

SUMMARY
Despite the central role PSD-95 plays in anchoring postsynaptic AMPARs, how PSD-95 itself is tethered to postsynaptic sites is not well understood. Here we show that the F-actin binding protein α-actinin binds to the very N-terminus of PSD-95. Knock-down (kd) of α-actinin phenocopies kd of PSD-95. Mutating lysine at position 10 or lysine at position 11 of PSD-95 to glutamate, or glutamate at position 53 or glutamate and aspartate at positions 213 and 217 of α-actinin, respectively, to lysine impairs in parallel PSD-95 binding to α-actinin and postsynaptic localization of PSD-95 and AMPARs. These experiments identify α-actinin as a critical PSD-95 anchor tethering the AMPAR - PSD-95 complex to postsynaptic sites.
Keywords: PSD-95, α-actinin, hippocampus, synapse, dendritic spines, AMPA receptors
eTOC
Matt et al. introduce α-actinin as critical postsynaptic docking protein for PSD-95 and AMPARs. They found that disruption of the PSD-95 – α-actinin interaction by point-mutating the essential residues on either protein leads to reduced synaptic localization of PSD-95 and AMPARs.
INTRODUCTION
The vast majority of synapses in the brain are glutamatergic (Micheva et al., 2010). Postsynaptic AMPA-type glutamate receptors (AMPARs) have to be precisely juxtaposed to presynaptic release sites for fast and efficient synaptic transmission (Tang et al., 2016; Sinnen et al., 2017). The localization of postsynaptic AMPAR critically depends on PSD-95 (El-Husseini et al., 2002; El-Husseini et al., 2000; Elias et al., 2006; Schluter et al., 2006). PSD-95 consists of three PDZ domains followed by an SH3 domain and a guanylate kinase homology domain (GK; Figure 1A). PSD-95 recruits AMPARs to postsynaptic sites by interacting with its first and second PDZ domains with the intracellular C-termini of auxiliary subunits known as transmembrane AMPAR regulatory proteins (TARPs), including stargazin (Stg, γ2) and its homologs γ3, γ4, and γ8 (Chen et al., 2000; Opazo et al., 2010; Schnell et al., 2002). Palmitoylation of Cys3 and Cys5 near the N-terminus of PSD-95 is critical for its postsynaptic accumulation, which, in turn, is important for postsynaptic targeting of AMPARs (El-Husseini et al., 2002; Fukata et al., 2013). The palmitoyl acyltransferase (PAT) DHHC2 accumulates at postsynaptic sites when basal neuronal activity is reduced. This DHHC2 accumulation leads to an increase in PSD-95 palmitoylation especially at postsynaptic sites (Noritake et al., 2009). Thus, palmitoylation that is localized to postsynaptic sites augments postsynaptic PSD-95 targeting. However, other PATs including DHHC3, which is mostly associated with the Golgi apparatus rather than postsynaptic sites, also contribute to postsynaptic localization of PSD-95 (Fukata et al., 2013; Noritake et al., 2009) and palmitoylation is not a tagging mechanism that would specifically anchor proteins at the postsynaptic plasma membrane but is a rather general mechanism for association with membranes. For instance, palmitoylation of the postsynaptic A kinase anchor protein AKAP150 preferentially targets it to endosomes rather than plasma membrane (Woolfrey et al., 2015). It is largely unknown, which proteins function to specifically anchor and stabilize PSD-95 at the postsynaptic plasma membrane despite the identification of many PSD-95 – interacting proteins (Sheng and Kim, 2011). For example, GK-domain associated proteins (GKAPs) bind to the PSD-95 GK domain (Kim et al., 1997) linking PSD-95 to Shank and thereby Homer and cortactin, the latter binding to F-actin (Naisbitt et al., 1999; Tu et al., 1999). Although GKAP up- and downregulation affected synaptic transmission accordingly, it is unclear whether GKAP exerted these effects by interacting with PSD-95 (Shin et al., 2012). Similarly, Shank1 knock-out decreased synapse density, size and strength but the mechanism of this change is unknown (Hung et al., 2008). Recent evidence now suggests that ephrin-B3 is a postsynaptic anchoring protein for PSD-95 (Hruska et al., 2015). PSD-95 can bind to the cytosolic C-terminus of this integral plasma membrane protein and the point mutations L293A and S332D in the C-terminus of ephrin-B3 impair PSD-95 binding and postsynaptic localization of PSD-95 and also of ephrin-B3 itself. However, L293A also affects ERK1/2 binding while S332 is an ERK phosphorylation site. Therefore, it is unclear whether these mutations reduce postsynaptic PSD-95 by decreasing its binding to ephrin-B3 or indirectly, possibly by altering ERK signaling. In fact, the decrease in postsynaptic PSD-95 anchoring seen upon ephrin-B3 knock down can be rescued by pharmacological inhibition of MEK-ERK signaling. This finding indicates that there must be another postsynaptic protein that can anchor PSD-95 independent of the presence of ephrin-B3. In addition, it is unclear how abundant ephrin-B3 is, and how well it is stoichiometrically matched with the highly abundant PSD-95.
Figure 1. α-actinin-1 CH1, CH2 and EF-hand domains interact with PSD-95.

(A) Schematic of PSD-95 and α-actinin domain structure. Horizontal lines: pull-down constructs.
(B) Co-immunoprecipitation (Co-IP) of α-actinin (pan antibody) with PSD-95 (JH62092) from Triton X-100 brain lysates.
(C) Immunoblots (IBs) of total brain lysates identify specific bands for PSD-93, PSD-95, SAP97, and SAP102 (all antibodies from NeuroMAB).
(D) IP of PSD-93, PSD-95, SAP97, and SAP102 from Triton X-100 brain lysates yielded the respective IB band(s) without cross-reactivity or co-IP among PSD-95 homologues. Pan α-actinin Co-IPed only with PSD-95.
(E) Top: Pull-down of MBP-tagged α-actinin-1 by GST-tagged PSD-95 domains (see 1A) detected by IB with anti-MBP. Bottom: IB with anti-GST indicates that comparable amounts of the PSD-95 fusion proteins were present. Right: Densitometric analysis of MBP intensities shows significantly stronger binding of α-actinin-1 to the N-terminal portion of PSD-95 (NT-PDZ1/2) than any other construct (n = 6).
(F) Top: Pull-down of MBP-tagged α-actinin domains but not MBP alone by immobilized PSD-95 NT-PDZ1/2. Bottom: IB with anti-GST indicates that comparable amounts of PSD-95 NT-PDZ1/2 were present in all samples. Right: Densitometric MBP quantification (n = 8).
(G) Top: Pull-down of MBP-tagged α-actinin CH1 and CH2 domains but not MBP alone by PSD-95 NT-PDZ1/2. Bottom: comparable amounts of PSD-95 NT-PDZ1/2 were present in all samples. Right: Densitometric quantification (n = 4).
(H) Summary IB run with equal amount of sample as panels F and G illustrates that comparable amounts of the MBP-tagged α-actinin constructs were present.
(I) Top: Pull-down of MBP-tagged α-actinin EF region with (EF) and without the last 4 residues that bind PDZ domains (EF-ΔPDZ) but not of MBP alone by PSD-95 NT-PDZ1/2. Bottom: comparable amounts of PSD-95 NT-PDZ1/2 were present in all samples. Right: Densitometric MBP quantification (n = 8).
Statistical significance was tested by 1-way ANOVA with Dunnett’s Multiple Comparison Test.
The postsynaptic abundance of AMPARs defines synaptic strength, which is usually stable at basal activity levels but can persistently be altered by heightened activity patterns that induce LTP and LTD (Huganir and Nicoll, 2013). LTP and LTD are thought to be important mechanisms for storage of information during learning and memory formation (Morris, 2013; Whitlock et al., 2006). Related forms of synaptic plasticity underlie different brain pathologies such as PTSD (Clem and Huganir, 2010) and drug addiction (Wolf and Tseng, 2012). AMPAR dysfunction and dysregulation also contribute to numerous diseases of the brain such as depression (Skolnick et al., 2009), autism (Ebert and Greenberg, 2013; Etherton et al., 2011), epilepsy (McNamara et al., 2006), and stroke-induced neuronal damage (Liu et al., 2004; Noh et al., 2005). Thus understanding the mechanisms that determine postsynaptic AMPAR localization and function are of high relevance.
Here we identify α-actinin as a major postsynaptic anchoring protein for PSD-95. α-actinin is best known for cross-linking F-actin and for anchoring F-actin to cell–cell and cell–matrix junctions (Foley and Young, 2014; Otey and Carpen, 2004; Sjoblom et al., 2008). Overexpression as well as KD of α-actinin affects spine density and structure (Hodges et al., 2014; Hoe et al., 2009; Kalinowska et al., 2015; Nakagawa et al., 2004). We now report that α-actinin binds to the very N-terminus of PSD-95 thereby stabilizing it at postsynaptic locations. Systematic extensive knock-down, rescue, and point mutation experiments unequivocally demonstrate a central role of α-actinin in postsynaptic localization of PSD-95 and thereby of AMPARs.
RESULTS
α-actinin is associated with PSD-95
We recently identified α-actinin as the critical anchoring protein for the L-type Ca2+ channel Cav1.2 in dendritic spines (Hall et al., 2013). Therefore and because of its prominent enrichment in spines (Wyszynski et al., 1998) and postsynaptic densities (PSDs; Racz and Weinberg, 2004; Walikonis et al., 2000), we evaluated whether α-actinin is involved in anchoring PSD-95 and, in turn, AMPARs. We immunoprecipitated (IP) PSD-95 and probed immunoblots (IB) with a pan-specific antibody against α-actinin. The four isoforms encoded by the four different α-actinin genes have near identical relative molecular mass (MR; Sjoblom et al., 2008) and migrate as a single band on SDS-PAGE (Figure 1B, lysate; Hall et al., 2013). This band was present in PSD-95 but not control IgG IPs.
To investigate if α-actinin specifically interacts with PSD-95 or perhaps also with one or more of the less abundant PSD-95 homologues PSD-93, SAP97, and SAP102, we first established a set of antibodies that specifically recognize one of each of these four proteins. Each antibody detected distinct bands in brain lysate of the approximated MR (Figure 1C; note that SAP97 runs as a doublet at ~120 kDa, which is often not resolved; Valtschanoff et al., 2000) with PSD-95 showing three different bands that are due to differential splicing and posttranslational modification such as palmitoylation, as described previously (El-Husseini et al., 2002; El-Husseini et al., 2000; Schluter et al., 2006; Topinka and Bredt, 1998). The specificity of each antibody was confirmed by IB after IP of each individual PSD-95 homologue, the IP only isolating the respective immunoreactive protein bands that were recognized by the individual antibody in total lysate (Figure 1D), as well as by IB of purified GST-tagged proteins (Figure S1A). The additional band for PSD-93 in the IP lane and the broad main band likely reflects the existence of six or more splice variants (Kruger et al., 2013; Parker et al., 2004). Remarkably, co-IP of α-actinin was only observed with PSD-95 (Figure 1D, bottom panel).
α-actinin CH1, CH2 and EF domains interact with the N-terminus of PSD-95
To assess the possibility of a direct interaction between PSD-95 and α-actinin, domains of PSD-95 (Figure 1A) were expressed in E. coli as GST fusion proteins and immobilized on glutathione Sepharose beads for use in pull-down assays. Of those, only the fragment consisting of the N-terminal 64 residues, PDZ1, and PDZ2 domains (NT-PDZ1/2) robustly and consistently pulled down recombinant full-length α-actinin-1 fused to maltose binding protein (MBP; Figure 1E, upper left panel), confirming a specific and direct interaction between the two proteins. PSD-95 NT-PDZ1/2 pulled down MBP-tagged CH1/2, SR3, and EF-hand of α-actinin-1 (Figure 1F) and individual CH1 and CH2 domains (Figure 1G). The additional lower weight band observed for MBP-tagged constructs is due to limited proteolysis of a fraction of the isolated protein. Similar quantities of all MBP-tagged α-actinin-1 domains were used in pull-down experiments (Figure 1H). PSD-95 NT-PDZ1/2 also pulled down the EF hand domain lacking the C-terminal PDZ ligand residues ESDL (EF-ΔPDZ; Kim and Sheng, 2004; Walikonis et al., 2001; Xia et al., 1997) indicating that binding between the EF-hand and PDZ-1/2 is not mediated by a PDZ interaction (Figure 1I). Similar results were obtained with full length GST-PSD-95 (Figures S1B–D). A GST construct containing only the first 71 residues of PSD-95, which are N-terminal to PDZ1, was equally effective in pulling down CH1, CH2, CH1/2, SR3, EF, and EF-ΔPDZ domains (Figure S1E) identifying the N-terminus of PSD-95 as a binding site for different α-actinin domains.
Knock-down of α-actinin reduces synapse density but not structure
We previously developed four shRNAs, each of which specifically knocks down (kd) one of the four α-actinins (Hall et al., 2013; Schnizler et al., 2009). Neurons express α-actinin-1, -2, and -4, but not -3 (Hall et al., 2013; Nakagawa et al., 2004; Schnizler et al., 2009; Wyszynski et al., 1998; Matt and Hell, unpublished). We transfected hippocampal cultures with parental vector as control or a combination of shRNA-expressing vectors against α-actinin-1, -2, and -4 to kd all three isoforms (kd124). An α-actinin-1 construct rendered shRNA-insensitive by silent mutations (rescue; Hall et al., 2013) served as control for the specificity of the kd. A validated shRNA against α-actinin-3 (kd3; Hall et al., 2013; Schnizler et al., 2009) constituted an additional control to rule out side effects of transfection per se with a hairpin-forming shRNA. Sholl analysis did not reveal any effect on dendritic arborization (Figure S2A). Kd124 did, however, lead to an ~40% reduction in density of spines (Figure S2G) and of immunofluorescent (IF) puncta for all synaptic proteins we studied (α-actinin, bassoon, F-actin, GluA1, GluA2, GluN1, PSD-93, PSD-95, SAP97, SAP102, Shank, synapsin; Figures 2 and S3). Kd124 decreased not only α-actinin puncta density but also intensity by ~50% vs vector control (Figure 2A), illustrating α-actinin kd. Accordingly, kd124 not only reduced synapse number but also α-actinin content of the remaining synapses by half. Remarkably, a morphometric study of individual spines did not show any detectable changes in the size, shape, or normal distribution of types upon kd124 (Figure S2C–F). This finding was somewhat surprising as α-actinin is an effective F-actin crosslinking protein (Sjoblom et al., 2008) and is highly enriched at postsynaptic sites (Racz and Weinberg, 2004; Walikonis et al., 2000; Wyszynski et al., 1998). In support of lack of a profound effect on postsynaptic structure by kd124, puncta intensity of F-actin staining with fluorescently labeled phalloidin (Figure 2C) or of IF of Shank did not change either upon kd124 (Figure S3B). Puncta intensity of the presynaptic markers synapsin (Figure 2A) and bassoon (Figure S3A) was also unaltered by kd124, indicating that postsynaptic α-actinin kd does not affect presynaptic structures.
Figure 2. α-actinin knockdown reduces synapse density and postsynaptic PSD-95 content.

Primary rat hippocampal neuronal cultures were transfected at 4 days in vitro (DIV), fixed, and stained at 18 DIV. Parental pSilencer vector encoding eGFP and kd3 served as controls. α-actinin-1, -2, and -4 were knocked-down with specific shRNAs (kd124) while shRNA-insensitive α-actinin-1* was co-expressed for rescue of kd124.
Representative images for each synaptic protein examined are shown on the left. Bar diagrams on the right represent quantifications of the density and mean intensity (in arbitrary units, AU) of immunofluorescent (IF) puncta.
(A) Puncta density of α-actinin and synapsin was reduced by kd124. Puncta intensity of α-actinin (but not synapsin) was reduced by kd124 (n >50 images from > 25 cells from 5 independent experiments).
(B) Puncta density of PSD-95 and extracellular GluA1EC IF was reduced by kd124. Puncta intensity of PSD-95 was reduced by ~ 50% by kd124. GluA1EC showed a small, non-significant reduction (n > 33 images from > 16 cells from 3 independent experiments).
(C) Puncta density but not intensity of F-actin staining with Alexa555-labeled phalloidin was reduced by kd124 (n > 33 images from > 16 cells from 4 independent experiments).
(D) Puncta density but not intensity of PSD-93 and extracellular GluA2EC IF was reduced by kd124 (n > 36 images of > 18 cells from 6 independent experiments).
(E) Puncta density but not intensity of extracellular GluN1EC IF was reduced by kd124 (n > 35 images of > 17 cell from 4 independent experiments).
(F) Puncta density but not intensity of SAP102 was reduced by kd124 (n > 34 images of > 17 cells from 3 independent experiments).
(G) Puncta density of SAP97 was insignificantly reduced by kd124. Puncta intensity of SAP97 was not affected by kd124 (n > 19 images of > 9 cells from 3 independent experiments).
Statistical significance was tested by 1-way ANOVA with Bonferroni’s Multiple Comparison Test.
See Table S2 for values. See also Figure S2 and S3.
α-actinin kd reduces postsynaptic PSD-95 but not AMPAR or NMDAR content
Mirroring its effect on α-actinin, kd124 also reduced the IF puncta intensity of PSD-95 by 50%, but had no effect on PSD-93, SAP102, or SAP97 (Figure 2B,D,F,G). Intensity of surface staining for the prevalent AMPAR subunits GluA1 (GluA1EC; Figure 2B) and GluA2 (GluA2EC; Figure 2D) and obligatory NMDAR subunit GluN1 (GluN1EC; Figure 2E) were minimally if at all affected in synapses that survived kd124. Consistently, kd124 reduced frequency but not amplitude of miniature excitatory postsynaptic currents (mEPSCs; Figure S3C). These findings show that α-actinin kd phenocopies PSD-95 kd and knock-out, which also lead to a reduction in synapse number with unaltered AMPAR content in remaining synapses (Beique et al., 2006; Ehrlich et al., 2007; Elias et al., 2006; Levy et al., 2015; see Discussion).
α-actinin interacts with residues 1-13 of PSD-95
A library of fluorescein-labeled peptides covering the N-terminus of PSD-95 (Figure 3A) were titrated with increasing amounts of α-actinin-1 (Zhang et al., 2014). Only the peptide spanning residues 1-13 displayed saturable binding (Figure 3B). Peptide 1-13 also showed saturable binding to CH1/2 and EF, but not the SR3 domain of α-actinin-1 (Figure 3C). GST fusion proteins of full length and N-terminal fragments of PSD-95 pulled down the MBP-tagged SR3 domain of α-actinin but not MBP alone (Figure 1F, S1B, S1E). To reconcile the negative peptide and positive pull down data we loaded glutathione Sepharose with plain GST, which pulled down MBP-SR3 but not MBP alone (Figure S1F). Accordingly, the MBP-tagged SR3 domain binds to GST alone and this interaction does not reflect SR3 binding to PSD-95. Neither full length MBP - α-actinin nor its CH1, CH2, and EF domains bound to GST alone confirming specificity of their PSD-95 interactions (Figure S1F).
Figure 3. K10 and K11 of PSD-95 are essential for α-actinin binding.

(A) Sequence of the PSD-95 N-terminus and the 4 peptides covering its length (black bars on bottom).
(B) Fluorescence polarization (FP) of 1 μM FITC-labeled peptides given in A. Titration with full length MBP-α-actinin showed binding only for peptide 1-13 (n = 3; mean ± S.D.).
(C) FP of FITC-labeled peptide 1-13. Titration with α-actinin-1 domains showed binding with CH1/2 and EF-hand, but not SR3 (n = 3; mean ± S.D.).
(D) Co-IP of α-actinin with PSD-95 from brain lysates was disrupted by adding PSD-95 peptide 1-13 and GST-PSD-95 1-64 (N-term) but not 15-36. Addition of Ca2+ plus CaM also disrupted the co-IP (similar results in 2 other experiments).
(E) Overlay of peptide spot arrays shift-scanning through the PSD-95 N-terminus with MBP-tagged full-length α-actinin-1, α-actinin-2 and CH1/2 domain of α-actinin-1. Residues with strong effects on binding are shaded orange (K10, K11) (similar results in 1–2 other experiments).
(F) Overlay of a triple alanine scanning peptide spot array of the PSD-95 N-terminus with MBP-CH1/2 domain of α-actinin-1. Residues with strong effect on binding are shaded orange (K10, K11) (similar results in 2 other experiments).
(G,H) FP of FITC-labeled, point-mutated PSD-95 1-13 peptides. Titration with full length MBP-α-actinin-1 (G) showed strongest reduction in binding for peptides K10E, K11E, and KK10,11EE. Titration with Ca2+/CaM (H) was strongly affected by all mutations (n = 3; mean ± S.D.).
Peptide 1-13 but not 15-36 could competitively prevent co-IP of α-actinin with PSD-95 from brain lysate (Figure 3D). Thus this peptide mimics the binding site on PSD-95 for α-actinin. α-actinin could also be displaced from PSD-95 by the N-terminal 64 residues of PSD-95. Accordingly, PSD-95 residues 1-13 constitute the physiologically relevant binding site for α-actinin, whose interaction with PSD-95 is mediated by its CH1/2 or EF domain or both.
Lys10 and Lys11 of PSD-95 are necessary for α-actinin binding
Binding of α-actinin-1, -2 and the CH1/2 domain of α-actinin-1 in overlay assays to spot synthesized peptide arrays of 13-residue-long peptides shifted through the N-terminus of PSD-95 (Figure 3E), was weakened as T9 (peptide 11) and lost as K10 (peptide 12) were shifted out of the peptide. Consistently, a triple alanine scan of peptide 1-13 showed the strongest reduction in CH1/2 binding when K10 or K11 were altered (Figure 3F). Scanning peptide 1-13 by replacing individual residues with glutamate for titration with MBP-α-actinin showed that binding was only slightly affected when T8, T9, Y12, or R13 were changed (T8E, T9E, Y12E, R13E) while changing K10 or K11 (K10E, K11E, KK10,11EE) resulted in a strong reduction in binding (Figure 3G). All of these mutations affected Ca2+/CaM binding (Figure 3H; Zhang et al., 2014) suggesting that Ca2+/CaM interacts with all of these residues including those important for α-actinin binding. Thus binding of Ca2+/CaM is predicted to antagonize α-actinin binding, which was indeed observed when Ca2+ and CaM was added during IP of PSD-95 (Figure 3D, right lane; see also Figure 7).
Figure 7. NMDA-induced Ca2+ influx causes disruption of the PSD-95 - α-actinin interaction by Ca2+/CaM.
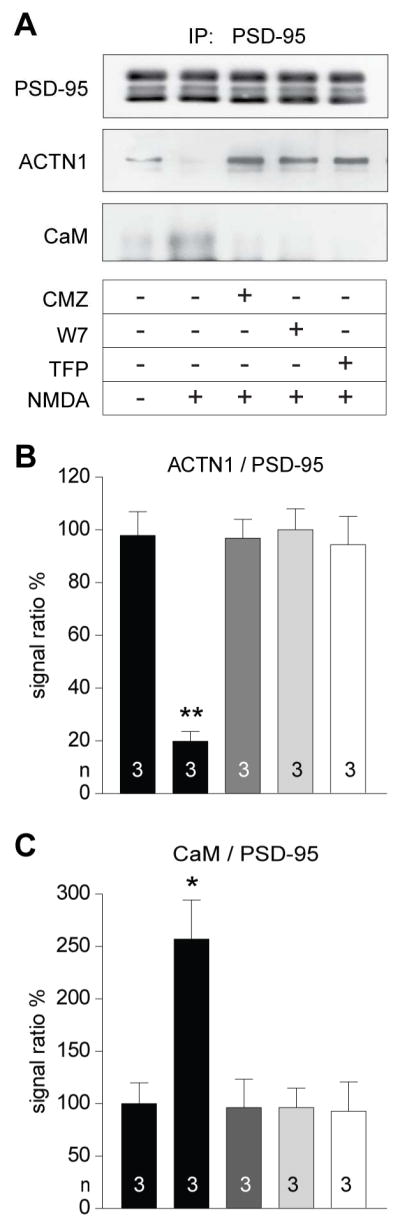
(A) Acute forebrain slices were pre-treated with the CaM antagonists calmidazolium (CMZ; 30 μM), W7 (20 μM), and trifluoperazine (TFP; 20 μM) before treatment with vehicle (CT) or 100 μM NMDA for 5 min, lysed, and analyzed by IP of PSD-95 and IB for PSD-95, α-actinin, and CaM.
(B,C) NMDA-induced Ca2+ influx decreased coIP of α-actinin and increased coIP of CaM with PSD-95. Both effects were blocked by all three CaM antagonists.
Statistical significance was tested by 1-way ANOVA with Dunnett’s Multiple Comparison Test. See Table S6 for values.
See also Figure S7.
α-actinin binding-deficient PSD-95 shows reduced synaptic enrichment
As a first evaluation of the synaptic role of α-actinin binding to PSD-95, RFP-tagged WT and mutant PSD-95 was expressed in cultured neurons. Expression levels of all PSD-95 mutants were comparable to those of RFP and RFP-PSD-95WT except for increased expression of RFP-PSD-95YRY12,13,14EEE (Figure S4B,F; see also Figure 4B). Deletion of the whole N-terminus (Δ64) abrogated postsynaptic enrichment of PSD-95 (Figure S4A–D) consistent with the requirement of PSD-95 palmitoylation at C3 and C5 (El-Husseini et al., 2002). Mutating residues 10-12 (KKY) to glutamate (PSD-95KKY10,11,12EEE) decreased postsynaptic PSD-95 targeting to the same degree as Δ64 and to alanine (PSD-95KKY10,11,12AAA) had a partial effect while other triple mutants showed little if any reduction in synaptic intensity. Consistent with the above binding studies (Figure 3G), individual mutations of K10, K11 and Y12 identified K10 and K11 as critical for postsynaptic targeting, with Y12 playing a minor if any role (Figure S4E–H). A new set of transfection experiments focused on K10 and K11 and refined analysis by confocal microscopy confirmed these initial results (Figure 4). Palmitoylation of α-actinin binding-deficient mutants PSD-95K10E, PSD-95K11E, and PSD-95KK10,11EE did not differ from that of PSD-95WT (Figure S5A). Accordingly, these mutations did not affect postsynaptic PSD-95 binding through a palmitoylation-related mechanism but rather by decreasing α-actinin binding (Figure 3G).
Figure 4. α-actinin binding-deficient PSD-95 displays reduced synaptic enrichment.

(A) Hippocampal cultures were transfected with GFP and RFP alone or C-terminally RFP-tagged PSD-95WT, PSD-95K10E, PSD-95K11E, and PSD-95KK10,11EE at 7 DIV and fixed at 18 DIV.
(B) Expression of WT and mutant PSD-95 was comparable as reflected by cell-wide RFP intensity normalized to cell-wide GFP intensity.
(C) Synaptic intensities for all three PSD-95 mutants were lower vs PSD-95WT and higher vs RFP (except for PSD-95KK10,11EE).
(D) Synaptic enrichment was significantly less for all three PSD-95 mutants vs PSD-95WT.
(B–D) n > 36 images of > 18 cells per condition of 9 independent experiments.
Statistical significance was tested by 1-way ANOVA with Bonferroni’s Multiple Comparison Test.
α-actinin binding-deficient PSD-95 fails to promote postsynaptic AMPAR accumulation
PSD-95 kd reduces mEPSC frequency but not amplitude of surviving synapses (Ehrlich et al., 2007; Elias et al., 2006) due to homeostatic mechanisms that allow functionally surviving synapses to maintain full AMPAR quota when most other synapses lose all AMPARs (Levy et al., 2015; see also Bian et al., 2015). The engagement of those homeostatic mechanisms complicate interpretation of PSD-95 kd effects. On the other hand, overexpressed PSD-95WT accumulates in the postsynaptic site and increases not only the number of AMPAR containing synapses and mEPSC frequency but also postsynaptic AMPAR content and mEPSC amplitude (Beique and Andrade, 2003; Ehrlich and Malinow, 2004; El-Husseini et al., 2000). Thus we chose this approach to evaluate whether the α-actinin binding-deficient PSD-95K10E, PSD-95K11E, and PSD-95KK10,11EE mutants exhibit deficiencies in their ability to increase postsynaptic AMPAR content. All mutants showed overall RFP intensities that were comparable to the parental vector (RFP) and especially PSD-95WT (Figure 4B). We found no differences in the spine length, width, volume, or distribution of spine types between neurons expressing these mutants or control RFP or RFP-PSD-95WT (Figure S5B–F). Accordingly no overt off-target effects were evident. In accordance with the literature (Beique and Andrade, 2003; Ehrlich and Malinow, 2004; El-Husseini et al., 2000), overexpression of PSD-95WT led to significant increases in spine density (Figure S5G), GluA1EC puncta density and intensity (Figure 5A), and mEPSC frequency and amplitude (Figure 5B). Strikingly, overexpression of all α-actinin binding-deficient mutants failed completely to increase spine density (Figure S5G), GluA1EC puncta density and intensity (Figure 5A), and mEPSC frequency and amplitude (Figure 5B).
Figure 5. α-actinin binding-deficient PSD-95 fails to promote synaptic enrichment of AMPAR.

Hippocampal cultures were transfected as in Figure 4, fixed and stained at 18 DIV for extracellular GluA1 (GluA1EC; A) or, after permeabilization, total GluN1 (GluN1total; C) or used for mEPSC recordings at 11–14 DIV (B). Mean IF intensities (right bar diagrams) were quantified as above.
(A) All three PSD-95 mutations abrogated the increase in GluA1EC puncta density and intensity induced by ectopic PSD-95WT expression (n > 12 images from > 6 cells from 3 independent experiments).
(B) Mini EPSC sample traces (left) and averaged individual events from a single cell (right) to scale (top) and normalized to peak (bottom). All three PSD-95 mutations abrogated the increase in frequency and amplitude induced by ectopic PSD-95WT expression. The average mEPSC decay time was unchanged (n = 5–9 cells per condition).
(C) GluN1total puncta density was unaltered by PSD-95WT or any of the PSD-95 mutants vs RFP. GluN1total synaptic IF intensity was unaltered by PSD-95WT and PSD-95K10E vs RFP but decreased without reaching statistical significance by PSD-95K11E and PSD-95KK10,11EE vs PSD-95WT (n > 28 images of > 14 cells from 3 independent experiments).
Statistical significance was tested by 1-way ANOVA with Bonferroni’s Multiple Comparison Test.
Again in accordance with previously published data (Ehrlich and Malinow, 2004; El-Husseini et al., 2000), overexpression of WT and mutant PSD-95 did not affect GluN1 puncta density and intensity except for a modest decrease in puncta intensity upon overexpression of PSD-95K11E and PSD-95K10,11EE (Figure 5C).
PSD-95 residue K10 interacts with E213 and D217 in CH2 and K11 with E53 in CH1
To identify residues in α-actinin that bind to K10 and K11 in PSD-95, we generated a structural model (Figure 6A) by docking our published structure of the first 19 residues of the PSD-95 N-terminus (Zhang et al., 2014) to a published structure of the CH1/2 domain of α-actinin-1 (PDB-ID: 2EYI; Borrego-Diaz et al., 2006) using the program HADDOCK (de Vries et al., 2010). This model predicted that PSD-95 residue K10 interacts with E213 and D217 in CH2, and K11 with E53 in CH1, respectively. To test this prediction, we mutated the negatively charged E53 or E213 and D217 in MBP-tagged CH1/CH2 to positively charged lysine (CH1/2E53K and CH1/2ED213/217KK). As with full length α-actinin-1 (Figure 3G), titration of N-terminal PSD-95 peptides with MBP-tagged WT CH1/CH2 showed decreased affinity for 1-13K10E, 1-13K11E, 1-13KK10,11EE versus WT peptide 1-13 (Figure S6A). Importantly, affinity for WT peptide 1-13 was strongly reduced for MBP-tagged CH1/2E53K and CH1/2ED213,217KK (Figure S6B). Pull down experiments of purified WT and mutant full length MBP-α-actinin-1 with purified full length WT and mutant GST-PSD-95 confirmed that the E53K and ED213,217KK point mutations in α-actinin-1 as well as the K10E and K11E point mutations in PSD-95 strongly impaired the binding (Figure S6C).
Figure 6. Molecular replacement of α-actinin with α-actinin-1E53K or α-actinin-1ED213,217KK and of PSD-95 with PSD-95K10E and PSD-95K11E impairs synaptic localization of PSD-95 and AMPARs.

(A) Structural model of the CH1/2 domain of α-actinin-1 (purple, PDB ID: 2EYI) docked onto residues 1-19 of PSD-95 (red, PDB ID: 2MES; see Supplemental Material). K10 and K11 of PSD-95 are predicted to be in close proximity to E213/D217 and E53, respectively, of α-actinin (interactions indicated by red lines).
(B) Hippocampal cultures were transfected at 7 DIV with α-actinin kd124 plasmids and sh-resistant plasmids for replacement of endogenous α-actinin by α-actinin-1*WT, α-actinin-1*E53K, or α-actinin-1*ED213,217KK.
(C–F) Replacement of α-actinin by α-actinin-1*E53K, or α-actinin-1*ED213,217KK (ED_KK) decreased intensity (C) and enrichment (D) of endogenous PSD-95 in spines as well as surface-labeled GluA2 spine intensity (E) and puncta density (F) versus replacement by α-actinin-1*WT.
(G) Hippocampal cultures were infected at 7 DIV with shRNA against PSD-95 and sh-resistant cDNA for replacement of endogenous PSD-95 by GFP-PSD-95*WT, GFP-PSD-95*K10E, or GFP-PSD-95*K11E.
(H–K) Replacement of endogenous PSD-95 by GFP-PSD-95*K10E, or GFP-PSD-95*K11E decreases intensity (H) and enrichment (I) of GFP-PSD-95 in spines as well as surface-labeled GluA2 spine intensity (J) and puncta density (K) versus replacement by GFP-PSD-95*WT
(C–F) n>10 images from 2 independent cultures
(H–K) n>18 images from 2 independent cultures
Statistical significance was tested by Kruskal-Wallis test followed by Mann-Whitney U test with Bonferroni correction.
Molecular replacement of α-actinin with α-actinin-1 deficient in PSD-95 binding impairs postsynaptic PSD-95 and AMPAR targeting
We created full length shRNA-insensitive α-actinin-1*E53K and α-actinin-1*ED213,217KK for a kd – replacement approach, as simple overexpression of α-actinin in neurons leads to a number of well documented morphological defects (Hodges et al., 2014; Hoe et al., 2009; Kalinowska et al., 2015; Nakagawa et al., 2004; Tseng and Hell, unpublished). Co-transfection of our combined sh124 plasmids to kd α-actinin-1, -2, and -4 with shRNA-insensitive α-actinin-1*WT allowed for rescue with substantial though not complete replacement of endogenous α-actinin in cultured neurons (Figure 2). Co-transfection with α-actinin-1*E53K (E53K) or α-actinin-1*ED213,217KK (ED_KK), however, resulted in an ~50% reduction in IF signal intensity of endogenous PSD-95 and GluA2, in PSD-95 spine enrichment, and in the density of GluA2-positive puncta (Figure 6B–F). The point mutations did not affect the total levels of PSD-95 (Figure S6D) or α-actinin (Figure S6E). There was, however, a modest but statistically significant reduction in spine levels for α-actinin-1*E53K but not for α-actinin-1*ED213,217KK (Figure S6F). Because this reduction could affect F-actin content and thereby have indirect effects on PSD-95 localization, we quantified F-actin after labeling with phalloidin by STED microscopy. Importantly, neither α-actinin-1*E53K nor α-actinin-1*ED213,217KK affected F-actin content in spines (Figure S6G,H).
Molecular replacement of PSD-95 with α-actinin-1 binding-deficient PSD-95 impairs postsynaptic PSD-95 and AMPAR targeting
We performed analogous molecular replacement experiments with PSD-95 using an established kd/rescue plasmid (Schluter et al., 2006). The shRNA strongly decreased endogenous PSD-95 and IB indicated that ectopically expressed GFP-PSD-95 matched the level of endogenous PSD-95 in untransfected neurons (Chowdhury et al., 2017). Furthermore, IF analysis indicated that total PSD-95 levels and specifically the levels of ectopically expressed GFP-PSD-95 were comparable between WT and mutant PSD-95 isoforms (Figure S6I). Importantly, PSD-95 replacement with GFP-PSD-95K10E as well as GFP-PSD-95K11E showed an ~ 50% decrease in spine intensity and enrichment for GFP-PSD-95 signals (Figure 6G–I) as well as sGluA2 spine intensity and puncta density (Figure 6G,J,K).
Ca2+ influx disrupts α-actinin association with PSD-95 via Ca2+/CaM
Ca2+ influx through the NMDAR leads to a reduction in PSD-95 spine content by capping of the very N-terminus of PSD-95 with Ca2+/CaM and thus prevents re-palmitoylation of PSD-95 (Zhang et al., 2014). Given that α-actinin also binds to the first 13 residues of PSD-95 we hypothesized that Ca2+ influx disrupts this interaction via Ca2+/CaM. As mentioned above, addition of Ca2+/CaM during IP of PSD-95 resulted in loss of α-actinin co-IP (Figure 3D, right lane). Most remarkable, treatment of forebrain slices with NMDA nearly abolished co-IP of α-actinin with PSD-95 (Figure 7). In parallel, co-IP of CaM with PSD-95 was strongly increased. Both of these effects were blocked by three structurally different CaM antagonists (Figure 7).
DISCUSSION
Collectively, our biochemical, cell biological, and electrophysiological data clearly demonstrate that K10 and K11 in the PSD-95 N-terminus are critical for α-actinin binding and postsynaptic targeting of PSD-95 and AMPARs. Two different point mutations on PSD-95 independently affect α-actinin binding and postsynaptic PSD-95 and AMPAR accumulation upon both, overexpression and its molecular replacement. Moreover, complementary analysis of two point mutations in α-actinin-1 that impaired PSD-95 binding showed analogous results with molecular α-actinin replacement experiments.
The postsynaptic content for the PSD-95 homologues PSD-93, SAP97, and SAP102 is not affected by α-actinin kd when PSD-95 is reduced by ~50% (Figure 2). Given that SAP97, SAP102, and four of the six known N-terminal splice variants of PSD-93 (Kruger et al., 2013; Parker et al., 2004) do not contain sequences that are homologous to the N-terminal 13 residues of PSD-95, those PSD-95 homologues must be postsynaptically anchored in a way different from PSD-95 and apparently independent of α-actinin. In fact, whereas co-IP of α-actinin with PSD-95 was prominent, no such co-IP was detectable for PSD-93, SAP97, or SAP102 (Figure 1D).
Notably, α-actinin kd did not overtly affect AMPARs nor NMDARs at surviving synapses as indicated by surface labeling of GluA1, GluA2, and GluN1 and mEPSC analysis. This finding provides further strong evidence for α-actinin’s role as a PSD-95 docking protein as it phenocopies the remarkable effect of PSD-95 kd that similarly results in a comparable (40%) decrease of functional synapses with surviving synapses maintaining their full AMPAR and NMDAR activity (Elias et al., 2006; Levy et al., 2015). Although a compensatory up-regulation of PSD-93, SAP97, or SAP102, which contributes to postsynaptic AMPAR and NMDAR targeting (Beique et al., 2006; Ehrlich et al., 2007; Elias et al., 2006; Levy et al., 2015; Schluter et al., 2006), is not detectable by our IF labeling following α-actinin1/2/4 kd (Figure 2F and G), it would have been conceivable that each of these proteins experiences a small 5–10% increase, which could mediate a good portion of compensation. Arguing against compensation of loss of PSD-95 upon α-actinin1/2/4 kd by PSD-93, SAP97, and SAP102, combined kd of PSD-95, PSD-93, and SAP102 results in synapses that maintain full strength even in SAP97 knock-out neurons although PSD-95/93/102 kd did not completely remove all protein (Levy et al., 2015).
In contrast to plain α-actinin KD (Figure 2A), molecular α-actinin replacement by KD combined with rescue by shRNA-resistant α-actinin-1 results in a clear reduction in postsynaptic GluA2 for E53K and E213K/D217K mutant α-actinin (Figure 6B,E,F). Plain KD reduces α-actinin by 46% in surviving spines, which is well matched by the 39% reduction in PSD-95 content in spines (Figure 2B). Upon plain KD the remaining α-actinin is all WT and competent to bind postsynaptic PSD-95, which in turn anchors postsynaptic AMPARs. Upon replacement the remaining endogenous α-actinin is diluted out by recombinant α-actinin. Accordingly, in addition to the reduction in endogenous α-actinin, which is PSD-95 binding-competent, ectopically expressed α-actinin that is PSD-95 binding-impaired dilutes out the remaining endogenous α-actinin. As a result the decrease in postsynaptic PSD-95 is larger (52% rather than 39%) than upon simple α-actinin KD and a reduction in postsynaptic GluA2 localization becomes obvious upon molecular replacement when it is not upon plain KD of α-actinin. Of note, we do see a 25% reduction in postsynaptic GluA1 content upon plain α-actinin KD although this reduction did not reach statistical significance (Figure 2B). In addition one needs to be taken into account that NMDARs and some other postsynaptic proteins have a much higher affinity for the PDZ domains of PSD-95 than TARPs (Lim et al., 2002; Zhu et al., 2016). Thus NMDARs and other proteins will tie up a substantial pool of PSD-95 before PSD-95 is available for AMPAR/TARP anchoring. This notion is reflected by the observation that KD of PSD-95, PSD-93, or SAP102 only affects postsynaptic AMPAR and not NMDAR responses (Elias et al., 2006; Schlüter et al., 2006) unless all three of these MAGUKs are knocked down or eliminated otherwise (Elias et al., 2008; Levy et al., 2015).”
Maintenance of the strength of surviving synapses after PSD-95 kd depended on L-type Ca2+ channels and CaMKK/CaMKIV signaling (Levy et al., 2015). Inhibition of this signaling cascade resulted in a near normal number of functionally active synapses but at much reduced strength. A similar mechanism that allows some synapses to survive with full strength and eliminates most of the others is likely at work in our α-actinin1/2/4 kd experiments. α-actinin-1/2/4 kd also reduces Cav1.2 in spines (Hall et al., 2013). However, this reduction was only 25% and thus 75 % of it is maintained.
How does α-actinin link PSD-95 to postsynaptic sites? F-actin determines spine size, which is correlated with PSD size (Harris and Stevens, 1989) and thereby AMPAR content (Takumi et al., 1999) and synaptic strength (Matsuzaki et al., 2001). In the most parsimonious model, F-actin content and thereby spine size translates into α-actinin/PSD-95/AMPAR docking to F-actin. In fact it appears that the bulk of spine α-actinin is docked to F-actin as dissociation of F-actin also reduces synaptic α-actinin (Allison et al., 2000) and the bulk of both, α-actinin and F-actin, is in the spine interior (Korobova and Svitkina, 2010; Racz and Weinberg, 2004). However, we are not convinced that F-actin is the main docking site for the α-actinin subpopulation that anchors PSD-95 because dissociation of F-actin reduces postsynaptic PSD-95 modestly but postsynaptic α-actinin strongly (Allison et al., 2000). In addition, AMPAR anchoring is only one of many α-actinin functions likely mediated by a rather small subpopulation of α-actinin. We rather favor a model whereby α-actinin links PSD-95 at postsynaptic sites to other critical organizers of the PSD. In fact, there is a clear zone of modest enrichment of α-actinin immediately cytosolic to the PSD (Racz and Weinberg, 2004) and α-actinin has been detected multiple times in PSD preparations (e.g., Peng et al., 2004). α-actinin also binds to other postsynaptic proteins including NMDARs, densin 180, and CaMKII (Merrill et al., 2007; Robison et al., 2005; Walikonis et al., 2001; Wyszynski et al., 1998), all of which could participate in anchoring PSD-95 via α-actinin under various conditions. In addition, α-actinin is linked to cadherin via α- and β-catenin (Nieset et al., 1997). Cadherin promotes postsynaptic strength (Tai et al., 2008) and growth of individual spines at the costs of their neighbors (Bian et al., 2015), which could explain how PSD-95 kd and α-actinin1/2/4 kd strongly reduces synapse number but not strength. A synapse with a small advantage with respect to activity might augment their strength by diverting postsynaptic building blocks including AMPARs from neighboring spines.
Ca2+ influx triggers postsynaptic displacement of PSD-95 by inducing Ca2+/CaM binding to the depalmitoylated N-terminus of PSD-95, thereby preventing re-palmitoylation (Zhang et al., 2014). We now show that NMDA-induced Ca2+ influx also disrupts the association of PSD-95 with α-actinin in a Ca2+/CaM-dependent manner (Figure 7). Consistent with a competitive mechanism, the N-terminal PSD-95 1-13 peptides K10E and K11E exhibit impaired binding for both α-actinin and Ca2+/CaM (Figure 3G,H). Decreasing the affinity for Ca2+/CaM by the K10E and K11E mutations could increase postsynaptic PSD-95 localization if basal levels of Ca2+ influx are strong enough to engage Ca2+/CaM binding. However, the clear decrease in postsynaptic PSD-95 and GluA2 content seen with PSD-95 K10E and K11E matches the decreases seen with the α-actinin E53K and ED213/217KK mutants. Thus, it seems that the impairment of Ca2+/CaM binding has no major effect in postsynaptic PSD-95 targeting under basal conditions. In fact, Ca2+/CaM might only be minimally engaged in the absence of neuronal stimulations. In any case, reducing Ca2+/CaM by the K10E and K11E mutations should augment postsynaptic PSD-95 localization when we see the opposite. Thus this reduction in postsynaptic PSD-95 content is due to impaired α-actinin and not Ca2+/CaM binding.
The above data show that upon Ca2+ influx, binding of Ca2+/CaM to the PSD-95 N-terminus antagonizes two key mechanisms of regulating postsynaptic PSD-95 anchoring, palmitoylation and association with α-actinin. The PSD-95 mutation E17R impairs binding of Ca2+/CaM to PSD-95 (Chowdhury et al., 2017). This mutation abrogates LTD (Schlüter et al., 2006) as well as homeostatic synaptic down scaling during increased network activity (Chowdhury et al., 2017). Accordingly, displacement of PSD-95 by Ca2+/CaM from α-actinin as described for the first time here is a central molecular event in homeostatic down scaling and LTD.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Johannes W. Hell (jwhell@ucdavis.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Dissociated hippocampal cultures were prepared from E18 embryonic rats of both sexes.
All animal procedures had been approved by the University of California at Davis Animal Care and Use Committee and followed NIH guidelines.
METHOD DETAILS
Antibodies and other Reagents
All primary antibodies used in the present study are listed in Star Methods Table S1. Secondary horseradish peroxidase (HRP)-conjugated goat anti-mouse and anti-rabbit antibodies were from BioRad. All secondary antibodies for immunofluorescence were from goat host, conjugated to Alexa-dyes (Life Technologies). Exclusively isotype-specific secondary antibodies were used against monoclonal mouse antibodies. Alexa555-tagged Phalloidin was from Life Technologies.
Synthetic peptides were purchased from ChinaPeptides (Shanghai, China). Calmidazolium and W7 were bought from Tocris, and TFP from MP Biomedicals. All reagents used for cloning were obtained from New England Biolabs (NEB). If not specifically stated, all other reagents were of standard quality and from the usual vendors.
DNA constructs
Maltose binding protein (MBP) tagged α-actinin domains CH1/2 (residues 1-262), CH1 (1-139), CH2 (140-262), SR1 (263-388), SR2 (389-504), SR3 (505-624), SR4 (625-739), EF (740-892), EF-ΔPDZ (740-888) were generated like previously described for MBP-tagged full length α-actinin-1 and -2 (Hall et al., 2013). In brief, required domains were PCR-amplified with oligonucleotide primers containing EcoRI and HindIII sites (all oligonucleotides used in this study are listed in Star Methods Table S4). The PCR-products were digested and ligated into pMalE-c2 (NEB) using EcoRI and HindIII. GST-tagged PSD-95 domains N-term (residues 1-71), NT-PDZ1/2 (1–248), PDZ2 (156–248), PDZ3 (302–402), SH3 (431–500), and GK (534–724) were previously described (Joiner et al., 2010; Seabold et al., 2003; Zhang et al., 2014). GST-tagged full length PSD-95 and SAP97 were kind gifts from C.C. Garner (Müller et al., 1996 and Wu et al., 2000). Full length Mus musculus PSD-93α cDNA was a kind gift from Morgan Sheng, and was cloned in frame into the EcoRI restriction site of pGEX-4T1. To correct for the missing N-terminal 133 amino acids in the SAP102-GST construct (Leonard et al., 1998), we subcloned a 2.1 kb BamHI fragment from the full length SAP102 pCMV (an expression construct obtained from C.C. Garner) into the 7.5 kb BamHI fragment of the truncated SAP102-GST construct. All GST tags are located at the N-terminus of the fusion proteins. Full length calmodulin (Zhang et al., 2014) consists of Xenopus calmodulin cDNA inserted in pET3a (no tag). The Xenopus calmodulin amino acid sequence is identical to that of vertebrate calmodulins (Chien and Dawid 1984).
Small hairpin RNAs targeting α-actinin isoforms 1 through 4 in pSilencer 1.0-Ub6 (Ambion) were previously described (Schnizler et al., 2009).
To replace dsRed with eGFP, the CMV-eGFP cassette from pEGFP (Clontech) was PCR-amplified with oligonucleotide primers containing KpnI sites and ligated into the respective KpnI-digested pSilencer vectors.
The previously described (Hall et al., 2013) shRNA-insensitive rescue α-actinin-1* was modified to remove superfluous residues from the C-terminus by PCR-amplification of the C-terminal part of the cDNA with a reverse primer containing a stop codon and an EcoRI restriction site. The PCR product was cloned back into the original template using EcoRI and SbfI resulting in a shRNA-insensitive α-actinin-1 cDNA featuring the original C-terminal sequence (ESDL).
RFP-tagged PSD-95 (PSD-95WT) was previously described (Zhang et al., 2014). Briefly, PSD-95 cDNA (gratefully received from David Bredt, UCSF) was first inserted into pEGFP using HindIII and EcoRI and subsequently shuttled into pTagRFP-N (Evrogen, Moscow, Russia) using NheI and KpnI restriction sites.
Mutated α-actinin and PSD-95 constructs were created with complementary mutagenic nucleotides for PCR amplification of the whole plasmid using Q5 high-fidelity DNA polymerase (NEB) and subsequent digestion with DpnI to remove non-mutated, methylated template DNA before transformation of the nicked PCR-product into NEB 5-alpha competent E. coli.
PSD-95 lacking the first 64 residues (PSD-95 Δ64) was created by PCR-amplification using a forward-priming oligonucleotide to introduce an NheI restriction site and new start codon. The PCR product was cloned back into the original template using NheI and EcoNI.
Lentivial transfer vector construct (FHUG+W) expressing shRNA against PSD-95 and shRNA-insensitive rescue PSD-95 (with eGFP fused to its C terminus; Schluter et al., 2006) was kindly provided by Drs. O. M. Schlüter and R. C. Malenka. To generate lentiviral construct expressing rescue PSD-95 containing K10E or K11E mutation, the N terminus of PSD-95 in FHUG+W vector was removed by digestion with XbaI (5′ external)/BsmBI (internal), and corresponding region containing mutation generated by digestion with Nhe/BsmBI from pTagRFP was ligated into FHUG+W construct.
Immunoprecipitation and Immunoblotting
All procedures were previously described in detail (Hall et al., 2006; Hell et al., 1993; Zhang et al., 2014). In brief, 1 g adult rat brain was homogenized in 10 ml ice-cold buffer A (in mM; 150 NaCl, 10 HEPES, 10 EDTA, 10 EGTA, pH 7.4) containing protease inhibitors (in μg/ml; 1 phenylmethanesulfonyl fluoride, 1 pepstatin A, 10 leupeptin, 20 aprotinin) and 1% Triton-X-100 with a glass-teflon homogenizer. The homogenate was cleared by ultracentrifugation for 30 min at 250,000 × g. A Bicinchoninic Acid (BCA) plate assay (Thermo Scientific) was used to measure protein concentration. 1 mg of dissolved protein was incubated with 1 μg antibody (JH62092, α-actinin (pan), PSD-93, PSD-95, SAP97, SAP102, or non-immune IgG control) and 20 μl protein G-Sepharose beads (GE Healthcare) for 4 h at 4 °C under tilting before washing 3 times at 4 °C in buffer A containing protease inhibitors as above and 1% Triton-X-100, extraction with SDS sample buffer at 95 °C for 5 min, separation by electrophoresis in 7. 5 % polyacrylamide gels, transfer to polyvinylidene fluoride (PVDF) membranes, probing with the indicated primary antibodies, and detection with secondary, horseradish peroxidase-coupled antibodies. Immunosignals were visualized using chemiluminescent peroxidase substrate (Merck Millipore) and photographic film (Denville). Multiple exposures ensured linear range of the signals which were quantified through densitometry using Photoshop CS3 software (Adobe; Hall et al., 2006).
In vitro pull-down assays
Expression and purification of GST fusion proteins (Hall et al., 2007; Leonard et al., 1998; Lim et al., 2002) and MBP fusion proteins (Hall et al., 2013; Merrill et al., 2007) from BL21(DE3) E. coli (NEB) was performed as cited. Briefly, transformed bacteria were incubated in 500 ml medium at 37 °C until reaching an OD600 of 1 and then induced for 2–4 h with 100 μM IPTG. Bacteria were harvested by centrifugation and lysed first by incubation with 0.1 mg/ml lysozyme in 50 ml TBS (150 mM NaCl, 15 mM Tris-Cl, pH 7.4) before sonication and, for GST fusion proteins, by addition of 1.5% Sarcosyl, which was neutralized by addition of one volume TBS and Triton X-100 to a final concentration of 4%. Insoluble material was removed by centrifugation. For purification, 100 ml GST fusion protein extracts were bound to 2 ml glutathione Sepharose 4B (Amersham) and 50 ml MBP fusion protein to 2 ml amylose resin (NEB), washed with 100 column volumes TBS containing 0.1% Triton X-100 (TBS-T), and eluted with 10–15 mM glutathione and 10 mM maltose, respectively. For pull down assays, one partner (0.1–1 ml lysate) was immobilized on the respective resin (30 μl), washed three times with TBS-T, incubation with purified binding partners for 90–120 min at 4 °C in TBS-T, and washed tree times with TBS-T before immunoblotting with anti-GST or anti-MBP antibodies. For re-probing, PVDF membranes were stripped with SDS and dithiothreitol (Hall et al., 2013; Leonard et al., 1998).
CMZ, W7, TFP
Fluorescence polarization (FP) assays
Synthetic peptides with N-terminal fluorescein label were purchased from ChinaPeptides. 100 nM peptide was added to multiple aliquots of 50 μl FP buffer (in mM; 50 HEPES, pH 7.4, 100 KCl, 1 MgCl2, 0.05 EGTA, 5 mM nitrilotriacetic acid) in 96-well black polystyrene plates (Corning) for titration with increasing amounts of purified recombinant proteins. Raw FP values were determined with a Synergy 2 plate reader (BioTek) with 485/20λ excitation and 528/20λ emission filters using the manufacturer’s proprietary Gen5 software. Data were analyzed in GraphPad Prism as described (Lim et al., 2002). For each titration the strongest signal was normalized to 100% after subtraction of baseline.
Peptide array
PEG-cellulose SPOT Synthesis Peptide Arrays were obtained from the Biopolymers & Proteomics Laboratory at MIT. Membranes with peptides were blocked with 10% milk powder in TBS (TBS-M), incubated with MBP fusion proteins (see above) in TBS-M, and washed with TBS. Bound fusion proteins were electrophoretically transferred onto PVDF membranes, which were probed with the anti-MBP antibody.
Primary embryonic hippocampal cultures
Dissociated hippocampal cells were prepared as follows (Chen et al., 2014; Zhang et al., 2014). Hippocampi from E18 embryonic rats of both sexes (Sprague-Dawley, Charles River Laboratories) were treated with 0.03% trypsin (Sigma) in Hank’s balanced salt solution (HBSS; Life Technologies) for 10 min at 37°C. After inactivation of trypsin with neuronal medium (NeuralQ basal medium (GlobalStem) supplemented with NS21 (Chen et al., 2008), 0.5 mM glutamine, 10 mM HEPES, 1 mM sodium pyruvate) plus 2.5% Horse serum (HS; Gemini), the tissue was washed twice with HBSS, and triturated with a fire-polished Pasteur pipette. After non-dissociated pieces of tissue settled, cells in the supernatant were collected by centrifugation (200 × g, 3.5 min), re-suspended in neuronal medium plus 2.5% HS, counted, and plated on coverslips (Warner Instruments) coated with 0.1% (w/v) poly-L-lysine at a cell density of 3×104 cm−2. After 4 h, the medium was replaced with serum-free neuronal medium. Cells were grown in a humidified environment with 5% CO2 at 37 °C. One third of the medium was changed after 5 days in vitro (DIV) and weekly thereafter. Transfection was performed at 4 DIV using Lipofectamine 2000 (Life Technologies) or calcium phosphate at 7 DIV.
Virus preparation and infection
For production of lentivirus, the transfer vector, the HIV-1 packaging vector expressing gag and pol, and the VSVG envelope glycoprotein vector were cotransfected into HEK293 cells using the calcium phosphate method. Culture media collected at 48 and 72 hours after transfection was centrifuged at 64,000 × g to concentrate virus. Dissociated hippocampal neurons were infected with lentivirus at 10 DIV and fixed after 8 days.
Immunofluorescence
18 DIV neurons were fixed in PBS containing 4 % paraformaldehyde (PFA) and 4% (w/v) sucrose at room temperature (RT) for 5 min, washed 3 × 5 min with PBS before permeablilization for 10 min with 0.25% Triton-X-100 in PBS at RT. Cells were subsequently washed 3 × 5 min in washing buffer (PBS, 0.01% Triton-X-100), and incubated with blocking solution (PBS containing 50 mM NH4Cl, 2% glycerol, 2% normal goat serum (Jackson ImmunoResearch), 5% fetal bovine serum (Atlanta Biologicals)) for 2 h at RT before primary antibodies were applied in blocking solution overnight at 4 °C. After washing 2 × 5 min and 1 × 15 min in washing buffer, cells were incubated in blocking solution for 45 min at RT before secondary antibodies were applied in blocking solution for 2 h at RT. After washing 2 × 5 min and 1 × 15 min with washing buffer, 1 × 5min with PBS, and 2 × 5 min with H2O, coverslips were mounted onto microscope slides with Permafluor (Thermo Fisher).
To stain for surface epitopes, the following additional steps were performed before permeabilization: Coverslips were incubated in blocking solution for 2 h at RT before primary antibodies against extracellular epitopes were applied in blocking solution for 36 h at 4 °C. After washing 2 × 5 min and 1 × 15 min in PBS, cells were subjected to a second fixation step with 4% PFA and 4% (w/v) sucrose in PBS for 5 min at RT. Cells were then washed 3 × 5 min with PBS before the staining was continued as described above.
For visualization of F-actin in STED microscopy (Figure S6G), coverslips were incubated in PBS containing Alexa488-conjugated phalloidin (Invitrogen, 1:20) for 30 min at RT before mounted onto microscope slides with Prolong Gold Antifade Reagent (Invitrogen).
Imaging
See Star Methods Table S6 for all combinations of fluorescent probes used in this study. Images for the Sholl analysis of α-actinin kd (Figure S2) and the initial screening of PSD-95 mutants (Figure S4) were taken using an IX-70 inverted microscope from Olympus (Shinjuku, Japan) with fluorescent optics and a cooled CCD camera (Hammamatsu) controlled by Metamorph software (Molecular Devices). Objective lenses were a 100x/1.35 NA and a 10x/0.25 NA for Sholl analysis. Within one experiment, the same exposure time was kept constant for all images from one channel.
For analysis of α-actinin kd (Figures 2, S2, and S3) and the expression of mutant PSD-95 and α-actinin1 (Figures 4, 5, 6, and S5), images were taken using a Zeiss LSM 700 confocal laser scanning microscope controlled by Zeiss Zen software using a 100x/1.3 NA objective lens and 512 × 512 pixel resolution. For all images within an experiment, laser output (below 10%) and detector gain (below 850) were kept at a constant level within the detector’s dynamic range. Pinhole diameter was set to 1 Airy unit for the 639 nm laser while pinhole diameters for the 405 nm, 488 nm, and 555 nm lasers were set to conserve optical slice thickness. The slice thickness was optimized by the manufacturer’s software (usually around 0.3 μm) to accommodate the selected Z-range. Images were acquired as Z-stacks (4–8 optical sections).
STED images of F-actin (Figure S6G) were obtained with a Leica TCS SP8 STED 3X microscope using a 100x HC PL APO CS2 oil immersion objective lens (1.4 NA) with 1248 × 1248 resolution. F-actin was visualized with Alexa488-conjugated phalloidin using 499 nm excitation and 592 nm STED depletion laser. DsRed fill from pSilencer vector was used to visualize spine shape using 554 nm excitation.
Image analysis
Characterization of α-actinin kd
Image quantification was performed by a sample blinded operator in ImageJ (Schneider et al., 2012) using custom macros. At least 6 images were used for every condition from a minimum of 3 independent experiments. For analysis, two segments of dendrite (typically 30 – 60 μm long) were cropped from every image and their length was measured using ImageJ’s segmented line tool. Within every experiment the threshold for each channel was determined as the average of the mean pixel intensities of all cropped segments plus two standard deviations above the mean. Using this threshold, a binary mask including all pixels with intensity above the threshold was created for every channel. Puncta density was calculated as the number of triple co-localization between GFP fill, synaptic marker (typically either synapsin or bassoon) and protein of interest, divided by the length of the segment. Intensity of the protein of interest was measured as the average of the mean intensities measured under all the areas of a synaptic mask prepared from the combined binary images of GFP fill and synaptic marker. The intensity was normalized by the respective channel’s threshold to combine results from several experiments.
Characterization of PSD-95 mutants
Images were quantified in a blinded manner using custom scripts for ImageJ. At least 6 images from a minimum of 3 different experiments (i.e., 3 different cultures) were taken for each condition. For each image, the total cell area was defined by a binary mask created from the GFP channel using the thresholding method described above. Synaptic areas encompass all pixels, where the total cell mask overlaps with a binary mask created from the synaptic marker channel (Shank, synapsin). The shaft area was produced by subtracting the synaptic marker mask from the total cell area. These binary masks were then used to measure the intensity of the RFP fluorescence in the respective areas. To estimate the relative RFP-PSD-95 expression levels of the individual PSD-95 mutants, mean RFP fluorescence was measured under the total area mask (total RFP) and normalized to total GFP.
The synaptic RFP intensity represents the mean RFP intensity under the synaptic mask normalized for expression levels by the mean GFP intensity under the total mask.
The synaptic enrichment factor is calculated as the quotient of mean synaptic RFP and GFP intensity divided by the quotient of mean shaft RFP and GFP intensities.
Normalization with volume filler was omitted from the equations above for calculation of spine intensity and spine enrichment factor in Figure 6 because no channel was available for imaging a volume filler in the PSD-95 replacement experiments. Density and intensity of GluA1EC, GluN1total, GluA2total, and α-actinin-1 staining was evaluated analogous to the analysis of α-actinin kd.
Sholl analysis
Epifluorescent images were analyzed in a blinded manner by drawing 6 circles with radii ranging from 20 μm to 120 μm around the soma using the ImageJ extension ConcentricCircles. The number of dendrites crossing each circle was counted manually using ImageJ’s Cell Counter plugin.
Spine density and morphology
This analysis was performed in a blinded manner. To analyze the spine density, two segments of dendrite (typically 30 – 60 μm long) were cropped from maximum intensity projections of confocal Z-stacks. The density was calculated by dividing the number of visible spines per segment counted using ImageJ’s Cell Counter plugin by the length as measured using ImageJ’s segmented line tool. To analyze the morphology of individual spines, segments of dendrites were cropped from single Z-slices. ImageJ was used to measure the length and width of every visible spine. The volume of an individual spine was calculated as follows: A polygon selection was drawn around the spine and the integrated intensity of the GFP signal was measured in that selection. Background fluorescence was assessed using a selection of the same size to measure integrated intensity in the background area next to the spine and subtracted. The result was normalized by the mean intensity of an area at the proximal dendrite of the same cell to correct for GFP expression levels across multiple cells and experiments. Additionally, each spine was assigned to one of three classes by the experimenter (mushroom: spine neck thinner than spine head; thin: no discernable neck or head and longer than wide; stubby: no discernable neck or head and wider than long).
Miniature excitatory postsynaptic currents (mEPSC)
Recording of mEPSCs was performed using slightly modified protocols published earlier (Chen et al., 2014; Matt et al., 2011). Coverslips with 11–14 DIV neurons were transferred into a submerged-type recording chamber. Bath solution was (in mM): 135 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, 0.01 tetrodotoxin (Tocris), 0.05 bicuculline (Sigma), pH7.4, 280–300 mosm/kg). Identification of transfected neurons and visual guidance of the patch pipette were performed using an Olympus BX50WI upright microscope and an Olympus LumPlanFL 40x water-immersion objective with fluorescence optics and IR-DIC contrast through a Hamamatsu C2400 CCD camera. Patch micropipettes (2.5 – 5 MΩ) were filled with intracellular solution (in mM; 145 CsCl, 0.1 CaCl2, 2 MgCl2, 1 EGTA, 5 HEPES, pH 7.4, 280–300 mosm/kg). The osmolalities of extracellular and intracellular solutions were exactly matched by adding NaCl or CsCl, respectively. Miniature EPSCs were recorded in whole-cell configuration using Clampex 10 to control an Axopatch 200B patch-clamp amplifier (Molecular Devices) through a Digidata 1322A digitizer. Cell capacitance and access resistance were monitored but not compensated throughout experiments. Cells with access resistances of more than 20 MΩ or changes in access resistance exceeding 20% were excluded from analysis. Cells were held at −70 mV and miniature events were sampled for up to 20 min at 2 kHz and filtered with a 1 kHz low-pass filter. Events were identified with Clampfit 10 using its built-in template-based event detection function. Statistics were performed using GraphPad Prism.
Acyl-Biotin Exchange (ABE)
Analysis of palmitoylation by ABE was as previously described (Zhang et al., 2014). In brief, HEK293T cells were transfected with WT and mutant RFP-tagged PSD-95, harvested in PBS (150 mM NaCl, 10 mM Na-phosphate, pH 7.2), and lysed in ABE buffer (PBS, 5 mM EDTA and protease inhibitors), which was supplemented with 1% Triton-X-100 and 25 mM N-ethylmaleimide (NEM) on ice to block free thiol groups on cysteine residues. Protein was extracted by chloroform-methanol (CM) precipitation: 4 volumes methanol, 1.5 volumes chloroform, and 3 volumes water were added to samples. Phases were separated by centrifugation; aqueous phase discarded. The pellet was washed with 3 volumes of methanol, the supernatant after centrifugation discarded, and the pellet dissolved at 37 °C in ABE2 buffer (ABE with 4% SDS and 25 mM NEM), diluted 5-fold with ABE containing 0.2% Triton-X-100 and incubated overnight at 4°C. Excess NEM was removed by CM precipitation and the protein pellet solubilized in ABE2 followed by five fold dilution with ABE containing 0.2% Triton-X-100. Acyl moieties and especially palmitoyl moieties linked to cysteine residues were cleaved with 0.7 M NH2OH. Newly created SH groups on previously palmitoylated cysteine residues were biotinylated in ABE with 1 mM EZ-link HPDP-biotin (Thermo Fisher) for 1 h at RT. Omission of NH2OH served as specificity control for the depalmitoylation and subsequent biotinylation reaction. Unreacted HPDP-biotin was removed by CM precipitation, and the protein pellets solubilized in ABE2 containing 2% SDS before ten-fold dilution with ABE (0.2% Triton-X-100). Biotinylated protein was pulled-down with NeutrAvidin-agarose beads (Thermo Fisher), separated by SDS-PAGE, and analyzed by immunoblotting using indicated antibodies as described above.
Docking calculation
The program HADDOCK (de Vries et al., 2010) was used to calculate a structural model of PSD-95 residues 1-19 binding to a previously published x-ray structure of the CH1/2 domain of α-actinin-1 (PDB ID: 2EYI. Borrego-Diaz et al., 2006). For HADDOCK docking, the PSD-95 fragment formed by residues 1-16 was assumed to have an α-helical structure as in Zhang et al. (2014). All residues were set as active residues and defined as fully flexible. For α-actinin-1, the residues 53, 56, 57, 58, 210, 213, 217, 223, 225, 226 were selected as active residues, as these negatively-charged amino acids are all surface-exposed in the F-actin-binding compact conformation of CH1/2 (Borrego-Diaz et al., 2006) and might interact with positively charged residues on the PSD-95 fragment. Docking calculations were performed on the HADDOCK online server (http://haddock.science.uu.nl/services/HADDOCK/haddock.php) using default parameters and following the standard Haddock protocol (de Vries et al., 2010; Wassenaar et al., 2012). By defining the ambiguous interaction restraints (AIRs) generated from the chosen active residues, 1000 structures were calculated during rigid body energy minimization, and the 200 best solutions based on the intermolecular energy were used for semi-flexible, simulated annealing, followed by an explicit water refinement. These calculations yielded a cluster of 37 structures with root mean squared deviation (RMSD) of 0.5 Å. The best structure shows the salt-bridge between PSD-95 K11and α-actinin E53, PSD-95 K10 and α-actinin E213 and D217. As the direct interactions between E53 and K11, E213/D217 and K10 were confirmed by mutagenesis, they were set as unambiguous restraints for a second round of docking calculations together with the AIR from the first round. Docking calculation with HADDOCK generated 200 structures in a single cluster with RMSD of 0.5 Å of which the one with the lowest energy was selected for the model.
QUANTIFICATION AND STASTICAL ANALYSIS
All bar graphs are presented as the means ± SEM except Fig S6C (means ± SD). The sample size and statistical methods of each experiment is provided in the relevant figure legend. Significance is shown as *p < 0.05, **p < 0.01, ***p < 0.001.
Supplementary Material
Highlights.
α-Actinin directly binds to the N-terminus of PSD-95
Knock-down of α-actinin phenocopies knock-down of PSD-95
Four different point mutations show that α-actinin anchors PSD-95
α-actinin postsynaptically anchors AMPARs via PSD-95
Acknowledgments
This work was supported by AHA 11POST7020009 (LM), Brain & Behavior Research Foundation NARSAD Young Investigator Grant 20748 (LM), T32 GM099608 (PBH), AHA 14PRE19900021 (PBH), NIH R01 EY012347 (JBA), R01 MH097887 (JWH), R01 NS078792 (JWH), and R01 AG055357 (JWH).
Footnotes
Author Contribution
LM, KK, AEH, DM, MCH, and JWH designed experiments, LM, KK, AEH, TP, ZAM, DKP, DC, ORB, PBH, ÇGS, YZ, and DM performed experiments. LM, KK, AEH, TP, ZAM, DKP, ZAM, PBH, YZ, DM, MCH, JBA, and JWH analyzed data. LM, KK, YZ, MCH, JBA, and JWH wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allison DW, Chervin AS, Gelfand VI, Craig AM. Postsynaptic scaffolds of excitatory and inhibitory synapses in hippocampal neurons: maintenance of core components independent of actin filaments and microtubules. J Neurosci. 2000;20:4545–4554. doi: 10.1523/JNEUROSCI.20-12-04545.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beique JC, Andrade R. PSD-95 regulates synaptic transmission and plasticity in rat cerebral cortex. J Physiol. 2003;546:859–867. doi: 10.1113/jphysiol.2002.031369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beique JC, Lin DT, Kang MG, Aizawa H, Takamiya K, Huganir RL. Synapse-specific regulation of AMPA receptor function by PSD-95. Proc Natl Acad Sci USA. 2006;103:19535–19540. doi: 10.1073/pnas.0608492103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian WJ, Miao WY, He SJ, Qiu Z, Yu X. Coordinated Spine Pruning and Maturation Mediated by Inter-Spine Competition for Cadherin/Catenin Complexes. Cell. 2015;162:808–822. doi: 10.1016/j.cell.2015.07.018. [DOI] [PubMed] [Google Scholar]
- Borrego-Diaz E, Kerff F, Lee SH, Ferron F, Li Y, Dominguez R. Crystal structure of the actin-binding domain of alpha-actinin 1: evaluating two competing actin-binding models. J Strc Biol. 2006;155:230–238. doi: 10.1016/j.jsb.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Chen CY, Matt L, Hell JW, Rogawski MA. Perampanel inhibition of AMPA receptor currents in cultured hippocampal neurons. PloS one. 2014;9:e108021. doi: 10.1371/journal.pone.0108021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- Chen Y, Stevens B, Chang J, Milbrandt J, Barres BA, Hell JW. NS21: re-defined and modified supplement B27 for neuronal cultures. J Neurosci Meth. 2008;171:239–247. doi: 10.1016/j.jneumeth.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenaux G, Matt L, Hill TC, Kaur I, Liu XB, Kirk LM, Speca DJ, McMahon SA, Zito K, Hell JW, Diaz E. Loss of SynDIG1 Reduces Excitatory Synapse Maturation But Not Formation In Vivo. eNeuro. 2016;3:0130–16. doi: 10.1523/ENEURO.0130-16.2016. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien YH, Dawid IB. Isolation and characterization of calmodulin genes from Xenopus laevis. Mol Cell Biol. 1984;4:507–513. doi: 10.1128/mcb.4.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury D, Turner M, Patriarchi T, Hergarden AC, Anderson D, Zhang Y, Sun J, Chen CY, Ames JB, Hell JW. Ca(2+)/calmodulin binding to PSD-95 mediates homeostatic synaptic scaling down. EMBO J. 2017 doi: 10.15252/embj.201695829. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RL, Huganir RL. Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science. 2010;330:1108–1112. doi: 10.1126/science.1195298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davare MA, Hell JW. Increased phosphorylation of the neuronal L-type Ca(2+) channel Ca(v)1.2 during aging. Proc Natl Acad Sci USA. 2003;100:16018–16023. doi: 10.1073/pnas.2236970100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Andrade GB, Kunzelman L, Merrill MM, Fuerst PG. Developmentally dynamic colocalization patterns of DSCAM with adhesion and synaptic proteins in the mouse retina. Mol Vis. 2014;20:1422–1433. [PMC free article] [PubMed] [Google Scholar]
- de Vries SJ, van Dijk M, Bonvin AM. The HADDOCK web server for data-driven biomolecular docking. Nat Protoc. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- Ebert DH, Greenberg ME. Activity-dependent neuronal signalling and autism spectrum disorder. Nature. 2013;493:327–337. doi: 10.1038/nature11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Klein M, Rumpel S, Malinow R. PSD-95 is required for activity-driven synapse stabilization. Proc Natl Acad Sci USA. 2007;104:4176–4181. doi: 10.1073/pnas.0609307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. J Neurosci. 2004;24:916–927. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseini A, Schnell E, Dakoji S, Sweeney N, Zhou Q, Prange O, Gauthier C, Aquilera A, Nicoll RA, Bredt DS. Synaptic strength regulated by palmitate cycling on PSD-95. Cell. 2002;108:849–863. doi: 10.1016/s0092-8674(02)00683-9. [DOI] [PubMed] [Google Scholar]
- El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA, Bredt DS. PSD-95 involvement in maturation of excitatory synapses. Science. 2000;290:1364–1368. [PubMed] [Google Scholar]
- Elias GM, Funke L, Stein V, Grant SG, Bredt DS, Nicoll RA. Synapse-specific and developmentally regulated targeting of AMPA receptors by a family of MAGUK scaffolding proteins. Neuron. 2006;52:307–320. doi: 10.1016/j.neuron.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Elias GM, Elias LA, Apostolides PF, Kriegstein AR, Nicoll RA. Differential trafficking of AMPA and NMDA receptors by SAP102 and PSD-95 underlies synapse development. Proc Natl Acad Sci USA. 2008;105:20953–20958. doi: 10.1073/pnas.0811025106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etherton MR, Tabuchi K, Sharma M, Ko J, Sudhof TC. An autism-associated point mutation in the neuroligin cytoplasmic tail selectively impairs AMPA receptor-mediated synaptic transmission in hippocampus. EMBO J. 2011;30:2908–2919. doi: 10.1038/emboj.2011.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferron L, Nieto-Rostro M, Cassidy JS, Dolphin AC. Fragile X mental retardation protein controls synaptic vesicle exocytosis by modulating N-type calcium channel density. Nat Commun. 2014;5:3628. doi: 10.1038/ncomms4628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley KS, Young PW. The non-muscle functions of actinins: an update. Biochem J. 2014;459:1–13. doi: 10.1042/BJ20131511. [DOI] [PubMed] [Google Scholar]
- Frank RA, Komiyama NH, Ryan TJ, Zhu F, O’Dell TJ, Grant SG. NMDA receptors are selectively partitioned into complexes and supercomplexes during synapse maturation. Nat Commun. 2016;7:11264. doi: 10.1038/ncomms11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikawa T, Petralia RS, Fitzgerald TS, Wang YX, Millis B, Morgado-Diaz JA, Kitamura K, Kachar B. Localization of kainate receptors in inner and outer hair cell synapses. Hear Res. 2014;314:20–32. doi: 10.1016/j.heares.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata Y, Dimitrov A, Boncompain G, Vielemeyer O, Perez F, Fukata M. Local palmitoylation cycles define activity-regulated postsynaptic subdomains. J Cell Biol. 2013;202:145–161. doi: 10.1083/jcb.201302071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DD, Dai S, Tseng PY, Malik Z, Nguyen M, Matt L, Schnizler K, Shephard A, Mohapatra DP, Tsuruta F, et al. Competition between a-actinin and Ca2+-calmodulin controls surface retention of the L-type Ca2+ channel Ca(V)1.2. Neuron. 2013;78:483–497. doi: 10.1016/j.neuron.2013.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DD, Davare MA, Shi M, Allen ML, Weisenhaus M, McKnight GS, Hell JW. Critical role of cAMP-dependent protein kinase anchoring to the L-type calcium channel Cav1.2 via A-kinase anchor protein 150 in neurons. Biochem. 2007;46:1635–1646. doi: 10.1021/bi062217x. [DOI] [PubMed] [Google Scholar]
- Hall DD, Feekes JA, Arachchige AS, Shi M, Hamid J, Chen L, Strack S, Zamponi GW, Horne MC, Hell JW. Binding of protein phosphatase 2A to the L-type calcium channel Cav1.2 next to Ser1928, its main PKA site, is critical for Ser1928 dephosphorylation. Biochem. 2006;45:3448–3459. doi: 10.1021/bi051593z. [DOI] [PubMed] [Google Scholar]
- Harris KM, Stevens JK. Dendritic spines of CA 1 pyramidal cells in the rat hippocampus: serial electron microscopy with reference to their biophysical characteristics. J Neurosci. 1989;9:2982–2997. doi: 10.1523/JNEUROSCI.09-08-02982.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol. 1993;123:949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges JL, Vilchez SM, Asmussen H, Whitmore LA, Horwitz AR. alpha-Actinin-2 mediates spine morphology and assembly of the post-synaptic density in hippocampal neurons. PloS one. 2014;9:e101770. doi: 10.1371/journal.pone.0101770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoe HS, Lee JY, Pak DT. Combinatorial morphogenesis of dendritic spines and filopodia by SPAR and alpha-actinin2. Biochem Biophys Res Commun. 2009;384:55–60. doi: 10.1016/j.bbrc.2009.04.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruska M, Henderson NT, Xia NL, Le Marchand SJ, Dalva MB. Anchoring and synaptic stability of PSD-95 is driven by ephrin-B3. Nature Neurosci. 2015;18:1594–1605. doi: 10.1038/nn.4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung AY, Futai K, Sala C, Valtschanoff JG, Ryu J, Woodworth MA, Kidd FL, Sung CC, Miyakawa T, Bear MF, et al. Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J Neurosci. 2008;28:1697–1708. doi: 10.1523/JNEUROSCI.3032-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joiner ML, Lise MF, Yuen EY, Kam AY, Zhang M, Hall DD, Malik ZA, Qian H, Chen Y, Ulrich JD, et al. Assembly of a beta2-adrenergic receptor--GluR1 signalling complex for localized cAMP signalling. EMBO J. 2010;29:482–495. doi: 10.1038/emboj.2009.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinowska M, Chavez AE, Lutzu S, Castillo PE, Bukauskas FF, Francesconi A. Actinin-4 Governs Dendritic Spine Dynamics and Promotes Their Remodeling by Metabotropic Glutamate Receptors. J Biol Chem. 2015;290:15909–15920. doi: 10.1074/jbc.M115.640136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Naisbitt S, Hsueh YP, Rao A, Rothschild A, Craig AM, Sheng M. GKAP, a novel synaptic protein that interacts with the guanylate kinase-like domain of the PSD-95/SAP90 family of channel clustering molecules. J Cell Biol. 1997;136:669–678. doi: 10.1083/jcb.136.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- Korobova F, Svitkina T. Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol Biol Cell. 2010;21:165–176. doi: 10.1091/mbc.E09-07-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger JM, Favaro PD, Liu M, Kitlinska A, Huang X, Raabe M, Akad DS, Liu Y, Urlaub H, Dong Y, et al. Differential roles of postsynaptic density-93 isoforms in regulating synaptic transmission. J Neurosci. 2013;33:15504–15517. doi: 10.1523/JNEUROSCI.0019-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AS, Davare MA, Horne MC, Garner CC, Hell JW. SAP97 is associated with the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor GluR1 subunit. J Biol Chem. 1998;273:19518–19524. doi: 10.1074/jbc.273.31.19518. [DOI] [PubMed] [Google Scholar]
- Levy JM, Chen X, Reese TS, Nicoll RA. Synaptic Consolidation Normalizes AMPAR Quantal Size following MAGUK Loss. Neuron. 2015;87:534–548. doi: 10.1016/j.neuron.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim IA, Hall DD, Hell JW. Selectivity and promiscuity of the first and second PDZ domains of PSD-95 and synapse-associated protein 102. J Biol Chem. 2002;277:21697–21711. doi: 10.1074/jbc.M112339200. [DOI] [PubMed] [Google Scholar]
- Liu S, Lau L, Wei J, Zhu D, Zou S, Sun HS, Fu Y, Liu F, Lu Y. Expression of Ca(2+)-permeable AMPA receptor channels primes cell death in transient forebrain ischemia. Neuron. 2004;43:43–55. doi: 10.1016/j.neuron.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Matsuzaki M, Ellis-Davies GC, Nemoto T, Miyashita Y, Iino M, Kasai H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nature Neurosci. 2001;4:1086–1092. doi: 10.1038/nn736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matt L, Michalakis S, Hofmann F, Hammelmann V, Ludwig A, Biel M, Kleppisch T. HCN2 channels in local inhibitory interneurons constrain LTP in the hippocampal direct perforant path. Cell Mol Life Sci. 2011;68:125–137. doi: 10.1007/s00018-010-0446-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxeiner S, Luo F, Tan A, Schmitz F, Sudhof TC. How to make a synaptic ribbon: RIBEYE deletion abolishes ribbons in retinal synapses and disrupts neurotransmitter release. EMBO J. 2016;35:1098–1114. doi: 10.15252/embj.201592701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara JO, Huang YZ, Leonard AS. Molecular signaling mechanisms underlying epileptogenesis. Sci STKE. 2006;2006:re12. doi: 10.1126/stke.3562006re12. [DOI] [PubMed] [Google Scholar]
- Merrill MA, Malik Z, Akyol Z, Bartos JA, Leonard AS, Hudmon A, Shea MA, Hell JW. Displacement of alpha-actinin from the NMDA receptor NR1 C0 domain By Ca2+/calmodulin promotes CaMKII binding. Biochem. 2007;46:8485–8497. doi: 10.1021/bi0623025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheva KD, Busse B, Wiler NC, O’Rourke N, Smith SJ. Single-synapse analysis of a diverse synapse population: proteomic imaging methods and markers. Neuron. 2010;68:639–653. doi: 10.1016/j.neuron.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG. NMDA receptors and memory encoding. Neuropharmacol. 2013;74:32–40. doi: 10.1016/j.neuropharm.2013.04.014. [DOI] [PubMed] [Google Scholar]
- Müller BM, Kistner U, Kindler S, Chung WJ, Kuhlendahl S, Fenster SD, Lau LF, Veh RW, Huganir RL, Gundelfinger ED, Garner CC. SAP102, a Novel Postsynaptic Protein That Interacts with NMDA Receptor Complexes In Vivo. Neuron. 1996;17:255–265. doi: 10.1016/s0896-6273(00)80157-9. [DOI] [PubMed] [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M. Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron. 1999;23:569–582. doi: 10.1016/s0896-6273(00)80809-0. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Engler JA, Sheng M. The dynamic turnover and functional roles of alpha-actinin in dendritic spines. Neuropharmacol. 2004;47:734–745. doi: 10.1016/j.neuropharm.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Nieset JE, Redfield AR, Jin F, Knudsen KA, Johnson KR, Wheelock MJ. Characterization of the interactions of alpha-catenin with alpha-actinin and beta-catenin/plakoglobin. J Cell Sci. 1997;110(Pt 8):1013–1022. doi: 10.1242/jcs.110.8.1013. [DOI] [PubMed] [Google Scholar]
- Noh KM, Yokota H, Mashiko T, Castillo PE, Zukin RS, Bennett MV. Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proc Natl Acad Sci USA. 2005;102:12230–12235. doi: 10.1073/pnas.0505408102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noritake J, Fukata Y, Iwanaga T, Hosomi N, Tsutsumi R, Matsuda N, Tani H, Iwanari H, Mochizuki Y, Kodama T, et al. Mobile DHHC palmitoylating enzyme mediates activity-sensitive synaptic targeting of PSD-95. J Cell Biol. 2009;186:147–160. doi: 10.1083/jcb.200903101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opazo P, Labrecque S, Tigaret CM, Frouin A, Wiseman PW, De Koninck P, Choquet D. CaMKII triggers the diffusional trapping of surface AMPARs through phosphorylation of stargazin. Neuron. 2010;67:239–252. doi: 10.1016/j.neuron.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Otey CA, Carpen O. Alpha-actinin revisited: a fresh look at an old player. Cell Motil Cytoskeleton. 2004;58:104–111. doi: 10.1002/cm.20007. [DOI] [PubMed] [Google Scholar]
- Parker MJ, Zhao S, Bredt DS, Sanes JR, Feng G. PSD93 regulates synaptic stability at neuronal cholinergic synapses. J Neurosci. 2004;24:378–388. doi: 10.1523/JNEUROSCI.3865-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Kim MJ, Cheng D, Duong DM, Gygi SP, Sheng M. Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J Biol Chem. 2004;279:21003–21011. doi: 10.1074/jbc.M400103200. [DOI] [PubMed] [Google Scholar]
- Racz B, Weinberg RJ. The subcellular organization of cortactin in hippocampus. J Neurosci. 2004;24:10310–10317. doi: 10.1523/JNEUROSCI.2080-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison AJ, Bass MA, Jiao Y, Macmillan LB, Carmody LC, Bartlett RK, Colbran RJ. Multivalent Interactions of Calcium/Calmodulin-dependent Protein Kinase II with the Postsynaptic Density Proteins NR2B, Densin-180, and {alpha}-Actinin-2. The Journal of biological chemistry. 2005;280:35329–35336. doi: 10.1074/jbc.M502191200. [DOI] [PubMed] [Google Scholar]
- Sans N, Petralia RS, Wang YX, Blahos J, 2nd, Hell JW, Wenthold RJ. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci. 2000;20:1260–1271. doi: 10.1523/JNEUROSCI.20-03-01260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter OM, Xu W, Malenka RC. Alternative N-terminal domains of PSD-95 and SAP97 govern activity-dependent regulation of synaptic AMPA receptor function. Neuron. 2006;51:99–111. doi: 10.1016/j.neuron.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature Meth. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell E, Sizemore M, Karimzadegan S, Chen L, Bredt DS, Nicoll RA. Direct interactions between PSD-95 and stargazin control synaptic AMPA receptor number. Proc Natl Acad Sci USA. 2002;99:13902–13907. doi: 10.1073/pnas.172511199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnizler MK, Schnizler K, Zha XM, Hall DD, Wemmie JA, Hell JW, Welsh MJ. The cytoskeletal protein alpha-actinin regulates acid-sensing ion channel 1a through a C-terminal interaction. J Biol Chem. 2009;284:2697–2705. doi: 10.1074/jbc.M805110200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seabold GK, Burette A, Lim IA, Weinberg RJ, Hell JW. Interaction of the tyrosine kinase Pyk2 with the N-methyl-D-aspartate receptor complex via the Src homology 3 domains of PSD-95 and SAP102. J Biol Chem. 2003;278:15040–15048. doi: 10.1074/jbc.M212825200. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim E. The postsynaptic organization of synapses. Cold Spring Harb Perspect Biol. 2011:3. doi: 10.1101/cshperspect.a005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SM, Zhang N, Hansen J, Gerges NZ, Pak DT, Sheng M, Lee SH. GKAP orchestrates activity-dependent postsynaptic protein remodeling and homeostatic scaling. Nature Neurosci. 2012;15:1655–1666. doi: 10.1038/nn.3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnen BL, Bowen AB, Forte JS, Hiester BG, Crosby KC, Gibson ES, Dell’Acqua ML, Kennedy MJ. Optogenetic Control of Synaptic Composition and Function. Neuron. 2017;93:646–660. e645. doi: 10.1016/j.neuron.2016.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjoblom B, Salmazo A, Djinovic-Carugo K. Alpha-actinin structure and regulation. Cell Mol Life Sci. 2008;65:2688–2701. doi: 10.1007/s00018-008-8080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skolnick P, Popik P, Trullas R. Glutamate-based antidepressants: 20 years on. Trends Pharmacol Sci. 2009;30:563–569. doi: 10.1016/j.tips.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Sugiyama Y, Kawabata I, Sobue K, Okabe S. Determination of absolute protein numbers in single synapses by a GFP-based calibration technique. Nature Meth. 2005;2:677–684. doi: 10.1038/nmeth783. [DOI] [PubMed] [Google Scholar]
- Tai CY, Kim SA, Schuman EM. Cadherins and synaptic plasticity. Curr Op Cell Biol. 2008;20:567–575. doi: 10.1016/j.ceb.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Takumi Y, Ramirez-Leon V, Laake P, Rinvik E, Ottersen OP. Different modes of expression of AMPA and NMDA receptors in hippocampal synapses. Nature Neurosci. 1999;2:618–624. doi: 10.1038/10172. [DOI] [PubMed] [Google Scholar]
- Tang AH, Chen H, Li TP, Metzbower SR, MacGillavry HD, Blanpied TA. A trans-synaptic nanocolumn aligns neurotransmitter release to receptors. Nature. 2016;536:210–214. doi: 10.1038/nature19058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topinka JR, Bredt DS. N-terminal palmitoylation of PSD-95 regulates association with cell membranes and interaction with K+ channel Kv1.4. Neuron. 1998;20:125–134. doi: 10.1016/s0896-6273(00)80440-7. [DOI] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, et al. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23:583–592. doi: 10.1016/s0896-6273(00)80810-7. [DOI] [PubMed] [Google Scholar]
- Valtschanoff JG, Burette A, Davare MA, Leonard AS, Hell JW, Weinberg RJ. SAP97 concentrates at the postsynaptic density in cerebral cortex. Eur J Neurosci. 2000;12:3605–3614. doi: 10.1046/j.1460-9568.2000.00256.x. [DOI] [PubMed] [Google Scholar]
- Walikonis RS, Jensen ON, Mann M, Provance DW, Jr, Mercer JA, Kennedy MB. Identification of proteins in the postsynaptic density fraction by mass spectrometry. J Neurosci. 2000;20:4069–4080. doi: 10.1523/JNEUROSCI.20-11-04069.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walikonis RS, Oguni A, Khorosheva EM, Jeng CJ, Asuncion FJ, Kennedy MB. Densin-180 forms a ternary complex with the (alpha)-subunit of Ca2+/calmodulin-dependent protein kinase II and (alpha)-actinin. J Neurosci. 2001;21:423–433. doi: 10.1523/JNEUROSCI.21-02-00423.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassenaar T, van Dijk M, Loureiro-Ferreira N, van der Schot G, de Vries S, Schmitz C, van der Zwan J, Boelens R, Giachetti A, Ferella L, et al. WeNMR: Structural Biology on the Grid. J Grid Computing. 2012;10:743–767. [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Wolf ME, Tseng KY. Calcium-permeable AMPA receptors in the VTA and nucleus accumbens after cocaine exposure: when, how, and why? Front Mol Neurosci. 2012;5:72. doi: 10.3389/fnmol.2012.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfrey KM, Sanderson JL, Dell’Acqua ML. The palmitoyl acyltransferase DHHC2 regulates recycling endosome exocytosis and synaptic potentiation through palmitoylation of AKAP79/150. J Neurosci. 2015;35:442–456. doi: 10.1523/JNEUROSCI.2243-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Reissner C, Kuhlendahl S, Coblentz B, Reuver S, Kindler S, Gundelfinger ED, Garner CC. Intramolecular interactions regulate SAP97 binding to GKAP. EMBO J. 2000;19:5740–5751. doi: 10.1093/emboj/19.21.5740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyszynski M, Kharazia V, Shanghvi R, Rao A, Beggs AH, Craig AM, Weinberg R, Sheng M. Differential regional expression and ultrastructural localization of alpha-actinin-2, a putative NMDA receptor-anchoring protein, in rat brain. J Neurosci. 1998;18:1383–1392. doi: 10.1523/JNEUROSCI.18-04-01383.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H, Winokur ST, Kuo WL, Altherr MR, Bredt DS. Actinin-associated LIM protein: identification of a domain interaction between PDZ and spectrin-like repeat motifs. J Cell Biol. 1997;139:507–515. doi: 10.1083/jcb.139.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi N, Fukata Y, Sekiya A, Murakami T, Kobayashi K, Fukata M. Identification of PSD-95 Depalmitoylating Enzymes. J Neurosci. 2016;36:6431–6444. doi: 10.1523/JNEUROSCI.0419-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Matt L, Patriarchi T, Malik ZA, Chowdhury D, Park DK, Renieri A, Ames JB, Hell JW. Capping of the N-terminus of PSD-95 by calmodulin triggers its postsynaptic release. EMBO J. 2014;33:1341–1353. doi: 10.1002/embj.201488126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wu S, Liang C, Lin Y, Zou Y, Li K, Lu B, Shu M, Huang Y, Zhu W, et al. Transcriptional upregulation of microtubule-associated protein 2 is involved in the protein kinase A-induced decrease in the invasiveness of glioma cells. Neuro Oncol. 2015;17:1578–1588. doi: 10.1093/neuonc/nov060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Shang Y, Zhang M. Mechanistic basis of MAGUK-organized complexes in synaptic development and signalling. Nature Rev Neurosci. 2016;17:209–223. doi: 10.1038/nrn.2016.18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.