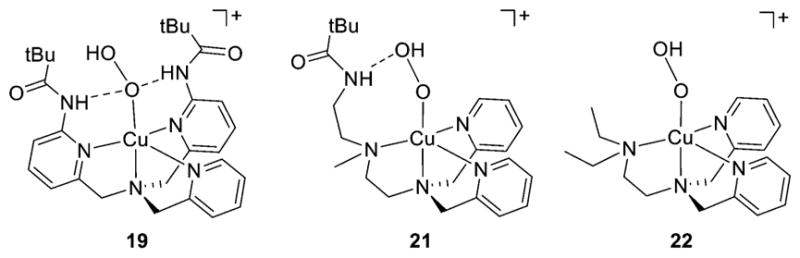
[CuOOH]+ complexes illustrating hydrogen bonding to the proximal O atom (19, supported by L41e) (refs 198 and 211), distal O atom (21, supported by L43a; ref 207), and with no hydrogen bonding (22, supported by L43b; ref 207).
Hydrogen bonding from a secondary amine group to the proximal O atom in a [CuOOH]+ complex of L41h (23, Figure 22) also inhibits N-dealkylation reactions (see below) as well as reactions with exogenous substrates.188 Interestingly, this complex forms a (trans-1,2-peroxo)dicopper(II) species upon warming (Figure 22). In addition, it yields 1 equiv. H2O2 upon treatment with HClO4, a reaction that can be reversed by subsequent addition of Et3N over multiple cycles. Hydrogen bonding was also postulated to stabilize a [CuOOH]+ complex of L70b, here involving the hydroperoxo O–H interacting with a ligand phenoxide O atom.193 This complex was proposed to be an intermediate in the catalytic oxidations of cyclohexane and toluene by H2O2 in the presence of HNO3.
Figure 22.
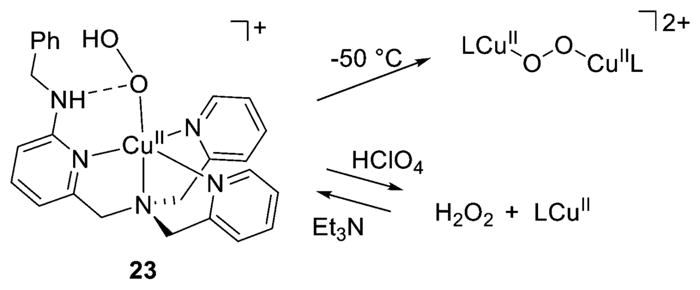
Reactivity of [CuOOH]+ complex 23 (L = L41h) (refs 188 and 193).
Intramolecular hydroxylation of supporting ligand aryl groups was observed upon decay of several [CuOOR]+ complexes. 180,181,184 Warming of the [CuOOH]+ complex 24 supported by L41f in acetone from −80 °C to room temperature followed by aqueous workup yielded the phenol shown in Figure 23a, which was labeled with 18O when H218O2 was used.181 The involvement of a bis(μ-oxo)dicopper species was ruled out by independent synthesis of such a species from a Cu(I) complex of L41f and O2 and determination that it did not yield hydroxylated ligand. Mechanisms involving either direct attack at the aryl group of the hydroperoxo moiety or O–O bond homolysis to yield a reactive copper-oxyl were proposed.
Figure 23.

Aryl group hydroxylations by [CuOOR]+ complexes. (a) Reaction of complex 24 supported by L41f (ref 181). (b) Reaction of 2-hydroxy-2-peroxypropane complexes 13, highlighting the proposed mechanism. X = NO2, Cl, H, Me, OMe (refs 180 and 184).
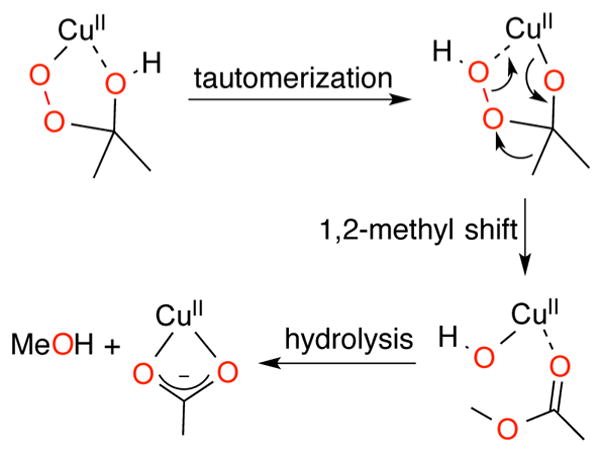
Intramolecular aryl group hydroxylation was also observed upon warming of the 2-hydroxy-2-peroxypropane complex 13 (Figures 18 and 23b).180,184 The final product was the phenoxide complex 26 (Figure 23b), which was isolated and characterized by X-ray crystallography.180 The reaction followed first-order kinetics to yield an intermediate 25, and studies of the series with X = NO2, Cl, H, Me, OMe gave a Hammett ρ = −2.2 consistent with electrophilic attack at the aryl group. The KIE for the perdeuterated aryl analog was negligible (0.9 ± 0.02). The structure of 25 shown in Figure 23b was proposed on the basis of the combined experimental data and DFT calculations, and the indicated mechanism involving general acid–base catalysis by HNEt3+ and its conjugate (used in the synthesis of 13) was suggested. The analog of 13 lacking the aryl substituents (i.e., complex supported by L18a) decomposed to yield a Cu(II)-acetate complex, in which one of the O atoms in the acetate ligand was shown to derive from H2O2 (determined from isotopic labeling). A mechanism was proposed on the basis of DFT calculations involving tautomerization of the 2-hydroxy-2-peroxypropane ligand, a Baeyer–Villiger-type 1,2-methyl shift, and hydrolysis of the resulting ester complex (Figure 24).184
Figure 24.
Proposed mechanism for the conversion of the 2-hydroxy-2-peroxypropane complex of L18a to a Cu(II)-acetate complex (ref 184).
The [CuOOR]+ unit has also been implicated as an oxidant of pendant N-alkyl amine groups,175,182,186,187,189 including N-dealkylations that model the function of PHM.41 In one set of studies,182,186,189 the warming and subsequent demetalation of [CuOOH]+ complexes supported by L41g and L41i–k yielded unperturbed ligand, mono-N-dealkylated ligand, and the respective aldehyde as predominant products (>40% yield each), with smaller amounts of overoxidized coproducts (Figure 25). An intermediate copper(II)-alkoxide complex 28 was identified by ESI-MS,182,186 the O atom of which derived from the H2O2 used to prepare the [CuOOH]+ unit according to the results of isotope labeling experiments. Initial mechanistic hypotheses invoked O–O bond homolysis of the [CuOOH]+ complex to yield a reactive [CuO]+ species that cleaves the weak C–H bond adjacent to the amine N atom to yield an iminium radical cation. Subsequent “rebound” would yield the alkoxide intermediate, which upon aqueous workup decomposes to the N-dealkylated amine and the aldehyde. Indirect support for the initial O–O bond homolysis route included the observations that (a) N-dealkylation did not occur to the same extent when bis(μ-oxo)dicopper species of the same ligands were examined (ruling out such species as potential intermediates) and (b) the same alkoxide intermediate 28 was observed upon treatment of Cu(I) precursors of the intact ligand with PhIO. In addition, ESI-MS evidence consistent with the [CuO]+ intermediate was obtained.
Figure 25.
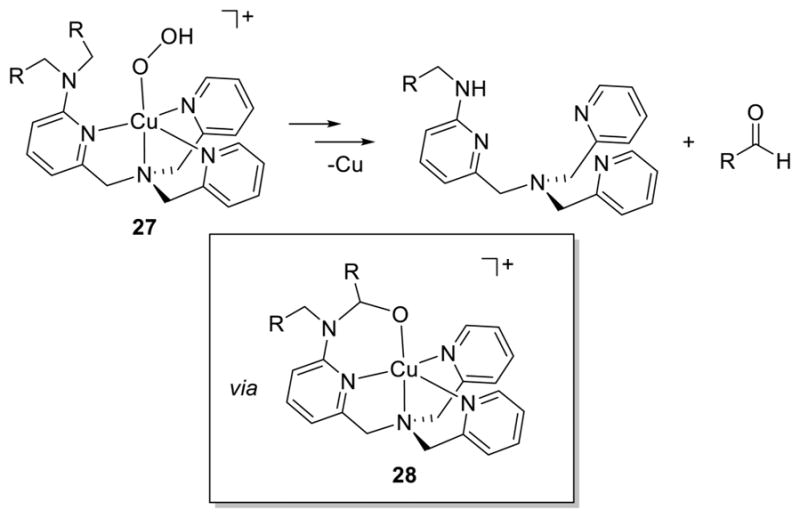
N-dealkylation reactions of complexes (27) of L41g (R = H) and L41i–k (R = aryl) (refs 182, 186, and 189).
A similar pathway was proposed to rationalize the formation of the copper(II)-alkoxide 30 upon reaction of the 1:1 Cu:O2 adduct 2 with phenols or TEMPOH (Figure 26).175 In these reactions, the 1:1 Cu:O2 adduct abstracts an H atom from the phenol or TEMPOH to generate a [CuOOH]+ complex 29, that then was proposed to undergo the O–O bond homolysis process. Supporting evidence included observation of the same alkoxide complex 30 upon treatment of a Cu(II) precursor with H2O2 (consistent with a [CuOOH]+ intermediate) or reaction of a Cu(I) precursor with PhIO (consistent with a copper-oxyl intermediate). A DFT study proposed a 15 kcal/mol barrier for abstraction of the H atom of the methyl group of the amine by the distal O atom of the [CuOOH]+ complex, with concomitant O–O bond scission.212
Figure 26.
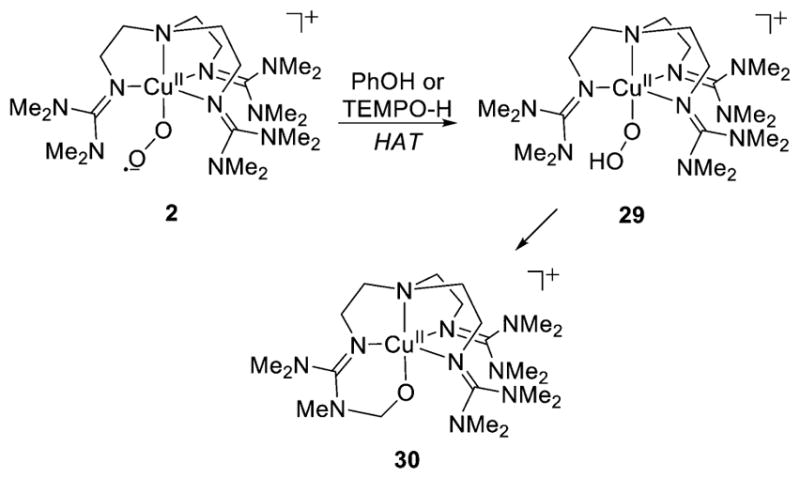
Proposed conversion of 1:1 Cu:O2 complex 2 to copper(II)-alkoxide 30 upon reaction with H atom donor reagents (ref 175).
These mechanistic hypotheses for N-dealkylation reactions of [CuOOH]+ complexes have been called into question in more recent work.189 In a detailed mechanistic investigation of the system (27, Figure 25), with R = para-X-phenyl (X = Cl, H, and OMe), DFT calculations revealed high-energy barriers (27–34 kcal/mol) inconsistent with measured reaction rates for mechanisms involving (a) direct HAT by the distal oxygen (like that proposed for 2), (b) prior O–O bond homolysis to yield a copper-oxyl, or (c) a pathway involving concerted Cu–O bond homolysis and HAT (to give Cu(I) and H2O2). Upon deuteration of the ligand, no KIE was observed, further arguing against the direct HAT pathway. Instead, a mechanism involving Cu–O bond homolysis to yield Cu(I) and the hydroperoxyl radical was proposed, which was found to have a reasonably low barrier of 14.8 kcal/mol (Figure 27). Subsequent Fenton-like chemistry involving reaction of the Cu(I) complex with H2O2 was suggested to yield a Cu(II)-hydroxide and hydroxyl radical. HAT by this radical followed by “rebound” from the Cu(II)-hydroxide would afford the requisite carbinolamine that undergoes N-dealkylation.
Figure 27.
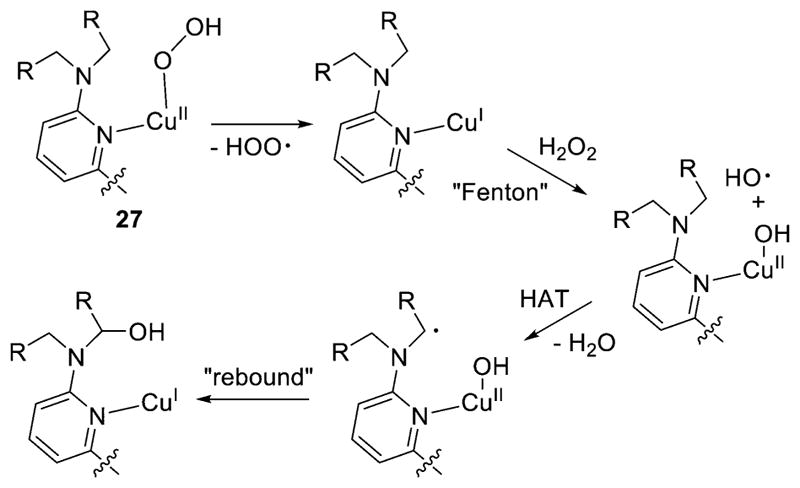
Hypothesized mechanism for N-dealkylation of 27, with only the attacked arm of the L41i–k ligand shown. All copper species have an overall charge of +1 (ref 189).
Evidence in favor of O–O bond homolysis in a [CuOOR]+ (R = C(Me)2Ph) complex was observed in 31 (Figure 28).185 Decomposition yielded a bis(hydroxo)dicopper(II) complex and acetophenone. Oxidation of exogenous substrates 10-methyl-9,10-dihydroacridine or 1,4-cyclohexadiene was observed, with a large KIE of 19.2 at −40 °C for the 9,9-dideuterated derivative of the former indicating rate-determining C–H(D) attack. In the presence of the radical trap, 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), hydroxylation to yield a complex assigned as 32 occurred. Acetophenone was a coproduct in all of the reactions. A stepwise mechanism involving rapid pre-equilibrium formation of a [CuO]+ species 33 (Keq) followed by HAT or radical trapping steps was proposed, although it was noted that the kinetic data are also consistent with a concerted process.
Figure 28.
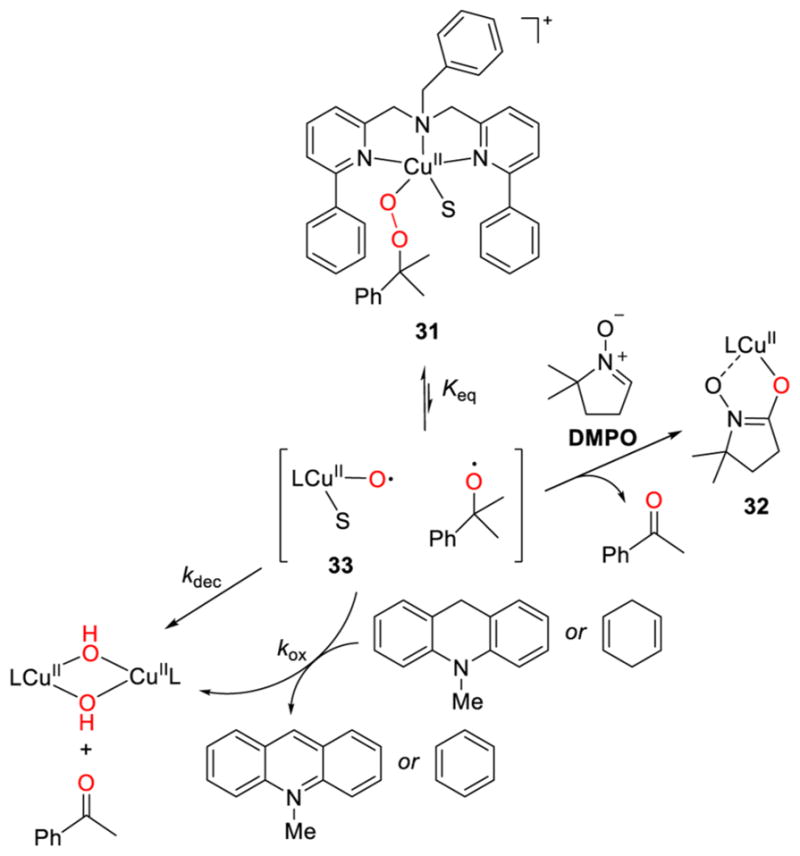
Reactivity of [CuOOR]+ (R = Cm) complex 31 with proposed mechanism involving O–O bond homolysis (ref 185).
Heterolytic O–O bond scission was implicated in reactions of Cu(I) complexes of L18a (34) and L69 (35) with cumyl hydroperoxide (Figure 29).191,192 In both systems, the reaction proceeded to give cumyl alcohol (CmOH) as the predominant product (90–98%), with only minor amounts of acetophenone observed. These results are consistent with 2-electron reduction of the peroxide moiety. However, the stoichiometry for the reactions involving the two ligands differed; for 34, a 2:1 Cu:HOOR stoichiometry was observed (50% yield of CmOH), whereas for 35, the yield of CmOH was ~100% (1:1 Cu:HOOR stoichiometry). In addition, upon workup of the reaction with 35, the sulfoxide form of the ligand was isolated. Presumably, and on the basis of analogy to results for a dicopper(I) complex (section 3), the pathway for 34 involves dicopper intermediates [1 electron from each Cu(I)]. For 35, a mechanism involving formation of a [CuOOR]+ intermediate was proposed, with the second necessary electron coming from the sulfur donor to give the intermediate 36. Subsequent heterolytic cleavage of the O–O bond generates CmOH and the Cu(I) complex 38 of the sulfoxide, possibly via the intermediacy of a species such as 37. It is worth noting that heterolytic O–O bond scission and involvement of a ligand donor atom was also observed for 15 (Figure 19).194
Figure 29.
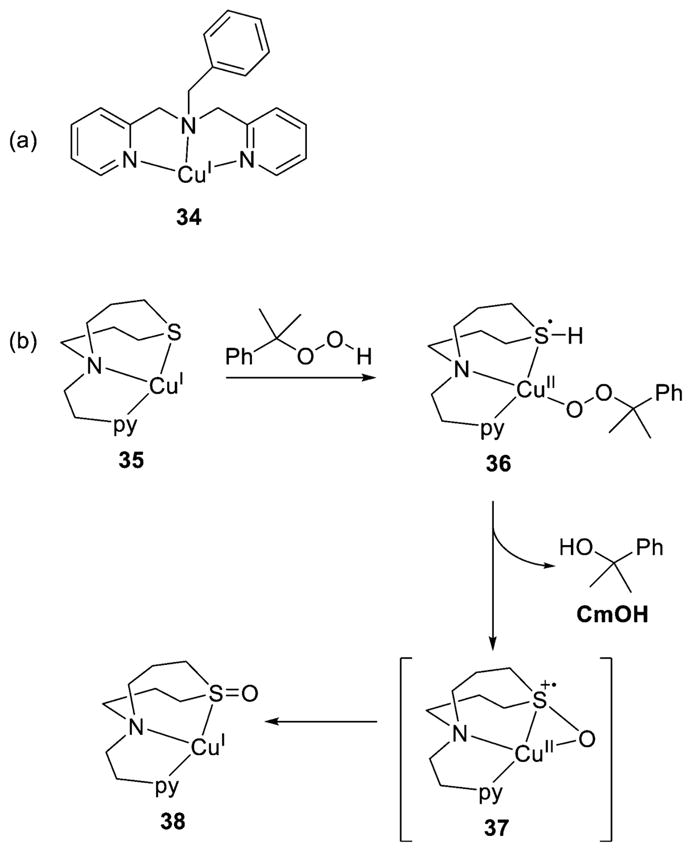
Copper(I) complexes (a) 34 (supported by L18a) which proceeds via a 2:1 stoichiometry (not shown) and (b) the proposed pathway for reaction of cumyl hydroperoxide with 35 (supported by L69) to yield CmOH and the Cu(I) complex of the oxidized ligand 38 (refs 191 and 192).
The unusual [CuOOR]+ radical complex 18194 (Figure 19) cleanly oxidized various para-substituted benzylic alcohols to benzaldehydes (substituents: OMe, Me, F, Cl) in a 2-electron process reminiscent of the copper(II)-phenoxyl unit in galactose oxidase (GAO)213 and model complexes.214,215 In the presence of excess substrate, the reaction followed pseudo first-order kinetics, and a Hammett plot of the second-order rate constants had a ρ value of −0.42 ± 0.08, similar to that reported for GAO (−0.09 ± 0.32).216 On the basis of this similarity to the enzyme, a mechanism was proposed involving substitution of the peroxide ligand by the alcohol (to yield ROOH), followed by intramolecular HAT by the anilino radical (vs the phenoxyl radical in GAO).
Finally, in chemistry relevant to biomolecule oxidation by reactive nitrogen species,217 the [CuOOH]+ complex 23 (Figure 22) supported by L41h was found to react with NO according to eq 2.201 A Cu(I)-peroxynitrite complex was postulated as an intermediate, with support coming from observation of nitration of 2,4-di-tert-butylphenol after treatment of 23 with the phenol followed by addition of NO. The finding of N2O as a coproduct in the reaction of 23 with NO was rationalized by proposing disproportionation of NO by a Cu(I) intermediate(s).
| (2) |
2.3. [CuO]+ and [CuOH]2+ Species
Of the monocopper–oxygen intermediates proposed to be involved in catalytic oxidations, species which contain the [CuO]+ unit (“copper-oxyl” species) have proven to be particularly elusive. Proposals for the intermediacy of such species in reactions of copper complexes in solution go back more than two decades.120,218,219 Yet, while a number of computational studies have probed their properties and led to proposals that intermediates of this type are potent oxidants, such species have only been observed experimentally in the gas phase and only indirect evidence exists for their involvement in homogeneous systems. Examples of such cases involving reactions of [CuOOR]+ complexes were discussed in section 2.2. The following discussion will briefly summarize the computational predictions concerning the properties of the [CuO]+ moiety and some other experimental examples that hint at the involvement of the [CuO]+ unit in homogeneous oxidation reactions. The discussion will then shift toward recent examples of [CuOH]2+ species, which may be considered to be the conjugate acid of the [CuO]+ moiety and have also been suggested as relevant species in biological oxidations.
2.3.1. [CuO]+
Numerous computational studies have evaluated the [CuO]+ unit within gas-phase ions,57,61,220–226 a protein environment,17,59 and complexes in solution. 121,122,227,228 Detailed evaluation of the bare [CuO]+ ion supports a triplet ground state with the configuration (1σ)2(2σ)2(1πx)2(1πy)2(1δ)4(3σ*)2(2πx*)1(2πy*)1(4σ*)0 [Figure 30 (left)],57,220,222 which has been noted to be analogous to the 3Σg− ground state of dioxygen.229 But rather than having biradical spin density equally distributed between the two atoms like in O2, in [CuO]+ the singly occupied 2π* orbitals have predominant oxygen p character, as reflected by the spin densities of 1.68 on O and 0.32 on Cu.222 Analogous triplet ground states were found for the [CuO]+ unit in various ligand environments, albeit sometimes with different orbital descriptions. For example, in the distorted trigonal bipyramidal environment of the PHM active site, one electron occupies what is essentially a nonbonding px (O) orbital and the other occupies a σ-type molecular orbital comprising antibonding dz2 (Cu) and pz (O) orbitals [Figure 30 (right)]. This situation has been contrasted with the much more strongly bonding interactions involved in the FeIVO unit.17 Indeed, the Cu–O bond in [CuO]+ is weak, as reflected in low bond dissociation energies determined from experiment (31.1 ± 2.8 kcal/mol)230 and theory (~25 kcal/mol).57
Figure 30.
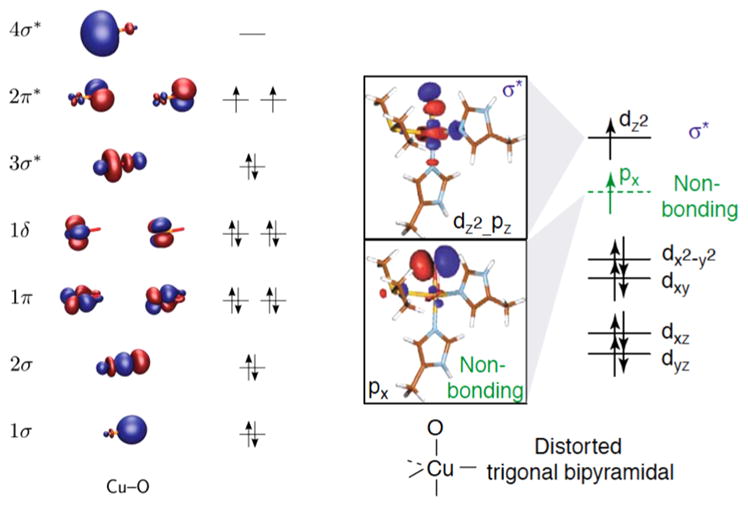
(left) Qualitative molecular orbital (MO) scheme for [CuO]+. Reprinted with permission from ref 57. Copyright 2011 AIP Publishing). (right) Orbital scheme for [CuO]+ unit in PHM. Reprinted with permission from ref 17. Copyright 2005 Elsevier Ltd.
Consistent with its biradical character and a weak Cu–O bond, the [CuO]+ unit by itself, or in ligated form, has been predicted to be highly reactive. As noted previously (section 1), computations predict that reaction barriers for substrate attack by [CuO]+ in enzymes such as DβM,59,60 PHM,17,46 or LPMO61 are significantly lower than that for other intermediates such as 1:1 Cu:O2 adducts or [CuOOH]+. Similar predictions have been made for synthetic systems.183 Additionally, the product O–H bonds formed in HAT reactions mediated by these species are generally strong (~90–99 kcal mol−1 in some cases).227 Experiments have shown that in the gas phase, the ion [(phen)CuO]+ attacks a variety of hydrocarbon C–H bonds221,231 and the even more reactive [CuO]+ ion readily attacks the strong C–H bond of methane.222,223,231 Full discussion of this extensive work is beyond the scope of this review, which focuses primarily on complexes in condensed phase. We note here, however, that a key feature of many of these reactions is spin-inversion from the triplet potential energy surface to the singlet surface, which generally occurs after the initial oxidation step (either HAT or O atom transfer).59,183,226 The subsequent steps in these reactions (either radical rebound in the case of the HAT reactions or ring closure in the case of epoxidation reactions) generally involve the one electron reduction from copper(II) to copper(I). The reduction is more favorable for the singlet state than the triplet state which is why spin-inversion generally happens after the initial oxidation step but before the second transition state.
Postulates of [CuO]+ as an intermediate in reactions of [CuOOR]+ complexes were discussed in section 2.2, where it was noted that most supporting evidence is indirect (with the exception of ESI-MS data for the reaction of 28 with PhIO).186 Another example drawing inspiration from nonheme iron enzymes232–234 involved the reaction of copper(I)-α-ketocarboxylate complexes (39) supported by L17 with O2 (Figure 31).228 Demetalation and workup of the reaction mixtures revealed that aromatic hydroxylation of the ligand had taken place. DFT calculations predicted a pathway involving nucleophilic attack on the α-ketocarboxylate ligand by a 1:1 Cu:O2 intermediate followed by decarboxylation. The resulting peracid species can then attack the ring directly via a very “oxolike” peracid transition state [“TS-peracid”, path (b)] or form a [CuO]+ type intermediate that then attacks the ring [“TS-oxo”, path (a)]. The latter was found to be the more kinetically favorable pathway. In line with other studies, theory indicated that the [CuO]+ species in path (a) has a triplet ground state and that spin crossover from the triplet to the singlet potential energy surfaces should be efficient.235
Figure 31.

Reaction of 39 (supported by L17) that results in hydroxylation of the ligand and the mechanism proposed on the basis of DFT calculations. Adapted from ref 228.
In a more direct attempt to access a [CuO]+ complex, a set of Cu(I) complexes of bidentate N-donor ligands were treated with oxo transfer reagents Me3NO, pyridinium N-oxides, or PhIO.236 In several cases, stable Cu(I)-N-oxide adducts formed, attesting to the energetic cost of accessing a [CuO]+ species. With ligand L2d, a bis(μ-oxo)dicopper complex was generated in the reaction with Me3NO, which might have derived from dimerization of a [CuO]+ precursor. However, alternative pathways such as that involving dimerization of a Me3NO adduct followed by amine loss could not be ruled out.
2.3.2. [CuOH]2+ Complexes
Protonation of the [CuO]+ unit would yield a [CuOH]2+ core, which may be envisioned as a (hydroxo)copper(III) species that could exhibit significant reactivity with C–H bonds. Such species 40–43 (Figure 32) have been prepared using strongly electron-donating dicarboxamido ligands,237–239 which are related to other amide-containing ligands that had been used previously to stabilize Cu(III) complexes.240–244 These complexes were prepared by 1-electron oxidation of [CuOH]+ precursors and were formulated as Cu(III) compounds on the basis of X-ray absorption spectroscopy, EPR spectroscopy, and TD-DFT analysis of UV–vis spectra. Key spectroscopic features for the [CuOH]2+ core include (a) an X-ray absorption edge energy ~1.7 eV higher than that of the precursor Cu(II) complex and average Cu–O(N) distances shorter by ~0.1 Å than the Cu(II) precursor by EXAFS, (b) EPR silence consistent with a S = 0 Cu(III) formulation, and (c) identification of the intense absorption feature ~500–570 nm assigned by TD-DFT calculations as a ligand-to-metal charge transfer transition from the π system of the flanking aryl rings to the [CuOH]2+ core for 40–43.
Figure 32.
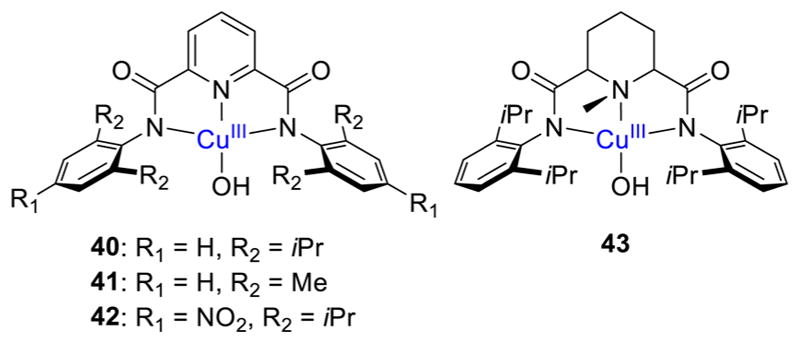
Complexes with a [CuOH]2+ core supported by L28a–c and L25, respectively (refs 237–239).
In the initial report describing 40, high rates for H atom abstraction from 9,10-dihydroanthracene (DHA) were found (i.e., k = 1.1(1) M−1 s−1 at −80 °C).237 The products observed were anthracene and the corresponding complex with a [Cu(OH2)]2+ core. Kinetic studies using deuterated substrate revealed a high H/D KIE of 40 at −60 °C, clearly reflecting C– H bond scission in the rate-determining step and suggestive of a significant tunneling contribution. Since this first report, more detailed studies of the properties and reactivities of 40, 42, and 43 were performed.238,239 The differing degrees of electron donation by the ligands across the series were reflected in spectroscopic properties and oxidation potential differences. For example, a 400 mV range in E1/2 values for the CuIII/CuII redox couple was observed [43 (−260 mV) < 40 (−74 mV) < 42 (+124 mV), all versus Fc+/Fc in 1,2-difluorobenzene (DFB)]. The redox behavior is inversely correlated to the basicity of the hydroxide in the Cu(II) precursors, which spans a range of ~4 pKa units (16–20), and together these effects result in the formation of strong O–H bonds in the aquo complexes that are products of HAT reactions (bond dissociation enthalpies (BDEs) = 88–91 kcal mol−1; 43 < 40 < 42).238,239 The complexes 40, 42, and 43 attack substrates with C–H bond enthalpies ranging from 76 (DHA) to 99 kcal/mol (cyclohexane). A plot of the log of the second-order rate constants (k) versus the difference in BDEs between the substrate C–H bonds and the product aquo complex O–H bonds was linear (Figure 33), indicating a common HAT mechanism across the series of substrates and complexes. The results are also consistent with a rate-dependence on the thermodynamic driving forces, in line with results observed for PCET reactions of other metal oxo/hydroxo compounds.245,246 In computations evaluating the pathway of the reactions with DHA, transition state structures were defined and significant corrections to account for proton tunneling were necessary to obtain activation parameters that agreed with experimental values.
Figure 33.
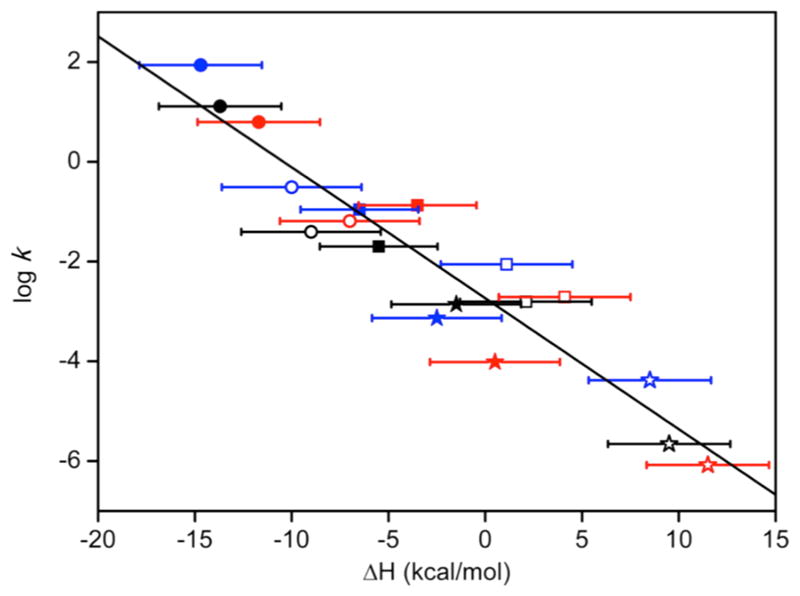
Plot of log(k) vs ΔH (equivalent to the ΔBDE between the aqua complexes and the C–H bonds of the substrates) for reactions of 40 (black), 43 (red), and 42 (blue) with the substrates DHA (filled circles), cyclohexene (open circles), diphenylmethane (filled squares), THF (open squares), toluene (filled stars), and cyclohexane (open stars) at −25 °C in 1,2-DFB. Reprinted from ref 239. Copyright 2016 American Chemical Society.
More recently, stopped-flow kinetics studies of the fast reactions of 40 and 42 with a range of para-substituted phenols were performed (para-substituents X = NMe2, OMe, Me, H, Cl, NO2, and CF3).247 The data were interpreted to indicate that concerted PCET occurred across the series, except for the most acidic case (X = NO2), for which a pathway involving proton transfer prior to electron transfer (PT/ET) was implicated. Importantly, the high reactivity of 40–43 with C–H and O–H bonds provides key precedence for the notion that the [CuOH]2+ unit could be involved in copper-catalyzed oxidations and might be a more viable intermediate than the more elusive [CuO]+ core.31
3. DICOPPER COMPOUNDS
As noted in section 1, dicopper–oxygen species have been identified as intermediates in the CB-PPO enzymes such as tyrosinase and catechol oxidase and have been under intense discussion as possible reactive species in pMMO (Figure 1, panels c and d). Most commonly, 2:1 Cu:O2 complexes have been prepared by reaction of Cu(I) complexes with O2 at low temperature, via trapping of an initially formed 1:1 Cu/O2 adduct by an additional Cu(I) center. Multiple isomeric structures for 2:1 Cu:O2 complexes are possible (Figure 34). Of these, the (trans-1,2-peroxo), (μ-η2:η2-peroxo), and bis(μ-oxo)dicopper cores are the most well-studied, and their diagnostic spectroscopic properties, structural features, and typical reactivity patterns have been well-documented in previous reviews.15,16,32,34,66,248,249 More recent work on complexes with these cores, other ones shown in Figure 34, and additional moieties comprising single oxo, hydroxo, and hydroperoxo bridges are described below.
Figure 34.
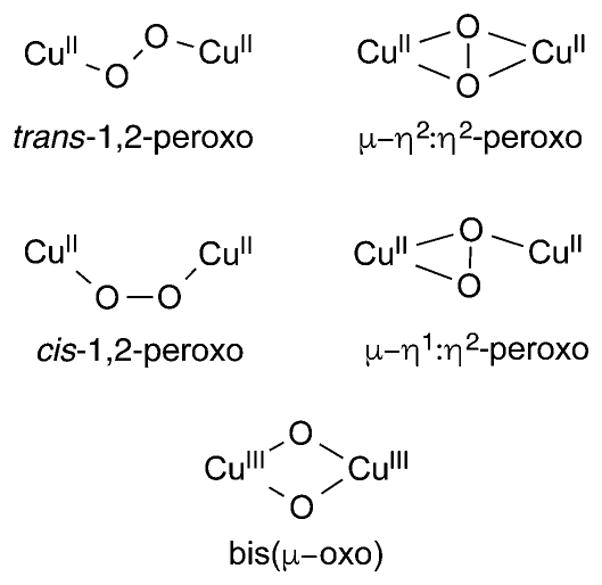
Isomeric cores of 2:1 Cu:O2 complexes.
3.1. (μ-η2:η2-Peroxo)- and Bis(μ-oxo)dicopper Complexes
In view of the evidence that the (μ-η2:η2-peroxo)- and bis(μ-oxo)dicopper cores can readily interconvert, any discussion of the reactivity of one must acknowledge the possible involvement of the other. Nonetheless, the respective cores are differentially stabilized as a result of ligand structural and other influences, such that, in many cases, one or the other is observed as the sole or predominant product of oxygenations of Cu(I) complexes. Thus, in the following discussion we consider complexes of each core in turn and then turn to new insights into the factors that affect their interconversions.
3.1.1. Reactivity of (μ-η2:η2-Peroxo)dicopper Complexes
Since 2004, several new (μ-η2:η2-peroxo)dicopper complexes have been identified and their reactivity examined, with a particular view toward understanding the details of aromatic hydroxylation relevant to tyrosinase function.32 An especially stable (μ-η2:η2-peroxo)dicopper complex was prepared using the extremely hindered ligand L20c (t1/2 = 14 h in MeOH, 9.6 days in aqueous Na2HPO4), and its X-ray crystal structure was determined (Figure 35).250 It exhibits a high ν(O–O) of 773 cm−1 indicative of a strong O–O bond and as a result of the high degree of steric bulk of the supporting ligand does not coexist with a bis(μ-oxo)dicopper isomer, like the system supported by L20b comprising iPr rather than tBu ligand substituents.68,69 The complex effects the catalytic aerobic oxidation of 3,5-di-tert-butylcatechol to 3,5-di-tert-butylquinone and oxidation of benzyl alcohol to benzaldehyde.
Figure 35.
X-ray crystal structure of the (μ-η2:η2-peroxo)dicopper complex dication supported by L20c. Selected interatomic distances (Å): O1a-O1b = 1.475(4), Cu···Cu = 3.6349(8). Reprinted from ref 250. Copyright 2016 American Chemical Society.
Reaction of the Cu(I) complex of L1a with O2 rapidly yielded a (μ-η2:η2-peroxo)dicopper complex 44 identified on the basis of UV–vis and resonance Raman spectroscopy and EXAFS (Figure 36).251,252 Formation of the product followed first-order kinetics, indicative of rate-determining generation of a 1:1 Cu:O2 complex followed by rapid trapping by an additional Cu(I) precursor. Subtle but clear differences in spectroscopic properties among the various complexes 44 with variable counteranions (X) were traced to different counteranion interactions with the dicopper core. It is noteworthy that the formation of 44 only using L1a contrasts with the generation of a mixture of (μ-η2:η2-peroxo)- and bis(μ-oxo)dicopper complexes when the N-methylated variant L1b was used. Preferential stabilization of the μ-η2:η2-peroxo complex by the weaker σ-donating 2° amine ligands in L1a was suggested as a rationale for this difference.
Figure 36.
Reaction of (μ-η2:η2-peroxo)dicopper complexes 44 (supported by L1a) with 2,4-di-tert-butylphenolate, with proposed mechanism based on spectroscopy and theory (refs 251–253).
Importantly, unlike what is typically seen for bis(μ-oxo)-dicopper compounds, 44 did not abstract an H atom from 2,4-di-tert-butylphenol. Moreover, in a reaction directly relevant to tyrosinase function, treatment of 44 with 2,4-di-tert-butylphenolate followed by warming of the reaction mixture results in the formation of a 1:1 mixture of catechol and quinone (Figure 36).251,252 When the reaction was performed at −125 °C, a long-lived (~3 h) intermediate formed which was identified on the basis of UV–vis and resonance Raman spectroscopy and DFT computations as 45, a bis(μ-oxo)dicopper complex in which the phenolate has displaced an arm of L1a to bind in an equatorial position.253 Compound 45 decays via first-order kinetics to an intermediate proposed to be the catecholate adduct 46. Consistent with an electrophilic aromatic substitution pathway, the rate of decay was slowed by electron-withdrawing substituents on the phenolate (Hammett ρ = −2.2) and an inverse 2° kinetic isotope effect was observed upon deuteration at the (hydroxylated) ortho position. Protonation then yields the final products, (semiquinonato)Cu(II) (47) and (aquo)Cu(I) (48) complexes. This work demonstrated that a pathway involving isomerization of the (μ-η2:η2-peroxo)-to a bis(μ-oxo)dicopper species that then attacks bound deprotonated substrate is a viable mechanism that may also occur in tyrosinase.251–253 This reactivity has been effectively exploited in effecting catalytic oxidations of alcohols and phenols.254–256
Assembly of the (μ-η2:η2-peroxo)dicopper core using simple monodentate ligands has been accomplished257–259 by performing synthetic reactions at very low temperature (−125 to −145 °C). This research builds upon earlier work performed under different conditions that did not allow for conclusive identification of the product (Figure 37).260,261 The (μ-η2:η2-peroxo)dicopper core was formed in reactions of simple [(Im)3Cu(I)]+ (Im = 2- or 4,5-alkyl-substituted imidazoles) complexes with O2 in 2-MeTHF at −125 °C.257 The resulting (μ-η2:η2-peroxo)dicopper complexes 49 ligated only by the indicated monodentate imidazoles are stable at −125 °C but decay upon warming (t1/2 = 25 min at −105 °C). These species are highly reactive with sodium phenolates which yield predominantly catechols and lesser amounts of quinones (hydrogen atom abstraction not observed) without an observable intermediate. Addition of excess L42a to the (μ-η2:η2-peroxo)dicopper complex supported by 1,2-dimethylimidazole at −125 °C yields the (trans-1,2-peroxo)dicopper complex 50 bound by L42a, a ligand-exchange process (“core capture”)259 with precedent in copper–oxygen chemistry261 that here also involves isomerization of the Cu2O2 unit. In more recent work,258 the previous failure to observe a (μ-η2:η2-peroxo)dicopper complex using 1- or 4-methylimidazole or unsubstituted imidazole was obviated by an alternate synthesis involving reaction of the known262 bis(μ-oxo)dicopper complex 51 of tetramethylpropylenediamine (L10a) with an excess of the imidazole at −145 °C, a temperature attained by using a 4:1 2-MeTHF:THF eutectic mixture as solvent. Addition of 4 equiv of sodium 15-crown-5,2-tert-butyl-4-cyano phenolate to the resulting (μ-η2:η2-peroxo)dicopper complex 49 at −145 °C yielded a phenolate-bound bis(μ-oxo)dicopper species akin to 45 (Figure 36), but here in a more biomimetic and sterically unencumbered ligand environment comprising imidazoles coordinating via their Nτ positions.
Figure 37.
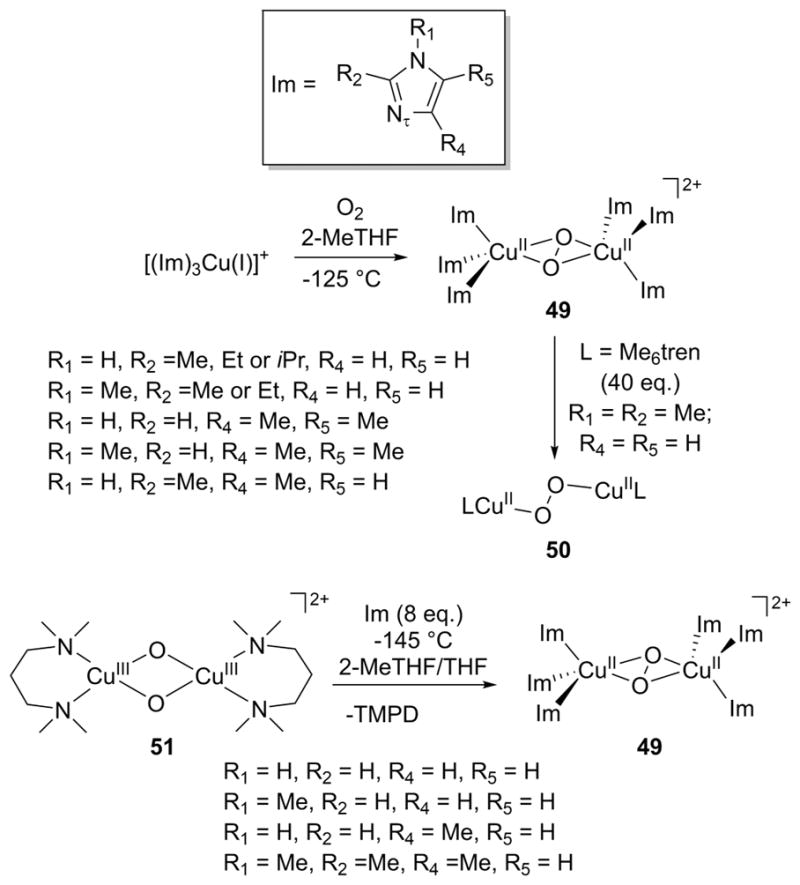
Synthesis of (μ-η2:η2-peroxo)dicopper complexes with simple imidazole ligands (refs 257–259).
A relatively stable (μ-η2:η2-peroxo)dicopper complex (t1/2 ~ 30 min at room temperature) prepared using L37 as supporting ligand was found to convert exogenous phenolates to catechols.263 Kinetics of the stoichiometric reactions of sodium phenolates revealed saturation behavior interpreted to indicate a pre-equilibrium binding step, with studies of phenolate substituent effects on the rate constant giving a Hammett ρ of −0.99. More complicated phenols such as estrone or 8-hydroxyquinoline are also hydroxylated. The stability of the (μ-η2:η2-peroxo)dicopper complex enabled its use as a hydroxylation catalyst, with 25 equiv p-methoxyphenol and 50 equiv NEt3 as well as 10 equiv of quinone formed in 1 h and 15 equiv formed in 24 h. A similar type of reactivity is also observed for copper complexes supported by L15 which effects catalytic conversion of 2,4-di-tert-butylphenol to the corresponding quinone in the presence of trimethylamine with a turnover number of 22.264 Additionally other similar ligand frameworks comprising L9, L46, L16a, or L16b have also been used to promote similar catalytic oxidations.265–267
Oxygenation of a dicopper(I) complex of the hexadentate ligand L58c in acetone at −80 °C yields a UV–vis spectrum consistent with the formation of a (μ-η2:η2-peroxo)dicopper complex (52, Figure 38).268 The kinetics and thermodynamics of O2 binding to form this complex were determined and comparisons to those previously reported for other analogous systems of xylyl-bridged hexadentate dinucleating ligands were drawn.269,270 Importantly, unlike those systems that decayed to give products resulting from intramolecular hydroxylation of the bridging xylyl group, decomposition of 52 yielded a bis(μ-hydroxo)dicopper product with no xylyl hydroxylation observed. Reaction of 52 with sodium phenolates resulted in hydroxylation to yield catechols, and the observed saturation kinetics for the reactions were interpreted to indicate weak preequilibrium binding of the phenolate (ΔH° = −1.9 ± 0.1 kcal mol−1, ΔS° = −2.1 ± 1.5 cal K−1 mol−1) prior to rate-determining electrophilic attack (supported by Hammett ρ = −1.8). No evidence for bis(μ-oxo)dicopper core formation was observed, suggesting that in this system the μ-η2:η2-peroxo unit is responsible for the phenolate hydroxylation. In a separate study, 52 was shown to effect the oxidation of thioanisole to the sulfoxide.271 The reaction is slow, however [k = (12 ± 1) × 10−5 s−1], so the more reactive system supported by L58a272,273 was examined. Catalytic sulfoxidation was observed upon treatment of a dicopper(II) complex of L58a with reductant and O2, and it was proposed that a reactive intermediate 53 was involved (Figure 38). In a more recent study using L58d, which employs chiral alkyl substituents on the benzimidazole instead of the achiral methyl group in L58a, the corresponding copper complexes were used to affect stereoselective oxidation reactions of enantiomeric mixtures of catechols and thioansioles.274
Figure 38.
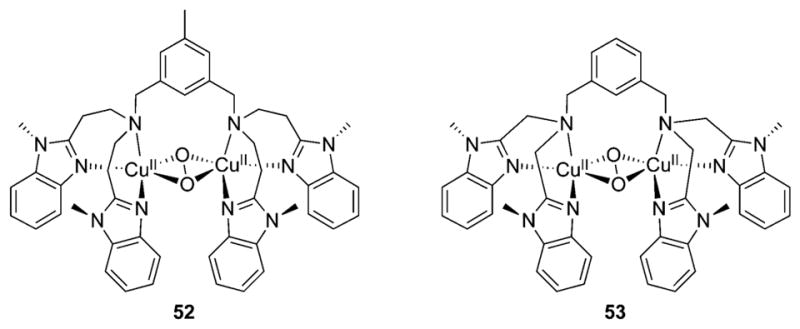
Proposed (μ-η2:η2-peroxo)dicopper complexes supported by L58c (52; ref 268) and L58a (53; refs 272 and 273).
In contrast to what was observed for the system supported by L58c, oxygenation of dicopper(I) complexes of dinucleating ligands featuring aromatic bridges often results in hydroxylation of those bridges, as originally described for xylyl complexes (Figure 39).275,276 In several cases, no oxygenated intermediate was observed,181,277 and in some of these instances a (μ-η2:η2-peroxo)dicopper species was proposed on the basis of precedent or DFT calculations.278,279 In other cases, a (μ-η2:η2-peroxo)-dicopper was observed by spectroscopy and insights into the kinetics and thermodynamics of its formation and/or intramolecular hydroxylation reactivity were attained.280–282 For example, kinetic studies of the oxygenation and subsequent intramolecular hydroxylation reactions of Cu(I) complexes of L58g and L58h and the asymmetric ligand L50a were performed.280 A low ΔH‡ for the oxygenation of the L58g and L58h complexes was rationalized by postulating a left-lying pre-equilibrium formation of a 1:1 Cu:O2 adduct [i.e., CuICuII(O2−) species] that slowly evolved to a (μ-η2:η2-peroxo)dicopper complex (Figure 40). Both the rates of oxygenation and the intramolecular hydroxylation increased as the ligand was rendered more electron-donating (L58h > L58g > L58f). Slow ligand hydroxylation for the system supported by L50a was proposed to reflect “a less than ideal proximity or orientation of the complex’s electrophilic peroxo group toward the arene pi system.”
Figure 39.
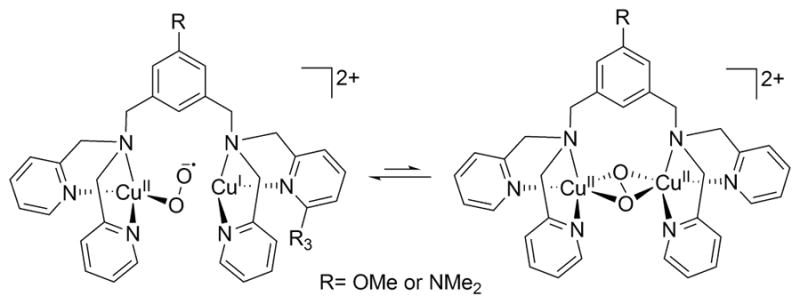
Ligand hydroxylation reactions of (μ–η2:η2-peroxo)-dicopper complexes supported by L58f–h (54; refs 275 and 277) or L58i (R3 = Me, R = H, OMe, tBu, and NO2) (55; ref 281).
Figure 40.
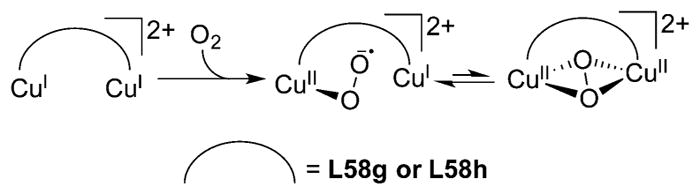
Proposed formation of an intermediate CuICuII(O2−•) species (ref 280).
In another study of a related system supported by L58i (Figure 39, R3 = Me; R = H, OMe, tBu, NO2), the (μ-η2:η2-peroxo)dicopper intermediate 54, implicated in the hydroxylation of the bridging arene ring to yield 55, was detected by UV–vis and resonance Raman spectroscopy.281 Once again, a Hammett ρ value of −1.9 was observed, consistent with electrophilic attack at the ring and in agreement with the value measured for tyrosinase. DFT calculations benchmarked by Cu L-edge XAS for R = NO2 corroborated this conclusion (via comparison to the bis(μ-oxo)dicopper complex of L1a).282,283 The computationally proposed mechanism involves transfer of π electrons from the bridging arene to the peroxo σ* orbital and O–O bond scission in the key reaction step, rather than prior isomerization to a bis(μ-oxo)dicopper isomer. In novel transformations for the (μ-η2:η2-peroxo)dicopper moiety, for R = H, trapping of 54 by excess styrene and solvent THF was observed to compete with intramolecular hydroxylation, to yield styrene oxide or 2-hydroxy-THF, respectively. In addition, the (μ-η2:η2-peroxo)dicopper core has been implicated in hydrogen atom abstractions, reactions that are unusual for this type of core. This reactivity has been seen for a range of substrates with C–H bond dissociation enthalpies between 75 and 92 kcal/mol, with key evidence being a linear log k vs C–H bond BDE plot and large H/D kinetic isotope effects.282 Additional studies focused on variation of para-substituents on the pyridine rings with the same ligand backbone, revealing that the rate of O2 binding increases with increasing electron-donation.280 Radical chemistry was implicated in reactions of several other (μ-η2:η2-peroxo)dicopper complexes.284–287 Conclusive evidence for the formation of a (μ-η2:η2-peroxo)dicopper complex upon reaction of a Cu(I) complex of L8 ligands was reported on the basis of UV–vis and resonance Raman spectroscopy, XAS, magnetic susceptibility measurements, and DFT calculations.284 Hydrogen atom abstraction without hydroxylation was observed upon reaction of the complex with R = tBu with 2,4-di-tert-butylphenol.
Radical coupling of phenols was observed for the (μ-η2:η2-peroxo)dicopper complex of L52, which was characterized by UV–vis spectroscopy and XAS.285 Spectroscopic evidence indicated that peroxo complexes of ill-defined structure formed upon reaction of Cu(II) complexes of L32 and L51a-c with H2O2 in water,286 and trapping experiments suggested that hydroxyl radicals formed upon their decay that were implicated in DNA cleavage reactions.287
A mechanistic study probed the involvement of the (μ-η2:η2-peroxo)dicopper complex (58) of L51a in the catalytic reduction of O2 to H2O by a Cu(II) complex (56) using decamethylferrocene (Fc*) and CF3CO2H as reductant and proton source, respectively (Figure 41).288 The proposed mechanism involves rapid electron transfer from Fc* to 56 to yield the dicopper(I) complex 57, the kinetics of which were analyzed by Marcus theory. Subsequent oxygenation of 57 yields 58, a known reaction.289 The rate of reduction of 58 by Fc* is unaffected by CF3CO2H, supporting the stepwise reduction/protonation sequence. Because of the possibility of rapid equilibration of 58 with a bis(μ-oxo)dicopper isomer 59, the possibility that reduction/protonation occurs from 59 was examined. Analysis of activation parameters for reduction of 58 involving comparison to those obtained for the bis(μ-oxo)-dicopper complex of L18a (see below) led to the conclusion that direct reduction of 58 occurred.
Figure 41.

Proposed mechanisms for the catalytic reduction of O2 to H2O by 56 (L51a) in the presence of exogenous Fc* as a reductant (ref 288).
3.1.2. Reactivity of Bis(μ-oxo)dicopper Complexes
With the aim of understanding the effects of supporting ligands on the properties of the bis(μ-oxo)dicopper core and accessing accurate and functional models of purported biological intermediates, new examples of bis(μ-oxo)dicopper complexes have been prepared and their reactivity probed. One approach has centered on using ever-simpler and less sterically encumbered N-donor supporting ligands, with the goal of enhancing reactivity of this core with exogenous substrates and better mimicking the putative active site of pMMO that features the “histidine brace” (Figure 1d). In a systematic study building upon earlier work,290,291 a set of N-peralkylated diamine ligands (L6a–e, L1d–f), as well as tridentate polyazacyclononane ligands (L20a and L84), studied for comparative purposes, were used to prepare nine different bis(μ-oxo)dicopper complexes by reacting monocopper(I) starting complexes with dioxygen in various solvents.292 The formulations of the products were confirmed by EPR, UV–vis, and resonance Raman spectroscopy, as well as an X-ray structure in one case (supported by L6c; Figure 42). Among the key findings was that the rate of oxygenation of the Cu(I) complex of L6d was ~300 times slower than others with methyl substituents. This was interpreted to indicate an associative mechanism for initial 1:1 Cu:O2 adduct formation that is slowed by the larger ethyl groups in L6d. The least hindered complex of L6c was the most stable, a discovery that was noted to have positive implications for future studies of reactivity with external substrates that might have greater access to the bis(μ-oxo)dicopper core if smaller supporting ligands were used.
Figure 42.
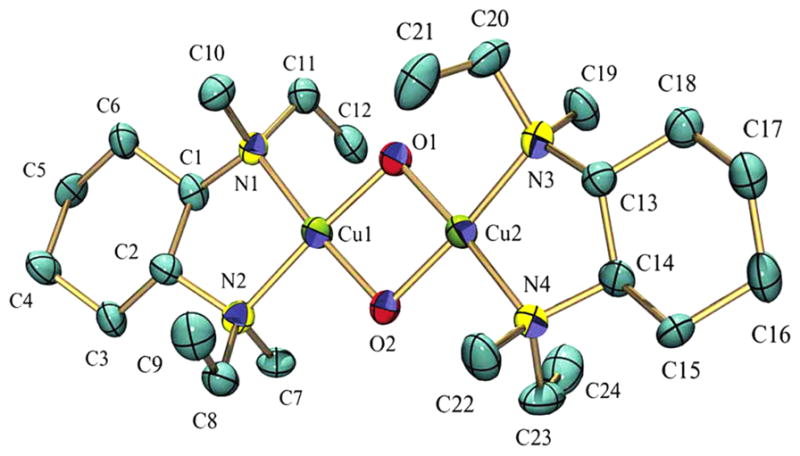
X-ray crystal structure of the bis(μ-oxo)dicopper complex supported by L6c. Reprinted from ref 292. Copyright 2005 American Chemical Society. Selected interatomic distances (Å): Cu(1)–O(1), 1.809(6); Cu(1)–O(2), 1.808(6); Cu(2)–O(1), 1.795(5); Cu(2)–O(2), 1.799(6); Cu(1)···Cu(2), 2.744(1); and O(1) ···O(2), 2.334(1).
Support for this idea came from studies of the system supported by N,N,N′,N′-tetramethylethylenediamine (L1c).293 It had been found previously that oxygenation of the Cu(I) complex of this sterically unencumbered ligand yielded a bis(μ-oxo)tricopper(II,II,III) complex arising from reaction of an initially formed bis(μ-oxo)dicopper intermediate with a [(L1c)-Cu(I)]+ moiety.291,294 By performing the oxygenation at low concentrations of Cu(I) (<2 mM), the dinuclear complex was prepared preferentially (Figure 43). Importantly, the complex was found to be particularly stable toward decomposition, enabling the oxidation of benzyl alcohol to benzaldehyde, a new reaction for bis(μ-oxo)complexes supported by N-peralkylated diamines.
Figure 43.
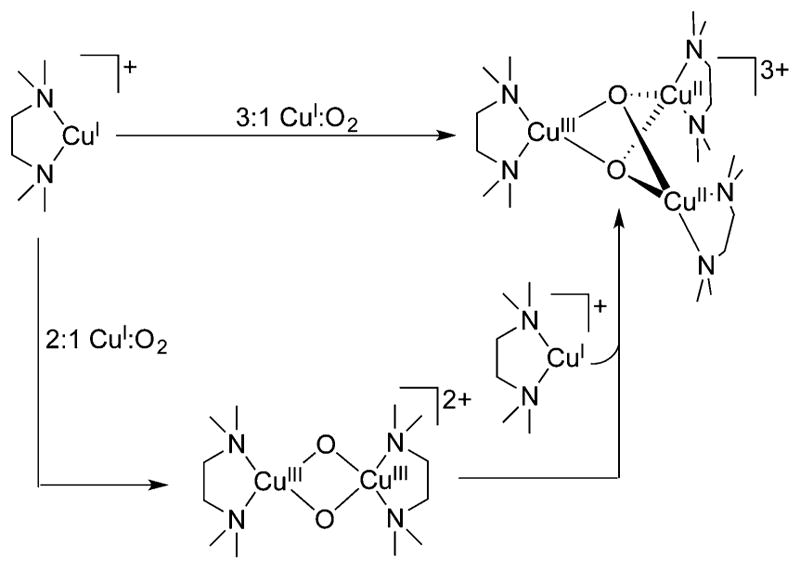
Controlled formation of di- and tricopper complexes, [(L1c)Cu2O2]2+ and [(L1c)Cu3O2]3+, via selective addition of dioxygen (refs 291 and 294).
Even less sterically hindered complexes comprising primary amine donors were then targeted, a key goal being to model the RNH2 coordination found in the proposed active site of pMMO. Direct oxygenation of Cu(I) complexes of primary amines failed to yield isolable products, so the “core capture” method was used as described in section 3.1.1.295,296 Thus, the bis(μ-oxo)dicopper complex (51) of L10a was prepared by reaction of a Cu(I) precursor with O2, and then this complex was treated with another ligand (2 equiv) at −125 °C to rapidly yield new bis(μ-oxo)dicopper products 61–63 (Figure 44). The shown thermodynamic stability order was determined through mixing experiments and DFT calculations. The stabilization of the complexes by primary amine and histamine ligands arises from stronger metal–ligand interactions, as reflected by blue-shifted LMCT features in UV–vis spectra caused by higher energies of acceptor orbitals. These strengthened interactions were proposed to arise not from greater ligand basicity but from decreased hindrance that facilitates shorter metal–ligand bonds. The histamine ligation in 62 and 63 is notable with respect to its similarity to the histidine brace in the proposed active sites of pMMO and LPMO. Compounds 61–63 were found to be capable of HAT from substrates with weak C–H bonds (74–76 kcal/mol) even at −125 °C, with steric accessibility to the bis(μ-oxo)dicopper core being key for substrate access.
Figure 44.

Stability order of bis(μ-oxo)dicopper complexes. Adapted from ref 296.
The involvement of a bis(μ-oxo)dicopper unit in catalytic oxidations has been proposed in both homogeneous297 and heterogeneous systems.298 Oxygenation of solutions of Cu(I) complexes of β-diketiminate ligands L2a and L2b or treatment of the corresponding Cu(II)-acetato complexes with H2O2/NEt3 afforded bis(μ-oxo)dicopper complexes as shown by UV–vis and resonance Raman spectroscopy. Oxidation of cyclohexane (2.5 M) to cyclohexanol (~20% yield) and cyclohexanone (~6% yield) was effected by the Cu(II)-acetato complexes (0.83 mM) and H2O2 (83 mM). Catalysis did not proceed when ligands with Me groups in the position adjacent to the N-donors were used (L2c and L2h), and with these ligands bis(μ-oxo)dicopper complexes were not formed upon treatment with H2O2/NEt3, presumably for precedented steric reasons.149 These results were interpreted to support a mechanistic hypothesis that the bis(μ-oxo)dicopper core (activated by the electron-withdrawing ligand substituents) was responsible for attacking the cyclohexane substrate.
Catalytic oxidation of toluene to benzaldehyde was performed by a Cu(II) complex of ligand L34 immobilized within the nanochannels of functionalized mesoporous silica nanoparticles. 298 The involvement of a bis(μ-oxo)dicopper complex was inferred on the basis of the results of experiments wherein the immobilized complex was first reduced by ascorbate and then exposed to O2. UV–vis spectroscopy and XAS data were consistent with formation of a bis(μ-oxo)dicopper core. Confinement of this core and O2 within the nanoparticles was argued to be critical for the high levels of catalytic activity observed in what was determined by kinetics to be a consecutive process: toluene → benzyl alcohol → benzaldehyde.
The catalytic reduction of O2 to H2O by Fc* and CF3CO2H was examined using a Cu(II) complex of L18a (64), and a mechanism involving initial reduction to a Cu(I) species that then reacts with O2 to yield a bis(μ-oxo)dicopper core (65) was proposed (Figure 45).288 Consistent with this pathway, reaction of the independently prepared bis(μ-oxo)dicopper complex with Fc* occurred rapidly upon mixing at a rate that was not influenced by added CF3CO2H.
Figure 45.
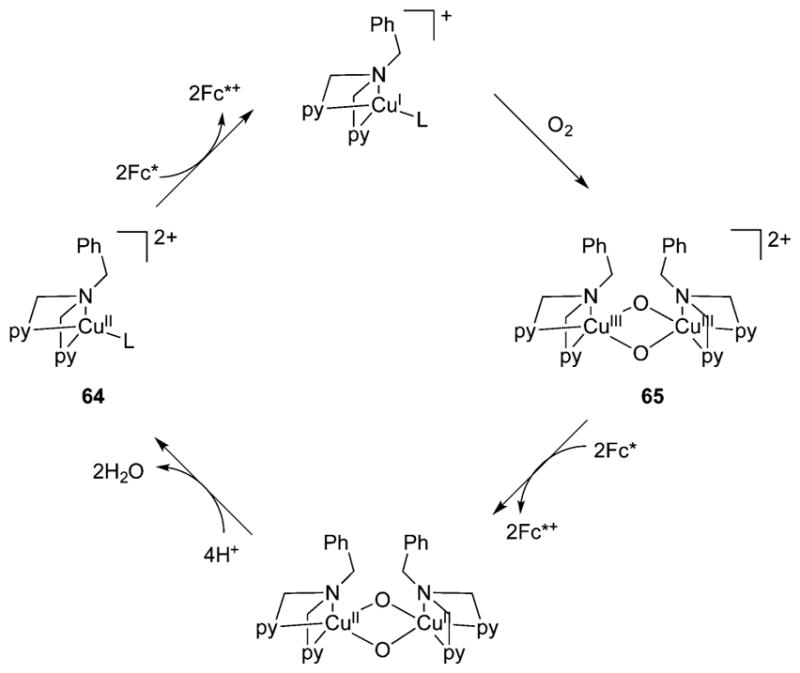
Proposed mechanism for the reduction of O2 to H2O invoking the intermediacy of the bis(μ-oxo)dicopper (65) complex supported by L18a (ref 288).
Using a ligand that incorporates elements of previously studied systems comprising bis[2-(pyridin-2-yl)ethyl]amine derivatives and amines, a hybrid ligand L24 was used to prepare a bis(μ-oxo)dicopper complex.299 This product was identified by UV–vis spectroscopy and found to generate radicals or derived coupling products upon reaction with phenols.
In work following up to the previously reported discovery that oxygenation of a Cu(I) complex of L5 leads to hydroxylation of its appended arene group via the intermediacy of a bis(μ-oxo)dicopper complex,300 DFT calculations of this system and studies of a related and synthetically more readily accessible ligand L7 were performed.301 Oxygenation of a Cu(I) complex of L7 also resulted in arene hydroxylation, with subsequent hydrolysis yielding aldehydes as the final products (Figure 46). The formation of a bis(μ-oxo)dicopper intermediate was supported by the observation of an optical absorption at 400 nm in low-temperature stopped-flow kinetic experiments. Importantly, within the context of the viability of bis(μ-oxo)dicopper core as an oxidant capable of tyrosinase activity, DFT calculations supported rapid conversion of an initially formed (μ-η2:η2-peroxo)dicopper complex to the more stable bis(μ-oxo)dicopper isomer, which then performed the electrophilic attack at the arene.
Figure 46.
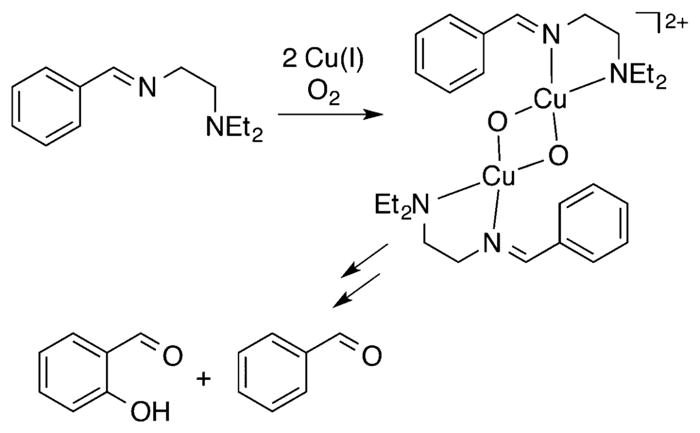
Proposed pathway for hydroxylation of an appended arene of the L7 ligand (ref 301).
In a related strategy, the appended phenol in ligand L80a was oxidized to a quinone upon reaction of its Cu(I) complex with O2.302 The same quinone product was formed upon oxygenation of the Cu(I) complex of L80b, supporting initial catechol formation in the overall reaction of L80a. By analogy to the finding that the system supported by a ligand analog comprising a phenyl (L18b–f) instead of a phenol appendage yields a bis(μ-oxo)dicopper complex, a similar intermediate was invoked, with support from DFT calculations (Figure 47).
Figure 47.
DFT geometry-optimized structure for the bis(μ-oxo)-dicopper intermediate proposed in the oxidation of the appended phenol in L80a. Reprinted with permission from ref 302. Copyright 2015 the Royal Society of Chemistry.
The diimine ligand L11,303 bis(guanidine) ligands L14 and L13,304,305 and the hybrid guanidine-amine ligand L27306 were found to support formation of bis(μ-oxo)dicopper complexes upon oxygenation of Cu(I) precursors. The complex supported by L14 decayed to yield alkoxo-bridged products derived from hydroxylation of ligand methyl groups.305 A comparison of the reactivity of the bis(μ-oxo)dicopper complexes 66, 67, and 51 supported by L14, L27, and L10a,262 respectively, with 2,4-ditert-butylphenol and -phenolate revealed intriguing differences (Figure 48). Complex 66 was unreactive, 51 gave radical coupling products upon reaction with both the phenol and phenolate, and 67 coupled 2,4-di-tert-butylphenol and hydroxylated 2,4-di-tert-butylphenolate. These results were rationalized by invoking the greater basicity and stronger σ-donating power of the guanidate. With two such guanidate groups in 66, stabilization of the bis(μ-oxo)dicopper core is sufficient to shut down oxidative reactivity, whereas in the hybrid ligand system 67, this effect is attenuated and both radical and hydroxylation reactions are observed. As in other systems that hydroxylate phenolates, saturation kinetics were observed, consistent with association of the phenolate to the bis(μ-oxo)dicopper core prior to electrophilic attack. It is also noteworthy that the reactions of 51, 66, and 67 with 2 equiv. FcCO2H (an electron and proton donor) results in conversion to a bis(μ-hydroxo)-dicopper(II) complex, with an intermediate (unobserved) (μ-oxo)(μ-hydroxo)Cu(II)Cu(III) complex proposed on the basis of DFT calculations.306
Figure 48.
Comparison of the reactivity of set of bis(μ-oxo)dicopper complexes 51 (L10a), 66 (L14), and 67 (L27) with 2,4-di-tertbutylphenol (“phenol”) and 2,4-di-tert-butylphenolate (“phenolate”) (ref 306).
Identification of a bis(μ-oxo)dicopper intermediate that decays via hydroxylation of a bridging aryl unit in a dinucleating ligand was reported for the system supported by L47.307 This finding stands in contrast to the more common observation of (μ-η2:η2-peroxo)dicopper intermediates with related xylylbridged ligands (see section 3.1.1). A related hydroxylation where an unobserved bis(μ-oxo)dicopper species is a possible intermediate has been reported.308 It is noteworthy that in addition to attacking its bridging aryl unit, the bis(μ-oxo)-dicopper complex of L47 also reacts with 2,4-di-tert-butylphenolate to give both radical coupling and ring hydroxylation products (catechol plus trace quinone).307
A different xylyl-bridged dinucleating ligand was found to support formation of a bis(μ-oxo)dicopper complex that hydroxylates phenolates.39,309,310 Comparison of the reactivity of dicopper(I) complexes of ligands L57 and L48 revealed different behavior, wherein only L48 supported formation of a copper–oxygen intermediate, identified as 68 on the basis of UV–vis and resonance Raman spectroscopy (Figure 49).309 Treatment of 68 with various para-substituted phenolates resulted in clean catechol formation (with no complications from intramolecular arene hydroxylation), and in the case of the reaction with p-chlorophenolate performed at −95 °C, the bis(μ-oxo)dicopper phenolate adduct 69 was identified as an intermediate by resonance Raman spectroscopy.310 The properties and key kinetic parameters for the decay of 69 (i.e., Hammett ρ = −1.9) are similar to those reported for 45 supported by L1a (Figure 36), corroborating their similar structures.
Figure 49.
Reaction of 68 (L48) with phenolates (69) identified for X = Cl (refs 309 and 310).
Finally, we note that an earlier report of the synthesis of a bis(μ-oxo)dicopper complex supported by L23a,311 an open chain analog of the L20 framework, was followed by a low-temperature stopped-flow study of the kinetics of its formation.164 A comparison to a series of other related oxygenations revealed the reaction of the Cu(I) complex of L23a to be significantly faster, as illustrated by a kon for the initial binding step for L23a being ~107 greater than for L20b.68,269
3.1.3. Interconversions of (μ-η2:η2-Peroxo)- and Bis(μ-oxo)dicopper Cores
Since 2004, a number of studies have further examined the factors that influence the relative stabilities of these isomeric cores and the possible interconversions between them. The roles of ligand structural variations on the reactivity of Cu(I) complexes of a large set of pyridyl-amine ligands have been examined particularly extensively over several decades and the results reviewed.312,313 In work reported after 2004, the ligand L38b was used to draw comparisons with the properties and O2 reactivity of previously studied Cu(I) complexes of L38a and L38c (illustrated for R = CH2CH2Ph in Figure 50).314 The Cu(I) complex of L38b exhibited an oxidation potential and rate of reaction with O2 intermediate between those of L38a and L38c. Upon oxygenation it yielded a (μ-η2:η2-peroxo)dicopper complex like that formed using L38a315,316 but with a weaker O–O bond as reflected by a lower ν(O–O) (Δν = ~20 cm−1). This result contrasts with the formation of a bis(μ-oxo)dicopper complex that had been reported previously using L38c.317 Thus, the 6-methyl substituents reduce the electron-donating power of the ligand framework that L38b and L38c share, apparently by weakening the Cu–N(pyridine) bonds through steric repulsions, thus inhibiting O–O bond scission. Related steric effects caused by quinolyl groups also led oxygenation of the Cu(I) complex of L35b to yield a (μ-η2:η2-peroxo)dicopper complex.318
Figure 50.
Comparison of results of oxygenation of Cu(I) complexes of the indicated ligands (R = CH2CH2Ph), which yield either the indicated (μ-η2:η2-peroxo)- or bis(μ-oxo)dicopper cores (refs 314–317).
Building upon previous work probing solvent effects on the (μ-η2:η2-peroxo)/bis(μ-oxo)dicopper equilibrium,69,291 this equilibrium was examined in detail as a function of solvent for the system involving L21a.319 The operation of a rapid equilibrium between the two isomers supported by L21a was confirmed by low-temperature stopped-flow kinetics. UV–vis and resonance Raman spectra showed the proportion of bis(μ-oxo)dicopper isomer formed followed the order CH2Cl2 < Et2O ~ acetone < THF, consistent with greater stabilization of this isomer by more strongly coordinating solvents. Low-frequency features associated with Cu–Neq and Cu---Cu modes in resonance Raman spectra for the (μ-η2:η2-peroxo)dicopper isomer shifted as a function of solvent similarly, but the Cu2O2 core vibration of the bis(μ-oxo)dicopper core was invariant. These results were interpreted to indicate that the solvent coordinates to the (μ-η2:η2-peroxo)dicopper core, but how the overall thermodynamics favoring the other isomer are influenced by the solvent remained unclear.
Electronic effects on the equilibrium were examined in a subsequent comparative study of the series L21 (R = NMe2, OMe, H, and Cl).320 Varying the para substituents had negligible effects on the oxidation potentials of their Cu(I) complexes and on the ν(C–O) stretches in their Cu(I)-carbonyl complexes, but the bis(μ-oxo):(μ-η2:η2-peroxo) ratio formed upon oxygenation of the Cu(I) complexes was significantly influenced (in contrast to more minor effects on that ratio reported previously for the L32 system).321,322 Thus, this ratio increased as the supporting ligand became more electron-donating, consistent with enhanced stabilization of the more oxidized copper sites in the bis(μ-oxo)dicopper core (Figure 51). This trend was seen using both noncoordinating CH2Cl2 and coordinating THF as solvent, but for CH2Cl2, it was attenuated for R = OMe, H, and Cl, such that the proportions of the (μ-η2:η2-peroxo)dicopper isomer were increased in that solvent. Only the bis(μ-oxo)dicopper isomer was seen for R = NMe2 using either solvent, indicating that the solvent effects are secondary to the electronic ones propagated by the para substituents on the ligand.
Figure 51.
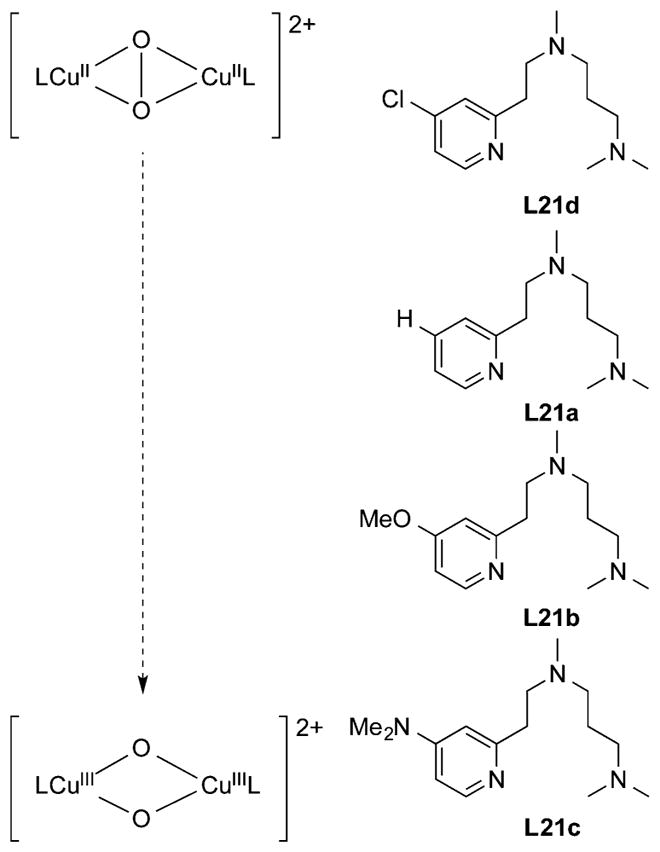
Variation in ratio of isomers formed as a function of the substituent in the L21 supporting ligand (ref 320).
In the above study, enhanced electron donation by the supporting ligand results in reductive cleavage of the O–O bond, which has been postulated to occur via “backbonding” into the μ-η2:η2-peroxo σ* orbital. Weakening of the O–O bond without such reductive bond scission was defined in an X-ray structure of the (μ-η2:η2-peroxo)dicopper complex of L23b,323 which had been shown previously to have a low ν(O–O) of 721 cm−1.324 A long O–O bond of 1.540(5) Å was measured, and EXAFS and resonance Raman data confirmed that no bis(μ-oxo)dicopper isomer was present, ruling out compositional disorder as the reason for observation of the long O–O distance. This observation was rationalized using DFT calculations by invoking a trans-influence of the supporting ligand that “decreases the O22− π*σ-to-Cu charge transfer (which) results in more electron density in the π antibonding orbitals of the peroxide and thus the weaker O–O bond,”323 a process distinct from the backbonding to the σ* orbital that induces formation of the bis(μ-oxo) dicopper core.
A detailed study of the reactivity of the mixture of (μ-η2:η2-peroxo) and bis(μ-oxo)dicopper complexes supported by L32 (R = H, MeO, and Me2N) provided insights into redox behavior, mechanisms of attack at various exogenous substrates, and mechanisms of PCET reactions.325 Among the findings was the discovery that for reactions with exogenous substrates like THF or dimethylaniline, pre-equilibrium binding of substrate occurs prior to oxidation. Using the mechanistic probes Ncyclopropyl-N-methylaniline (CMA) and (p-methoxyphenyl)-2,2-dimethylpropanol (MDP), it was concluded that the systems supported by L32 (R = H, MeO) reacted by a CPET (concerted proton electron transfer) pathway, whereas for R = NMe2, a consecutive ET/PT (electron transfer/proton transfer) mechanism is followed. Complicating the interpretation of the results of these studies is the presence of an equilibrium between (μ-η2:η2-peroxo)- and bis(μ-oxo)dicopper isomers, either or both of which may be the reactant in each case.
Subtle ligand geometric factors had significant effects on the course of oxygenations of Cu(I) complexes of the ligand series L22, L26, L29, and L33, including on the ratios of (μ-η2:η2-peroxo)- and bis(μ-oxo)dicopper isomers (Figure 52).326 The Cu(I) complex of the 6-membered ring ligand L26 was reactive with O2, however an intermediate was not observed. The complex with the 7-membered ring L29 yielded a bis(μ-oxo)dicopper core, the complex with the 8-membered ring L33 formed a 1:1 Cu:O2 adduct, and the complex of the noncyclic ligand L22 yielded a mixture of (μ-η2:η2-peroxo)- and bis(μ-oxo)dicopper products. These differences were rationalized using electrochemical data and analysis of X-ray crystal structures of the Cu(I) complexes. Bite angle and Cu–N distance constraints associated with the macrocycles in L26, L29, and L33 were deemed responsible for the observed Cu(I)/O2 reactivity. While L33 and L22 contain similar propyl linkers between ligand N-donors, the rigidity of the former prevented attainment of proper geometries to support (μ-η2:η2-peroxo)- or bis(μ-oxo)dicopper isomers, while flexibility in the latter enabled formation of both cores as a mixture. In another example of subtle ligand geometry changes influencing the stability of these isomers, DFT calculations indicated that simply changing one methylene linker in L49a to an ethyl linker (L49b) shifted the preference for formation of a bis(μ-oxo)dicopper complex to the (μ-η2:η2-peroxo)dicopper congener. 327
Figure 52.
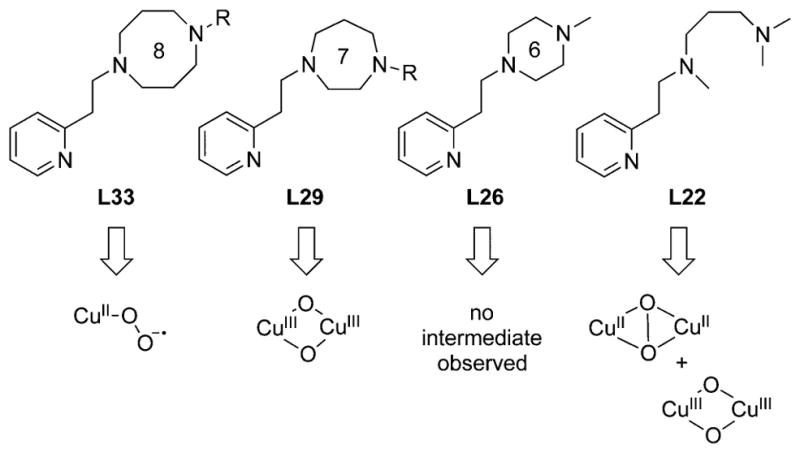
Results of the reactions of the Cu(I) complexes of the indicated ligands with O2 (ref 326).
In addition to solvent and ligand electronic, steric, and geometric influences, counterions also were found to affect the relative stability of the (μ-η2:η2-peroxo)- or bis(μ-oxo)dicopper cores.328 Oxygenation of the complex [(L10b)Cu(CH3CN)]X in THF, CH2Cl2, or acetone yielded an equilibrating mixture of the two cores, the ratio of which depended on the identity of X. With a relatively noncoordinating anion such as SbF6−, the bis(μ-oxo)dicopper isomer is favored. More basic anions like CH3SO3− or PhCO2− favor the (μ-η2:η2-peroxo)dicopper form, with titration data for the most basic ones indicating association of one anion per dicopper complex. EXAFS supported by DFT calculations indicated bidentate bridging coordination of the anion to the syn axial positions of the (μ-η2:η2-peroxo)dicopper unit, thus rationalizing stabilization relative to its bis(μ-oxo)dicopper isomer. Analogous stabilization of a (μ-η2:η2-peroxo)dicopper core was proposed in a study of decarboxylation of α-ketocarboxylates, where binding of benzoylformate or benzoate were proposed to convert the bis(μ-oxo)dicopper complex of L10a to the carboxylate-bridged (μ-η2:η2-peroxo)-dicopper unit.329
The presence of an appropriate thioether appendage on a diamine ligand was found to change the preference for formation of the respective [Cu2O2]2+ cores (Figure 53).330 Thus, oxygenation of a Cu(I) complex of L73 proceeds similarly to that of L10a262 to give a bis(μ-oxo)dicopper complex, whereas the alkylthioether group in L72 induces generation of a (μ-η2:η2-peroxo)dicopper core through metal coordination (thus acting as a tridentate ligand analogous to L23b,324 which gives the same isomer). Previous studies of the O2 reactions with Cu(I) complexes of ligands similar to L72 and L73, but with pyridyl arms (L76 and L77), resulted in sulfoxidations with no copper–oxygen intermediates observed.331
Figure 53.
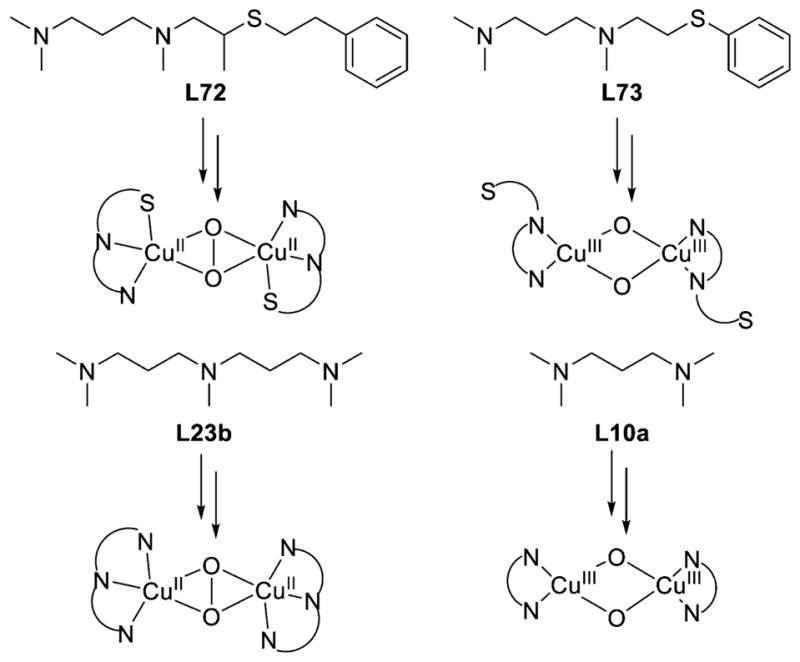
Comparison of the results of oxygenations of Cu(I) complexes of the indicated ligands (refs 262, 324, 330, and 331).
Predicting the relative stability of the (μ-η2:η2-peroxo) and bis(μ-oxo)dicopper cores by theory has been pursued by many investigators but with widely varying results as a function of methodology used. A full elaboration of these difficulties is beyond the scope of the current discussion, so we point the interested reader to two insightful and comprehensive discussions.332,333
3.1.4. Heterobinuclear Bis(μ-oxo) Complexes
In efforts to expand the pallet of bi(μ-oxo)dimetal complexes with the aim of discovering new reactivity of potential relevance to catalytic oxidations, several synthetic strategies toward such species containing a copper ion have been pursued.167,168,334–336 In one approach, a 1:1 metal/O2 adduct was mixed with a second metal reagent (Figures 54 and 55).167,334–336 Thus, reaction of the 1:1 Cu:O2 adduct 3a (see Figure 5) with Ni(I) complex 70 (L81) generated a mixture of 71 (major) and 72 (minor) (Figure 54), formulated as having the indicated isomeric cores on the basis of UV–vis, EPR, and resonance Raman spectroscopy [ν(O–O) and [CuNi(μ-O)2]2+ core vibration at 847 and 625 cm−1, respectively].167 An opposite route was taken in preparing compounds 73a–h and 74, whereby a 1:1 M:O2 adduct [for example, M = (Ph3P)2Pd or Pt,334 or (L2d)Ni335] was treated with a Cu(I) complex of ligands L23c, L2d, L10a, L6a, or L20b (Figure 55). UV–vis and resonance Raman data supported the indicated formulations of the products (Table 6), with additional XAS/EXAFS and computational results provided for 74 (Cu–Ni = 2.81 Å).335
Figure 54.

Synthesis of Cu-containing heterobimetallic complexes using 3a (L2d,e) and 70 (L81) as the starting materials (ref 167).
Figure 55.
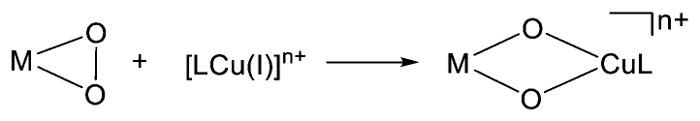
General scheme showing the synthesis of Cu-containing heterobimetallic complexes using 1:1 M:O2 adducts as starting materials (see Table 6 for specific M and L combinations).
Table 6.
Properties of Heterobimetallic LCu-(μ-O)2-M Complexes
| compound | L | M | solvent | UV–vis λmax (nm) (ε, M−1 cm−1) | Raman (exp, cm−1) ν(CuMO2) (18O) | ref |
|---|---|---|---|---|---|---|
| 73a | L4c | Pd(PPh3)2 | THF | 448 (5900) | 660 (631) | 334 |
| 73b | L4c | Pt(PPh3)2 | 4:1 CH2Cl2:THF | 450 (5600) | 628, 613 (594) | 334 |
| 73c | L10a | Pd(PPh3)2 | CH2Cl2 | 472 (2800) | 610 (580) | 334 |
| 73d | L10a | Pt(PPh3)2 | CH2Cl2 | 483 (2700) | 595 (569) | 334 |
| 73e | L6a | Pd(PPh3)2 | CH2Cl2 | 463 (3500) | 640, 616 (600) | 334 |
| 73f | L6a | Pt(PPh3)2 | CH2Cl2 | 457 (3600) | 647, 616 (603) | 334 |
| 73g | L20b | Pd(PPh3)2 | CH2Cl2 | 458 (2500) | 630 | 334 |
| 73h | L20b | Pt(PPh3)2 | CH2Cl2 | 462 (3000) | 628 (601, 585) | 334 |
| 71 | L2d | Ni(L81) | THF | 499 (8300) | 625 (595) | 167 |
| 74 | L23b | Ni(L2d) | THF | 895 (5000) | 625 (595) | 335 |
| 75 | L4c | Ge(N(SiMe3)2)2 | toluene | 440 (4400) | 578 (559, 546) | 168 |
Reactivity distinct from that typical of bis(μ-oxo)dicopper complexes was seen in several studies of the heterobimetallic complexes. For example, [NH4][PF6] protonated 73b (ESIMS), while reaction with CO2 led to a (PPh3)2PtII–CO3 adduct. No reaction of 73a–h was observed with DHA, thioanisole, or 1-decene, but a coupled biphenol was observed upon treatment with 2,4-di-tert-butylphenol.334 Taken together, the reactivity of compounds 73a–h is indicative of nucleophilic character that contrasts with what is generally seen for bis(μ-oxo)dicopper complexes but is in line with the norm for bis(μ-oxo)diplatinum complexes.337,338 Studies of 74 also show its O atoms to act as nucleophiles.336 Thus, reaction of 74 with benzoyl chloride led to formation of benzoic acid, and examination of the kinetics of the reactions with a series of para-substituted benzoyl chlorides revealed a Hammett ρ = 2.5. On the other hand, examination of the kinetics of the reactions of 74 with phenols showed for R = H on the supporting ligand L23c, HAT occurred like what was seen for the bis(μ-oxo)dicopper species. For R = Me, both HAT and PCET mechanisms were followed depending on the phenol, highlighting the subtle effect ligand substituents can have on mechanisms of reactions with exogenous substrates.
In a different synthetic route to heterobimetallic complexes, oxygenation of Cu(I)–Ge(II) complexes 76 and 77 was explored (Figure 56).168 For the system 76 ligated by N(SiMe3)2− ligands, UV–vis and resonance Raman spectroscopy indicated that 75 was produced (Table 6). The analogous complexes supported by L2d and L4c were also prepared by reaction of the corresponding transient 1:1 Cu:O2 adduct with Ge[N(SiMe3)2]2. Reaction of 77 with O2 proceeded differently, giving products indicative of loss of the Ge(II) fragment and formation of a transient 1:1 Cu:O2 adduct. In support of this formulation for the transient species, reaction with Ge[N-(SiMe3)2]2 yielded 75.
Figure 56.

Course of oxygenations of Cu(I)–Ge(II) complexes (ref 168).
3.2. Other Peroxo Complexes
3.2.1. (1,2-Peroxo)dicopper Complexes
Ever since the report in 1988 of the first X-ray structure of a Cu/O2 complex that showed it to be a (trans-1,2-peroxo)dicopper(II) species,339 many examples of this type of core have been characterized, including by X-ray crystallography.145,340 Key spectroscopic properties of such complexes, mostly reported since 2004, are presented in Table 7.128,144,200,341–346 They have in common the typical, previously analyzed347 signatures comprising (1) peroxide π*→ Cu(II) d LMCT features at 530–550 nm (~10,000 M−1 cm−1) and 600 nm (sh) and (2) characteristic resonance Raman stretching frequencies for ν(O–O) and ν(Cu–O) at ~800–850 cm−1 and ~550 cm−1, respectively. Slight variations evaluated through detailed comparisons with the parent L41a system have provided insights into geometric differences or donor atom effects. For example, in 78 (L67), respective ν(O–O) and ν(Cu–O) values were found to be 10 and 16 cm−1 lower than those for the L41a complex (Figure 57). These shifts were interpreted to indicate increased electron donation by the thioether donor in L67 that reduces peroxide-to-Cu π* donation, weakening both the Cu–O and O–O bonds.342 Similar arguments were used to rationalize why L82 is a weaker donor than L67.343 The relative absorption intensities for the complexes supported by the thioether-containing ligands L82 and L67 are inverted (extinction coefficient at ~610 nm greater than that at ~550 nm) relative to the more typical pattern (extinction coefficient at ~550 nm greater than that at ~610 nm), which was attributed to a geometric distortion toward square pyramidal in the thioether donor cases that inverts the energy order of the π*σ and π*v orbitals.342,343 A similar geometric distortion was invoked to explain weaker Cu–O and O–O bonding in the (trans-1,2-peroxo)dicopper complex of L40a.344
Table 7.
Properties of (1,2-Peroxo)dicopper Complexes
| structure | ligand | solvent | UV–vis | Raman (exp, cm−1) | ref | |
|---|---|---|---|---|---|---|
|
|
|
|
|
|
|
|
| λmax (nm) (ε, M−1 cm−1) | ν(O–O) (Δ18O) | ν(Cu–O) (Δ18O) | ||||
| trans | L41a | EtCN | 525 (11,300), 615 (5800) | 832 (44) | 561 (26) | 341 |
| trans | L41a | MeTHF | 525 (11,500), 615 (5800) | 827 | 561 | 342 |
| trans | L78a | THF | 540 (9550), 610 (6500) | 848 (47) | 550 (26) | 341 |
| trans | L42a | acetone | 552 (13,500), 600 | 825 (48) | – | 144 |
| trans | L28a/L41a | DMF/THF | 624 (8300), 550 | 832 (44) | – | 128 |
| trans | L31a | MeTHF | 618, 520, 450 | 811 (45), 801 (35) | 547 (25) | 200 |
| trans | L31a | acetone | 816 (48), 804 (36) | 550 (20) | ||
| trans | L67 | MeTHF | 445 (2150), 521 (8640), 615 (10,850) | 817 (46) | 545 (26) | 342 |
| trans | L82 | MeTHF | 442 (1500), 530 (8600), 605 (10,400) | 828 (48) | 547 (23) | 343 |
| trans | L40a | MeTHF | 445 (2500), 535 (11,000), 610 (8100) | 822 (46) | 539 (26) | 344 |
| trans | L40d | THF | 545, 620 | 835 (42), 821 (44) | 542 (21), 504 (16) | 345 |
| trans | L60 | acetone | 478 (7800), 575–700(sh) | 832 (45) | 520 (22) | 346 |
| cis/trans | L65b | EtCN | 506 (4800), 600 (2800) | 803 (54) | 512 (22) | 354 |
| cis | L65a | acetone | 500 (3000) | 799 (45) | 437 (19) | 353 |
| trans | L30 | CH2Cl2 | 508 (2000), 630 (1250) | 837 (45) | 571 (26) | 348 |
| trans | L30 | toluene | 513 (8300), 628 (5200) | – | – | 348 |
Figure 57.
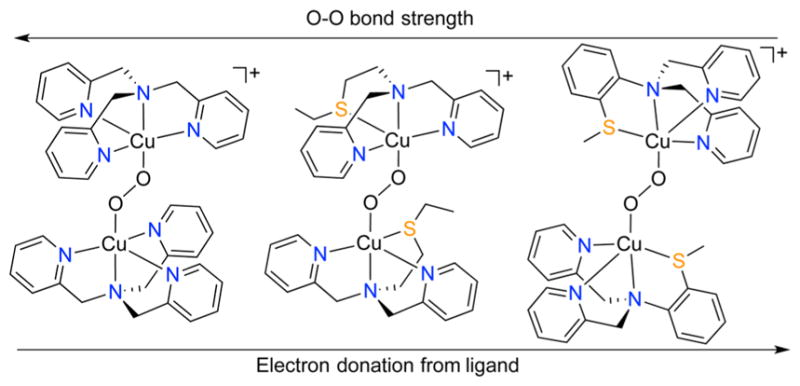
Relationship of O–O bond strength and electron donation between complexes [(L41a)Cu2O2]+ (left), 78 (L67) (middle), and [(L82)Cu2O2]+ (right) (ref 342).
In general, (trans-1,2-peroxo)dicopper compounds are supported by tetradentate ligands, which typically inhibit adoption of coordination numbers >5 and thus prevent formation of (μ-η2:η2-peroxo)- or bis(μ-oxo)dicopper cores. Yet, different cores can be accessed through variation of ligand steric influences or donor types. Thus, while oxygenation of the Cu(I) complex of L82 yielded a (trans-1,2-peroxo)dicopper complex, replacement of the thioether S with an ether O (L78b) resulted in formation of a bis(μ-oxo)dicopper core.343 The bis(pyridylmethyl)amine derivatives L30 and L38d are similarly divergent in their oxygenation chemistry, with the former yielding a (trans-1,2-peroxo)dicopper core proposed to involve anisole O coordination to the metal ions and the latter yielding a bis(μ-oxo)dicopper complex because of the noncoordinating nature of the benzyl group.348 A comparison of a series of derivatives of L41a that are modified at the 6-position of one pyridyl arm showed that for R = Me, a (trans-1,2-peroxo)-dicopper core forms, but if that substituent is more sterically encumbered (i.e., R = aryl or secondary amine), a bis(μ-oxo)dicopper complex is favored (Figure 58).341 These findings were rationalized by positing that the large substituent weakens the Cu–N interaction with the substituted pyridyl donor, favoring the lower coordination number typical for bis-(pyridylmethyl)amine ligands suitable for bis(μ-oxo)dicopper complex formation.317 Differences in steric effects also were proposed to underlie the different course of oxygenations of Cu(I) complexes of bispidine derivatives L31a and L31b; only 1:1 Cu:O2 adduct formation was seen for L31b, whereas L31a supported a (trans-1,2-peroxo)dicopper complex.200
Figure 58.
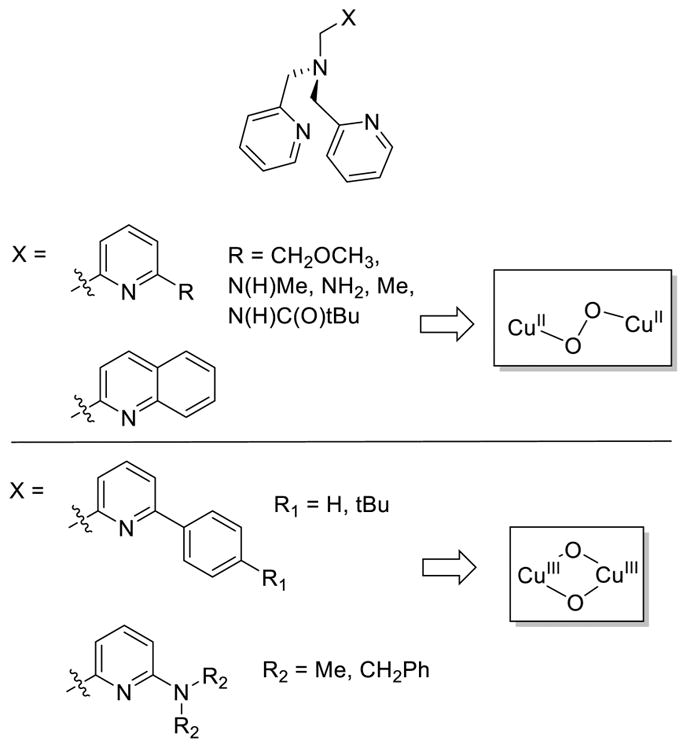
Ligand derivatives that yielded indicated copper–oxygen cores upon reaction of their Cu(I) complexes with O2 (refs 341, 343, and 348).
An equilibrium between (trans-1,2-peroxo)- and bis(μ-oxo)dicopper cores was reported,345 with possible implications for understanding novel reactivity ascribed to the former.346 Following previous studies of the oxygenation of [(L40d)Cu]-PF6 in EtCN,349,350 it was found that oxygenation of its B(C6F5)4− salt in THF initially yielded UV–vis spectra consistent with (trans-1,2-peroxo)dicopper species but then evolved to a final spectrum that also contained features indicative of the bis(μ-oxo)dicopper core. The presence of two sets of ν(O–O) and ν(Cu–O) stretching frequencies (in addition to the Cu2O2 mode at 584 cm−1) was attributed to the presence of two (trans-1,2-peroxo)dicopper isomers differing with respect to the disposition of the L40d ligands (C1 vs Ci symmetry species; Figure 59). Kinetic and thermodynamic parameters for the equilibrium were reported. Inspired by the finding of this equilibrium, it was suggested on the basis of DFT calculations that a bis(μ-oxo)dicopper isomer might also be energetically accessible for another system previously reported to yield a (trans-1,2-peroxo)dicopper complex. This complex supported by L60 had been found to bind and hydroxylate exogenous phenolates in what was cited to be a novel reactivity for the typically nucleophilic (trans-1,2-peroxo)dicopper core.346 It was proposed that a bis(μ-oxo)dicopper isomer was energetically inaccessible; however, this was challenged using the calibrated DFT method, which suggests that the phenolate oxidation might actually be performed by a bis(μ-oxo)dicopper isomer.345
Figure 59.

Proposed equilibrium between Ci (left) and C1 (right) isomers of (trans-1,2-peroxo)dicopper complexes with bis(μ-oxo)dicopper isomer (ref 345).
In recent studies of the reactivity of (trans-1,2-peroxo)-dicopper complexes supported by L30348 or L42a,340 oxidation of toluene to predominantly benzaldehyde was observed. It was noted, however,348 that the involvement of other copper–oxygen intermediates could not be ruled out. Protonation of the (trans-1,2-peroxo)dicopper core typically results in formation of H2O2344,351 but can also yield a (1,1-hydroperoxo)dicopper species (see section 3.3).
Inspired by a theoretical analysis of the mechanism of formation of the (μ-η2:η2-peroxo)dicopper core in hemocyanin, 352 a (cis-1,2-peroxo)dicopper moiety was targeted for synthesis and characterization.38,353,354 In accordance with the computations, the process of formation of the antiferromagnetically coupled singlet (μ-η2:η2-peroxo)dicopper unit from triplet O2 and two Cu(I) ions requires an initial activation event followed by intersystem crossing. This initial activation was proposed to involve an electron transfer from each Cu(I) ion into orthogonal O2 π* orbitals to lead to a triplet (1,2-peroxo)dicopper unit. The first example of a (cis-1,2-peroxo)-dicopper complex was prepared using the pyrazolate-bridged ligand L65a (Figure 60).353 The X-ray structure of the stable complex 79 revealed cis binding of the peroxide ligand with a Cu–O–O–Cu torsion angle of 65.2°. A sodium ion binds to the peroxide in the crystals and in solution. Detailed characterization by spectroscopy and magnetism studies indicated weak binding of the peroxide to the two Cu(II) ions (ν(Cu–O) = 437 cm−1, ν(O–O) = 799 cm−1) that are only weakly antiferromagnetically coupled (–2J = 144 cm−1). This weak coupling was ascribed to the torsion angle intermediate between the extremes expected for strong antiferromagnetic (0°) or ferromagnetic coupling (90°). With the aim of driving the geometry toward that which would favor a triplet ground state, ligand L65b featuring an additional methylene linker was examined.354 Indeed, the resulting (1,2-peroxo)dicopper complex 80 exhibited a shorter Cu–Cu separation of 3.68 Å and a 104.5° torsion angle (thus, denoted as a “trans” geometry) and a triplet ground state arising from noninteracting orthogonal magnetic orbitals (Figure 60). Thus, complex 80 represents a unique model of the intermediate species proposed along the pathway of O2 activation by hemocyanin and related enzymes.
Figure 60.
(top) X-ray crystal structures of the (1,2-peroxo)dicopper complexes 79 and 80 and (bottom) orthogonal molecular orbitals in 80 that give rise to its S = 1 ground state. Reprinted with permission from ref 38 (top) and ref 354 (bottom). Copyright 2015 Wiley-VCH.
3.2.2. (1,1-Hydroperoxo)dicopper Complexes
Several dicopper complexes featuring 1,1-hydroperoxo ligands have been prepared, typically via protonation of a (peroxo)dicopper precursor or reaction of a Cu(II) complex with H2O2.355–359 For those complexes characterized since 2004, spectroscopic data are presented in Table 8; X-ray crystal structures of complexes 81 and 82 (Figure 61) were determined. Common features include intense UV–vis absorptions assigned as LMCT transitions at ~350–400 nm, ν(O–O) ~860–890 cm−1 that are higher than typically seen for (peroxo)dicopper complexes (vide infra), and EPR data indicative of weak antiferromagnetic coupling between the Cu(II) ions.
Table 8.
Properties of (1,1-hydroperoxo)dicopper Complexes
| ligand | solvent | UV–vis | Raman (exp, cm−1) | ref | |
|---|---|---|---|---|---|
|
|
|
|
|
|
|
| λmax (nm) (ε, M−1cm−1) | ν(O–O) (Δ18O) | ν(Cu–O) (Δ18O) | |||
| L54 | EtCN | 407 (2700), 488 (1600), 622 (600) | 870 (50) | – | 355 |
| L40f | MeCN | 356 (6300), 580 (240), 664 (sh ~190) | 868 (45) | 572 (16) | 357 |
| L40e | MeCN | 341 (~7000), 581 (~170), 770 (sh ~80) | 883 (50) | 562 (23) | 357 |
| L58b | MeCN | 342 (8600), 444 (850), 610 (400) | – | – | 358 |
| L65b | MeCN | 416 (5700), 373 (3300) | 860 (46) | – | 359 |
| L53 | acetone | 370 (3700), 650 (300) | 881 (49) | – | 356 |
Figure 61.
(1,1-Hydroperoxo)dicopper complexes supported by L40f and L65b, respectively, that have been structurally characterized by Xray crystallography (refs 357 and 359).
The synthesis of 82 is notable insofar as it is a unique case where protonation of a (peroxo)dicopper species (80) to generate a (1,1-hydroperoxo)dicopper complex is reversible.359 The protonation of 80 to yield 82 proceeded without a detectable intermediate (stopped-flow, −20 °C) and was readily reversed by treatment with 1,8-diazabicyclo-undec-7-ene (DBU). The pKa for 82 in CH3CN was determined to be 22.2 ± 0.3.
Several examples of hydroxylation reactions have been observed for (1,1-hydroperoxo)dicopper complexes. The (1,1-hydroperoxo)dicopper complex resulting from reaction of the dicopper(I) complex of L54 with O2 was found to react with the nitrile solvent to yield an aldehyde and cyanide (found as a bridging ligand in a tetracopper(II) product).355 A mechanism involving hydroxylation of the α-C–H bond of the nitrile by the hydroperoxo moiety was proposed. In a separate report, the same (1,1-hydroperoxo)dicopper complex was implicated in the oxidation of guanine in reactions with DNA.360 Intramolecular hydroxylation of a ligand arm methylene group was observed for complex 81 upon its decomposition in the solid state.357 A (1,1-hydroperoxo)dicopper complex was identified at low temperature as an intermediate in the double hydroxylation of the bridging arene in L58b upon reaction of its dicopper(II) complex with H2O2,358 a reaction reminiscent of an earlier report of the system supported by L49a.361
(1,1-Hydroperoxo)dicopper complexes have also been implicated in dioxygen reduction reactions, with mechanistic differences seen under different reaction conditions.362,363 Catalytic 2-electron reduction of O2 to H2O2 was observed upon reaction of 83 with HOTf and Fc* in acetone, but 4-electron reduction of O2 to H2O occurred when HClO4 was used with Fc* or weaker reductants such as Fc (Figure 62). Mechanistic studies led to the proposal that when the stronger acid HClO4 is used, protonation of both the hydroxide and the phenoxide occurs, resulting in decomplexation of the latter and more facile reduction of the dicopper(I) intermediate (less powerful reductant needed). The weaker acid HOTf does not protonate the phenoxide bridge, making reduction more thermodynamically difficult. Importantly, in both cases a (1,1-hydroperoxo)dicopper intermediate is involved; but with HOTf, protonation and loss of H2O2 occurs (2-electron reduction pathway), whereas with HClO4, PCET reductive cleavage of the hydroperoxide is favored (4-electron reduction pathway).
Figure 62.
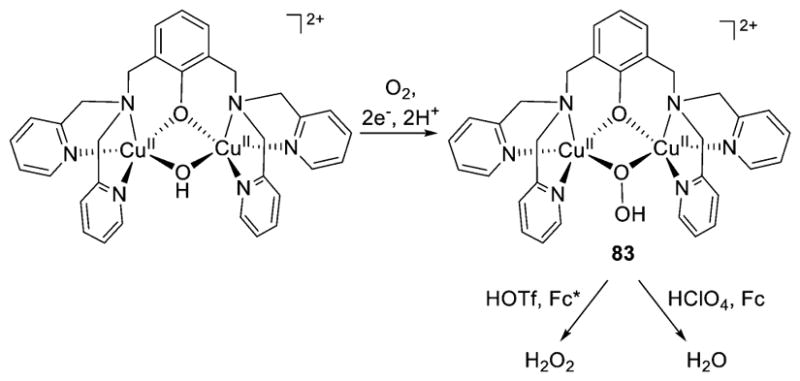
Oxygen reduction reactions involving intermediate 83 (refs 362 and 363).
3.2.3. (μ-η1:η2-Peroxo)dicopper Complexes
Two examples of this binding mode have been proposed as products of oxygenation of dicopper(I) complexes of pentadentate ligands L63 and L64 (see Figure 34).364,365 Unfortunately, confirmation of this unusual bonding mode via X-ray crystallography has not been reported, and the UV–vis, resonance Raman, and EPR spectroscopic data for these complexes do not differ significantly from that typical for (1,2-peroxo)dicopper complexes. Still, it is reasonable to suggest that the bis(pyridylmethyl)amine fragment in L63 and L64 would favor η2 coordination, and precedent exists for other metal ions for (μ-η1:η2-peroxo) coordination (Figure 63).352,364,365
Figure 63.
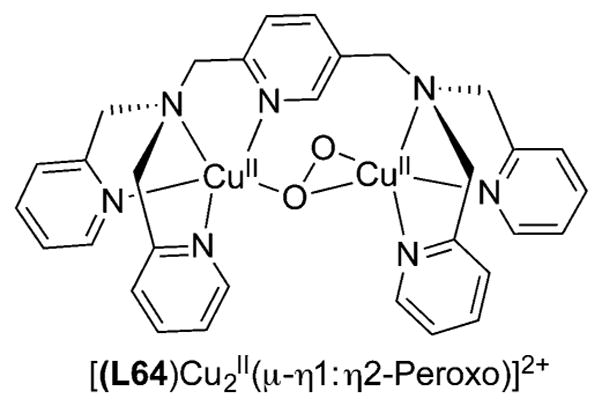
Proposed structure of a (μ-η1:η2-peroxo)dicopper complex supported by L64 (ref 364).
3.3. Mono(μ-oxo/hydroxo)dicopper Complexes
Inspired by the various postulates of oxo-and hydroxo-bridged dicopper cores as intermediates in catalytic oxidations by pMMO and Cu-doped zeolites (section 1), significant recent effort has been focused on understanding the properties of synthetic analogs. The relatively few synthetic complexes with (μ-oxo)dicopper(II) cores have been reviewed recently.29 Thus, herein, we only briefly survey selected examples. In early work, complexes 84,29,366 85,367 86,368–370 and 87218,371 supported by mononucleating ligands were prepared (Figure 64), with their formulations indicated by spectroscopy and their accessibility from multiple routes described (cf. reactions of Cu(I) complexes with O2, PhIO, and/or NO). An X-ray crystal structure was reported for 85, but interpretation was hindered by disorder involving the chemically inequivalent O atoms in the core.367 In general, the oxo ligands in these complexes are nucleophilic, readily protonated, and transferable to oxophilic substrates like PPh3. (μ-Oxo)dicopper(II) units were also identified in the complexes 88 (postulated, but not identified conclusively), 372,373,374 89, and 90 (Figure 65).375 Complex 89 was characterized by X-ray crystallography, but charge balance considerations led to the postulate that the crystals contained a 1:1 mixture of 89 and its protonated (μ-hydroxo)dicopper(II) congener. Complex 90 was characterized by 1H NMR spectroscopy, EXAFS, and ESI-MS, and was EPR silent. EXAFS and DFT calculations show a Cu–Cu distance of 2.91 and 2.844 Å, respectively. In addition to converting Ph3P to Ph3PO, 90 also oxidizes di-tert-butylphenol to yield products of radical coupling and further oxidation (quinone and 2,4,7,9-tetra-tert-butyloxepino[2,3-b]-benzofuran). Reaction of 91 with O2 or PhIO at low temperature also yielded (μ-oxo)dicopper-(II) units, although as a mixture of intramolecular, dimeric, and oligomeric species.376
Figure 64.
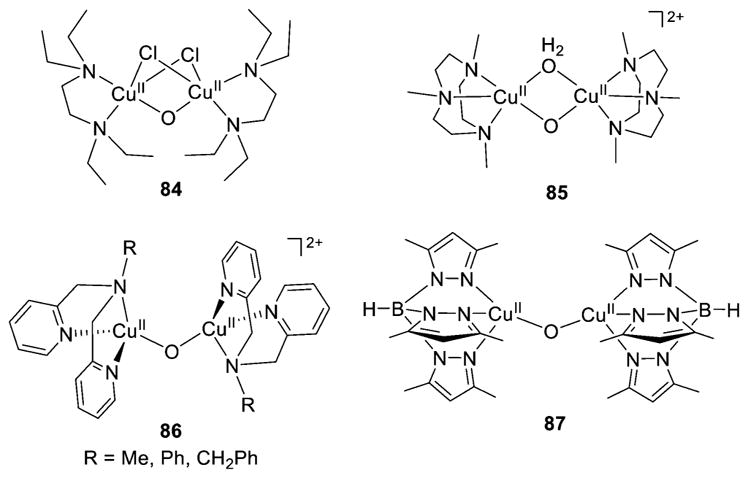
Examples of (μ-oxo)dicopper(II) complexes (references cited in text).
Figure 65.
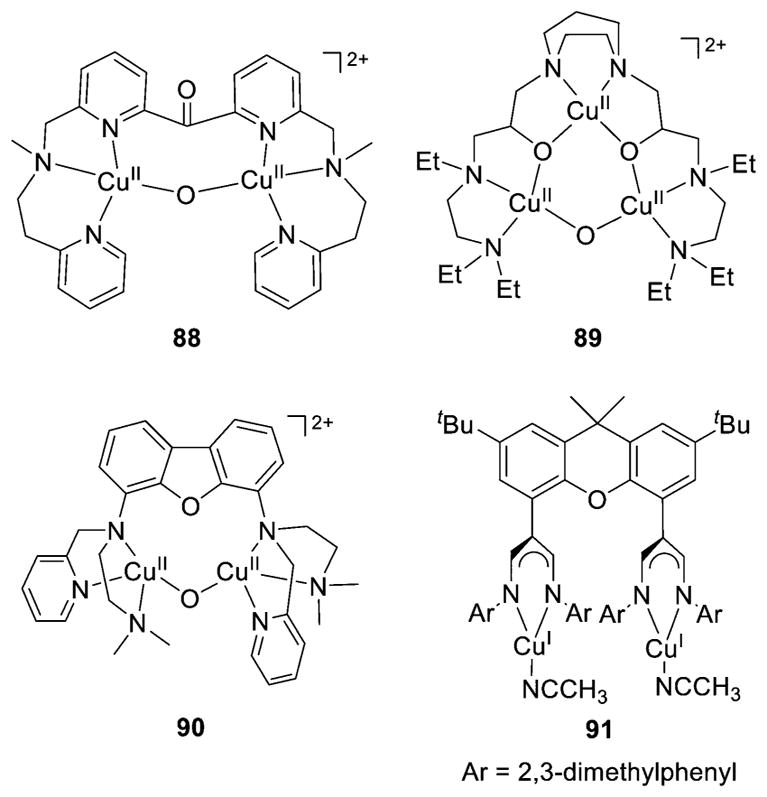
While dicopper(II) complexes with hydroxide bridges are common,377 higher valent examples relevant to proposed pMMO or Cu-zeolite intermediates have only been examined recently.378,379 The structurally defined dicopper(II) complexes 92 and 93 (Figure 66) were oxidized by 1-electron to yield species formulated as Cu(II)Cu(III) complexes on the basis of spectroscopic data. Both complexes exhibited axial EPR spectra consistent with a localized mixed-valent ground state, with additional support provided by DFT calculations. Subtle differences in the UV–vis data were interpreted to indicate Robin-Day380 class II behavior for 93.379 For the complex derived from 92, an additional 1-electron oxidation was proposed to yield a dicopper(III) species on the basis of Cu K-edge XAS and UV–vis redox titration results.378 DFT calculations supported retention of the hydroxo bridges in the oxidized complexes.
Figure 66.

X-ray crystal structures of hydroxo-bridged dicopper(II) complexes 92 (L59) and 93 (L55) that served as starting materials for the preparation of higher valent species. (left) Only anion shown; Cu–Cu = 2.6596(15) Å. Reprinted from ref 378. Copyright 2014 American Chemical Society. (right) Cation and one counterion shown; Cu–Cu = 2.7511(12) Å. Reprinted from ref 379. Copyright 2016 American Chemical Society.
4. TRICOPPER COMPOUNDS
Interest in the properties of tricopper–oxygen complexes has been stimulated by the role such species play in the reduction of O2 to H2O by the multicopper oxidases (MCOs) and by the postulate of tricopper species as active intermediates in pMMO. With respect to the MCOs, particular attention has been paid to identifying the so-called “peroxo” and “native” intermediates in these enzymes through spectroscopy, as described in several reviews (Figure 1e).5,6,381 Importantly, detailed studies of relevant tricopper-oxgyen compounds have provided fundamental information useful for delineation of the structures of these enzyme intermediates.382 In addition, the controversial postulate of a tricopper active site in pMMO76,86 has stimulated intriguing studies of the biological reactivity of tricopper complexes with C–H bonds.
4.1. Bis(μ-oxo)tricopper Complexes
Since the first report of the [Cu3O2]3+ core in complexes supported by L6a294 and others described in the previous review,383,384 two other examples have been identified.385,386 In one case, oxygenation of a Cu(I) complex of a sterically unencumbered L4a with O2 generated a novel neutral complex proposed to contain the [Cu3O2]3+ core on the basis of UV–vis and EPR spectroscopy (signal indicative of an S = 1 ground state), ESI-MS, and a 3:1 Cu:O2 reaction stoichiometry.385 Decreased electrophilic reactivity relative to other examples and the observation of oxidation of PPh3 to OPPh3 were traced to the overall neutral charge of the complex and the strong electron-donating characteristics of the L4a supporting ligands.
In a second example, ligands comprising bis(pyridylmethyl)-amine (L61a) or mono(pyridylmethyl)amine (L61b) chelates were preorganized to bind three Cu(I) ions by Y(III) binding to a heptadentate L61 donor set (Figure 67).386 Low-temperature oxygenation at low concentrations (0.05 mM) yielded [Cu3O2]3+ cores as indicated by UV–vis spectroscopy and the Cu:O2 stoichiometry. These findings contrast with the results of oxygenations of Cu(I) complexes of simple N-donor analogs, highlighting the key role of the ligand preorganization of the Ytemplate in driving the formation of the tricopper unit. The complexes were not reactive with 2,4-di-tert-butylphenol or DHA, but HAT was seen from TEMPOH and oxygen atom transfer was seen to PPh3.
Figure 67.

Oxygenation of a tricopper(I) complex of a templated, preorganized ligand (L61a,b). Adapted from ref 386.
4.2. Other Tricopper–Oxygen Complexes
Numerous unsuccessful attempts to prepare tricopper–oxygen intermediates via oxygenation reactions of tricopper(I) complexes supported by multidentate ligands have been reported, with the formation of stable tricopper(II) motifs or undesired dicopper–oxygen units being common outcomes. 278,387,388 In addition, many examples of trinuclear copper(II) complexes have been characterized, with particular interest focused on their use as catalysts for hydrocarbon oxidations.389–395 A full discussion of such complexes is beyond the scope of the current review. Instead, we focus on select examples of complexes of particular relevance or use in understanding the nature of the “native” and “peroxo” intermediates in MCOs or the active center in pMMO.
While oxygenation of the Cu(I) complexes of L1a at low temperature yielded the peroxo complex 44 (Figure 36), reaction of [(L1a)Cu(CH3CN)]X (X = ClO4−) with O2 at room temperature yielded the tris(hydroxo)tricopper complex 94 that models a proposed structure for the native intermediate in MCOs (Figure 68).396 For X = CF3SO3−, or when excess (Bu4N) (CF3SO3) was added to 94, a bis(hydroxo)dicopper complex 95 formed instead, and the preference for the species formed was traced to hydrogen bonding and ion-pairing interactions in both the solid state and THF solution. Complex 94 exhibited spin-frustration and interesting magnetic properties that were examined in detail using variable-field magnetic circular dichroism and EPR spectroscopy.397 Subsequent detailed comparisons were made between the properties of 94 and those of an alternative structure for the native intermediate,398,399 namely a (μ3-oxo)tricopper cluster which lacked additional bridging ligands.400 It was concluded that the properties of the (μ3-oxo)tricopper cluster best matched those of the native intermediate; the driving force for the formation of the stable (μ3-oxo)tricopper core was shown to be critical for the overall O2 reduction process catalyzed by the enzymes.399
Figure 68.
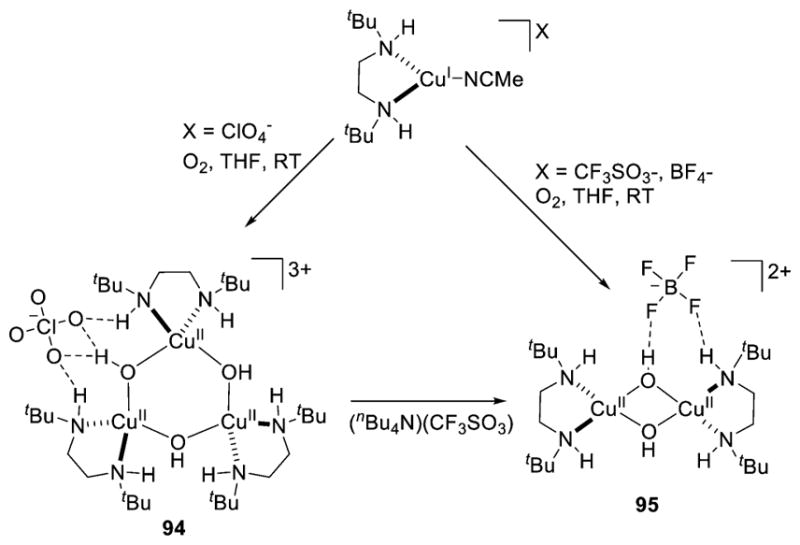
Results of room temperature oxygenation of Cu(I) complexes of L1a (ref 396).
Various tricopper complexes supported by ligands L79a–f have been shown to participate in the catalytic oxidations of hydrocarbons,374,401,402 including the conversion of methane to methanol.403 These results have been cited in favor of the proposition that the active site of pMMO contains a tricopper cluster.86 A general mechanism for dioxygen activation by tricopper(I) complexes of these ligands has been proposed which involves initial formation of a (peroxo)dicopper intermediate (96) that undergoes O–O bond scission to yield a highly reactive mixed-valent species 97 (Figure 69). DFT calculations led to the suggestion of a mechanism for substrate oxidation by this intermediate involving “singlet oxene transfer.” 85 While a provocative proposal, such a pathway remains speculative because intermediate 97 has not been identified definitively by experiment. Within this context, we note the identification of a [Cu3(μ-O)3]2+ core that hydroxylates methane in the zeolite mordenite (Figure 2).118
Figure 69.
(top) Proposed mechanism for generation of the hypothesized reactive intermediate in hydrocarbon oxidations by complexes of ligands L79a–f. Supporting ligands not shown. (bottom) Space-filling and ball-and-stick drawing of calculated structure for intermediate 97 supported by L79f. Reprinted with permission from ref 403. Copyright 2013 Wiley-VCH. *Corresponding Author: wtolman@umn.edu.
5. SUMMARY AND CONCLUSIONS
Since the publication of the previous articles published in this journal on this subject in 2004,15,16 significant further advances have been made in our understanding of the nature of copper–oxygen complexes relevant to catalytic intermediates. New motifs have been discovered, including heterbimetallic bis(μ-oxo), [CuOH]2+, (cis-1,2-peroxo)dicopper, and mixed-valent (μ-hydroxo)dicopper(II,III) cores. In addition, as described above, new examples of previously known cores have been characterized with variable supporting ligands, including simple ones like imidazoles, which more closely mimic biological donors. Moreover, new insights into the reactivity of a variety of copper–oxygen species have been obtained that have changed the way we think about their role in catalytic oxidations.
No doubt, the field has matured and the questions being addressed are ever more focused on details of spectroscopy and mechanism. Many important challenges remain, however, that continue to stimulate research. For example, while identified in the gas phase and evaluated by theory, complexes with the [CuO]+ core have yet to be isolated and characterized. Postulates of novel copper-containing structures as active species in important oxidations, such as the hydroxylation of methane or the oxidative cleavage of DNA (by the ATCUN motif, for example),98 await unequivocal verification. Indeed, the nature of the oxidant in LPMO and pMMO remains a mystery, and efforts continue to be made to synthesize and characterize relevant compounds that are capable of attacking strong C–H bonds at rapid rates, such as those with oxo/hydroxo bridges between copper ions at various oxidation levels. New catalytic oxidations using copper compounds as catalysts continue to be discovered, but firm identification of intermediates and mechanisms often is lacking. Examples span oxidations of water404,405 and hydrocarbons,406 reactions of particularly keen interest, because of their relevance to energy transformations. The need to understand how such reactions proceed provides ample impetus for further study using approaches like those described herein that involve the clever use of supporting ligands to enable the detailed characterization of novel copper–oxygen compounds.
Acknowledgments
We thank all the co-workers and collaborators who contributed to the work cited in this review. Financial support for this work was provided by the National Institutes of Health (R37GM47365).
ABBREVIATIONS
- ATCUN
amino terminal Cu(II)- and Ni(II)-binding
- ATP
adenosine triphosphate
- BDE
bond dissociation enthalpy
- BNAH
1-benzyl-1,4-dihydronicotinamide
- BzIm
1,3-dimethyl-2,3-dihydrobenzimidazole
- CB-PPO
coupled binuclear polyphenol oxidases
- CcO
cytochrome c oxidase
- Cm
cumyl
- CMA
N-cyclopropyl-N-methylamine
- CPET
concerted proton electron transfer
- DβM
dopamine β-monooxygenase
- DBU
1,8-diazabicyclo-undec-7-ene
- DFB
1,2-difluorobenzene
- DFT
density functional theory
- DHA
9,10-dihydroanthracene
- DMPO
5,5-dimethyl-1-pyrroline N-oxide
- DNA
deoxyribonucleic acid
- EPR
electron paramagnetic resonance
- ESI-MS
electrospray ionization-mass spectrometry
- EXAFS
extended X-ray absorption fine structure
- Fc
ferrocene
- Fc*
decamethylferrocene
- GAO
galactose oxidase
- Gly
glycine
- HAT
hydrogen atom transfer
- HIPT
3,5-bis(2,4,6-triisopropylphenyl)phenyl
- His
histidine
- Im
imidazole
- KIE
kinetic isotope effect
- LMCT
ligand-to-metal charge transfer
- LPMO
lytic polysaccharide monooxygenase
- MCD
magnetic circular dichroism
- MCO
multicopper oxidase
- MDP
(p-methoxyphenyl)-2,2-dimethylpropanol
- Met
methionine
- MO
molecular orbital
- NIH
National Institutes of Health
- NMR
nuclear magnetic resonance
- ORR
oxygen reduction reaction
- OTf
trifluoromethanesulfonate
- PCET
proton-coupled electron transfer
- Phen
phenanthroline
- PHM
peptidylglycine α-hydroxylating monooxygenase
- pMMO
particulate methane monooxygenase
- PT/ET
proton transfer/electron transfer
- pz
3,5-diphenylpyrazole
- ROS
reactive oxygen species
- RT
room temperature
- SCE
standard calomel electrode
- TβM
tyramine β-monooxygenase
- TD-DFT
time dependent-density functional theory
- TEMPO
2,2,6,6-tetramethyl-1-piperidinyloxy radical
- THF
tetrahydrofuran
- TIPT
3,5-bis(2,6-diisopropylphenyl)phenyl
- TMAO
trimethylamine-N-oxide
- TMPA
tris(2-methylpyridyl)amine
- TS
transition state
- UV–vis
ultraviolet–visible
- XAS
X-ray absorption spectroscopy
- ZSM
zeolite socony mobile
Biographies
Courtney E. Elwell grew up in upstate New York and obtained her B.S. in Chemistry from Union College (Schenectady, NY) in 2014. There she worked under the direction of Laurie A. Tyler in the synthesis of thiazole-derived copper complexes and the study of their biological activity with DNA and serum proteins. She is currently a doctoral candidate advised by William B. Tolman at the University of Minnesota working on the synthesis of ligand frameworks that demonstrate how electronics and overall charge influence reactivity of monocopper and dicopper-oxygen cores.
Nicole L. Gagnon grew up in Lino Lakes, MN, and received a B.A. degree from the College of St. Benedict, MN, in 2010 under the guidance of Dr. Richard White. During her undergraduate work, she studied the simultaneous determination of partition coefficient and acid-dissociation constant of benzoic acid in water. After studying at the University of Arizona, she joined the laboratory of W. B. Tolman at the University of Minnesota in 2012. She is currently a Ph.D. candidate working on the synthesis of dicopper-hydroxide complexes for C–H bond activation using a naphthyridine-based ligand.
Benjamin D. Neisen received his B.S. in chemistry and biochemistry from the University of Minnesota–Duluth in 2011. While there, he studied ferrocene-substituted porphyrin systems under Professor Viktor Nemykin. He then moved to the University of Minnesota–Twin Cities where he earned his M.S. in chemistry and is now a chemistry Ph.D. candidate performing research under Professor William B. Tolman studying the synthesis and reactivity of high valent copper-oxygen species.
Debanjan Dhar grew up in Kolkata, India. After the completion of his B.Sc. in Chemistry from Presidency College, Kolkata, in 2009, he moved to Indian Institute of Technology Kanpur, India, to pursue a M.Sc. in Chemistry. While in Kanpur he carried out research under the supervision of R. N. Mukherjee, where he worked on the chemistry of metal complexes of redox non-innocent ligand frameworks. He joined the Department of Chemistry, University of Minnesota, as a graduate student in 2012, where he is currently working under the guidance of William B. Tolman. His current research focus is on the chemistry of mononuclear copper-oxygen complexes and their role in C–H bond activation.
Andrew D. Spaeth grew up in Kenosha, WI. He received his B.S. from Michigan Technological University working with B. Török and E. Urnezius. He pursued graduate studies with M. V. Barybin at the University of Kansas and obtained a Ph.D. in 2014, focusing on linear azulenic organometallics. He is currently a postdoctoral research associate with William B. Tolman at the University of Minnesota. His research interests are at the intersection of computational and experimental inorganic chemistry of biological significance.
Gereon M. Yee, hailing from northern California, attended Santa Rosa Junior college before transferring to UC Davis, where he received his B.S. in chemistry in 2010. There he did undergraduate research in inorganic chemistry with Louise Berben before moving to the University of Minnesota to pursue a Ph.D. under the advisement of William B. Tolman. In 2013, he received his M.S. in chemistry, and currently, he is a 5th year Ph.D. candidate studying the effects of ligand modification on the HAT reactivity of mononuclear copper(III)-hydroxide complexes.
William B. Tolman obtained a B.A. degree from Wesleyan University, CT, in 1983, and a Ph.D. from the University of California, Berkeley, in 1987. He then was a postdoctoral associate at the Massachusetts Institute of Technology (1987–1990). Appointed as Assistant Professor in the Department of Chemistry at the University of Minnesota in 1990, he is now Distinguished McKnight University Professor. He is a member of the Centers for Metals in Biocatalysis and Sustainable Polymers and has served as Chair of the Department of Chemistry since 2009. He has received the Searle Scholars, NSF National Young Investigator, Camille & Henry Dreyfus Foundation Teacher-Scholar, and Alfred P. Sloan Foundation Awards, the Buck-Whitney Medal from the American Chemical Society (ACS), a Research Award from the Humboldt Foundation, and the 2017 ACS Award for Distinguished Service in the Advancement of Inorganic Chemistry. He is a Fellow of the American Association for the Advancement of Science and the American Chemical Society. He served as Associate Editor (2009–2012) and now as Editor-in-Chief of the ACS journal Inorganic Chemistry (from 2013 to present), sits on a number of governing and advisory boards, and was Chair of the Gordon Research Conferences on Inorganic Reaction Mechanisms (2005) and Metals in Biology (2011). Research in his group focuses on synthetic bioinorganic and organometallic/polymer chemistry.
Footnotes
NOTE ADDED IN PROOF
A useful review published in July, 2016, was mistakenly not included, see ref 407. Additional ideas about the nature of the active intermediate in PHM were provided in the following paper, which we had also neglected to cite, see ref 408.
Notes
The authors declare no competing financial interest.
References
- 1.Punniyamurthy T, Velusamy S, Iqbal J. Recent Advances in Transition Metal Catalyzed Oxidation of Organic Substrates with Molecular Oxygen. Chem Rev. 2005;105:2329–2364. doi: 10.1021/cr050523v. [DOI] [PubMed] [Google Scholar]
- 2.Newhouse T, Baran PS. If C-H Bonds Could Talk: Selective CH Bond Oxidation. Angew Chem, Int Ed. 2011;50:3362–3374. doi: 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wencel-Delord J, Droge T, Liu F, Glorius F. Towards mild metal-catalyzed C-H bond activation. Chem Soc Rev. 2011;40:4740–4761. doi: 10.1039/c1cs15083a. [DOI] [PubMed] [Google Scholar]
- 4.Yee GM, Tolman WB. Transition Metal Complexes and the Activation of Dioxygen. In: Kroneck PMH, Sosa Torres ME, Sigel A, Sigel H, Sigel RKO, editors. Sustaining Life on Planet Earth: Metalloenzymes Mastering Dioxygen and Other Chewy Gases. Vol. 15. Springer; Cham, Switzerland: 2015. pp. 131–204. Metal Ions in Life Sciences Series. [Google Scholar]
- 5.Solomon EI, Heppner DE, Johnston EM, Ginsbach JW, Cirera J, Qayyum M, Kieber-Emmons MT, Kjaergaard CH, Hadt RG, Tian L. Copper Active Sites in Biology. Chem Rev. 2014;114:3659–3853. doi: 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solomon EI, Sarangi R, Woertink JS, Augustine AJ, Yoon J, Ghosh S. O2 and N2O Activation by Bi-, Tri-, and Tetranuclear Cu Clusters in Biology. Acc Chem Res. 2007;40:581–591. doi: 10.1021/ar600060t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ali ME, Rahman MM, Sarkar SM, Hamid SBA. Heterogeneous Metal Catalysts for Oxidation Reactions. J Nanomater. 2014;2014:1–23. [Google Scholar]
- 8.Ren Y, Wang M, Chen X, Yue B, He H. Heterogeneous Catalysis of Polyoxometalate Based Organic–Inorganic Hybrids. Materials. 2015;8:1545–1567. doi: 10.3390/ma8041545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van der Vlugt JI, Meyer F. Homogeneous Copper-Catalyzed Oxidations. In: Meyer F, Limberg C, editors. Organometallic Oxidation Catalysis. Vol. 22. Springer-Verlag; Berlin: 2007. pp. 191–240. Topics in Organometallic Chemistry Series. [Google Scholar]
- 10.Allen SE, Walvoord RR, Padilla-Salinas R, Kozlowski MC. Aerobic Copper-Catalyzed Organic Reactions. Chem Rev. 2013;113:6234–6458. doi: 10.1021/cr300527g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ryland BL, Stahl SS. Practical Aerobic Oxidations of Alcohols and Amines with Homogeneous Copper/TEMPO and Related Catalyst Systems. Angew Chem, Int Ed. 2014;53:8824–8838. doi: 10.1002/anie.201403110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hematian S, Garcia-Bosch I, Karlin KD. Synthetic Heme/Copper Assemblies: Toward an Understanding of Cytochrome c Oxidase Interactions with Dioxygen and Nitrogen Oxides. Acc Chem Res. 2015;48:2462–2474. doi: 10.1021/acs.accounts.5b00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thorseth MA, Tornow CE, Tse ECM, Gewirth AA. Cu Complexes that Catalyze the Oxygen Reduction Reaction. Coord Chem Rev. 2013;257:130–139. [Google Scholar]
- 14.Singh A, Spiccia L. Water Oxidation Catalysts Based on Abundant 1st Row Transition Metals. Coord Chem Rev. 2013;257:2607–2622. [Google Scholar]
- 15.Mirica LM, Ottenwaelder X, Stack TDP. Structure and Spectroscopy of Copper–Dioxygen Complexes. Chem Rev. 2004;104:1013–1046. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- 16.Lewis EA, Tolman WB. Reactivity of Dioxygen–Copper Systems. Chem Rev. 2004;104:1047–1076. doi: 10.1021/cr020633r. [DOI] [PubMed] [Google Scholar]
- 17.Decker A, Solomon EI. Dioxygen Activation by Copper, Heme and Non-Heme Iron Enzymes: Comparison of Electronic Structures and Reactivities. Curr Opin Chem Biol. 2005;9:152–163. doi: 10.1016/j.cbpa.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 18.De A, Mandal S, Mukherjee R. Modeling Tyrosinase Activity. Effect of Ligand Topology on Aromatic Ring Hydroxylation: An Overview. J Inorg Biochem. 2008;102:1170–1189. doi: 10.1016/j.jinorgbio.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 19.Smirnov VV, Brinkley DW, Lanci MP, Karlin KD, Roth JP. Probing Metal-Mediated O2 Activation in Chemical and Biological System. J Mol Catal A: Chem. 2006;251:100–107. [Google Scholar]
- 20.Tolman WB. Using Synthetic Chemistry to Understand Copper Protein Active Sites: A Personal Perspective. JBIC, J Biol Inorg Chem. 2006;11:261–271. doi: 10.1007/s00775-006-0078-9. [DOI] [PubMed] [Google Scholar]
- 21.Itoh S. Mononuclear copper active-oxygen complexes. Curr Opin Chem Biol. 2006;10:115–122. doi: 10.1016/j.cbpa.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 22.Chufán EE, Puiu SC, Karlin KD. Heme–Copper/Dioxygen Adduct Formation, Properties, and Reactivity. Acc Chem Res. 2007;40:563–572. doi: 10.1021/ar700031t. [DOI] [PubMed] [Google Scholar]
- 23.Hatcher LQ, Karlin KD. Ligand Influences in Copper-Dioxygen Complex-Formation and Substrate Oxidations. Adv Inorg Chem. 2006;58:131–184. [Google Scholar]
- 24.Collman JP, Decreau RA. Functional Biomimetic Models for the Active Site in the Respiratory Enzyme Cytochrome c Oxidase. Chem Commun. 2008:5065–5076. doi: 10.1039/b808070b. [DOI] [PubMed] [Google Scholar]
- 25.Que L, Jr, Tolman WB. Biologically Inspired Oxidation Catalysis. Nature. 2008;455:333–340. doi: 10.1038/nature07371. [DOI] [PubMed] [Google Scholar]
- 26.Karlin KD, Itoh S, Rokita S, editors. Copper-Oxygen Chemistry. Wiley Series of Reactive Intermediates in Chemistry and Biology. Vol. 4 John Wiley & Sons, Inc; Hoboken, NJ: 2011. [Google Scholar]
- 27.Solomon EI, Ginsbach JW, Heppner DE, Kieber-Emmons MT, Kjaergaard CH, Smeets PJ, Tian L, Woertink JS. Copper Dioxygen (Bio)inorganic Chemistry. Faraday Discuss. 2011;148:11–39. doi: 10.1039/c005500j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ray K, Heims F, Pfaff FF. Terminal Oxo and Imido Transition-Metal Complexes of Groups 9–11. Eur J Inorg Chem. 2013;2013:3784–3807. [Google Scholar]
- 29.Haack P, Limberg C. Molecular CuII-O-CuII Complexes: Still Waters Run Deep. Angew Chem, Int Ed. 2014;53:4282–4293. doi: 10.1002/anie.201309505. [DOI] [PubMed] [Google Scholar]
- 30.Sala X, Maji S, Bofill R, García-Antón J, Escriche L, Llobet A. Molecular Water Oxidation Mechanisms Followed by Transition Metals: State of the Art. Acc Chem Res. 2014;47:504–516. doi: 10.1021/ar400169p. [DOI] [PubMed] [Google Scholar]
- 31.Gagnon N, Tolman WB. [CuO]+ and [CuOH]2+ Complexes: Intermediates in Oxidation Catalysis? Acc Chem Res. 2015;48:2126–2131. doi: 10.1021/acs.accounts.5b00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rolff M, Schottenheim J, Tuczek F. Monooxygenation of External Phenolic Substrates in Small-Molecule Dicopper Complexes: Implications on the Reaction Mechanism of Tyrosinase. J Coord Chem. 2010;63:2382–2399. [Google Scholar]
- 33.Koval IA, Gamez P, Belle C, Selmeczi K, Reedijk J. Synthetic Models of the Active Site of Catechol Oxidase: Mechanistic Studies. Chem Soc Rev. 2006;35:814–840. doi: 10.1039/b516250p. [DOI] [PubMed] [Google Scholar]
- 34.Rolff M, Schottenheim J, Decker H, Tuczek F. Copper–O2 Reactivity of Tyrosinase Models Towards External Monophenolic Substrates: Molecular Mechanism and Comparison with the Enzyme. Chem Soc Rev. 2011;40:4077–4098. doi: 10.1039/c0cs00202j. [DOI] [PubMed] [Google Scholar]
- 35.Lee JY, Karlin KD. Elaboration of Copper–Oxygen Mediated C–H Activation Chemistry in Consideration of Future Fuel and Feedstock Generation. Curr Opin Chem Biol. 2015;25:184–193. doi: 10.1016/j.cbpa.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ray K, Heims F, Schwalbe M, Nam W. High-Valent Metal-Oxo Intermediates in Energy Demanding Processes: From Dioxygen Reduction to Water Splitting. Curr Opin Chem Biol. 2015;25:159–171. doi: 10.1016/j.cbpa.2015.01.014. [DOI] [PubMed] [Google Scholar]
- 37.Yamaguchi S, Masuda H. Basic Approach to Development of Environment-Friendly Oxidation Catalyst Materials. Mononuclear Hydroperoxo Copper(II) Complexes. Sci Technol Adv Mater. 2005;6:34–47. [Google Scholar]
- 38.Dalle KE, Meyer F. Modelling Binuclear Metallobiosites: Insights from Pyrazole-Supported Biomimetic and Bioinspired Complexes. Eur J Inorg Chem. 2015;2015:3391–3405. [Google Scholar]
- 39.Serrano-Plana J, Garcia-Bosch I, Company A, Costas M. Structural and Reactivity Models for Copper Oxygenases: Cooperative Effects and Novel Reactivities. Acc Chem Res. 2015;48:2397–2406. doi: 10.1021/acs.accounts.5b00187. [DOI] [PubMed] [Google Scholar]
- 40.Li J, Farrokhnia M, Rulíšek L, Ryde U. Catalytic Cycle of Multicopper Oxidases Studied by Combined Quantum- and Molecular-Mechanical Free-Energy Perturbation Methods. J Phys Chem B. 2015;119:8268–8284. doi: 10.1021/acs.jpcb.5b02864. [DOI] [PubMed] [Google Scholar]
- 41.Klinman JP. The Copper-Enzyme Family of Dopamine β-Monooxygenase and Peptidylglycine α-Hydroxylating Monooxygenase: Resolving the Chemical Pathway for Substrate Hydroxylation. J Biol Chem. 2006;281:3013–3016. doi: 10.1074/jbc.R500011200. [DOI] [PubMed] [Google Scholar]
- 42.Hess CR, Wu Z, Ng A, Gray EE, McGuirl MA, Klinman JP. Hydroxylase Activity of Met471Cys Tyramine β-Monooxygenase. J Am Chem Soc. 2008;130:11939–11944. doi: 10.1021/ja800408h. [DOI] [PubMed] [Google Scholar]
- 43.Hess C, Klinman J, Blackburn N. The Copper Centers of Tyramine β-Monooxygenase and its Catalytic-Site Methionine Variants: An X-ray Absorption Study. JBIC, J Biol Inorg Chem. 2010;15:1195–1207. doi: 10.1007/s00775-010-0677-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vendelboe TV, Harris P, Zhao Y, Walter TS, Harlos K, El Omari K, Christensen HEM. The crystal structure of human dopamine β-hydroxylase at 2. 9 Å resolution. Science Advances. 2016;2:1–10. doi: 10.1126/sciadv.1500980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chufán EE, Prigge ST, Siebert X, Eipper BA, Mains RE, Amzel LM. Differential Reactivity between Two Copper Sites in Peptidylglycine α-Hydroxylating Monooxygenase. J Am Chem Soc. 2010;132:15565–15572. doi: 10.1021/ja103117r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rudzka K, Moreno DM, Eipper B, Mains R, Estrin DA, Amzel LM. Coordination of Peroxide to the CuM Center of Peptidylglycine α-Hydroxylating Monooxygenase (PHM): Structural and Computational Study. JBIC, J Biol Inorg Chem. 2013;18:223–232. doi: 10.1007/s00775-012-0967-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hemsworth GR, Davies GJ, Walton PH. Recent Insights Into Copper-Containing Lytic Polysaccharide Mono-Oxygenases. Curr Opin Struct Biol. 2013;23:660–668. doi: 10.1016/j.sbi.2013.05.006. [DOI] [PubMed] [Google Scholar]
- 48.Kjaergaard CH, Qayyum MF, Wong SD, Xu F, Hemsworth GR, Walton DJ, Young NA, Davies GJ, Walton PH, Johansen KS, Hodgson KO, Hedman B, Solomon EI. Spectroscopic and computational insight into the activation of O2 by the mononuclear Cu center in polysaccharide monooxygenases. Proc Natl Acad Sci U S A. 2014;111:8797–8802. doi: 10.1073/pnas.1408115111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gudmundsson M, Kim S, Wu M, Ishida T, Momeni MH, Vaaje-Kolstad G, Lundberg D, Royant A, Ståhlberg J, Eijsink VGH, et al. Structural and Electronic Snapshots during the Transition from a Cu(II) to Cu(I) Metal Center of a Lytic Polysaccharide Monooxygenase by X-ray Photoreduction. J Biol Chem. 2014;289:18782–18792. doi: 10.1074/jbc.M114.563494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hemsworth GR, Henrissat B, Davies GJ, Walton PH. Discovery and Characterization of a New Family of Lytic Polysaccharide Monooxygenases. Nat Chem Biol. 2013;10:122–126. doi: 10.1038/nchembio.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lo Leggio L, Simmons TJ, Poulsen JCN, Frandsen KEH, Hemsworth GR, Stringer MA, von Freiesleben P, Tovborg M, Johansen KS, De Maria L, et al. Structure and Boosting Activity of a Starch-Degrading Lytic Polysaccharide Monooxygenase. Nat Commun. 2015;6:5961–5969. doi: 10.1038/ncomms6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Culpepper MA, Rosenzweig AC. Architecture and Active Site of Particulate Methane Monooxygenase. Crit Rev Biochem Mol Biol. 2012;47:483–492. doi: 10.3109/10409238.2012.697865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bollinger JM, Jr, Krebs C. Enzymatic C–H Activation by Metal–Superoxo Intermediates. Curr Opin Chem Biol. 2007;11:151–158. doi: 10.1016/j.cbpa.2007.02.037. [DOI] [PubMed] [Google Scholar]
- 54.Prigge ST, Eipper BA, Mains RE, Amzel LM. Dioxygen Binds End-On to Mononuclear Copper in a Precatalytic Enzyme Complex. Science. 2004;304:864–867. doi: 10.1126/science.1094583. [DOI] [PubMed] [Google Scholar]
- 55.Chen P, Bell J, Eipper BA, Solomon EI. Oxygen Activation by the Noncoupled Binuclear Copper Site in Peptidylglycine α-Hydroxylating Monooxygenase. Spectroscopic Definition of the Resting Sites and the Putative CuIIM–OOH Intermediate. Biochemistry. 2004;43:5735–5747. doi: 10.1021/bi0362830. [DOI] [PubMed] [Google Scholar]
- 56.Meliá C, Ferrer S, Řezáč J, Parisel O, Reinaud O, Moliner V, de la Lande A. Investigation of the Hydroxylation Mechanism of Noncoupled Copper Oxygenases by Ab Initio Molecular Dynamics Simulations. Chem - Eur J. 2013;19:17328–17337. doi: 10.1002/chem.201301000. [DOI] [PubMed] [Google Scholar]
- 57.Rezabal E, Gauss J, Matxain JM, Berger R, Diefenbach M, Holthausen MC. Quantum Chemical Assessment of the Binding Energy of CuO+ J Chem Phys. 2011;134:064304-1–064304-13. doi: 10.1063/1.3537797. [DOI] [PubMed] [Google Scholar]
- 58.Chen P, Solomon EI. O2 activation by Binuclear Cu Sites: Noncoupled Versus Exchange Coupled Reaction Mechanisms. Proc Natl Acad Sci U S A. 2004;101:13105–13110. doi: 10.1073/pnas.0402114101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kamachi T, Kihara N, Shiota Y, Yoshizawa K. Computational Exploration of the Catalytic Mechanism of Dopamine β-Monooxygenase: Modeling of Its Mononuclear Copper Active Sites. Inorg Chem. 2005;44:4226–4236. doi: 10.1021/ic048477p. [DOI] [PubMed] [Google Scholar]
- 60.Yoshizawa K, Kihara N, Kamachi T, Shiota Y. Catalytic Mechanism of Dopamine β-Monooxygenase Mediated by Cu(III)-Oxo. Inorg Chem. 2006;45:3034–3041. doi: 10.1021/ic0521168. [DOI] [PubMed] [Google Scholar]
- 61.Kim S, Ståhlberg J, Sandgren M, Paton RS, Beckham GT. Quantum Mechanical Calculations Suggest that Lytic Polysaccharide Monooxygenases Use a Copper-Oxyl, Oxygen-Rebound Mechanism. Proc Natl Acad Sci U S A. 2014;111:149–154. doi: 10.1073/pnas.1316609111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Magnus KA, Ton-That H, Carpenter JE. Recent Structural Work on the Oxygen Transport Protein Hemocyanin. Chem Rev. 1994;94:727–735. [Google Scholar]
- 63.Matoba Y, Kumagai T, Yamamoto A, Yoshitsu H, Sugiyama M. Crystallographic Evidence That the Dinuclear Copper Center of Tyrosinase Is Flexible during Catalysis. J Biol Chem. 2006;281:8981–8990. doi: 10.1074/jbc.M509785200. [DOI] [PubMed] [Google Scholar]
- 64.Gerdemann C, Eicken C, Krebs B. The Crystal Structure of Catechol Oxidase: New Insight into the Function of Type-3 Copper Proteins. Acc Chem Res. 2002;35:183–191. doi: 10.1021/ar990019a. [DOI] [PubMed] [Google Scholar]
- 65.Ginsbach JW, Kieber-Emmons MT, Nomoto R, Noguchi A, Ohnishi Y, Solomon EI. Structure/Function Correlations Among Coupled Binuclear Copper Proteins Through Spectroscopic and Reactivity Studies of NspF. Proc Natl Acad Sci U S A. 2012;109:10793–10797. doi: 10.1073/pnas.1208718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Decker H, Schweikardt T, Nillius D, Salzbrunn U, Jaenicke E, Tuczek F. Similar Enzyme Activation and Catalysis in Hemocyanins and Tyrosinases. Gene. 2007;398:183–191. doi: 10.1016/j.gene.2007.02.051. [DOI] [PubMed] [Google Scholar]
- 67.Inoue T, Shiota Y, Yoshizawa K. Quantum Chemical Approach to the Mechanism for the Biological Conversion of Tyrosine to Dopaquinone. J Am Chem Soc. 2008;130:16890–16897. doi: 10.1021/ja802618s. [DOI] [PubMed] [Google Scholar]
- 68.Halfen JA, Mahapatra S, Wilkinson EC, Kaderli S, Young VG, Que L, Zuberbuhler AD, Tolman WB. Reversible Cleavage and Formation of the Dioxygen O-O Bond Within a Dicopper Complex. Science. 1996;271:1397–1400. doi: 10.1126/science.271.5254.1397. [DOI] [PubMed] [Google Scholar]
- 69.Cahoy J, Holland PL, Tolman WB. Experimental Studies of the Interconversion of μ-η2: η2-Peroxo- and Bis(μ-oxo)dicopper Complexes. Inorg Chem. 1999;38:2161–2168. doi: 10.1021/ic990095+. [DOI] [PubMed] [Google Scholar]
- 70.Himes RA, Karlin KD. Copper–Dioxygen Complex Mediated C–H Bond Oxygenation: Relevance for Particulate Methane Monooxygenase (pMMO) Curr Opin Chem Biol. 2009;13:119–131. doi: 10.1016/j.cbpa.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Culpepper MA, Cutsail GE, Hoffman BM, Rosenzweig AC. Evidence for Oxygen Binding at the Active Site of Particulate Methane Monooxygenase. J Am Chem Soc. 2012;134:7640–7643. doi: 10.1021/ja302195p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoshizawa K, Shiota Y. Conversion of Methane to Methanol at the Mononuclear and Dinuclear Copper Sites of Particulate Methane Monooxygenase (pMMO): A DFT and QM/MM Study. J Am Chem Soc. 2006;128:9873–9881. doi: 10.1021/ja061604r. [DOI] [PubMed] [Google Scholar]
- 73.Shiota Y, Yoshizawa K. Comparison of the Reactivity of Bis(μ-oxo)CuIICuIII and CuIIICuIII Species to Methane. Inorg Chem. 2009;48:838–845. doi: 10.1021/ic8003933. [DOI] [PubMed] [Google Scholar]
- 74.Itoyama S, Doitomi K, Kamachi T, Shiota Y, Yoshizawa K. Possible Peroxo State of the Dicopper Site of Particulate Methane Monooxygenase from Combined Quantum Mechanics and Molecular Mechanics Calculations. Inorg Chem. 2016;55:2771–2775. doi: 10.1021/acs.inorgchem.5b02603. [DOI] [PubMed] [Google Scholar]
- 75.Da Silva JCS, Pennifold RCR, Harvey JN, Rocha WR. A Radical Rebound Mechanism for the Methane Oxidation Reaction Promoted by the Dicopper Center of a pMMO Enzyme: A Computational Perspective. Dalton Trans. 2016;45:2492–2504. doi: 10.1039/c5dt02638e. [DOI] [PubMed] [Google Scholar]
- 76.Sirajuddin S, Rosenzweig AC. Enzymatic Oxidation of Methane. Biochemistry. 2015;54:2283–2294. doi: 10.1021/acs.biochem.5b00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Balasubramanian R, Rosenzweig AC. Structural and Mechanistic Insights into Methane Oxidation by Particulate Methane Monooxygenase. Acc Chem Res. 2007;40:573–580. doi: 10.1021/ar700004s. [DOI] [PubMed] [Google Scholar]
- 78.Balasubramanian R, Smith SM, Rawat S, Yatsunyk LA, Stemmler TL, Rosenzweig AC. Oxidation of Methane by a Biological Dicopper Centre. Nature. 2010;465:115–119. doi: 10.1038/nature08992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smith SM, Rawat S, Telser J, Hoffman BM, Stemmler TL, Rosenzweig AC. Crystal Structure and Characterization of Particulate Methane Monooxygenase from Methylocystis species Strain M. Biochemistry. 2011;50:10231–10240. doi: 10.1021/bi200801z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rosenzweig AC, Sazinsky MH. Structural Insights into Dioxygen-Activating Copper Enzymes. Curr Opin Struct Biol. 2006;16:729–735. doi: 10.1016/j.sbi.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 81.Vanelderen P, Vancauwenbergh J, Sels BF, Schoonheydt RA. Coordination Chemistry and Reactivity of Copper in Zeolites. Coord Chem Rev. 2013;257:483–494. [Google Scholar]
- 82.Vanelderen P, Hadt RG, Smeets PJ, Solomon EI, Schoonheydt RA, Sels BF. Cu-ZSM-5: A Biomimetic Inorganic Model for Methane Oxidation. J Catal. 2011;284:157–164. doi: 10.1016/j.jcat.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yumura T, Hirose Y, Wakasugi T, Kuroda Y, Kobayashi H. Roles of Water Molecules in Modulating the Reactivity of Dioxygen-Bound Cu-ZSM-5 toward Methane: A Theoretical Prediction. ACS Catal. 2016;6:2487–2495. [Google Scholar]
- 84.Lawton TJ, Rosenzweig AC. Methane-Oxidizing Enzymes: An Upstream Problem in Biological Gas-to-Liquids Conversion. J Am Chem Soc. 2016;138:9327–9340. doi: 10.1021/jacs.6b04568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen PPY, Chan SI. Theoretical Modeling of the Hydroxylation of Methane as Mediated by the Particulate Methane Monooxygenase. J Inorg Biochem. 2006;100:801–809. doi: 10.1016/j.jinorgbio.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 86.Chan SI, Yu SSF. Controlled Oxidation of Hydrocarbons by the Membrane-Bound Methane Monooxygenase: The Case for a Tricopper Cluster. Acc Chem Res. 2008;41:969–979. doi: 10.1021/ar700277n. [DOI] [PubMed] [Google Scholar]
- 87.Shao M, Chang Q, Dodelet JP, Chenitz R. Recent Advances in Electrocatalysts for Oxygen Reduction Reaction. Chem Rev. 2016;116:3594–3657. doi: 10.1021/acs.chemrev.5b00462. [DOI] [PubMed] [Google Scholar]
- 88.Yoshikawa S, Shimada A. Reaction Mechanism of Cytochrome c Oxidase. Chem Rev. 2015;115:1936–1989. doi: 10.1021/cr500266a. [DOI] [PubMed] [Google Scholar]
- 89.Kaila VRI, Verkhovsky MI, Wikström M. Proton-Coupled Electron Transfer in Cytochrome Oxidase. Chem Rev. 2010;110:7062–7081. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]
- 90.Kim E, Chufán EE, Kamaraj K, Karlin KD. Synthetic Models for Heme–Copper Oxidases. Chem Rev (Washington, DC, U S) 2004;104:1077–1134. doi: 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]
- 91.Karlin KD, Kim E. Ligand Influences in Heme–Copper O2-Chemistry as Synthetic Models for Cytochrome c Oxidase. Chem Lett. 2004;33:1226–1231. [Google Scholar]
- 92.Decreau RA, Collman JP, Hosseini A. Electrochemical Applications. How Click Chemistry Brought Biomimetic Models to the Next Level: Electrocatalysis Under Controlled Rate of Electron Transfer. Chem Soc Rev. 2010;39:1291–1301. doi: 10.1039/b901972n. [DOI] [PubMed] [Google Scholar]
- 93.Collman JP, Boulatov R, Sunderland CJ, Fu L. Functional Analogues of Cytochrome c Oxidase, Myoglobin, and Hemoglobin. Chem Rev. 2004;104:561–588. doi: 10.1021/cr0206059. [DOI] [PubMed] [Google Scholar]
- 94.Collman JP, Ghosh S. Recent Applications of a Synthetic Model of Cytochrome c Oxidase: Beyond Functional Modeling. Inorg Chem. 2010;49:5798–5810. doi: 10.1021/ic100472p. [DOI] [PubMed] [Google Scholar]
- 95.Chatterjee S, Sengupta K, Hematian S, Karlin KD, Dey A. Electrocatalytic O2-Reduction by Synthetic Cytochrome c Oxidase Mimics: Identification of a “Bridging Peroxo” Intermediate Involved in Facile 4e−/4H+ O2-Reduction. J Am Chem Soc. 2015;137:12897–12905. doi: 10.1021/jacs.5b06513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wende C, Lüdtke C, Kulak N. Copper Complexes of NDonor Ligands as Artificial Nucleases. Eur J Inorg Chem. 2014;2014:2597–2612. [Google Scholar]
- 97.Tegoni M, Valensin D, Toso L, Remelli M. Copper Chelators: Chemical Properties and Bio-medical Applications. Curr Med Chem. 2014;21:3785–3818. doi: 10.2174/0929867321666140601161939. [DOI] [PubMed] [Google Scholar]
- 98.Jin Y, Cowan JA. DNA Cleavage by Copper–ATCUN Complexes. Factors Influencing Cleavage Mechanism and Linearization of dsDNA. J Am Chem Soc. 2005;127:8408–8415. doi: 10.1021/ja0503985. [DOI] [PubMed] [Google Scholar]
- 99.Harford C, Sarkar B. Amino Terminal Cu(II)- and Ni(II)-Binding (ATCUN) Motif of Proteins and Peptides: Metal Binding, DNA Cleavage, and Other Properties. Acc Chem Res. 1997;30:123–130. [Google Scholar]
- 100.Yu Z, Han M, Cowan JA. Toward the Design of a Catalytic Metallodrug: Selective Cleavage of G-Quadruplex Telomeric DNA by an Anticancer Copper–Acridine–ATCUN Complex. Angew Chem, Int Ed. 2015;54:1901–1905. doi: 10.1002/anie.201410434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Joyner JC, Reichfield J, Cowan JA. Factors Influencing the DNA Nuclease Activity of Iron, Cobalt, Nickel, and Copper Chelates. J Am Chem Soc. 2011;133:15613–15626. doi: 10.1021/ja2052599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gaggelli E, Kozlowski H, Valensin D, Valensin G. Copper Homeostasis and Neurodegenerative Disorders (Alzheimer’s, Prion, and Parkinson’s Diseases and Amyotrophic Lateral Sclerosis) Chem Rev. 2006;106:1995–2044. doi: 10.1021/cr040410w. [DOI] [PubMed] [Google Scholar]
- 103.Barnham KJ, Bush AI. Metals in Alzheimer’s and Parkinson’s Diseases. Curr Opin Chem Biol. 2008;12:222–228. doi: 10.1016/j.cbpa.2008.02.019. [DOI] [PubMed] [Google Scholar]
- 104.Hureau C, Faller P. Aβ-mediated ROS Production by Cu Ions: Structural Insights, Mechanisms and Relevance to Alzheimer’s Disease. Biochimie. 2009;91:1212–1217. doi: 10.1016/j.biochi.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 105.Barnham KJ, Bush AI. Biological Metals and Metal-Targeting Compounds in Major Neurodegenerative Diseases. Chem Soc Rev. 2014;43:6727–6749. doi: 10.1039/c4cs00138a. [DOI] [PubMed] [Google Scholar]
- 106.McCann SD, Stahl SS. Mechanism of Copper/Azodicarboxylate-Catalyzed Aerobic Alcohol Oxidation: Evidence for Uncooperative Catalysis. J Am Chem Soc. 2016;138:199–206. doi: 10.1021/jacs.5b09940. [DOI] [PubMed] [Google Scholar]
- 107.Tsang ASK, Kapat A, Schoenebeck F. Factors That Control C–C Cleavage versus C–H Bond Hydroxylation in Copper-Catalyzed Oxidations of Ketones with O2. J Am Chem Soc. 2016;138:518–526. doi: 10.1021/jacs.5b08347. [DOI] [PubMed] [Google Scholar]
- 108.Alayon EMC, Nachtegaal M, Ranocchiari M, van Bokhoven JA. Catalytic Conversion of Methane to Methanol Using Cu-Zeolites. Chimia. 2012;66:668–674. doi: 10.2533/chimia.2012.668. [DOI] [PubMed] [Google Scholar]
- 109.Tomkins P, Mansouri A, Bozbag SE, Krumeich F, Park MB, Alayon EMC, Ranocchiari M, van Bokhoven JA. Isothermal Cyclic Conversion of Methane into Methanol over Copper-Exchanged Zeolite at Low Temperature. Angew Chem, Int Ed. 2016;55:5467–5471. doi: 10.1002/anie.201511065. [DOI] [PubMed] [Google Scholar]
- 110.Himes RA, Karlin KD. A New Copper-Oxo Player in Methane Oxidation. Proc Natl Acad Sci U S A. 2009;106:18877–18878. doi: 10.1073/pnas.0911413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Groothaert MH, Smeets PJ, Sels BF, Jacobs PA, Schoonheydt RA. Selective Oxidation of Methane by the Bis(μ-oxo)dicopper Core Stabilized on ZSM-5 and Mordenite Zeolites. J Am Chem Soc. 2005;127:1394–1395. doi: 10.1021/ja047158u. [DOI] [PubMed] [Google Scholar]
- 112.Woertink JS, Smeets PJ, Groothaert MH, Vance MA, Sels BF, Schoonheydt RA, Solomon EI. A [Cu2O]2+ Core in Cu-ZSM-5, the Active Site in the Oxidation of Methane to Methanol. Proc Natl Acad Sci U S A. 2009;106:18908–18913. doi: 10.1073/pnas.0910461106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vanelderen P, Snyder BER, Tsai ML, Hadt RG, Vancauwenbergh J, Coussens O, Schoonheydt RA, Sels BF, Solomon EI. Spectroscopic Definition of the Copper Active Sites in Mordenite: Selective Methane Oxidation. J Am Chem Soc. 2015;137:6383–6392. doi: 10.1021/jacs.5b02817. [DOI] [PubMed] [Google Scholar]
- 114.Alayon EMC, Nachtegaal M, Bodi A, Ranocchiari M, van Bokhoven JA. Bis(μ-oxo) Versus Mono(μ-oxo)dicopper Cores in a Zeolite for Converting Methane to Methanol: an in situ XAS and DFT Investigation. Phys Chem Chem Phys. 2015;17:7681–7693. doi: 10.1039/c4cp03226h. [DOI] [PubMed] [Google Scholar]
- 115.Smeets PJ, Hadt RG, Woertink JS, Vanelderen P, Schoonheydt RA, Sels BF, Solomon EI. Oxygen Precursor to the Reactive Intermediate in Methanol Synthesis by Cu-ZSM-5. J Am Chem Soc. 2010;132:14736–14738. doi: 10.1021/ja106283u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Alayon EM, Nachtegaal M, Ranocchiari M, van Bokhoven JA. Catalytic Conversion of Methane to Methanol over Cu–mordenite. Chem Commun. 2012;48:404–406. doi: 10.1039/c1cc15840f. [DOI] [PubMed] [Google Scholar]
- 117.Alayon EMC, Nachtegaal M, Bodi A, van Bokhoven JA. Reaction Conditions of Methane-to-Methanol Conversion Affect the Structure of Active Copper Sites. ACS Catal. 2014;4:16–22. [Google Scholar]
- 118.Grundner S, Markovits MAC, Li G, Tromp M, Pidko EA, Hensen EJM, Jentys A, Sanchez-Sanchez M, Lercher JA. Single-site Trinuclear Copper Oxygen Clusters in Mordenite for Selective Conversion of Methane to Methanol. Nat Commun. 2015;6:7546. doi: 10.1038/ncomms8546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kulkarni AR, Zhao ZJ, Siahrostami S, Nørskov JK, Studt F. Monocopper Active Site for Partial Methane Oxidation in Cu-Exchanged 8MR Zeolites. ACS Catal. 2016;6:6531–6536. [Google Scholar]
- 120.Reinaud O, Capdevielle P, Maumy M. Copper(II) Mediated Aromatic Hydroxylation by Trimethylamine N-oxide. J Chem Soc, Chem Commun. 1990:566–568. [Google Scholar]
- 121.Comba P, Knoppe S, Martin B, Rajaraman G, Rolli C, Shapiro B, Stork T. Copper(II)-Mediated Aromatic ortho-Hydroxylation: A Hybrid DFT and Ab Initio Exploration. Chem - Eur J. 2008;14:344–357. doi: 10.1002/chem.200700865. [DOI] [PubMed] [Google Scholar]
- 122.Conde A, Vilella L, Balcells D, Diaz-Requejo MM, Lledos A, Perez PJ. Introducing Copper as Catalyst for Oxidative Alkane Dehydrogenation. J Am Chem Soc. 2013;135:3887–3896. doi: 10.1021/ja310866k. [DOI] [PubMed] [Google Scholar]
- 123.Cramer CJ, Tolman WB. Mononuclear CuO2 complexes: Geometries, Spectroscopic Properties, Electronic Structures, and Reactivity. Acc Chem Res. 2007;40:601–608. doi: 10.1021/ar700008c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Itoh S. Developing Mononuclear Copper–Active-Oxygen Complexes Relevant to Reactive Intermediates of Biological Oxidation Reactions. Acc Chem Res. 2015;48:2066–2074. doi: 10.1021/acs.accounts.5b00140. [DOI] [PubMed] [Google Scholar]
- 125.Schatz M, Raab V, Foxon SP, Brehm G, Schneider S, Reiher M, Holthausen MC, Sundermeyer J, Schindler S. Combined Spectroscopic and Theoretical Evidence for a Persistent End-On Copper Superoxo Complex. Angew Chem, Int Ed. 2004;43:4360–4363. doi: 10.1002/anie.200454125. [DOI] [PubMed] [Google Scholar]
- 126.Würtele C, Gaoutchenova E, Harms K, Holthausen MC, Sundermeyer J, Schindler S. Crystallographic Characterization of a Synthetic 1:1 End-On Copper Dioxygen Adduct Complex. Angew Chem, Int Ed. 2006;45:3867–3869. doi: 10.1002/anie.200600351. [DOI] [PubMed] [Google Scholar]
- 127.Lee JY, Peterson RL, Ohkubo K, Garcia-Bosch I, Himes RA, Woertink J, Moore CD, Solomon EI, Fukuzumi S, Karlin KD. Mechanistic Insights into the Oxidation of Substituted Phenols via Hydrogen Atom Abstraction by a Cupric–Superoxo Complex. J Am Chem Soc. 2014;136:9925–9937. doi: 10.1021/ja503105b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Donoghue PJ, Gupta AK, Boyce DW, Cramer CJ, Tolman WB. An Anionic, Tetragonal Copper(II) Superoxide Complex. J Am Chem Soc. 2010;132:15869–15871. doi: 10.1021/ja106244k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kobayashi Y, Ohkubo K, Nomura T, Kubo M, Fujieda N, Sugimoto H, Fukuzumi S, Goto K, Ogura T, Itoh S. Copper(I)-Dioxygen Reactivity in a Sterically Demanding Tripodal Tetradentate tren Ligand: Formation and Reactivity of a Mononuclear Copper(II) End-On Superoxo Complex. Eur J Inorg Chem. 2012;2012:4574–4578. [Google Scholar]
- 130.Peterson RL, Himes RA, Kotani H, Suenobu T, Tian L, Siegler MA, Solomon EI, Fukuzumi S, Karlin KD. Cupric Superoxo-Mediated Intermolecular C–H Activation Chemistry. J Am Chem Soc. 2011;133:1702–1705. doi: 10.1021/ja110466q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kunishita A, Kubo M, Sugimoto H, Ogura T, Sato K, Takui T, Itoh S. Mononuclear Copper(II)–Superoxo Complexes that Mimic the Structure and Reactivity of the Active Centers of PHM and DβM. J Am Chem Soc. 2009;131:2788–2789. doi: 10.1021/ja809464e. [DOI] [PubMed] [Google Scholar]
- 132.Lanci MP, Smirnov VV, Cramer CJ, Gauchenova EV, Sundermeyer J, Roth JP. Isotopic Probing of Molecular Oxygen Activation at Copper(I) Sites. J Am Chem Soc. 2007;129:14697–14709. doi: 10.1021/ja074620c. [DOI] [PubMed] [Google Scholar]
- 133.Woertink JS, Tian L, Maiti D, Lucas HR, Himes RA, Karlin KD, Neese F, Würtele C, Holthausen MC, Bill E, et al. Spectroscopic and Computational Studies of an End-on Bound Superoxo-Cu(II) Complex: Geometric and Electronic Factors That Determine the Ground State. Inorg Chem. 2010;49:9450–9459. doi: 10.1021/ic101138u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Peterson RL, Ginsbach JW, Cowley RE, Qayyum MF, Himes RA, Siegler MA, Moore CD, Hedman B, Hodgson KO, Fukuzumi S, et al. Stepwise Protonation and Electron-Transfer Reduction of a Primary Copper-Dioxygen Adduct. J Am Chem Soc. 2013;135:16454–16467. doi: 10.1021/ja4065377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Ginsbach JW, Peterson RL, Cowley RE, Karlin KD, Solomon EI. Correlation of the Electronic and Geometric Structures in Mononuclear Copper(II) Superoxide Complexes. Inorg Chem. 2013;52:12872–12874. doi: 10.1021/ic402357u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Maiti D, Fry HC, Woertink JS, Vance MA, Solomon EI, Karlin KD. A 1:1 Copper–Dioxygen Adduct is an End-on Bound Superoxo Copper(II) Complex which Undergoes Oxygenation Reactions with Phenols. J Am Chem Soc. 2007;129:264–265. doi: 10.1021/ja067411l. [DOI] [PubMed] [Google Scholar]
- 137.Kim S, Lee JY, Cowley RE, Ginsbach JW, Siegler MA, Solomon EI, Karlin KD. A N3S(thioether)-Ligated CuII-Superoxo with Enhanced Reactivity. J Am Chem Soc. 2015;137:2796–2799. doi: 10.1021/ja511504n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sarangi R, Aboelella N, Fujisawa K, Tolman WB, Hedman B, Hodgson KO, Solomon EI. X-ray Absorption Edge Spectroscopy and Computational Studies on LCuO2 Species: Superoxide–CuII versus Peroxide–CuIII Bonding. J Am Chem Soc. 2006;128:8286–8296. doi: 10.1021/ja0615223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hill LMR, Gherman BF, Aboelella NW, Cramer CJ, Tolman WB. Electronic Tuning of β-Diketiminate Ligands with Fluorinated Substituents: Effects on the O2-Reactivity of Mononuclear Cu(I) Complexes. Dalton Trans. 2006:4944–4953. doi: 10.1039/b609939d. [DOI] [PubMed] [Google Scholar]
- 140.Reynolds AM, Gherman BF, Cramer CJ, Tolman WB. Characterization of a 1:1 Cu-O2 Adduct Supported by an Anilido Imine ligand. Inorg Chem. 2005;44:6989–6997. doi: 10.1021/ic050280p. [DOI] [PubMed] [Google Scholar]
- 141.Aboelella NW, Gherman BF, Hill LMR, York JT, Holm N, Young VG, Cramer CJ, Tolman WB. Effects of Thioether Substituents on the O2 reactivity of β-Diketiminate-Cu(I) complexes: Probing the Role of the Methionine Ligand in Copper Monooxygenases. J Am Chem Soc. 2006;128:3445–3458. doi: 10.1021/ja057745v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kjaergaard CH, Qayyum MF, Wong SD, Xu F, Hemsworth GR, Walton DJ, Young NA, Davies GJ, Walton PH, Johansen KS, et al. Spectroscopic and Computational Insight into the Activation of O2 by the Mononuclear Cu Center in Polysaccharide Monooxygenases. Proc Natl Acad Sci U S A. 2014;111:8797–8802. doi: 10.1073/pnas.1408115111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cramer CJ, Gour JR, Kinal A, Wloch M, Piecuch P, Shahi ARM, Gagliardi L. Stereoelectronic Effects on Molecular Geometries and State-Energy Splittings of Ligated Monocopper Dioxygen Complexes. J Phys Chem A. 2008;112:3754–3767. doi: 10.1021/jp800627e. [DOI] [PubMed] [Google Scholar]
- 144.Weitzer M, Schindler S, Brehm G, Schneider S, Hörmann E, Jung B, Kaderli S, Zuberbühler AD. Reversible Binding of Dioxygen by the Copper(I) Complex with Tris(2-dimethylaminoethyl)amine (Me6tren) Ligand. Inorg Chem. 2003;42:1800–1806. doi: 10.1021/ic025941m. [DOI] [PubMed] [Google Scholar]
- 145.Komiyama K, Furutachi H, Nagatomo S, Hashimoto A, Hayashi H, Fujinami S, Suzuki M, Kitagawa T. Dioxygen Reactivity of Copper(I) Complexes with Tetradentate Tripodal Ligands Having Aliphatic Nitrogen Donors: Synthesis, Structures, and Properties of Peroxo and Superoxo Complexes. Bull Chem Soc Jpn. 2004;77:59–72. [Google Scholar]
- 146.Chaudhuri P, Hess M, Weyhermüller T, Wieghardt K. Aerobic Oxidation of Primary Alcohols by a New Mononuclear CuII-Radical Catalyst. Angew Chem, Int Ed. 1999;38:1095–1098. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1095::AID-ANIE1095>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 147.Fujisawa K, Tanaka M, Moro-oka Y, Kitajima N. A Monomeric Side-On Superoxocopper(II) Complex: Cu(O2) (HB(3-tBu-5-iPrpz)3. J Am Chem Soc. 1994;116:12079–12080. [Google Scholar]
- 148.Chen P, Root DE, Campochiaro C, Fujisawa K, Solomon EI. Spectroscopic and Electronic Structure Studies of the Diamagnetic Side-On CuII-Superoxo Complex Cu(O2)[HB(3-R-5-iPrpz)3]: Anti-ferromagnetic Coupling versus Covalent Delocalization. J Am Chem Soc. 2003;125:466–474. doi: 10.1021/ja020969i. [DOI] [PubMed] [Google Scholar]
- 149.Spencer DJE, Aboelella NW, Reynolds AM, Holland PL, Tolman WB. β-Diketiminate Ligand Backbone Structural Effects on Cu(I)/O2 Reactivity: Unique Copper-Superoxo and Bis(μ-oxo) Complexes. J Am Chem Soc. 2002;124:2108–2809. doi: 10.1021/ja017820b. [DOI] [PubMed] [Google Scholar]
- 150.Aboelella NW, Lewis EA, Reynolds AM, Brennessel WW, Cramer CJ, Tolman WB. Snapshots of Dioxygen Activation by Copper: The Structure of a 1:1 Cu/O2 Adduct and Its Use in Syntheses of Asymmetric Bis(μ-oxo) Complexes. J Am Chem Soc. 2002;124:10660–10661. doi: 10.1021/ja027164v. [DOI] [PubMed] [Google Scholar]
- 151.Aboelella NW, Kryatov SV, Gherman BF, Brennessel WW, Young VG, Sarangi R, Rybak-Akimova EV, Hodgson KO, Hedman B, Solomon EI, et al. Dioxygen Activation at a Single Copper Site: Structure, Bonding, and Mechanism of Formation of 1:1 Cu–O2 Adducts. J Am Chem Soc. 2004;126:16896–16911. doi: 10.1021/ja045678j. [DOI] [PubMed] [Google Scholar]
- 152.Pantazis DA, McGrady JE. On the Nature of the Bonding in 1:1 Adducts of O2. Inorg Chem. 2003;42:7734–7736. doi: 10.1021/ic034867k. [DOI] [PubMed] [Google Scholar]
- 153.Gherman BF, Cramer CJ. Modeling the Peroxide/Superoxide Continuum in 1:1 Side-on Adducts of O2 with Cu. Inorg Chem. 2004;43:7281–7283. doi: 10.1021/ic049958b. [DOI] [PubMed] [Google Scholar]
- 154.Cramer CJ, Tolman WB, Theopold KH, Rheingold AL. Variable Character of O-O and M-O Bonding in Side-on (η2) 1:1 Metal Complexes of O2. Proc Natl Acad Sci U S A. 2003;100:3635–3640. doi: 10.1073/pnas.0535926100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lanci MP, Roth JP. Oxygen Isotope Effects upon Reversible O2-Binding Reactions: Characterizing Mononuclear Superoxide and Peroxide Structures. J Am Chem Soc. 2006;128:16006–16007. doi: 10.1021/ja0669326. [DOI] [PubMed] [Google Scholar]
- 156.Tian G, Klinman JP. Discrimination Between 16O and 18O in Oxygen Binding to the Reversible Oxygen Carriers Hemoglobin, Myoglobin, Hemerythrin, and Hemocyanin: A New Probe for Oxygen Binding and Reductive Activation by Proteins. J Am Chem Soc. 1993;115:8891–8897. [Google Scholar]
- 157.Harata M, Jitsukawa K, Masuda H, Einaga H. A Structurally Characterized Mononuclear Copper(II)-Superoxo Complex. J Am Chem Soc. 1994;116:10817–10818. [Google Scholar]
- 158.Berreau LM, Mahapatra S, Halfen JA, Young VG, Tolman WB. Independent Synthesis and Structural Characterization of a Mononuclear Copper–Hydroxide Complex Previously Assigned as a Copper–Superoxide Species. Inorg Chem. 1996;35:6339–6342. [Google Scholar]
- 159.Sanchez-Eguia BN, Flores-Alamo M, Orio M, Castillo I. Side-on Cupric-Superoxo Triplet Complexes as Competent Agents for H-abstraction Relevant to the Active Site of PHM. Chem Commun. 2015;51:11134–11137. doi: 10.1039/c5cc02332g. [DOI] [PubMed] [Google Scholar]
- 160.Saracini C, Liakos DG, Zapata Rivera JE, Neese F, Meyer GJ, Karlin KD. Excitation Wavelength Dependent O2 Release from Copper(II)–Superoxide Compounds: Laser Flash-Photolysis Experiments and Theoretical Studies. J Am Chem Soc. 2014;136:1260–1263. doi: 10.1021/ja4115314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lucas HR, Meyer GJ, Karlin KD. CO and O2 Binding to Pseudo-tetradentate Ligand–Copper(I) Complexes with a Variable NDonor Moiety: Kinetic/Thermodynamic Investigation Reveals Ligand-Induced Changes in Reaction Mechanism. J Am Chem Soc. 2010;132:12927–12940. doi: 10.1021/ja104107q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kunishita A, Ertem MZ, Okubo Y, Tano T, Sugimoto H, Ohkubo K, Fujieda N, Fukuzumi S, Cramer CJ, Itoh S. Active Site Models for the CuA Site of Peptidylglycine α-Hydroxylating Monooxygenase and Dopamine β-Monooxygenase. Inorg Chem. 2012;51:9465–9480. doi: 10.1021/ic301272h. [DOI] [PubMed] [Google Scholar]
- 163.Fry HC, Scaltrito DV, Karlin KD, Meyer GJ. The Rate of O2 and CO Binding to a Copper Complex, Determined by a “Flashand-Trap” Technique, Exceeds that for Hemes. J Am Chem Soc. 2003;125:11866–11871. doi: 10.1021/ja034911v. [DOI] [PubMed] [Google Scholar]
- 164.Astner J, Weitzer M, Foxon SP, Schindler S, Heinemann FW, Mukherjee J, Gupta R, Mahadevan V, Mukherjee R. Syntheses, Characterization, and Reactivity of Copper Complexes with Tridentate N-Donor Ligands. Inorg Chim Acta. 2008;361:279–292. [Google Scholar]
- 165.Smirnov VV, Roth JP. Evidence for Cu–O2 Intermediates in Superoxide Oxidations by Biomimetic Copper(II) Complexes. J Am Chem Soc. 2006;128:3683–3695. doi: 10.1021/ja056741n. [DOI] [PubMed] [Google Scholar]
- 166.Spencer DJE, Reynolds AM, Holland PL, Jazdzewski BA, Duboc-Toia C, Le Pape L, Yokota S, Tachi Y, Itoh S, Tolman WB. Copper Chemistry of β-Diketiminate Ligands: Monomer/Dimer Equilibria and a New Class of Bis(μ-oxo)dicopper Compounds. Inorg Chem. 2002;41:6307–6321. doi: 10.1021/ic020369k. [DOI] [PubMed] [Google Scholar]
- 167.Aboelella NW, York JT, Reynolds AM, Fujita K, Kinsinger CR, Cramer CJ, Riordan CG, Tolman WB. Mixed Metal Bis(μ-oxo) complexes with [CuM(μ-O)2]n+ (M = Ni(III) or Pd(II)) cores. Chem Commun. 2004:1716–1717. doi: 10.1039/b404640d. [DOI] [PubMed] [Google Scholar]
- 168.York JT, Young VG, Tolman WB. Heterobimetallic Activation of Dioxygen: Characterization and Reactivity of Novel Cu(I)-Ge(II) Complexes. Inorg Chem. 2006;45:4191–4198. doi: 10.1021/ic060050q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gherman BF, Heppner DE, Tolman WB, Cramer CJ. Models for Dioxygen Activation by the CuB Site of Dopamine β-Monooxygenase and Peptidylglycine α-Hydroxylating Monooxygenase. JBIC, J Biol Inorg Chem. 2006;11:197–205. doi: 10.1007/s00775-005-0066-5. [DOI] [PubMed] [Google Scholar]
- 170.Reynolds AM, Lewis EA, Aboelella NW, Tolman WB. Reactivity of a 1:1 Copper-Oxygen Complex: Isolation of a Cu(II)-o-Iminosemiquinonato Species. Chem Commun. 2005:2014–2016. doi: 10.1039/b418939f. [DOI] [PubMed] [Google Scholar]
- 171.Karlin KD, Cohen BI, Jacobson RR, Zubieta J. Dioxygen-Copper Reactivity: Hydroxylation-Induced Methyl Migration in a Copper Monooxygenase Model System. J Am Chem Soc. 1987;109:6194–6196. [Google Scholar]
- 172.Laitar DS, Mathison CJN, Davis WM, Sadighi JP. Copper(I) Complexes of a Heavily Fluorinated β-Diketiminate Ligand: Synthesis, Electronic Properties, and Intramolecular Aerobic Hydroxylation. Inorg Chem. 2003;42:7354–7356. doi: 10.1021/ic035056j. [DOI] [PubMed] [Google Scholar]
- 173.Sterckx H, De Houwer J, Mensch C, Caretti I, Tehrani KA, Herrebout WA, Van Doorslaer S, Maes BUW. Mechanism of the CuII-catalyzed benzylic oxygenation of (aryl) (heteroaryl)methanes with oxygen. Chem Sci. 2016;7:346–357. doi: 10.1039/c5sc03530a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tano T, Okubo Y, Kunishita A, Kubo M, Sugimoto H, Fujieda N, Ogura T, Itoh S. Redox Properties of a Mononuclear Copper(II)-Superoxide Complex. Inorg Chem. 2013;52:10431–10437. doi: 10.1021/ic401261z. [DOI] [PubMed] [Google Scholar]
- 175.Maiti D, Lee DH, Gaoutchenova K, Würtele C, Holthausen MC, Narducci Sarjeant AA, Sundermeyer J, Schindler S, Karlin KD. Reactions of a Copper(II) Superoxo Complex Lead to C–H and O–H Substrate Oxygenation: Modeling Copper-Monooxygenase C–H Hydroxylation. Angew Chem, Int Ed. 2008;47:82–85. doi: 10.1002/anie.200704389. [DOI] [PubMed] [Google Scholar]
- 176.Kakuda S, Rolle CJ, Ohkubo K, Siegler MA, Karlin KD, Fukuzumi S. Lewis Acid-Induced Change from Four-to Two-Electron Reduction of Dioxygen Catalyzed by Copper Complexes Using Scandium Triflate. J Am Chem Soc. 2015;137:3330–3337. doi: 10.1021/ja512584r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.McCrory CCL, Ottenwaelder X, Stack TDP, Chidsey CED. Kinetic and Mechanistic Studies of the Electrocatalytic Reduction of O2 to H2O with Mononuclear Cu Complexes of Substituted 1,10-Phenanthrolines. J Phys Chem A. 2007;111:12641–12650. doi: 10.1021/jp076106z. [DOI] [PubMed] [Google Scholar]
- 178.Fukuzumi S, Kotani H, Lucas HR, Doi K, Suenobu T, Peterson RL, Karlin KD. Mononuclear Copper Complex-Catalyzed Four-Electron Reduction of Oxygen. J Am Chem Soc. 2010;132:6874–6875. doi: 10.1021/ja100538x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Osako T, Nagatomo S, Kitagawa T, Cramer C, Itoh S. Kinetics and DFT Studies on the Reaction of Copper(II) Complexes and H2O2. JBIC, J Biol Inorg Chem. 2005;10:581–590. doi: 10.1007/s00775-005-0005-5. [DOI] [PubMed] [Google Scholar]
- 180.Kunishita A, Teraoka J, Scanlon JD, Matsumoto T, Suzuki M, Cramer CJ, Itoh S. Aromatic Hydroxylation Reactivity of a Mononuclear Cu(II)–Alkylperoxo Complex. J Am Chem Soc. 2007;129:7248–7249. doi: 10.1021/ja071623g. [DOI] [PubMed] [Google Scholar]
- 181.Mandal S, Mukherjee R. A New Tyrosinase Model with 1,3-Bis[(2-dimethylaminoethyl)iminomethyl]benzene: Binuclear Copper-(I) and Phenoxo/Hydroxo-Bridged Dicopper(II) Complexes. Inorg Chim Acta. 2006;359:4019–4026. [Google Scholar]
- 182.Maiti D, Narducci Sarjeant AA, Karlin KD. Copper(II)–Hydroperoxo Complex Induced Oxidative N-Dealkylation Chemistry. J Am Chem Soc. 2007;129:6720–6721. doi: 10.1021/ja0719024. [DOI] [PubMed] [Google Scholar]
- 183.Kamachi T, Lee YM, Nishimi T, Cho J, Yoshizawa K, Nam W. Combined Experimental and Theoretical Approach To Understand the Reactivity of a Mononuclear Cu(II)–Hydroperoxo Complex in Oxygenation Reactions. J Phys Chem A. 2008;112:13102–13108. doi: 10.1021/jp804804j. [DOI] [PubMed] [Google Scholar]
- 184.Kunishita A, Scanlon JD, Ishimaru H, Honda K, Ogura T, Suzuki M, Cramer CJ, Itoh S. Reactions of Copper(II)-H2O2 Adducts Supported by Tridentate Bis(2-pyridylmethyl)amine Ligands: Sensitivity to Solvent and Variations in Ligand Substitution. Inorg Chem. 2008;47:8222–8232. doi: 10.1021/ic800845h. [DOI] [PubMed] [Google Scholar]
- 185.Kunishita A, Ishimaru H, Nakashima S, Ogura T, Itoh S. Reactivity of Mononuclear Alkylperoxo Copper(II) Complex. O–O Bond Cleavage and C–H Bond Activation. J Am Chem Soc. 2008;130:4244–4245. doi: 10.1021/ja800443s. [DOI] [PubMed] [Google Scholar]
- 186.Maiti D, Narducci Sarjeant AA, Karlin KD. Copper–Hydroperoxo-Mediated N-Debenzylation Chemistry Mimicking Aspects of Copper Monooxygenases. Inorg Chem. 2008;47:8736–8747. doi: 10.1021/ic800617m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Chaloner L, Askari MS, Kutteh A, Schindler S, Ottenwaelder X. Formation and Reactivity of a Biomimetic Hydroperoxocopper(II) Cryptate. Eur J Inorg Chem. 2011;2011:4204–4211. [Google Scholar]
- 188.Kim S, Saracini C, Siegler MA, Drichko N, Karlin KD. Coordination Chemistry and Reactivity of a Cupric Hydroperoxide Species Featuring a Proximal H-Bonding Substituent. Inorg Chem. 2012;51:12603–12605. doi: 10.1021/ic302071e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kim S, Ginsbach JW, Lee JY, Peterson RL, Liu JJ, Siegler MA, Sarjeant AA, Solomon EI, Karlin KD. Amine Oxidative N-Dealkylation via Cupric Hydroperoxide Cu-OOH Homolytic Cleavage Followed by Site-Specific Fenton Chemistry. J Am Chem Soc. 2015;137:2867–2874. doi: 10.1021/ja508371q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Choi YJ, Cho KB, Kubo M, Ogura T, Karlin KD, Cho J, Nam W. Spectroscopic and Computational Characterization of CuIIOOR (R = H or cumyl) Complexes Bearing a Me6tren Ligand. Dalton Trans. 2011;40:2234–2241. doi: 10.1039/c0dt01036g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Tano T, Sugimoto H, Fujieda N, Itoh S. Heterolytic Alkyl Hydroperoxide O–O Bond Cleavage by Copper(I) Complexes. Eur J Inorg Chem. 2012;2012:4099–4103. [Google Scholar]
- 192.Tano T, Mieda K, Sugimoto H, Ogura T, Itoh S. A Copper Complex Supported by an N2S-Tridentate Ligand Inducing Efficient Heterolytic O–O Bond Cleavage of Alkylhydroperoxide. Dalton Trans. 2014;43:4871–4877. doi: 10.1039/c3dt52952e. [DOI] [PubMed] [Google Scholar]
- 193.Biswas S, Dutta A, Debnath M, Dolai M, Das KK, Ali M. A Novel Thermally Stable Hydroperoxo-Copper(II) Complex in a Cu(N2O2) Chromophore of a Potential N4O2 Donor Schiff Base Ligand: Synthesis, Structure and Catalytic Studies. Dalton Trans. 2013;42:13210–13219. doi: 10.1039/c3dt51359a. [DOI] [PubMed] [Google Scholar]
- 194.Paria S, Ohta T, Morimoto Y, Ogura T, Sugimoto H, Fujieda N, Goto K, Asano K, Suzuki T, Itoh S. Generation, Characterization, and Reactivity of a CuII–Alkylperoxide/Anilino Radical Complex: Insight into the O–O Bond Cleavage Mechanism. J Am Chem Soc. 2015;137:10870–10873. doi: 10.1021/jacs.5b04104. [DOI] [PubMed] [Google Scholar]
- 195.Das D, Lee YM, Ohkubo K, Nam W, Karlin KD, Fukuzumi S. Temperature-Independent Catalytic Two-Electron Reduction of Dioxygen by Ferrocenes with a Copper(II) Tris[2-(2-pyridyl)ethyl]amine Catalyst in the Presence of Perchloric Acid. J Am Chem Soc. 2013;135:2825–2834. doi: 10.1021/ja312523u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Osako T, Nagatomo S, Tachi Y, Kitagawa T, Itoh S. Low-Temperature Stopped-Flow Studies on the Reactions of Copper(II) Complexes and H2O2: The First Detection of a Mononuclear Copper(II)–Peroxo Intermediate. Angew Chem, Int Ed. 2002;41:4325–4328. doi: 10.1002/1521-3773(20021115)41:22<4325::AID-ANIE4325>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 197.Payeras AMi, Ho RYN, Fujita M, Que L., Jr The Reaction of [FeII(tpa)] with H2O2 in Acetonitrile and Acetone–Distinct Intermediates and Yet Similar Catalysis. Chem - Eur J. 2004;10:4944–4953. doi: 10.1002/chem.200400480. [DOI] [PubMed] [Google Scholar]
- 198.Wada A, Harata M, Hasegawa K, Jitsukawa K, Masuda H, Mukai M, Kitagawa T, Einaga H. Structural and Spectroscopic Characterization of a Mononuclear Hydroperoxo–Copper(II) Complex with Tripodal Pyridylamine Ligands. Angew Chem, Int Ed. 1998;37:798–799. doi: 10.1002/(SICI)1521-3773(19980403)37:6<798::AID-ANIE798>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 199.Kitajima N, Katayama T, Fujisawa K, Iwata Y, Morooka Y. Synthesis, Molecular Structure, and Reactivity of (Alkylperoxo)copper-(II) Complex. J Am Chem Soc. 1993;115:7872–7873. [Google Scholar]
- 200.Comba P, Haaf C, Helmle S, Karlin KD, Pandian S, Waleska A. Dioxygen Reactivity of New Bispidine-Copper Complexes. Inorg Chem. 2012;51:2841–2851. doi: 10.1021/ic2019296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kim S, Siegler MA, Karlin KD. Peroxynitrite Chemistry Derived from Nitric Oxide Reaction with a Cu(II)-OOH Species and a Copper Mediated NO Reductive Coupling Reaction. Chem Commun (Cambridge, U K) 2014;50:2844–2846. doi: 10.1039/c3cc47942k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Park GY, Lee JY, Himes RA, Thomas GS, Blackburn NJ, Karlin KD. Copper–Peptide Complex Structure and Reactivity When Found in Conserved His-Xaa-His Sequences. J Am Chem Soc. 2014;136:12532–12535. doi: 10.1021/ja505098v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Ohta T, Tachiyama T, Yoshizawa K, Yamabe T. Synthesis, Structure, and Catalytic Function of a Disulfide-Bridged Dicopper(I) Complex. Tetrahedron Lett. 2000;41:2581–2585. doi: 10.1021/ic000018a. [DOI] [PubMed] [Google Scholar]
- 204.Fujii T, Naito A, Yamaguchi S, Wada A, Funahashi Y, Jitsukawa K, Nagatomo S, Kitagawa T, Masuda H. Construction of a Square-Planar Hydroperoxo-Copper(II) Complex Inducing a Higher Catalytic Reactivity. Chem Commun. 2003:2700–2701. doi: 10.1039/b308073k. [DOI] [PubMed] [Google Scholar]
- 205.Fujii T, Yamaguchi S, Funahashi Y, Ozawa T, Tosha T, Kitagawa T, Masuda H. Mononuclear Copper(II)-Hydroperoxo Complex Derived from Reaction of Copper(I) Complex with Dioxygen as a Model of DβM and PHM. Chem Commun. 2006:4428–4430. doi: 10.1039/b609673e. [DOI] [PubMed] [Google Scholar]
- 206.Ohtsu H, Itoh S, Nagatomo S, Kitagawa T, Ogo S, Watanabe Y, Fukuzumi S. Characterization of Imidazolate-Bridged Dinuclear and Mononuclear Hydroperoxo Complexes. Inorg Chem. 2001;40:3200–3207. doi: 10.1021/ic001036v. [DOI] [PubMed] [Google Scholar]
- 207.Yamaguchi S, Nagatomo S, Kitagawa T, Funahashi Y, Ozawa T, Jitsukawa K, Masuda H. Copper Hydroperoxo Species Activated by Hydrogen-Bonding Interaction with Its Distal Oxygen. Inorg Chem. 2003;42:6968–6970. doi: 10.1021/ic035080x. [DOI] [PubMed] [Google Scholar]
- 208.Chen P, Fujisawa K, Solomon EI. Spectroscopic and Theoretical Studies of Mononuclear Copper(II) Alkyl- and Hydroperoxo Complexes: Electronic Structure Contributions to Reactivity. J Am Chem Soc. 2000;122:10177–10193. [Google Scholar]
- 209.Kodera M, Kita T, Miura I, Nakayama N, Kawata T, Kano K, Hirota S. Hydroperoxo–Copper(II) Complex Stabilized by N3S-Type Ligand Having a Phenyl Thioether. J Am Chem Soc. 2001;123:7715–7716. doi: 10.1021/ja010689n. [DOI] [PubMed] [Google Scholar]
- 210.Sanyal I, Ghosh P, Karlin KD. Mononuclear Copper(II)-Acylperoxo Complexes. Inorg Chem. 1995;34:3050–3056. [Google Scholar]
- 211.Yamaguchi S, Wada A, Nagatomo S, Kitagawa T, Jitsukawa K, Masuda H. Thermal Stability of Mononuclear Hydroperoxocopper(II) Species, Effects of Hydrogen Bonding and Hydrophobic Field. Chem Lett. 2004;33:1556–1557. [Google Scholar]
- 212.Poater A, Cavallo L. Probing the Mechanism of O2 Activation by a Copper(I) Biomimetic Complex of a C-H Hydroxylating Copper Monooxygenase. Inorg Chem. 2009;48:4062–4066. doi: 10.1021/ic802269v. [DOI] [PubMed] [Google Scholar]
- 213.Whittaker JW. Free Radical Catalysis by Galactose Oxidase. Chem Rev. 2003;103:2347–2363. doi: 10.1021/cr020425z. [DOI] [PubMed] [Google Scholar]
- 214.Jazdzewski BA, Tolman WB. Understanding the Copper-Phenoxyl Radical Array in Galactose Oxidase: Contributions from Synthetic Modeling Studies. Coord Chem Rev. 2000;200–202:633–685. [Google Scholar]
- 215.Itoh S, Taki M, Fukuzumi S. Active Site Models for Galactose Oxidase and Related Enyzmes. Coord Chem Rev. 2000;198:3–20. [Google Scholar]
- 216.Whittaker MM, Whittaker JW. Catalytic Reaction Profile for Alcohol Oxidation by Galactose Oxidase. Biochemistry. 2001;40:7140–7148. doi: 10.1021/bi010303l. [DOI] [PubMed] [Google Scholar]
- 217.Pacher P, Beckman JS, Liaudet L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Kitajima N, Koda T, Iwata Y, Moro-oka Y. Reaction Aspects of a μ-Peroxo Binuclear Copper(II) Complex. J Am Chem Soc. 1990;112:8833–8839. [Google Scholar]
- 219.Réglier M, Amadeï E, Tadayoni R, Waegell B. Pyridine Nucleus Hydroxylation with Copper Oxygenase Models. J Chem Soc, Chem Commun. 1989:447–450. [Google Scholar]
- 220.Fiedler A, Schröeder D, Shaik S, Schwarz H. Electronic Structures and Gas-Phase Reactivities of Cationic Late-Transition-Metal Oxides. J Am Chem Soc. 1994;116:10734–10741. [Google Scholar]
- 221.Schröder D, Holthausen MC, Schwarz H. Radical-Like Activation of Alkanes by the Ligated Copper Oxide Cation (Phenanthroline)CuO+ J Phys Chem B. 2004;108:14407–14416. [Google Scholar]
- 222.Dietl N, van der Linde C, Schlangen M, Beyer MK, Schwarz H. Diatomic [CuO]+ and Its Role in the Spin-Selective Hydrogen- and Oxygen-Atom Transfers in the Thermal Activation of Methane. Angew Chem, Int Ed. 2011;50:4966–4969. doi: 10.1002/anie.201100606. [DOI] [PubMed] [Google Scholar]
- 223.Dietl N, Schlangen M, Schwarz H. Thermal Hydrogen-Atom Transfer from Methane: The Role of Radicals and Spin States in Oxo-Cluster Chemistry. Angew Chem, Int Ed. 2012;51:5544–5555. doi: 10.1002/anie.201108363. [DOI] [PubMed] [Google Scholar]
- 224.Rijs NJ, Weiske T, Schlangen M, Schwarz H. On Divorcing Isomers, Dissecting Reactivity, and Resolving Mechanisms of Propane C-H and aryl C-X (X = halogen) Bond Activations Mediated by a Ligated Copper(III) Oxo Complex. Chem Phys Lett. 2014;608:408–424. [Google Scholar]
- 225.Schwarz H. Thermal Hydrogen-Atom Transfer from Methane: A Mechanistic Exercise. Chem Phys Lett. 2015;629:91–101. [Google Scholar]
- 226.Yoshizawa K. In: Methane Hydroxylation by First Row Transition Metal Oxides. Computational Modeling for Homogeneous and Enzymatic Catalysis: A Knowledge-Base for Designing Efficient Catalysts. Morokuma K, Musaev DG, editors. Wiley-VCH; Weinheim, Germany: 2008. pp. 317–335. [Google Scholar]
- 227.Gherman BF, Tolman WB, Cramer CJ. Characterization of the Structure and Reactivity of Monocopper-Oxygen Complexes Supported by β-Diketiminate and Anilido-Imine Ligands. J Comput Chem. 2006;27:1950–1961. doi: 10.1002/jcc.20502. [DOI] [PubMed] [Google Scholar]
- 228.Hong S, Huber SM, Gagliardi L, Cramer CC, Tolman WB. Copper(I)-α-Ketocarboxylate Complexes: Characterization and O2 Reactions That Yield Copper-Oxygen Intermediates Capable of Hydroxylating Arenes. J Am Chem Soc. 2007;129:14190–14192. doi: 10.1021/ja0760426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Shaik S, Danovich D, Fiedler A, Schröder D, Schwarz H. Two-State Reactivity in Organometallic Gas-Phase Ion Chemistry. Helv Chim Acta. 1995;78:1393–1407. [Google Scholar]
- 230.Rodgers MT, Walker B, Armentrout PB. Reactions of Cu+(1S and 3D) with O2, CO, CO2, N2, NO, N2O, and NO2 Studied by Guided Ion Beam Mass Spectrometry. Int J Mass Spectrom. 1999;182–183:99–120. [Google Scholar]
- 231.Rijs NJ, González-Navarrete P, Schlangen M, Schwarz H. Penetrating the Elusive Mechanism of Copper-Mediated Fluoromethylation in the Presence of Oxygen through the Gas-Phase Reactivity of Well-Defined [LCuO]+ Complexes with Fluoromethanes (CH(4−n)Fn, n = 1–3. J Am Chem Soc. 2016;138(9):3125–3135. doi: 10.1021/jacs.5b12972. [DOI] [PubMed] [Google Scholar]
- 232.Costas M, Mehn MP, Jensen MP, Que L., Jr Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes Models and Intermediates. Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 233.Abu-Omar MM, Loaiza A, Hontzeas N. Reaction Mechanisms of Mononuclear Non-Heme Iron Oxygenases. Chem Rev. 2005;105:2227–2252. doi: 10.1021/cr040653o. [DOI] [PubMed] [Google Scholar]
- 234.Purpero V, Moran GR. The Diverse and Pervasive Chemistries of the α-keto Acid Dependent Enzymes. JBIC, J Biol Inorg Chem. 2007;12:587–601. doi: 10.1007/s00775-007-0231-0. [DOI] [PubMed] [Google Scholar]
- 235.Huber SM, Ertem MZ, Aquilante F, Gagliardi L, Tolman WB, Cramer CJ. Generating CuII-Oxyl/CuIII-Oxo Species from CuI-α-Ketocarboxylate Complexes and O2: In Silico Studies on Ligand Effects and C-H-Activation Reactivity. Chem - Eur J. 2009;15:4886–4895. doi: 10.1002/chem.200802338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Hong S, Gupta AK, Tolman WB. Intermediates in Reactions of Copper(I) Complexes with N-Oxides: From the Formation of Stable Adducts to Oxo Transfer. Inorg Chem. 2009;48:6323–6325. doi: 10.1021/ic900435p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Donoghue PJ, Tehranchi J, Cramer CJ, Sarangi R, Solomon EI, Tolman WB. Rapid C–H Bond Activation by a Monocopper(III)–Hydroxide Complex. J Am Chem Soc. 2011;133:17602–17605. doi: 10.1021/ja207882h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Dhar D, Tolman WB. Hydrogen Atom Abstraction from Hydrocarbons by a Copper(III)-Hydroxide Complex. J Am Chem Soc. 2015;137:1322–1329. doi: 10.1021/ja512014z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Dhar D, Yee GM, Spaeth AD, Boyce DW, Zhang H, Dereli B, Cramer CJ, Tolman WB. Perturbing the Copper(III)-Hydroxide Unit through Ligand Structural Variation. J Am Chem Soc. 2016;138:356–368. doi: 10.1021/jacs.5b10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Anson FC, Collins TJ, Richmond TG, Santarsiero BD, Toth JE, Treco BGRT. Highly Stabilized Copper(III) Complexes. J Am Chem Soc. 1987;109:2974–2979. [Google Scholar]
- 241.McDonald MR, Fredericks FC, Margerum DW. Characterization of Copper(III)–Tetrapeptide Complexes with Histidine as the Third Residue. Inorg Chem. 1997;36:3119–3124. doi: 10.1021/ic9608713. [DOI] [PubMed] [Google Scholar]
- 242.Youngblood MP, Margerum DW. Reaction Entropies of Copper(III,II) Peptide and Nickel(III,II) Peptide Redox Couples and the Role of Axial Solvent Coordination. Inorg Chem. 1980;19:3068–3072. [Google Scholar]
- 243.Burke SK, Xu Y, Margerum DW. Cu(II)Gly2HisGly Oxidation by H2O2/Ascorbic Acid to the Cu(III) Complex and Its Subsequent Decay to Alkene Peptides. Inorg Chem. 2003;42:5807–5817. doi: 10.1021/ic0345774. [DOI] [PubMed] [Google Scholar]
- 244.Diaddario LL, Robinson WR, Margerum DW. Crystal and Molecular Structure of the Copper(III)-Tripeptide Complex of Tri-α-Aminoisobutyric Acid. Inorg Chem. 1983;22:1021–1025. [Google Scholar]
- 245.Warren JJ, Tronic TA, Mayer JM. Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem Rev. 2010;110:6961–7001. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Mayer JM. Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc Chem Res. 2011;44:36–46. doi: 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Dhar D, Yee GM, Markle TF, Mayer JM, Tolman WB. Reactivity of the Copper(III)-Hydroxide Unit with Phenols. Chem Sci. 2017 doi: 10.1039/C6SC03039D. published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Rolff M, Tuczek F. How Do Copper Enzymes Hydroxylate Aliphatic Substrates? Recent Insights from the Chemistry of Model Systems. Angew Chem, Int Ed. 2008;47:2344–2347. doi: 10.1002/anie.200705533. [DOI] [PubMed] [Google Scholar]
- 249.Decker H, Schweikardt T, Tuczek F. The First Crystal Structure of Tyrosinase: All Questions Answered? Angew Chem, Int Ed. 2006;45:4546–4550. doi: 10.1002/anie.200601255. [DOI] [PubMed] [Google Scholar]
- 250.Karahalis GJ, Thangavel A, Chica B, Bacsa J, Dyer RB, Scarborough CC. Synthesis and Catalytic Reactivity of a Dicopper(II) μ-η2:η2-Peroxo Species Supported by 1,4,7-Tri-tert-butyl-1,4,7-triazacyclononane. Inorg Chem. 2016;55:1102–1107. doi: 10.1021/acs.inorgchem.5b02205. [DOI] [PubMed] [Google Scholar]
- 251.Mirica LM, Vance M, Rudd DJ, Hedman B, Hodgson KO, Solomon EI, Stack TDP. Tyrosinase Reactivity in a Model Complex: An Alternative Hydroxylation Mechanism. Science. 2005;308:1890–1892. doi: 10.1126/science.1112081. [DOI] [PubMed] [Google Scholar]
- 252.Mirica LM, Rudd DJ, Vance MA, Solomon EI, Hodgson KO, Hedman B, Stack TDP. μ-η2:η2-Peroxodicopper-(II) Complex with a Secondary Diamine Ligand: A Functional Model of Tyrosinase. J Am Chem Soc. 2006;128:2654–2665. doi: 10.1021/ja056740v. [DOI] [PubMed] [Google Scholar]
- 253.Op’t Holt BT, Vance MA, Mirica LM, Heppner DE, Stack TDP, Solomon EI. Reaction Coordinate of a Functional Model of Tyrosinase: Spectroscopic and Computational Characterization. J Am Chem Soc. 2009;131:6421–6438. doi: 10.1021/ja807898h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Xu B, Lumb JP, Arndtsen BA. A TEMPO-Free Copper-Catalyzed Aerobic Oxidation of Alcohols. Angew Chem, Int Ed. 2015;54:4208–4211. doi: 10.1002/anie.201411483. [DOI] [PubMed] [Google Scholar]
- 255.Esguerra KVN, Fall Y, Lumb JP. A Biomimetic Catalytic Aerobic Functionalization of Phenols. Angew Chem, Int Ed. 2014;53:5877–5881. doi: 10.1002/anie.201311103. [DOI] [PubMed] [Google Scholar]
- 256.Esguerra KVN, Lumb JP. Adapting Melanogenesis to a Regioselective C–H Functionalization of Phenols. Synlett. 2015;26:2731–2738. [Google Scholar]
- 257.Citek C, Lyons CT, Wasinger EC, Stack TDP. Self-Assembly of the Oxy-Tyrosinase Core and the Fundamental Components of Phenolic Hydroxylation. Nat Chem. 2012;4:317–322. doi: 10.1038/nchem.1284. [DOI] [PubMed] [Google Scholar]
- 258.Chiang L, Keown W, Citek C, Wasinger EC, Stack TDP. Simplest Monodentate Imidazole Stabilization of the oxy-Tyrosinase Cu2O2 Core: Phenolate Hydroxylation through a CuIII Intermediate. Angew Chem, Int Ed. 2016;55:10453–10457. doi: 10.1002/anie.201605159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Citek C, Herres-Pawlis S, Stack TDP. Low Temperature Syntheses and Reactivity of Cu2O2 Active-Site Models. Acc Chem Res. 2015;48:2424–2433. doi: 10.1021/acs.accounts.5b00220. [DOI] [PubMed] [Google Scholar]
- 260.Sanyal I, Strange RW, Blackburn NJ, Karlin KD. Formation of a Copper-Dioxygen Complex (Cu2-O2)Using Simple Imidazole Ligands. J Am Chem Soc. 1991;113:4692–4693. [Google Scholar]
- 261.Sanyal I, Karlin KD, Strange RW, Blackburn NJ. Chemistry and Structural Studies on the Dioxygen-Binding Copper-1,2-Dimethylimidazole System. J Am Chem Soc. 1993;115:11259–11270. [Google Scholar]
- 262.Mahadevan V, DuBois JL, Hedman B, Hodgson KO, Stack TDP. Exogenous Substrate Reactivity with a [Cu(III)2O2]2+ Core: Structural Implications. J Am Chem Soc. 1999;121:5583–5584. [Google Scholar]
- 263.Hoffmann A, Citek C, Binder S, Goos A, Ruebhausen M, Troeppner O, Ivanović-Burmazović I, Wasinger EC, Stack TDP, Herres-Pawlis S. Catalytic Phenol Hydroxylation with Dioxygen: Extension of the Tyrosinase Mechanism beyond the Protein Matrix. Angew Chem, Int Ed. 2013;52:5398–5401. doi: 10.1002/anie.201301249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Schottenheim J, Fateeva N, Thimm W, Krahmer J, Tuczek F. Catalytic Conversion of Monophenols to Ortho-Quinones in a Tyrosinase-Like Fashion: Towards More Biomimetic and More Efficient Model Systems. Z Anorg Allg Chem. 2013;639:1491–1497. [Google Scholar]
- 265.Hamann JN, Tuczek F. New Catalytic Model Systems of Tyrosinase: Fine Tuning of the Reactivity with Pyrazole-Based NDonor Ligands. Chem Commun. 2014;50:2298–2300. doi: 10.1039/c3cc47888b. [DOI] [PubMed] [Google Scholar]
- 266.Rolff M, Schottenheim J, Peters G, Tuczek F. The First Catalytic Tyrosinase Model System Based on a Mononuclear Copper(I) Complex: Kinetics and Mechanism. Angew Chem, Int Ed. 2010;49:6438–6442. doi: 10.1002/anie.201000973. [DOI] [PubMed] [Google Scholar]
- 267.Schottenheim J, Gernert C, Herzigkeit B, Krahmer J, Tuczek F. Catalytic Models of Tyrosinase: Reactivity Differences between Systems Based on Mono- and Binucleating Ligands. Eur J Inorg Chem. 2015;2015:3501–3511. [Google Scholar]
- 268.Palavicini S, Granata A, Monzani E, Casella L. Hydroxylation of Phenolic Compounds by a Peroxodicopper(II) Complex: Further Insight into the Mechanism of Tyrosinase. J Am Chem Soc. 2005;127:18031–18036. doi: 10.1021/ja0544298. [DOI] [PubMed] [Google Scholar]
- 269.Mahapatra S, Kaderli S, Llobet A, Neuhold YM, Palanché T, Halfen JA, Young VG, Jr, Kaden TA, Que L, Jr, Zuberbühler AD, Tolman WB. Binucleating Ligand Structural Effects on (μ-Peroxo)-and Bis(μ-oxo)dicopper Complex Formation and Decay: Competition Between Arene Hydroxylation and Aliphatic C-H Bond Activation. Inorg Chem. 1997;36:6343–6356. [Google Scholar]
- 270.Becker M, Schindler S, Karlin KD, Kaden TA, Kaderli S, Palanché T, Zuberbühler AD. Intramolecular Ligand Dydroxylation: Mechanistic High-Pressure Studies on the Reaction of a Dinuclear Copper(I) Complex with Dioxygen. Inorg Chem. 1999;38:1989–1995. doi: 10.1021/ic981066m. [DOI] [PubMed] [Google Scholar]
- 271.Gamba I, Palavicini S, Monzani E, Casella L. Catalytic Sulfoxidation by Dinuclear Copper Complexes. Chem - Eur J. 2009;15:12932–12936. doi: 10.1002/chem.200902451. [DOI] [PubMed] [Google Scholar]
- 272.Battaini G, Casella L, Gullotti M, Monzani E, Nardin G, Perotti A, Randaccio L, Santagostini L, Heinemann FW, Schindler S. Structure and Reactivity Studies on Dinuclear Copper Complexes of the Ligand α,α′-Bis{bis[1-(1′-methyl-2′-benzimidazolyl)methyl]amino}-m-xylene. Eur J Inorg Chem. 2003;2003:1197–1205. [Google Scholar]
- 273.Granata A, Monzani E, Casella L. Mechanistic Insight into the Catechol Oxidase Activity by a Biomimetic Dinuclear Copper Complex. JBIC, J Biol Inorg Chem. 2004;9:903–913. doi: 10.1007/s00775-004-0595-3. [DOI] [PubMed] [Google Scholar]
- 274.Perrone ML, Lo Presti E, Dell’Acqua S, Monzani E, Santagostini L, Casella L. Synthesis, Characterization, and Stereoselective Oxidations of the Dinuclear Copper(II) Complex Derived from a Chiral Diamino-m-xylenetetra(benzimidazole) Ligand. Eur J Inorg Chem. 2015;2015:3493–3500. [Google Scholar]
- 275.Karlin KD, Hayes JC, Gultneh Y, Cruse RW, McKown JW, Hutchinson JP, Zubieta J. Copper-Mediated Hydroxylation of an Arene: Model System for the Action of Copper Monooxygenases. Structures of a Binuclear Cu(I) Complex and Its Oxygenated Product. J Am Chem Soc. 1984;106:2121–2128. [Google Scholar]
- 276.Pidcock E, Obias HV, Zhang CX, Karlin KD, Solomon EI. Investigation of the Reactive Oxygen Intermediate in An Arene Hydroxylation Reation Performed by Xylyl-Bridged Binuclear Copper Complexes. J Am Chem Soc. 1998;120:7841–7847. [Google Scholar]
- 277.Foxon SP, Utz D, Astner J, Schindler S, Thaler F, Heinemann FW, Liehr G, Mukherjee J, Balamurugan V, Ghosh D, et al. Reaction Behaviour of Dinuclear Copper(I) Complexes with m-Xylyl-Based Ligands Towards Dioxygen. Dalton Trans. 2004:2321–2328. doi: 10.1039/B406329P. [DOI] [PubMed] [Google Scholar]
- 278.Ohi H, Tachi Y, Itoh S. Modeling the Mononuclear, Dinuclear, and Trinuclear Copper(I) Reaction Centers of Copper Proteins Using Pyridylalkylamine Ligands Connected to 1,3,5-Triethylbenzene Spacer. Inorg Chem. 2006;45:10825–10835. doi: 10.1021/ic061509j. [DOI] [PubMed] [Google Scholar]
- 279.Sander O, Henβ A, Näther C, Würtele C, Holthausen MC, Schindler S, Tuczek F. Aromatic Hydroxylation in a Copper Bis(imine) Complex Mediated by a μ-η2:η2 Peroxo Dicopper Core: A Mechanistic Scenario. Chem - Eur J. 2008;14:9714–9729. doi: 10.1002/chem.200800799. [DOI] [PubMed] [Google Scholar]
- 280.Karlin KD, Zhang CX, Rheingold AL, Galliker B, Kaderli S, Zuberbühler AD. Reversible Dioxygen Binding and Arene Hydroxylation Reactions: Kinetic and Thermodynamic Studies Involving Ligand Electronic and Structural Variations. Inorg Chim Acta. 2012;389:138–150. doi: 10.1016/j.ica.2012.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Matsumoto T, Furutachi H, Kobino M, Tomii M, Nagatomo S, Tosha T, Osako T, Fujinami S, Itoh S, Kitagawa T, et al. Intramolecular Arene Hydroxylation versus Intermolecular Olefin Epoxidation by (μ-η2:η2-Peroxo)dicopper(II) Complex Supported by Dinucleating Ligand. J Am Chem Soc. 2006;128:3874–3875. doi: 10.1021/ja058117g. [DOI] [PubMed] [Google Scholar]
- 282.Matsumoto T, Ohkubo K, Honda K, Yazawa A, Furutachi H, Fujinami S, Fukuzumi S, Suzuki M. Aliphatic C-H Bond Activation Initiated by a (μ-η2:η2-Peroxo)dicopper(II) Complex in Comparison with Cumylperoxyl Radical. J Am Chem Soc. 2009;131:9258–9267. doi: 10.1021/ja809822c. [DOI] [PubMed] [Google Scholar]
- 283.Qayyum MF, Sarangi R, Fujisawa K, Stack TDP, Karlin KD, Hodgson KO, Hedman B, Solomon EI. L-Edge X-ray Absorption Spectroscopy and DFT Calculations on Cu2O2 Species: Direct Electrophilic Aromatic Attack by Side-on Peroxo Bridged Dicopper(II) Complexes. J Am Chem Soc. 2013;135:17417–17431. doi: 10.1021/ja4078717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Walli A, Dechert S, Bauer M, Demeshko S, Meyer F. BOX Ligands in Biomimetic Copper-Mediated Dioxygen Activation: A Hemocyanin Model. Eur J Inorg Chem. 2014;2014:4660–4676. [Google Scholar]
- 285.Sprakel VSI, Feiters MC, Meyer-Klaucke W, Klopstra M, Brinksma J, Feringa BL, Karlin KD, Nolte RJM. Oxygen Binding and Activation by the Complexes of PY2- and TPA-appended Diphenylglycoluril Receptors with Copper and Other Metals. Dalton Trans. 2005:3522–3534. doi: 10.1039/b506288h. [DOI] [PubMed] [Google Scholar]
- 286.Zhu Q, Lian Y, Thyagarajan S, Rokita SE, Karlin KD, Blough NV. Hydrogen Peroxide and Dioxygen Activation by Dinuclear Copper Complexes in Aqueous Solution: Hydroxyl Radical Production Initiated by Internal Electron Transfer. J Am Chem Soc. 2008;130:6304–6305. doi: 10.1021/ja800080z. [DOI] [PubMed] [Google Scholar]
- 287.Thyagarajan S, Murthy NN, Narducci Sarjeant AA, Karlin KD, Rokita SE. Selective DNA Strand Scission with Binuclear Copper Complexes: Implications for an Active Cu2O2 Species. J Am Chem Soc. 2006;128:7003–7008. doi: 10.1021/ja061014t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Tahsini L, Kotani H, Lee YM, Cho J, Nam W, Karlin KD, Fukuzumi S. Electron-Transfer Reduction of Dinuclear Copper Peroxo and Bis-μ-oxo Complexes Leading to the Catalytic Four-Electron Reduction of Dioxygen to Water. Chem - Eur J. 2012;18:1084–1093. doi: 10.1002/chem.201103215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Karlin KD, Tyeklar Z, Farooq A, Haka MS, Ghosh P, Cruse RW, Gultneh Y, Hayes JC, Toscano PJ, Zubieta J. Dioxygen-Copper Reactivity and Functional Modeling of Hemocyanins. Reversible Binding of O2 and Carbon Monoxide to Dicopper(I) Complexes [CuI2(L)]2+ (L = dinucleating ligand) and the Structure of a Bis(carbonyl) Adduct, [CuI2(L) (CO)2]2+ Inorg Chem. 1992;31:1436–1451. [Google Scholar]
- 290.Mahadevan V, Hou Z, Cole AP, Root DE, Lal TK, Solomon EI, Stack TDP. Irreversible Reduction of Dioxygen by Simple Peralkylated Diamine-Copper(I) Complexes: Characterization and Thermal Stability of a [Cu2(μ-O)2]2+ Core. J Am Chem Soc. 1997;119:11996–11997. [Google Scholar]
- 291.Stack TDP. Complexity with Simplicity: a Steric Continuum of Chelating Diamines with Copper(I) and Dioxygen. Dalton Trans. 2003:1881–1889. [Google Scholar]
- 292.Cole AP, Mahadevan V, Mirica LM, Ottenwaelder X, Stack TDP. Bis(μ-oxo)dicopper(III) Complexes of a Homologous Series of Simple Peralkylated 1,2-Diamines: Steric Modulation of Structure, Stability, and Reactivity. Inorg Chem. 2005;44:7345–7364. doi: 10.1021/ic050331i. [DOI] [PubMed] [Google Scholar]
- 293.Kang P, Bobyr E, Dustman J, Hodgson KO, Hedman B, Solomon EI, Stack TDP. Bis(μ-oxo) Dicopper(III) Species of the Simplest Peralkylated Diamine: Enhanced Reactivity toward Exogenous Substrates. Inorg Chem. 2010;49:11030–11038. doi: 10.1021/ic101515g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Cole AP, Root DE, Mukherjee P, Solomon EI, Stack TDP. A Trinuclear Intermediate in the Copper-Mediated Reduction of O2: Four Electrons from Three Coppers. Science. 1996;273:1848–1850. doi: 10.1126/science.273.5283.1848. [DOI] [PubMed] [Google Scholar]
- 295.Citek C, Lin BL, Phelps TE, Wasinger EC, Stack TDP. Primary Amine Stabilization of a Dicopper(III) Bis(μ-oxo) Species: Modeling the Ligation in pMMO. J Am Chem Soc. 2014;136:14405–14408. doi: 10.1021/ja508630d. [DOI] [PubMed] [Google Scholar]
- 296.Citek C, Gary JB, Wasinger EC, Stack TDP. Chemical Plausibility of Cu(III) with Biological Ligation in pMMO. J Am Chem Soc. 2015;137:6991–6994. doi: 10.1021/jacs.5b02157. [DOI] [PubMed] [Google Scholar]
- 297.Shimokawa C, Teraoka J, Tachi Y, Itoh S. A Functional Model for pMMO (Particulate Methane Monooxygenase): Hydroxylation of Alkanes with H2O2 Catalyzed by β-Diketiminatocopper(II) Complexes. J Inorg Biochem. 2006;100:1118–1127. doi: 10.1016/j.jinorgbio.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 298.Liu CC, Lin TS, Chan SI, Mou CY. A Room Temperature Catalyst for Toluene Aliphatic C-H Bond Oxidation: Tripodal Tridentate Copper Complex Immobilized in Mesoporous Silica. J Catal. 2015;322:139–151. [Google Scholar]
- 299.Mandal S, De A, Mukherjee R. Formation of {CuIII2(μ-O)2}2+ Core Due to Dioxygen Reactivity of a Copper(I) Complex Supported by a New Hybrid Tridentate Ligand: Reaction with Exogenous Substrates. Chem Biodiversity. 2008;5:1594–1608. doi: 10.1002/cbdv.200890147. [DOI] [PubMed] [Google Scholar]
- 300.Holland PL, Rodgers KR, Tolman WB. Is the Bis(μ-oxo)dicopper Core Capable of Hydroxylating an Arene? Angew Chem, Int Ed. 1999;38:1139–1142. doi: 10.1002/(SICI)1521-3773(19990419)38:8<1139::AID-ANIE1139>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 301.Becker J, Gupta P, Angersbach F, Tuczek F, Näther C, Holthausen MC, Schindler S. Selective Aromatic Hydroxylation with Dioxygen and Simple Copper Imine Complexes. Chem - Eur J. 2015;21:11735–11744. doi: 10.1002/chem.201501003. [DOI] [PubMed] [Google Scholar]
- 302.Hamann JN, Rolff M, Tuczek F. Monooxygenation of an Appended Phenol in a Model System of Tyrosinase: Implications on the Enzymatic Reaction Mechanism. Dalton Trans. 2015;44:3251–3258. doi: 10.1039/c4dt03010a. [DOI] [PubMed] [Google Scholar]
- 303.Petrovic D, Hill LMR, Jones PG, Tolman WB, Tamm M. Synthesis and Reactivity of Copper(I) Complexes with an Ethylene-Bridged Bis(imidazolin-2-imine) Ligand. Dalton Trans. 2008:887–894. doi: 10.1039/b715590e. [DOI] [PubMed] [Google Scholar]
- 304.Herres S, Heuwing AJ, Florke U, Schneider J, Henkel G. Hydroxylation of a Methyl Group: Synthesis of [Cu2(btmmO)2I]+ and of [Cu2(btmmO)2]2+ Containing the Novel Ligand Bis-(trimethylmethoxy)guanidino Propane (btmmO) by Copper-Assisted Oxygen Activation. Inorg Chim Acta. 2005;358:1089–1095. [Google Scholar]
- 305.Herres-Pawlis S, Flörke U, Henkel G. Tuning of Copper(I)–Dioxygen Reactivity by Bis(guanidine) Ligands. Eur J Inorg Chem. 2005;2005:3815–3824. [Google Scholar]
- 306.Herres-Pawlis S, Verma P, Haase R, Kang P, Lyons CT, Wasinger EC, Floerke U, Henkel G, Stack TDP. Phenolate Hydroxylation in a Bis(μ-oxo)dicopper(III) Complex: Lessons from the Guanidine/Amine Series. J Am Chem Soc. 2009;131:1154–1169. doi: 10.1021/ja807809x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Mandal S, Mukherjee J, Lloret F, Mukherjee R. Modeling Tyrosinase and Catecholase Activity Using New m-Xylyl-Based Ligands with Bidentate Alkylamine Terminal Coordination. Inorg Chem. 2012;51:13148–13161. doi: 10.1021/ic3013848. [DOI] [PubMed] [Google Scholar]
- 308.Martínez-Calvo M, Vázquez López M, Pedrido R, González-Noya AM, Bermejo MR, Monzani E, Casella L, Sorace L. Endogenous Arene Hydroxylation Promoted by Copper(I) Cluster Helicates. Chem - Eur J. 2010;16:14175–14180. doi: 10.1002/chem.201001285. [DOI] [PubMed] [Google Scholar]
- 309.Company A, Lamata D, Poater A, Solà M, Rybak-Akimova EV, Que L, Fontrodona X, Parella T, Llobet A, Costas M. O2 Chemistry of Dicopper Complexes with Alkyltriamine Ligands. Comparing Synergistic Effects on O2 Binding. Inorg Chem. 2006;45:5239–5241. doi: 10.1021/ic0602446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Company A, Palavicini S, Garcia-Bosch I, Mas-Ballesté R, Que L, Rybak-Akimova EV, Casella L, Ribas X, Costas M. Tyrosinase-Like Reactivity in a CuIII2(μ-O)2 Species. Chem - Eur J. 2008;14:3535–3538. doi: 10.1002/chem.200800229. [DOI] [PubMed] [Google Scholar]
- 311.Schindler S. Reactivity of Copper(I) Complexes Towards Dioxygen. Eur J Inorg Chem. 2000;2000:2311–2326. [Google Scholar]
- 312.Hatcher LQ, Karlin KD. Oxidant Types in Copper-Dioxygen Chemistry: The Ligand Coordination Defines the Cun-O2 Structure and Subsequent Reactivity. JBIC, J Biol Inorg Chem. 2004;9:669–683. doi: 10.1007/s00775-004-0578-4. [DOI] [PubMed] [Google Scholar]
- 313.Itoh S, Tachi Y. Structure and O2-Reactivity of Copper(I) Complexes Supported by Pyridylalkylamine Ligands. Dalton Trans. 2006:4531–4538. doi: 10.1039/b607964d. [DOI] [PubMed] [Google Scholar]
- 314.Osako T, Terada S, Tosha T, Nagatomo S, Furutachi H, Fujinami S, Kitagawa T, Suzuki M, Itoh S. Structure and Dioxygen-Reactivity of Copper(I) Complexes Supported by Bis(6-methylpyridin-2-ylmethyl)amine Tridentate Ligands. Dalton Trans. 2005:3514–3521. doi: 10.1039/b500202h. [DOI] [PubMed] [Google Scholar]
- 315.Karlin KD, Haka MS, Cruse RW, Meyer GJ, Farooq A, Gultneh Y, Hayes JC, Zubieta J. Dioxygen-Copper Reactivity. Models for hemocyanin: Reversible O2 and CO Binding to Structurally Characterized Dicopper(I) Complexes Containing Hydrocarbon-Linked Bis[2-(2-pyridyl)ethyl]amine Units. J Am Chem Soc. 1988;110:1196–1207. [Google Scholar]
- 316.Itoh S, Kumei H, Taki M, Nagatomo S, Kitagawa T, Fukuzumi S. Oxygenation of Phenols to Catechols by A (μ-η2:η2-Peroxo)dicopper(II) Complex: Mechanistic Insight into the Phenolase Activity of Tyrosinase. J Am Chem Soc. 2001;123:6708–6709. doi: 10.1021/ja015702i. [DOI] [PubMed] [Google Scholar]
- 317.Osako T, Ueno Y, Tachi Y, Itoh S. Structures and Redox Reactivities of Copper Complexes of (2-Pyridyl)alkylamine Ligands. Effects of the Alkyl Linker Chain Length. Inorg Chem. 2003;42:8087–8097. doi: 10.1021/ic034958h. [DOI] [PubMed] [Google Scholar]
- 318.Kunishita A, Osako T, Tachi Y, Teraoka J, Itoh S. Syntheses, Structures, and O2 Reactivities of Copper(I) Complexes with Bis(2-pyridylmethyl)amine and Bis(2-quinolylmethyl)amine Tridentate Ligands. Bull Chem Soc Jpn. 2006;79:1729–1741. [Google Scholar]
- 319.Liang HC, Henson MJ, Hatcher LQ, Vance MA, Zhang CX, Lahti D, Kaderli S, Sommer RD, Rheingold AL, Zuberbühler AD, et al. Solvent Effects on the Conversion of Dicopper(II) μ-η2:η2-Peroxo to Bis-μ-oxo Dicopper(III) Complexes: Direct Probing of the Solvent Interaction. Inorg Chem. 2004;43:4115–4117. doi: 10.1021/ic0498283. [DOI] [PubMed] [Google Scholar]
- 320.Hatcher LQ, Vance MA, Narducci Sarjeant AA, Solomon EI, Karlin KD. Copper–Dioxygen Adducts and the Sideon Peroxo Dicopper(II)/Bis(μ-oxo) Dicopper(III) Equilibrium: Significant Ligand Electronic Effects. Inorg Chem. 2006;45:3004–3013. doi: 10.1021/ic052185m. [DOI] [PubMed] [Google Scholar]
- 321.Zhang CX, Liang HC, Kim E-i, Shearer J, Helton ME, Kim E, Kaderli S, Incarvito CD, Zuberbühler AD, Rheingold AL, Karlin KD. Tuning Copper–Dioxygen Reactivity and Exogenous Substrate Oxidations via Alterations in Ligand Electronics. J Am Chem Soc. 2003;125:634–635. doi: 10.1021/ja028779v. [DOI] [PubMed] [Google Scholar]
- 322.Henson MJ, Vance MA, Zhang CX, Liang HC, Karlin KD, Solomon EI. Resonance Raman Investigation of Equatorial Ligand Donor Effects on the Cu2O22+ Core in End-On and Side-On μ-Peroxo-Dicopper(II) and Bis-μ-oxo-Dicopper(III) Complexes. J Am Chem Soc. 2003;125:5186–5192. doi: 10.1021/ja0276366. [DOI] [PubMed] [Google Scholar]
- 323.Park GY, Qayyum MF, Woertink J, Hodgson KO, Hedman B, Narducci Sarjeant AA, Solomon EI, Karlin KD. Geometric and Electronic Structure of [{Cu(MeAN)}2(μ-η2:η2(O22−))]2+ with an Unusually Long O–O Bond: O–O Bond Weakening vs. Activation for Reductive Cleavage. J Am Chem Soc. 2012;134:8513–8524. doi: 10.1021/ja300674m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Liang HC, Zhang CX, Henson MJ, Sommer RD, Hatwell KR, Kaderli S, Zuberbühler AD, Rheingold AL, Solomon EI, Karlin KD. Contrasting Copper–Dioxygen Chemistry Arising from Alike Tridentate Alkyltriamine Copper(I) Complexes. J Am Chem Soc. 2002;124:4170–4171. doi: 10.1021/ja0125265. [DOI] [PubMed] [Google Scholar]
- 325.Shearer J, Zhang CX, Zakharov LN, Rheingold AL, Karlin KD. Substrate Oxidation by Copper–Dioxygen Adducts: Mechanistic Considerations. J Am Chem Soc. 2005;127:5469–5483. doi: 10.1021/ja045191a. [DOI] [PubMed] [Google Scholar]
- 326.Abe T, Morimoto Y, Tano T, Mieda K, Sugimoto H, Fujieda N, Ogura T, Itoh S. Geometric Control of Nuclearity in Copper(I)/Dioxygen Chemistry. Inorg Chem. 2014;53:8786–8794. doi: 10.1021/ic501461n. [DOI] [PubMed] [Google Scholar]
- 327.Sander O, Näther C, Tuczek F. Chiral Dicopper Complexes with a Doubly Asymmetric Ligand as Models for the Tyrosinase Active Site: Synthesis, Structure, O2-Reactivity and Comparison with Their Symmetric Analogs. Z Anorg Allg Chem. 2009;635:1123–1133. [Google Scholar]
- 328.Ottenwaelder X, Rudd DJ, Corbett MC, Hodgson KO, Hedman B, Stack TDP. Reversible O-O Bond Cleavage in Copper-Dioxygen Isomers: Impact of Anion Basicity. J Am Chem Soc. 2006;128:9268–9269. doi: 10.1021/ja061132g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Gupta AK, Tolman WB. Copper/α-Ketocarboxylate Chemistry With Supporting Peralkylated Diamines: Reactivity of Copper(I) Complexes and Dicopper-Oxygen Intermediates. Inorg Chem. 2010;49:3531–3539. doi: 10.1021/ic100032n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Park GY, Lee Y, Lee DH, Woertink JS, Narducci Sarjeant AA, Solomon EI, Karlin KD. Thioether S-ligation in a Side-On μ-η2:η2-Peroxodicopper(II) Complex. Chem Commun. 2010;46:91–93. doi: 10.1039/b918616f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Lee Y, Lee DH, Narducci Sarjeant AA, Zakharov LN, Rheingold AL, Karlin KD. Thioether Sulfur Oxygenation from O2 or H2O2 Reactivity of Copper Complexes with Tridentate N2S thioether Ligands. Inorg Chem. 2006;45:10098–10107. doi: 10.1021/ic060730t. [DOI] [PubMed] [Google Scholar]
- 332.Lewin JL, Heppner DE, Cramer CJ. Validation of Density Functional Modeling Protocols on Experimental Bis(μ-oxo)/μ-η2:η2-Peroxo Dicopper Equilibria. JBIC, J Biol Inorg Chem. 2007;12:1221–1234. doi: 10.1007/s00775-007-0290-2. [DOI] [PubMed] [Google Scholar]
- 333.Liakos DG, Neese F. Interplay of Correlation and Relativistic Effects in Correlated Calculations on Transition-Metal Complexes: The (Cu2O2)2+ Core Revisited. J Chem Theory Comput. 2011;7:1511–1523. doi: 10.1021/ct1006949. [DOI] [PubMed] [Google Scholar]
- 334.York JT, Llobet A, Cramer CJ, Tolman WB. Heterobimetallic Dioxygen Activation: Synthesis and Reactivity of Mixed Cu–Pd and Cu–Pt Bis(μ-oxo) Complexes. J Am Chem Soc. 2007;129:7990–7999. doi: 10.1021/ja071744g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Kundu S, Pfaff FF, Miceli E, Zaharieva I, Herwig C, Yao S, Farquhar ER, Kuhlmann U, Bill E, Hildebrandt P, et al. A High-Valent Heterobimetallic [CuIII(μ-O)2NiIII]2+ Core with Nucleophilic Oxo Groups. Angew Chem, Int Ed. 2013;52:5622–5626. doi: 10.1002/anie.201300861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Kundu S, Miceli E, Farquhar ER, Ray K. Mechanism of Phenol Oxidation by Heterodinuclear Ni Cu Bis(μ-oxo) Complexes Involving Nucleophilic Oxo Groups. Dalton Trans. 2014;43:4264–4267. doi: 10.1039/c3dt52644e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Sharp PR. Oxo and Imido Ligands in Late Transition Metal Chemistry. J Chem Soc, Dalton Trans. 2000:2647–2657. [Google Scholar]
- 338.Li JJ, Li W, Sharp PR. Phosphine-Based Platinum(II) Hydroxo and Oxo Complexes. Inorg Chem. 1996;35:604–613. [Google Scholar]
- 339.Jacobson RR, Tyeklár Z, Farooq A, Karlin KD, Liu S, Zubieta J. A Copper-Oxygen (Cu2-O2) Complex. Crystal Structure and Characterization of a Reversible Dioxygen Binding System. J Am Chem Soc. 1988;110:3690–3692. [Google Scholar]
- 340.Würtele C, Sander O, Lutz V, Waitz T, Tuczek F, Schindler S. Aliphatic C–H Bond Oxidation of Toluene Using Copper Peroxo Complexes That Are Stable at Room Temperature. J Am Chem Soc. 2009;131:7544–7545. doi: 10.1021/ja902327s. [DOI] [PubMed] [Google Scholar]
- 341.Maiti D, Woertink JS, Narducci Sarjeant AA, Solomon EI, Karlin KD. Copper Dioxygen Adducts: Formation of Bis(μ-oxo)dicopper(III) versus (μ-1,2)Peroxodicopper(II) Complexes with Small Changes in One Pyridyl-Ligand Substituent. Inorg Chem. 2008;47:3787–3800. doi: 10.1021/ic702437c. [DOI] [PubMed] [Google Scholar]
- 342.Hatcher LQ, Lee DH, Vance MA, Milligan AE, Sarangi R, Hodgson KO, Hedman B, Solomon EI, Karlin KD. Dioxygen Reactivity of a Copper(I) Complex with a N3S Thioether Chelate; Peroxo–Dicopper(II) Formation Including Sulfur-Ligation. Inorg Chem. 2006;45:10055–10057. doi: 10.1021/ic061813c. [DOI] [PubMed] [Google Scholar]
- 343.Lee Y, Lee DH, Park GY, Lucas HR, Narducci Sarjeant AA, Kieber-Emmons MT, Vance MA, Milligan AE, Solomon EI, Karlin KD. Sulfur Donor Atom Effects on Copper(I)/O2 Chemistry with Thioanisole Containing Tetradentate N3S Ligand Leading to μ-1,2-Peroxo-Dicopper(II) Species. Inorg Chem. 2010;49:8873–8885. doi: 10.1021/ic101041m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Lee Y, Park GY, Lucas HR, Vajda PL, Kamaraj K, Vance MA, Milligan AE, Woertink JS, Siegler MA, Narducci Sarjeant AA, et al. Copper(I)/O2 Chemistry with Imidazole Containing Tripodal Tetradentate Ligands Leading to μ-1,2-Peroxo–Dicopper(II) Species. Inorg Chem. 2009;48:11297–11309. doi: 10.1021/ic9017695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Kieber-Emmons MT, Ginsbach JW, Wick PK, Lucas HR, Helton ME, Lucchese B, Suzuki M, Zuberbühler AD, Karlin KD, Solomon EI. Observation of a CuII2(μ-1,2-peroxo)/CuIII2(μ-oxo)2 Equilibrium and its Implications for Copper–Dioxygen Reactivity. Angew Chem, Int Ed. 2014;53:4935–4939. doi: 10.1002/anie.201402166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Garcia-Bosch I, Company A, Frisch JR, Torrent-Sucarrat M, Cardellach M, Gamba I, Güell M, Casella L, Que L, Ribas X, et al. O2 Activation and Selective Phenolate ortho Hydroxylation by an Unsymmetric Dicopper μ-η1:η1-Peroxido Complex. Angew Chem, Int Ed. 2010;49:2406–2409. doi: 10.1002/anie.200906749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Baldwin MJ, Ross PK, Pate JE, Tyeklar Z, Karlin KD, Solomon EI. Spectroscopic and Theoretical Studies of End-On Peroxide-Bridged Coupled Binuclear Copper(II) Model Complex of Relevance to the Active Sites in Hemocyanin and Tyrosinase. J Am Chem Soc. 1991;113:8671–8679. [Google Scholar]
- 348.Lucas HR, Li L, Sarjeant AAN, Vance MA, Solomon EI, Karlin KD. Toluene and Ethylbenzene Aliphatic C–H Bond Oxidations Initiated by a Dicopper(II)-μ-1,2-Peroxo Complex. J Am Chem Soc. 2009;131:3230–3245. doi: 10.1021/ja807081d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Wei N, Murthy NN, Chen Q, Zubieta J, Karlin KD. Copper(I)/Dioxygen Reactivity of Mononuclear Complexes with Pyridyl and Quinolyl Tripodal Tetradentate Ligands: Reversible Formation of Cu:O2 = 1:1 and 2:1 Adducts. Inorg Chem. 1994;33:1953–1965. [Google Scholar]
- 350.Karlin KD, Wei N, Jung B, Kaderli S, Niklaus P, Zuberbuehler AD. Kinetics and Thermodynamics of Formation of Copper-Dioxygen Adducts: Oxygenation of Mononuclear Copper(I) Complexes Containing Tripodal Tetradentate Ligands. J Am Chem Soc. 1993;115:9506–9514. [Google Scholar]
- 351.Paul PP, Tyeklár Z, Jacobson RR, Karlin KD. Reactivity Patterns and Comparisons in Three Classes of Synthetic Copper-Dioxygen {Cu2-O2}Complexes: Implication for Structure and Biological Relevance. J Am Chem Soc. 1991;113:5322–5332. [Google Scholar]
- 352.Metz M, Solomon EI. Dioxygen Binding to Deoxyhemocyanin: Electronic Structure and Mechanism of the Spin-Forbidden Two-Electron Reduction of O2. J Am Chem Soc. 2001;123:4938–4950. doi: 10.1021/ja004166b. [DOI] [PubMed] [Google Scholar]
- 353.Dalle KE, Gruene T, Dechert S, Demeshko S, Meyer F. Weakly Coupled Biologically Relevant CuII2(μ-η1:η1-O2) cis-Peroxo Adduct that Binds Side-On to Additional Metal Ions. J Am Chem Soc. 2014;136:7428–7434. doi: 10.1021/ja5025047. [DOI] [PubMed] [Google Scholar]
- 354.Kindermann N, Bill E, Dechert S, Demeshko S, Reijerse EJ, Meyer F. A Ferromagnetically Coupled (S = 1) Peroxodicopper(II) Complex. Angew Chem, Int Ed. 2015;54:1738–1743. doi: 10.1002/anie.201409709. [DOI] [PubMed] [Google Scholar]
- 355.Li L, Narducci Sarjeant AA, Vance MA, Zakharov LN, Rheingold AL, Solomon EI, Karlin KD. Exogenous Nitrile Substrate Hydroxylation by a New Dicopper-Hydroperoxide Complex. J Am Chem Soc. 2005;127:15360–15361. doi: 10.1021/ja054948a. [DOI] [PubMed] [Google Scholar]
- 356.Teramae S, Osako T, Nagatomo S, Kitagawa T, Fukuzumi S, Itoh S. Dinuclear Copper-Dioxygen Intermediates Supported by Polyamine Ligands. J Inorg Biochem. 2004;98:746–757. doi: 10.1016/j.jinorgbio.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 357.Itoh K, Hayashi H, Furutachi H, Matsumoto T, Nagatomo S, Tosha T, Terada S, Fujinami S, Suzuki M, Kitagawa T. Synthesis and Reactivity of a (μ-1,1-Hydroperoxo)(μ-hydroxo)-dicopper(II) Complex: Ligand Hydroxylation by a Bridging Hydroperoxo Ligand. J Am Chem Soc. 2005;127:5212–5223. doi: 10.1021/ja047437h. [DOI] [PubMed] [Google Scholar]
- 358.Battaini G, Monzani E, Perotti A, Para C, Casella L, Santagostini L, Gullotti M, Dillinger R, Näther C, Tuczek F. A Double Arene Hydroxylation Mediated by Dicopper(II)–Hydroperoxide Species. J Am Chem Soc. 2003;125:4185–4198. doi: 10.1021/ja0280776. [DOI] [PubMed] [Google Scholar]
- 359.Kindermann N, Dechert S, Demeshko S, Meyer F. Proton-Induced, Reversible Interconversion of a μ-1,2-Peroxo and a μ-1,1-Hydroperoxo Dicopper(II) Complex. J Am Chem Soc. 2015;137:8002–8005. doi: 10.1021/jacs.5b04361. [DOI] [PubMed] [Google Scholar]
- 360.Li L, Murthy NN, Telser J, Zakharov LN, Yap GPA, Rheingold AL, Karlin KD, Rokita SE. Targeted Guanine Oxidation by a Dinuclear Copper(II) Complex at Single Stranded/Double Stranded DNA Junctions. Inorg Chem. 2006;45:7144–7159. doi: 10.1021/ic0605930. [DOI] [PubMed] [Google Scholar]
- 361.Cruse RW, Kaderli S, Meyer CJ, Zuberbühler AD, Karlin KD. Copper-Mediated Hydroxylation of an Arene: Kinetics and Mechanism of the Reaction of a Dicopper(II) m-Xylyl-Containing Complex with H2O2 to Yield a Phenoxodicopper(II) Complex. J Am Chem Soc. 1988;110:5020–5024. [Google Scholar]
- 362.Fukuzumi S, Tahsini L, Lee YM, Ohkubo K, Nam W, Karlin KD. Factors That Control Catalytic Two-versus Four-Electron Reduction of Dioxygen by Copper Complexes. J Am Chem Soc. 2012;134:7025–7035. doi: 10.1021/ja211656g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Das D, Lee YM, Ohkubo K, Nam W, Karlin KD, Fukuzumi S. Acid-Induced Mechanism Change and Overpotential Decrease in Dioxygen Reduction Catalysis with a Dinuclear Copper Complex. J Am Chem Soc. 2013;135:4018–4026. doi: 10.1021/ja312256u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Tachi Y, Aita K, Teramae S, Tani F, Naruta Y, Fukuzumi S, Itoh S. Dicopper–Dioxygen Complex Supported by Asymmetric Pentapyridine Dinucleating Ligand. Inorg Chem. 2004;43:4558–4560. doi: 10.1021/ic049524g. [DOI] [PubMed] [Google Scholar]
- 365.Tachi Y, Matsukawa Y, Teraoka J, Itoh S. A Stable Cu2O2 Complex Supported by an Asymmetric Dinucleating Pentapyridine Ligand Involving an Amide Linkage. Chem Lett. 2009;38:202–203. [Google Scholar]
- 366.Davies G, El-Sayed MA, Henary M. Stoichiometry and Kinetics of the Low-Temperature Oxidation of L2Cu2Cl2 (L = N,N,N′,N′-Tetraethylethylenediamine) by Dioxygen in Methylene Chloride and Properties of the Peroxocopper Products. Inorg Chem. 1987;26:3266–3273. [Google Scholar]
- 367.Chaudhuri P, Ventur D, Wieghardt K, Peters EM, Peters K, Simon A. Preparation, Magnetism, and Crystal Structures of the Tautomers [LCu(μ2-OH)2CuL](ClO4)2 (Blue) and [LCu(μ2-OH2)-(μ2-O)CuL](ClO4)2 (Green): μ-Aqua-μ-oxo vs. Di-μ-hydroxo Linkage. Angew Chem, Int Ed Engl. 1985;24:57–59. [Google Scholar]
- 368.Karlin KD, Gultneh Y, Hayes JC, Zubieta J. Copper(I)-Dioxygen Reactivity. 2. Reaction of a Three-Coordinate Copper(I) Complex with Dioxygen, with Evidence for a Binuclear Oxo-Copper(II) Species: Structural Characterization of a Parallel-Planar Dihydroxo-Bridged Dimer. Inorg Chem. 1984;23:519–521. [Google Scholar]
- 369.Sanyal I, Mahroof-Tahir M, Nasir MS, Ghosh P, Cohen BI, Gultneh Y, Cruse RW, Farooq A, Karlin KD. Reactions of Dioxygen (O2) with Mononuclear Copper(I) Complexes: Temperature-Dependent Formation of Peroxo- or Oxo- (and Dihydroxo-) Bridged Dicopper(II) Complexes. Inorg Chem. 1992;31:4322–4332. [Google Scholar]
- 370.Obias HV, Lin Y, Murthy NN, Pidcock E, Solomon EI, Ralle M, Blackburn NJ, Neuhold YM, Zuberbühler AD, Karlin KD. Peroxo-, Oxo-, and Hydroxo-Bridged Dicopper Complexes: Observation of Exogenous Hydrocarbon Substrate Oxidation. J Am Chem Soc. 1998;120:12960–12961. [Google Scholar]
- 371.Kitajima N, Koda T, Moro-oka Y. Synthesis of a Novel μ-oxo Binuclear Copper(II) Complex Ligated by Hydrotris(3,5-dimethyl-1-pyrazolyl)borate. Chem Lett. 1988;17:347–350. [Google Scholar]
- 372.Lee DH, Murthy NN, Karlin KD. Copper(I)/Dioxygen Reactivity with Dinuclear Compounds: Catalytic Oxygenation and Oxo-Transfer to a Ketone. Inorg Chem. 1996;35:804–805. doi: 10.1021/ic951490c. [DOI] [PubMed] [Google Scholar]
- 373.Lee DH, Murthy NN, Karlin KD. Copper-Dioxygen Reactivity Using Dinucleating Ligands with Activated Methylene or Ketone Function. Bull Chem Soc Jpn. 2007;80:732–742. [Google Scholar]
- 374.Maji S, Lee JCM, Lu YJ, Chen CL, Hung MC, Chen PPY, Yu SSF, Chan SI. Dioxygen Activation of a Trinuclear CuICuICuI Cluster Capable of Mediating Facile Oxidation of Organic Substrates: Competition between O-Atom Transfer and Abortive Intercomplex Reduction. Chem - Eur J. 2012;18:3955–3968. doi: 10.1002/chem.201103075. [DOI] [PubMed] [Google Scholar]
- 375.Haack P, Kärgel A, Greco C, Dokic J, Braun B, Pfaff FF, Mebs S, Ray K, Limberg C. Access to a CuII–O–CuII Motif: Spectroscopic Properties, Solution Structure, and Reactivity. J Am Chem Soc. 2013;135:16148–16160. doi: 10.1021/ja406721a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Haack P, Limberg C, Ray K, Braun B, Kuhlmann U, Hildebrandt P, Herwig C. Dinuclear Copper Complexes Based on Parallel β-Diiminato Binding Sites and their Reactions with O2: Evidence for a Cu–O–Cu Entity. Inorg Chem. 2011;50:2133–2142. doi: 10.1021/ic101249k. [DOI] [PubMed] [Google Scholar]
- 377.Melník M. Study of the Relation Between the Structural Data and Magnetic nteraction in Oxo-Bridged Binuclear Copper(II) Compounds. Coord Chem Rev. 1982;42:259–293. [Google Scholar]
- 378.Halvagar MR, Solntsev PV, Lim H, Hedman B, Hodgson KO, Solomon EI, Cramer CJ, Tolman WB. Hydroxo-Bridged Dicopper(II,III) and -(III,III) Complexes: Models for Putative Intermediates in Oxidation Catalysis. J Am Chem Soc. 2014;136:7269–7272. doi: 10.1021/ja503629r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Isaac JA, Gennarini F, Lopez I, Thibon-Pourret A, David R, Gellon G, Gennaro B, Philouze C, Meyer F, Demeshko S, et al. Room-Temperature Characterization of a Mixed-Valent μ-Hydroxodicopper(II,III) Complex. Inorg Chem. 2016;55:8263–8266. doi: 10.1021/acs.inorgchem.6b01504. [DOI] [PubMed] [Google Scholar]
- 380.Robin MB, Day P. Mixed Valence Chemistry-A Survey and Classification. In: Emeléus HJ, Sharpe AG, editors. Advanced Inorganic Chemistry and Radiochemistry. Vol. 10. Academic Press; 1968. pp. 247–422. [Google Scholar]
- 381.Solomon EI. Dioxygen Binding, Activation, and Reduction to H2O by Cu Enzymes. Inorg Chem. 2016;55:6364–6375. doi: 10.1021/acs.inorgchem.6b01034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Yoon J, Solomon EI. Electronic Structures of Exchange Coupled Trigonal Trimeric Cu(II) Complexes: Spin Frustration, Antisymmetric Exchange, Pseudo-A Terms, and their Relation to O2 Activation in the Multicopper Oxidases. Coord Chem Rev. 2007;251:379–400. [Google Scholar]
- 383.Itoh S, Fukuzumi S. Dioxygen Activation by Copper Complexes. Mechanistic Insights into Copper Monooxygenases and Copper Oxidases. Bull Chem Soc Jpn. 2002;75:2081–2095. [Google Scholar]
- 384.Machonkin T, Mukherjee P, Henson M, Stack TDP, Solomon EI. The EPR Spectrum of a Cu(II/II/III) Cluster: Anisotropic Exchange in a Bent Cu(II)O2 Core. Inorg Chim Acta. 2002;341:39–44. [Google Scholar]
- 385.Gupta AK, Tolman WB. Cu(I)/O2 Chemistry Using a β-Diketiminate Supporting Ligand Derived from N,N-Dimethylhydrazine: A [Cu3O2]3+ Complex with Novel Reactivity. Inorg Chem. 2012;51:1881–1888. doi: 10.1021/ic202214c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Lionetti D, Day MW, Agapie T. Metal-Templated Ligand Architectures for Trinuclear Chemistry: Tricopper Complexes and Their O2 Reactivity. Chem Sci. 2013;4:785–790. doi: 10.1039/C2SC21758A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Maiti D, Woertink JS, Ghiladi RA, Solomon EI, Karlin KD. Molecular Oxygen and Sulfur Reactivity of a Cyclotriveratrylene Derived Trinuclear Copper(I) Complex. Inorg Chem. 2009;48:8342–8356. doi: 10.1021/ic900975y. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Tsui EY, Day MW, Agapie T. Trinucleating Copper: Synthesis and Magnetostructural Characterization of Complexes Supported by a Hexapyridyl 1,3,5-Triarylbenzene Ligand. Angew Chem, Int Ed. 2011;50:1668–1672. doi: 10.1002/anie.201005232. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 389.Kirillov AM, Kirillova MV, Pombeiro AJL. Homogeneous Multicopper Catalysts for Oxidation and Hydrocarboxylation of Alkanes. In: van Eldik R, Colin DH, editors. Advances in Inorganic Chemistry. Vol. 65. Academic Press; 2013. pp. 1–31. [Google Scholar]
- 390.Kirillov AM, Kirillova MV, Pombeiro AJL. Multicopper omplexes and Coordination Polymers for Mild Oxidative Functionalization of Alkanes. Coord Chem Rev. 2012;256:2741–2759. [Google Scholar]
- 391.Mutti FG, Zoppellaro G, Gullotti M, Santagostini L, Pagliarin R, Andersson KK, Casella L. Biomimetic Modelling of Copper Enzymes: Synthesis, Characterization, EPR Analysis and Enantioselective Catalytic Oxidations by a New Chiral Trinuclear Copper(II) Complex. Eur J Inorg Chem. 2009;2009:554–566. [Google Scholar]
- 392.Mutti FG, Gullotti M, Casella L, Santagostini L, Pagliarin R, Andersson KK, Iozzi MF, Zoppellaro G. A New Chiral, Poly-Imidazole N8-Ligand and the Related Di- and Tri-Copper(II) Complexes: Synthesis, Theoretical Modelling, Spectroscopic Properties, and Biomimetic Stereoselective Oxidations. Dalton Trans. 2011;40:5436–5457. doi: 10.1039/c0dt00669f. [DOI] [PubMed] [Google Scholar]
- 393.Gullotti M, Santagostini L, Pagliarin R, Granata A, Casella L. Synthesis and Characterization of New Chiral Octadentate Nitrogen Ligands and Related Copper(II) Complexes as Catalysts for Stereoselective Oxidation of Catechols. J Mol Catal A: Chem. 2005;235:271–284. [Google Scholar]
- 394.Mimmi MC, Gullotti M, Santagostini L, Saladino A, Casella L, Monzani E, Pagliarin R. Stereoselective Catalytic Oxidations of Biomimetic Copper Complexes with a Chiral Trinucleating Ligand Derived from 1,1-Binaphthalene. J Mol Catal A: Chem. 2003;204–205:381–389. [Google Scholar]
- 395.Mimmi MC, Gullotti M, Santagostini L, Battaini G, Monzani E, Pagliarin R, Zoppellaro G, Casella L. Models for Biological Trinuclear Copper Clusters. Characterization and Enantioselective Catalytic Oxidation of Catechols by the Copper(II) Complexes of a Chiral Ligand Derived from (S)-(–)-1,1′-Binaphthyl-2,2′-diamine. Dalton Trans. 2004:2192–2201. doi: 10.1039/B402539C. [DOI] [PubMed] [Google Scholar]
- 396.Mirica LM, Stack TDP. A Tris(μ-hydroxy)tricopper(II) Complex as a Model of the Native Intermediate in Laccase and Its Relationship to a Binuclear Analogue. Inorg Chem. 2005;44:2131–2133. doi: 10.1021/ic048182b. [DOI] [PubMed] [Google Scholar]
- 397.Yoon J, Mirica LM, Stack TDP, Solomon EI. Spectroscopic Demonstration of a Large Antisymmetric Exchange Contribution to the Spin-Frustrated Ground State of a D3 Symmetric Hydroxy-Bridged Trinuclear Cu(II) Complex: Ground-to-Excited State Superexchange Pathways. J Am Chem Soc. 2004;126:12586–12595. doi: 10.1021/ja046380w. [DOI] [PubMed] [Google Scholar]
- 398.Yoon J, Solomon EI. Ground-State Electronic and Magnetic Properties of a μ3-Oxo-Bridged Trinuclear Cu(II) Complex: Correlation to the Native Intermediate of the Multicopper Oxidases. Inorg Chem. 2005;44:8076–8086. doi: 10.1021/ic0507870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Yoon J, Mirica LM, Stack TDP, Solomon EI. Variable-Temperature, Variable-Field Magnetic Circular Dichroism Studies of Tris-Hydroxy- and μ3-Oxo-Bridged Trinuclear Cu(II) Complexes: Evaluation of Proposed Structures of the Native Intermediate of the Multicopper Oxidases. J Am Chem Soc. 2005;127:13680–13693. doi: 10.1021/ja0525152. [DOI] [PubMed] [Google Scholar]
- 400.Suh MP, Han MY, Lee JH, Min KS, Hyeon C. One- Pot Template Synthesis and Properties of a Molecular Bowl: Dodecaaza Macrotetracycle with μ3-Oxo and μ3-Hydroxo Tricopper- (II) Cores. J Am Chem Soc. 1998;120:3819–3820. [Google Scholar]
- 401.Chen PPY, Yang RBG, Lee JCM, Chan SI. Facile OAtom Insertion into C-C and C-H bonds by a Trinuclear Copper Complex Designed to Harness a Singlet Oxene. Proc Natl Acad Sci U S A. 2007;104:14570–14575. doi: 10.1073/pnas.0707119104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Chan SI, Chien CYC, Yu CSC, Nagababu P, Maji S, Chen PPY. Efficient Catalytic Oxidation of Hydrocarbons Mediated by Tricopper Clusters Under Mild Conditions. J Catal. 2012;293:186–194. [Google Scholar]
- 403.Chan SI, Lu YJ, Nagababu P, Maji S, Hung MC, Lee MM, Hsu IJ, Minh PD, Lai JCH, Ng KY, et al. Efficient Oxidation of Methane to Methanol by Dioxygen Mediated by Tricopper Clusters. Angew Chem, Int Ed. 2013;52:3731–3735. doi: 10.1002/anie.201209846. [DOI] [PubMed] [Google Scholar]
- 404.Barnett SM, Goldberg KI, Mayer JM. A Soluble Copper–Bipyridine Water-Oxidation Electrocatalyst. Nat Chem. 2012;4:498–502. doi: 10.1038/nchem.1350. [DOI] [PubMed] [Google Scholar]
- 405.Du J, Chen Z, Ye S, Wiley BJ, Meyer TJ. Copper as a Robust and Transparent Electrocatalyst for Water Oxidation. Angew Chem, Int Ed. 2015;54:2073–2078. doi: 10.1002/anie.201408854. [DOI] [PubMed] [Google Scholar]
- 406.A recent example: Garcia-Bosch I, Siegler MA. Copper- Catalyzed Oxidation of Alkanes with H2O2 under a Fenton-like Regime. Angew Chem, Int Ed. 2016;55:12873–12876. doi: 10.1002/anie.201607216.
- 407.Liu JJ, Diaz DE, Quist DA, Karlin KD. Copper(I)-Dioxygen Adducts and Copper Enzyme Mechanisms. Isr J Chem. 2016;56:738–755. doi: 10.1002/ijch.201600025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Abad E, Rommel JB, Kastner J. Reaction mechanism of the bicopper enzyme peptidylglycine alpha-hydroxylating monooxygenase. J Biol Chem. 2014;289:13726–13738. doi: 10.1074/jbc.M114.558494. [DOI] [PMC free article] [PubMed] [Google Scholar]