

ABSTRACT
The apical junctional complex (AJC), which includes tight junctions (TJs) and adherens junctions (AJs), determines the epithelial polarity, cell-cell adhesion and permeability barrier. An intriguing characteristic of a TJ is the dynamic nature of its multiprotein complex. Occludin is the most mobile TJ protein, but its significance in TJ dynamics is poorly understood. On the basis of phosphorylation sites, we distinguished a sequence in the C-terminal domain of occludin as a regulatory motif (ORM). Deletion of ORM and expression of a deletion mutant of occludin in renal and intestinal epithelia reduced the mobility of occludin at the TJs. ORM deletion attenuated Ca2+ depletion, osmotic stress and hydrogen peroxide-induced disruption of TJs, AJs and the cytoskeleton. The double point mutations T403A/T404A, but not T403D/T404D, in occludin mimicked the effects of ORM deletion on occludin mobility and AJC disruption by Ca2+ depletion. Both Y398A/Y402A and Y398D/Y402D double point mutations partially blocked AJC disruption. Expression of a deletion mutant of occludin attenuated collective cell migration in the renal and intestinal epithelia. Overall, this study reveals the role of ORM and its phosphorylation in occludin mobility, AJC dynamics and epithelial cell migration.
KEY WORDS: Tight junction, Occludin, Cell migration, Phosphorylation, Adherens junction, Cytoskeleton
Summary: A conserved sequence in occludin determines the dynamic property of tight junctions and adherens junctions in the renal and intestinal epithelia, and regulates collective cell migration.
INTRODUCTION
The epithelial apical junctional complex (AJC) consists of two well-organized junctions, zonula occludens (also known as tight junctions, TJs) and zonula adherens (also known as adherens junctions, AJs) (Vogelmann and Nelson, 2005). TJs are localized to the apical end of the lateral membrane of polarized epithelial cells and the AJs are localized just beneath the TJs. The close proximity of TJs and AJs is indicative of crosstalk between these junctional complexes. Cell-cell adhesion, maintenance of cell polarity and development of epithelial barriers are important functions of the AJC during embryogenesis (Sheth et al., 2000) and postnatal life (Vogelmann et al., 2004). The AJC forms a docking site for signaling elements that regulate cell proliferation and differentiation (Matter and Balda, 1999). A dysfunctional AJC leads to developmental defects as well as a variety of diseases of the different organ systems (Bruewer et al., 2006; Jang, 2014; Schlüter and Margolis, 2012).
TJs modulate the regulation of the paracellular permeability barrier and prevent apical-to-basolateral diffusion of membrane proteins (Anderson and Van Itallie, 1995; Shin et al., 2006). Structural organization of TJs involves interactions between transmembrane proteins such as occludin, tricellulin, marvel D3, the claudin subfamily, cytoplasmic scaffold proteins such as zonula occludens 1, 2 and 3 (ZO-1, ZO-2 and ZO-3, respectively) and the actin cytoskeleton (Anderson and Van Itallie, 2009; Shen et al., 2011). The intracellular TJ plaque recruits protein kinases, such as PKC, MAPK, c-Src and c-Yes, and protein phosphatases, such as PP2A and PP1, that mediate the fine-tuning of signaling cascades involved in the regulation of barrier function (Dörfel and Huber, 2012; Rao, 2008). Recent observations have demonstrated that TJ components are highly dynamic, with occludin being the most mobile among them (Shen et al., 2008). The structure of AJs involves interactions between E-cadherin, catenins and the actin cytoskeleton (Vogelmann and Nelson, 2005).
Occludin was the first transmembrane protein of TJ to be discovered (Furuse et al., 1993); however, its precise function in TJ assembly and regulation is unclear. Information from occludin knockout mice ruled out its requirement in TJ assembly, but the complex phenotypes of these mice are not completely understood (Saitou et al., 2000). Evidence suggests that occludin is associated with many cellular functions such as adhesion, differentiation, cell migration, apoptosis and Ca2+ homeostasis (Beeman et al., 2012; Rachow et al., 2013; Van Itallie and Anderson, 1997). Occludin is a tetraspanin with two extracellular loops, one intracellular loop, a large cytoplasmic C-terminal domain and a short N-terminal domain. The coiled-coil region of the C-terminal domain interacts with the guanylate kinase domain of ZO-1 (Cummins, 2012). Our previous studies have shown that occludin undergoes tyrosine (Tyr) phosphorylation in Caco-2 cell monolayers during TJ disruption by hydrogen peroxide, osmotic stress or other insults (Elias et al., 2009; Kale et al., 2003; Rao et al., 1997, 2002; Samak et al., 2011). Tyr phosphorylation of occludin on Y398 and Y402 regulates its interaction with ZO-1 and ZO-3, respectively (Elias et al., 2009). We further showed that PKCη and PKCζ-mediated phosphorylation of occludin on T403 and T404 facilitated its assembly into epithelial TJs (Jain et al., 2011; Suzuki et al., 2009). Other studies have demonstrated that S408 phosphorylation in occludin regulates Claudin-2 (Cldn-2) localization at TJs (Raleigh et al., 2011), reduces association of occludin with ZO-1 and ZO-2, and delays assembly of TJs (Dörfel et al., 2013). These studies highlight the presence of a highly conserved sequence with a cluster of phosphorylation sites (Y398-S408) in the occludin C-terminal domain, and we refer to this region as the ‘occludin regulatory motif’ (ORM).
In this study, we investigated the potential role of ORM in TJ dynamics by using renal and intestinal epithelial models and multiple models of TJ assembly and disassembly. We also investigated the potential role of ORM in directional cell migration. This study provides key insights into the role of occludin in the regulation of TJ dynamics, the crosstalk between TJs and AJs, and epithelial cell migration.
RESULTS
Deletion of ORM enhances assembly of occludin into TJs in MDCK cell monolayers
First, occludin deficient MDCK cell line (OD-MDCK) was generated by transfection of MDCK cells with shRNA for canine occludin and dilution cloning. The ORM sequence in human occludin (OCLNWT) was deleted to generate deletion mutant (DM) occludin (OCLNDM) (Fig. 1A). The pEGFP vector containing the OCLNWT or OCLNDM gene or the empty vector (Vec) was transfected into OD-MDCK cells. Stable clones were isolated and characterized. Immunoblot analysis showed the expression of EGFP-tagged OCLNWT and OCLNDM and EGFP in the corresponding clones (Fig. 1B). The interaction of C-terminal sequence (358-504) with ZO-1 has been shown to be required for assembly of chicken occludin at the TJ (Furuse et al., 1994). To determine the interaction of OCLNDM with ZO-1, EGFP was immunoprecipitated and immunoblotted for ZO-1 or EGFP. Results show that ZO-1 co-immunoprecipitates with OCLNDM, at a level similar to that of OCLNWT (Fig. 1C), indicating that ORM deletion does not prevent the interaction of occludin with ZO-1. Live-cell fluorescence imaging showed that EGFP-OCLNDM localized to the intercellular junctions (Fig. 1D). Densitometric fluorescence analysis indicated that the junctional localization of EGFP-OCLNDM was significantly greater than that of EGFP-OCLNWT (Fig. 1E). Z-section imaging (Fig. 1F) and z-profiling (Fig. 1G) of GFP fluorescence in these monolayers indicated that EGFP-OCLNDM is localized predominantly to the TJs, with a slightly broader distribution compared with that of EGFP-OCLNWT.
Fig. 1.
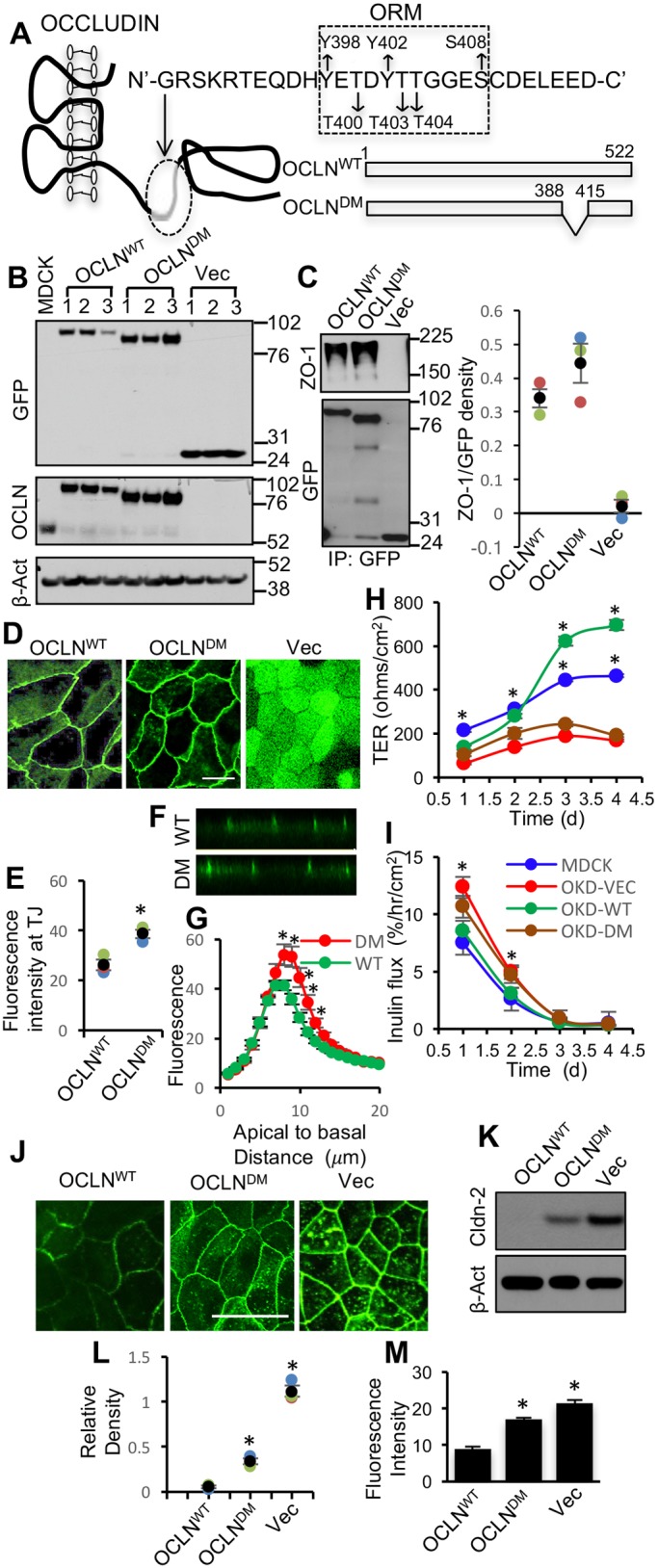
Deletion of ORM enhances occludin association with the TJ. (A) A part of the sequence (G388-D415) in the C-terminal domain of occludin including ORM (Y398-S408) was deleted from the wild-type human occludin (OCLNWT) to generate a deletion mutant of occludin (OCLNDM). (B) Stable clones of OD-MDCK cells expressing EGFP-OCLNWT, EGFP-OCLNDM and EGFP vector (Vec) were generated. Total protein extracts were immunoblotted for EGFP, occludin (OCLN) and β-actin (β-Act). (C) GFP was immunoprecipitated from the native extracts of OCLNWT, OCLNDM and Vec cells and immunoblotted for ZO-1. Density of ZO-1 was measured and normalized to the corresponding EGFP band density. Values presented in the graph are means±s.e.m. (n=3). (D,E) OCLNWT, OCLNDM and Vec cell monolayers were imaged live for EGFP (D). Junctional fluorescence was evaluated by densitometric analysis (E). Values, in arbitrary units of fluorescence intensity, are means±s.e.m. (n=3). Asterisks indicate that values are significantly (P<0.05) different from the OCLNWT value. (F,G) Z-section images of GFP fluorescence in OLCNWT (WT) and OCLNDM (DM) cell monolayers were captured (F), and Z-profiling of GFP fluorescence in these cell monolayers were analyzed (G). Values are means±s.e.m. (n=4). Asterisks indicate that values are significantly (P<0.05) different from corresponding values for WT cell monolayers. (H,I) Equal numbers of MDCK (blue), OCLNWT (OKD-WT; green), OCLNDM (OKD-DM; brown) and Vec (OKD-VEC; red) cells were seeded onto transwell inserts. TER (H) and FITC-inulin flux (I) were measured at various time points post seeding. Values presented in the graph are means±s.e.m. (n=3; three different clones for each group and the value for each clone is the average of six for 1 h, five for 2 h, four for 3 h and three for 4 h). The experiment was performed twice. Asterisks indicate MDCK and OCLNWT values that are significantly (P<0.05) different from corresponding values for Vec and OCLNDM groups. (K,L) Total protein extracts from OCLNWT, OCLNDM and Vec cells were immunoblotted for claudin-2 (Cldn-2) (K). Immunoblot bands were quantified by densitometric analysis (L). Values are means±s.e.m. (n=3). Asterisks indicate that values are significantly (P<0.05) different from the corresponding OCLNWT value. (J,M) Fixed cell monolayers were stained for Cldn-2 by immunofluorescence method (J). Fluorescence at the intercellular junctions was measured by densitometric analysis (M). Values are means±s.e.m. (n=3). Asterisks indicate that values are significantly (P<0.05) different from the corresponding OCLNWT value. Scale bars: 50 μm.
Barrier development was evaluated by measuring transepithelial electrical resistance (TER) and the unidirectional flux of fluoresceinyl isothiocyanate-inulin (FITC-inulin). On post-seeding days 3 and 4, TER in OCLNDM and Vec cell monolayers was significantly lower compared with those in MDCK and OCLNWT cell monolayers (Fig. 1H); however, the inulin flux was similar in all cell monolayers (Fig. 1I). To determine whether this was due to altered TJ pores, we examined the expression and distribution of Cldn-2, a major cationic pore-forming TJ protein. Immunoblot analysis and confocal microscopy showed that Cldn-2 levels (Fig. 1J,K) and junctional distributions (Fig. 1L,M) were greater in OCLNDM and Vec-cell monolayers compared with that in OCLNWT cell monolayers. These data indicate that the elevated expression and junctional localization of Cldn-2 might have contributed to low TER in OCLNDM and Vec cell monolayers.
ORM deletion augments association of occludin with the actin-rich cell fraction and diminishes its mobility in TJs
As fluorescence imaging showed a relatively high localization of EGFP-OCLNDM at the junctions, we examined the association of occludin with the actin-rich detergent-insoluble fraction of the cell. OCLNDM and Vec cell monolayers appear to have greater levels of F-actin at the perijunctional region compared with that in OCLNWT cell monolayers (Fig. 2A). Immunoblot analysis indicated that EGFP-OCLNWT and EGFP-OCLNDM were distributed in both Triton-insoluble (TI) and Triton-soluble (TS) fractions (Fig. 2B). Densitometric analysis showed that the TI fraction of EGFP-OCLNDM was significantly greater than that of EGFP-OCLNWT (Fig. 2C), suggesting that lack of ORM enhances the association of occludin with the actin cytoskeleton. The F-actin to G-actin ratio was calculated by measuring the amount of actin in TI and TS fractions by densitometric analysis of immunoblots. The TI/TS ratios for actin were 0.63±0.011 and 0.61±0.002 for OCLNWT and OCLNDM cell monolayers, suggesting that a difference in the rates of actin polymerization in these two monolayers is unlikely. To determine the rate of F-actin disassembly, we evaluated the effect of latrunculin A on the barrier function of monolayers prepared from different cell lines. Our data show that latrunculin induced a rapid disruption of barrier function in MDCK and OCLNWT cell monolayers, and the rates of disruption were significantly low in OCLNDM and Vec cell monolayers (Fig. S1A,B). Immunofluorescence staining for phospho-myosin light chain (pMLC) indicated that it was distributed mainly in the intracellular compartment in all cell lines, with no detectable distribution at the junctions (Fig. S1C,D).
Fig. 2.
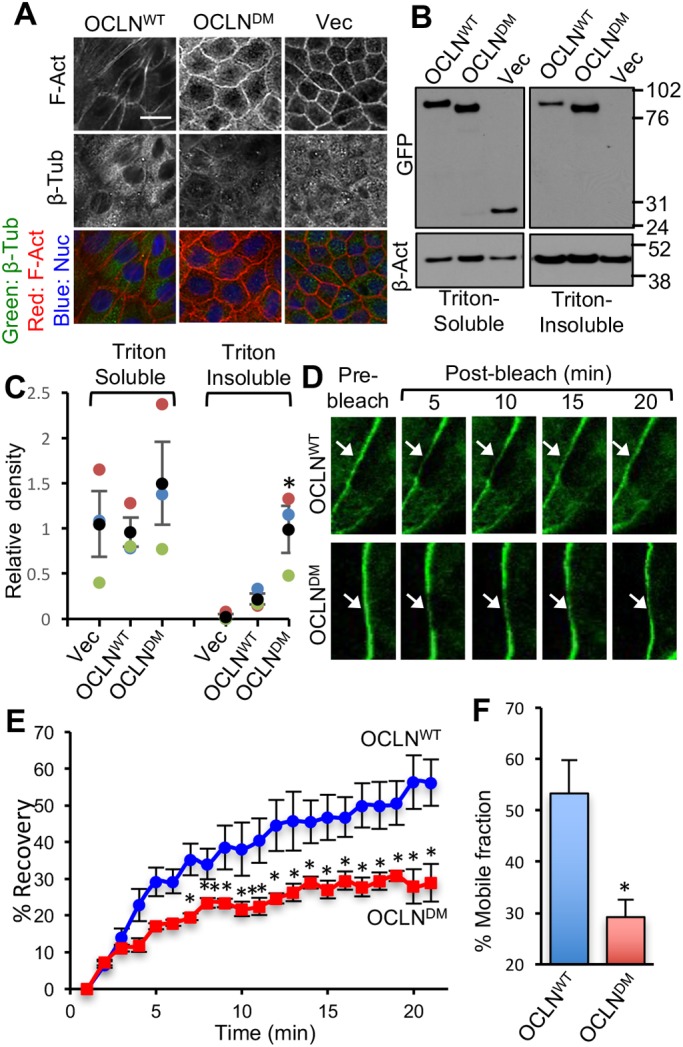
Deletion of ORM enhances the association of occludin with the actin-rich fraction of cells and decreases its mobility at TJs. (A) Two days after seeding, OCLNWT, OCLNDM and Vec cells were fixed and stained for F-actin (F-Act), β-tubulin (β-Tub) and nucleus (Nuc). Scale bar: 50 µm. (B,C) Triton-soluble and insoluble fractions prepared from OCLNWT, OCLNDM and Vec cells were immunoblotted for GFP and β-actin (β-Act) (B). GFP band densities were measured and normalized to corresponding β-actin band densities (C). Values are means±s.e.m. (n=3). Asterisks indicate the values that are significantly (P<0.05) different from corresponding value for OCLNWT cells. (D–F) FRAP analysis of EGFP was performed in OCLNWT and OCLNDM cell monolayers. Time-lapse images of several ROIs (regions of interest) at intercellular junctions were collected before and after photobleaching (D). Fluorescence intensity in the bleached area was measured (E) and percentage mobile fractions of OCLNWT and OCLNDM were calculated (F). Values in panels E and F are means±s.e.m. (n=8). Asterisks indicate the values that are significantly (P<0.05) different from the corresponding values for OCLNWT cells.
Fluorescence recovery after photobleaching (FRAP) analysis showed that EGFP-OCLNDM fluorescence was reduced compared with that of EGFP-OCLNWT (Fig. 2D,E). The mobile fraction of EGFP-OCLNDM (∼29.2%) was estimated to be significantly lower than that of EGFP-OCLNWT (∼53.3%) (Fig. 2F). A previous study showed that TJ proteins including occludin are highly dynamic and are in constant exchange with extra-junction pools (Shen et al., 2008). Our data suggest that ORM regulates the proportion of the mobile fraction of occludin in TJs.
ORM deletion attenuates Ca2+-depletion-mediated disruption of the AJC and barrier dysfunction in MDCK cell monolayers
Ca2+-switch assay was performed in MDCK, OCLNWT, OCLNDM and Vec cell monolayers. Live-cell fluorescence imaging indicated that incubation with a low-Ca2+ medium (LCM) for 16 h resulted in a redistribution of EGFP-OCLNWT from the intercellular junctions into the intracellular compartment. Interestingly, LCM failed to alter the junctional distribution of EGFP-OCLNDM (Fig. 3A). These results were confirmed in multiple clones of OCLNWT and OCLNDM cells (Fig. S2). Immunofluorescence staining of fixed cell monolayers showed that LCM failed to induce redistribution of EGFP-OCLN, ZO-1, E-cadherin and β-catenin in OCLNDM cell monolayers (Fig. 3B). Images for individual proteins are provided in the supplemental information (Fig. S3A,B). Reorganization of F-actin and β-tubulin was absent in LCM-treated OCLNDM cell monolayers (Fig. 3C). These observations suggest that ORM is required for Ca2+-depletion-mediated redistribution of occludin as well as other TJ and AJ proteins. LCM dramatically reduced TER (Fig. 3D) and increased inulin permeability (Fig. 3E) in MDCK and OCLNWT cell monolayers, but replacement of LCM with a normal-Ca2+ medium (NCM) gradually restored basal TER and inulin flux. These changes in TER and inulin flux indicate that barrier function is disrupted by Ca2+ depletion and restored by Ca2+ replacement in OCLNWT cell monolayers. However, LCM showed only minor effects on TER and inulin flux in OCLNDM and Vec cell monolayers, suggesting that ORM is required for Ca2+-depletion-mediated barrier dysfunction.
Fig. 3.

ORM deletion attenuates Ca2+-depletion-mediated disruption of the AJC and barrier dysfunction. (A–C) OCLNWT and OCLNDM cell monolayers were incubated with low-Ca2+ medium (LCM) or normal-Ca2+ medium (NCM) for 16 h. Live-cell images for EGFP fluorescence were captured before and after incubation (A). Fixed cell monolayers were stained for EGFP-occludin (GFP-OCLN), ZO-1, E-cadherin (E-Cad) and β-catenin (β-Cat) (B) or cytoskeletal proteins F-actin and β-tubulin (C). (D,E) MDCK (blue), OCLNWT (OKD-WT; green), OCLNDM (OKD-DM; brown) and Vec (OKD-VEC; red) cell monolayers on transwell inserts were incubated with LCM for 16 h followed by incubation with NCM for up to 3 h. TER (D) and FITC-inulin flux (E) were measured at various time points. Values are mean±s.e.m. (n=6). Asterisks indicate the values for MDCK and OCLNWT cell monolayers that are significantly (P<0.05) different from corresponding values for OCLNDM and Vec-cell monolayers. (F,G) MDCK (blue), Vec (red), OCLNWT (green) and OCLNDM (brown) cell monolayers on transwell inserts were incubated with (circles) or without (squares) 4 mM EGTA. TER (F) and FITC-inulin flux (G) were measured at various time points. Values are means±s.e.m. (n=6). Asterisks indicate the values for MDCK and OCLNDM monolayers that are significantly (P<0.05) different from corresponding values for Vec and OCLNWT monolayers. (H–J) Cell monolayers after EGTA treatment were co-stained for EGFP-occludin and ZO-1 (H), E-cadherin and β-catenin (I) or F-actin and β-tubulin (J). (K–M) Cell monolayers were incubated with LCM (L) or NCM (N) for 6 h. Phospho-threonine (p-Thr) was immunoprecipitated from denatured protein extracts. Immunoprecipitates and original extracts (Load) were immunoblotted for ZO-1 (K), E-cadherin (L), β-catenin and β-actin (M). Scale bars: 50 µm.
The effect of ORM deletion was also tested in an EGTA-mediated Ca2+ depletion model. In OCLNWT cell monolayers, EGTA rapidly reduced TER (Fig. 3F) and increased inulin flux (Fig. 3G), but its effect on TER and inulin flux was significantly low in OCLNDM cell monolayers. Confocal microscopy for EGFP-OCLN and ZO-1 showed that EGTA induces redistribution of these proteins from the junctions in MDCK and OCLNWT cell monolayers (Fig. 3H), but not in Vec and OCLNDM cell monolayers. Similarly, EGTA induced rapid redistribution of E-cadherin and β-catenin from the junctions in MDCK and OCLNWT cell monolayers, but had only minimal effect in Vec and OCLNDM cell monolayers (Fig. 3I); individual images for occludin, ZO-1, E-cadherin and β-catenin are presented in the supplemental information (Fig. S3C–F). These data together confirm that ORM deletion attenuates Ca2+-depletion-induced TJ and AJ disruption. EGTA treatment also induced remodeling of the actin cytoskeleton in MDCK and OCLNWT cell monolayers (Fig. 3J), whereas EGTA had only minimal effect in Vec cell monolayers and failed to modulate F-actin organization in OCLNDM cell monolayers.
Our previous studies have shown that dephosphorylation of TJ and AJ proteins occurs during Ca2+-depletion-mediated disruption of TJ and AJ (Seth et al., 2007). Therefore, we conducted a study to determine whether Ca2+-depletion-mediated dephosphorylation of E-cadherin, β-catenin and ZO-1 on threonine (Thr) residues occurs in OCLNWT and OCLNDM cell monolayers. Immunoprecipitation of phospho-Thr (p-Thr) followed by immunoblot analysis showed that LCM-induced Ca2+ depletion reduced Thr phosphorylation of E-cadherin, β-catenin and ZO-1 in MDCK and OCLNWT cell monolayers (Fig. 3K–M), whereas Thr dephosphorylation of E-cadherin, β-catenin and ZO-1 was low in LCM-treated Vec cell monolayers and absent in OCLNDN cell monolayers.
Deletion of ORM attenuates osmotic stress and hydrogen peroxide-mediated disruption of the AJC
The effect of ORM deletion on barrier function was examined in other models of TJ disruption. Osmotic stress induced an increase in inulin permeability in OCLNWT cell monolayers (Fig. 4A). OCLNDM and Vec cell monolayers were significantly resistant to osmotic stress-induced inulin permeability. Consistent with this, osmotic stress caused a redistribution of occludin and ZO-1 from the junctions in OCLNWT cell monolayers, but less so in OCLNDM cell monolayers (Fig. 4B). Inulin permeability in control cell monolayers (Fig. S4A) and images for individual proteins (Fig. S4B,C) are presented in the supplemental information. Osmotic stress-induced redistribution of E-cadherin and β-catenin was also minimal in OCLNDM cell monolayers (Fig. S5). Live-cell imaging revealed that exposure to hydrogen peroxide caused redistribution of EGFP-OCLNWT from the junctions, but it had minimal effect on EGFP-OCLNDM (Fig. 4C). Hydrogen peroxide caused a time-dependent increase in inulin flux (Fig. 4D), and redistribution of occludin/ZO-1 (Fig. 4E) and E-cadherin/β-catenin (Fig. 4F) in OCLNWT cell monolayers, but these effects were significantly low in OCLNDM and Vec cell monolayers. Inulin permeability in control cell monolayers (Fig. S6A) and images for individual proteins (Fig. S6B,C) are presented in the supplemental information. These data indicate that ORM is required for an effective disruption of AJC by osmotic stress and hydrogen peroxide.
Fig. 4.

ORM deletion attenuates disruption of the AJC and barrier dysfunction caused by osmotic stress and hydrogen peroxide. (A) MDCK (blue), OCLNWT (green), OCLNDM (brown) and Vec (red) cell monolayers on transwell inserts were incubated in DMEM (Control; data in Fig. S4A) or DMEM containing 0.3 M mannitol to induce osmotic stress (OS). FITC-inulin flux (A) was measured at various time points. Values are means±s.e.m. (n=6). Asterisks indicate the values for OCLNWT cell monolayers that are significantly (P<0.05) different from corresponding values for OCLNDM and Vec cell monolayers. (B) Fixed cell monolayers were stained for EGFP-occludin (GFP-OCLN) and ZO-1. (C) OCLNWT and OCLNDM cell monolayers were treated with hydrogen peroxide (100 µM) and live-cell images for EGFP fluorescence were collected at various time points. (D) MDCK (blue), OCLNWT (green), OCLNDM (brown) and Vec (red) cell monolayers on transwell inserts were treated with hydrogen peroxide (100 µM) in DMEM. FITC-inulin flux was measured at various time points. Values are means±s.e.m. (n=6). Asterisks indicate the values for OCLNWT cell monolayers that are significantly (P<0.05) different from corresponding values for OCLNDM and/or Vec cell monolayers. Control values are in Fig. S6A. (E,F) Fixed cell monolayers at different time points were stained for EGFP-occludin and ZO-1 (E) or E-cadherin and β-catenin (F).
Phosphorylation of ORM on residues T403/T404 and Y398/Y402 determines the dynamic property of TJs
To determine the role of ORM phosphorylation in TJ dynamics, point mutants of full-length occludin were generated and expressed in OD-MDCK cells. Live-cell fluorescence imaging (Fig. 5A) showed that LCM caused redistribution of EGFP-OCLNT403/404D from the intercellular junctions, similar to that of EGFP-OCLNWT, whereas the distribution of EGFP-OCLNT403/404A was not altered, similar to that of EGFP-OCLNDM. However, both EGFP-OCLNY398/402D and EGFP-OCLNY398/402A mutants showed significant resistance to LCM-mediated redistribution from the junctions (Fig. 5A). Confocal imaging of fixed cell monolayers showed that LCM induced redistribution of both EGFP-OCLN and ZO-1 from the intercellular junctions in OCLNWT and OCLNT403/404D cell monolayers, but not in OCLNDM, OCLNT403/404A, OCLNY398/402D and OCLNY398/402A cell monolayers (Fig. 5B). Images for the distribution of individual proteins are provided in the supplemental information (Fig. S7). Similarly, LCM caused redistribution of E-cadherin and β-catenin from the intercellular junctions in OCLNWT and OCLNT403/404D cell monolayers, but not in OCLNDM, OCLNT403/404A, OCLNY398/402D and OCLNY398/402A cell monolayers (Fig. S8). LCM also decreased TER (Fig. 5C) and increased inulin permeability (Fig. 5D) in OCLNWT and OCLNT403/404D cell monolayers; these effects of LCM were significantly attenuated in OCLNDM, OCLNT403/404A, OCLNY398/402D and OCLNY398/402A cell monolayers. FRAP analysis (Fig. 5E) demonstrated that the percentage mobile fraction of EGFP-OCLNT403/404D at the junctions was similar to that of EGFP-OCLNWT, whereas mobile fractions of EGFP-OCLNDM, EGFP-OCLNT403/404A, EGFP-OCLNY398/402D and EGFP-OCLNY398/402A were relatively low (Fig. 5F). The FRAP half-life (FRAP t1/2) values for different cell lines were not significantly different from that for EGFP-OCLNWT cell monolayers, except that FRAP t1/2 was significantly lower for EGFP-OCLNT403/404D cell monolayers (Fig. 5G).
Fig. 5.

Phosphorylation of ORM on T403/T404 and Y398/402 determines the dynamic properties of TJs. (A,B) OCLNWT (WT), OCLNDM (DM), OCLNT403/404A (T403/404A), OCLNT403/404D (T403/404D), OCLNY398/402A (Y398/402A) and OCLNY398/402D (Y398/402D) cell monolayers were incubated with low-Ca2+ medium (LCM) or normal-Ca2+ medium (NCM) for 1–24 h. Live-cell images for EGFP fluorescence were collected before and after incubation (A). Cell monolayers fixed at 1 h and 16 h were stained for EGFP-Occludin and ZO-1 (B). Scale bars: 50 µm. (C,D) OCLNWT, OCLNDM, OCLNT403/404A, OCLNT403/404D, OCLNY398/402A and OCLNY398/402D cell monolayers on transwell inserts were incubated with LCM for 16 h and TER (C) and FITC-inulin flux (D) were measured. Values are means± s.e.m. (n=6). Asterisks indicate the values that are significantly (P<0.05) different from corresponding values for OCLNWT cell monolayers. (E–G) FRAP analysis of EGFP was performed in OCLNWT (WT), OCLNDM (DM), OCLNT403/404A (T2A), OCLNT403/404D (T2D), OCLNY398/402A (Y2A) and OCLNY398/402D (Y2D) cell monolayers. Time-lapse images were collected before and after photobleaching of several ROIs at intercellular junctions. Fluorescence intensity was measured (E) and % mobile fraction (F) and t1/2 values (G) were calculated. Values are means±s.e.m. (n=4). Asterisks indicate the values that are significantly (P<0.05) different from the value for OCLNWT cell monolayers.
The effect of Y398A/Y402A and Y398D/Y402D mutations on LCM-mediated TJ disruption indicated a potential role of tyrosine kinases and protein Tyr phosphorylation of proteins during Ca2+-depletion-induced TJ disruption. To determine the role of Tyr phosphorylation in Ca2+-depletion-mediated TJ disruption, TER and inulin flux were measured in MDCK cell monolayers treated with genistein, a tyrosine kinase inhibitor, prior to LCM. Decrease in TER (Fig. 6A) and increase in inulin flux (Fig. 6B) in cells cultured in LCM was significantly blocked by genistein. Confocal imaging showed that genistein also blocked LCM-induced redistribution of occludin and ZO-1 (Fig. 6C–E) from the junctions into the intracellular compartment.
Fig. 6.
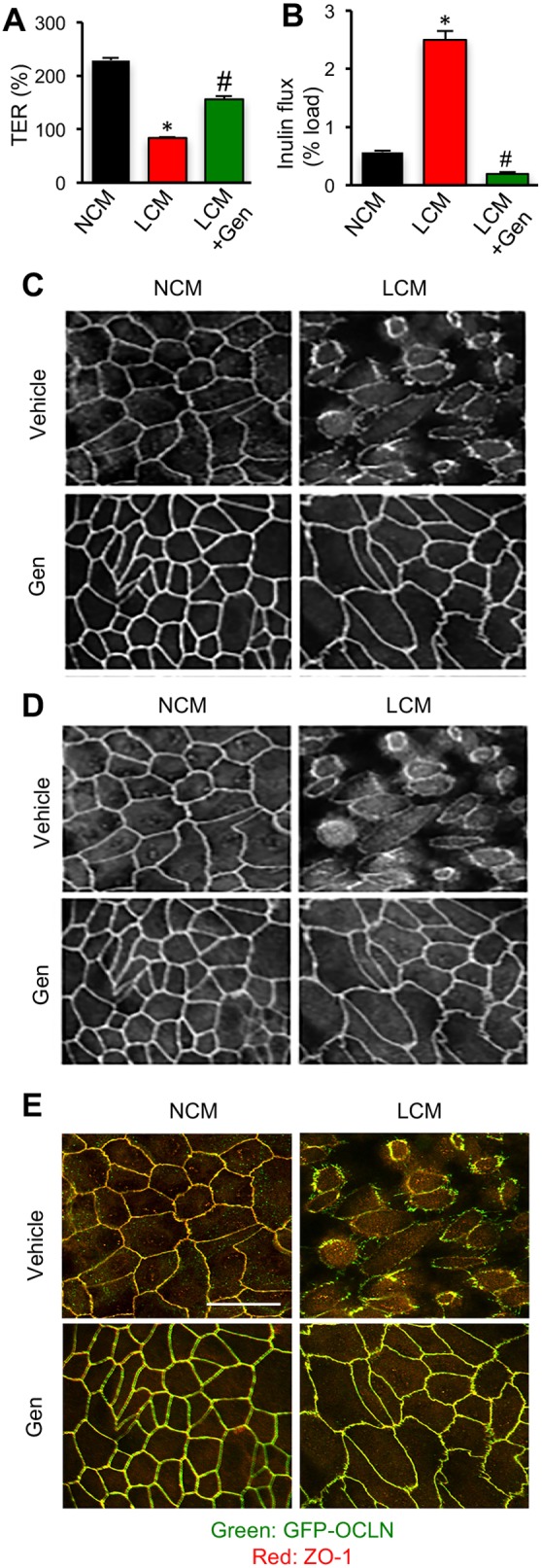
Inhibition of tyrosine kinase activity blocks Ca2+-depletion-mediated disruption of TJs and barrier function. (A,B) MDCK cell monolayers on transwell inserts were treated with 100 µM genistein (Gen) 30 min prior to LCM treatment. TER (A) and FITC-inulin flux (B) were measured after 1 h. Values are means±s.e.m. (n=6). Asterisks indicate the values that are significantly (P<0.05) different from corresponding value for OCLNWT cell monolayers and hash signs indicate the values that are significantly (P<0.05) different from that of OCLNDM cell monolayers. (C,D) Cell monolayers were co-stained for GFP-occludin (C) and ZO-1 (D). Merged images are presented in panel E. Scale bar: 50 µm.
Deletion of ORM reduces the mobile fraction of occludin and attenuates TJ dynamics in the intestinal epithelium
To investigate whether the phenotype rendered by ORM deletion is a general epithelial property, we evaluated the role of ORM in TJ dynamics in the intestinal epithelium. The OCLNWT or OCLNDM gene or Vec was expressed in the rat intestinal epithelial cell line, IEC-6. Immunoblot analysis showed a comparable expression of EGFP-OCLNWT and EGFP-OCLNDM, but there was no endogenous occludin detected in these cells (Fig. 7A). Live-cell imaging revealed that deletion of ORM did not prevent occludin localization at the junctions, confirming that ORM is not required for occludin assembly into TJs (Fig. 7B); however, junctional distribution of EGFP-OCLNWT was much greater than that of EGFP-OCLNDM. Densitometric analysis of fluorescence at the junctions showed a higher fluorescence intensity in EGFP-OCLNDM cell monolayers (41.57±5.69, n=3, each an average of three regions; arbitrary units) than that in EGFP-OCLNWT cell monolayers (31.20±2.02). FRAP analysis indicated that the fluorescence (Fig. 7C,D) and mobile fraction of EGFP-OCLNDM were significantly lower than those of EGFP-OCLNWT (Fig. 7E). Barrier function analysis showed the LCM-induced drop in TER (Fig. 7F) and rise in inulin-permeability (Fig. 7G) in OCLNWT cell monolayers, but the LCM effect was minimal in OCLNDM cell monolayers. Consistently, immunofluorescence microscopy showed that LCM induced redistribution of occludin and ZO-1 (Fig. 7H) as well as E-cadherin and β-catenin (Fig. 7I) from the junctions in OCLNWT-IEC-6 cell monolayers, but redistribution was minimal in EGFP-OCLNDM-IEC-6 cell monolayers, demonstrating that ORM is required for Ca2+-depletion-mediated disruption of TJs in the intestinal epithelium.
Fig. 7.

Deletion of ORM attenuates occludin mobility and Ca2+-depletion-mediated disruption of the AJC in the intestinal epithelium. (A,B) Total protein extracts from IEC-6 cells expressing EGFP-OCLNWT, EGFP-OCLNDM and EGFP vector (Vec) were immunoblotted for EGFP, occludin and β-actin (β-Act) (A). OCLNWT, OCLNDM and Vec cell monolayers were imaged live for EGFP (B). (C–E) FRAP analysis of EGFP was performed in OCLNWT (WT) and OCLNDM (DM) cell monolayers. Time-lapse images of several ROIs at intercellular junctions were collected before and after photobleaching (C). Fluorescence intensity in the bleached area was measured (D) and percentage mobile fractions of OCLNWT and OCLNDM were calculated (E). Values are means±s.e.m. (n=8). Asterisks indicate values that are significantly (P<0.05) different from the value for OCLNWT cells. (F,G) OCLNWT and OCLNDM cell monolayers on transwell inserts were incubated in LCM, and TER (F) and FITC-inulin flux (G) were measured at various time points. Values presented in the graph are means±s.e.m. (n=6). Asterisks indicate the values that are significantly (P<0.05) different from corresponding values for the OCLNWT group. (H,I) Fixed cell monolayers were stained for TJ proteins EGFP-occludin and ZO-1 (H) or AJ proteins E-cadherin and β-catenin (I). (J-L) Intestinal loops (∼2 cm long) from the mid small intestine of wild-type (WT) and occludin-deficient (OCLN−/−) mice were prepared and filled with saline containing FITC-inulin with or without 1 mM EGTA. Loss of inulin from the loops was measured after 30 min incubation in DMEM (J). Values are means±s.e.m. (n=3). The asterisk indicates the value for the OCLN−/− group that is significantly (P<0.05) different from the value for the WT group. Cryosections of the loops were immunostained for ZO-1, F-actin and nucleus (K) or E-cadherin and β-catenin (L). Scale bars: 50 µm.
To confirm the role of occludin in TJ dynamics in the intestinal tissue, we applied an ex vivo model of the intestinal epithelium by using the intestinal loops prepared from (wild-type) WT and occludin-deficient (OCLN−/−) mice and evaluated the effect of EGTA-mediated Ca2+ depletion. Mucosal barrier function in the intestinal loops was evaluated by measuring the uptake of FITC-inulin from the lumen. Inulin uptake from the lumen of OCLN−/− mouse intestine was significantly lower than that from WT mouse intestine (Fig. 7J). Confocal microscopy showed that EGTA induced redistribution of ZO-1 (Fig. 7K) and E-cadherin/β-catenin (Fig. 7L) from the junctions in WT mouse intestines. EGTA caused only a minimal effect on the junctional distributions of ZO-1, E-cadherin and β-catenin in OCLN−/− mouse intestines. These data suggest that lack of occludin confers resistance to AJC disruption in the intestinal tissue by depletion of Ca2+.
Deletion of ORM impairs collective cell migration in MDCK and IEC-6 cell monolayers
To determine the functional consequence of altered TJ dynamics caused by lack of ORM, we investigated the role of ORM in cell migration using OD-MDCK and IEC-6 cells that express EGFP-OCLNWT or EGFP-OCLNDM. Rates of cell migration following scrape wounding were significantly lower in Vec and EGFP-OCLNDM MDCK cell monolayers than in EGFP-OCLNWT cell monolayers (Fig. 8A,B). Similarly, Vec and EGFP-OCLNDM-IEC-6 cell monolayers showed lower rates of cell migration following scratch wounding than EGFP-OCLNWT-IEC-6 cell monolayers (Fig. 8C,D). Taken together, these data indicate that the absence of ORM significantly attenuates collective cell migration in both renal and intestinal epithelia. To determine whether lack of ORM affects single-cell migration, we evaluated transmigration of different lines of MDCK and IEC-6 cells. Transmigration of OD-MDCK cells expressing Vec or OCLNDM was significantly greater than migration of MDCK cells and OD-MDCK cells expressing OCLNWT (Fig. 8E). Similarly, migration of IEC-6 cells expressing Vec or OCLNDM was significantly greater than that of IEC-6 cells expressing OCLNWT (Fig. 8F).
Fig. 8.

Absence of ORM impairs directional cell migration in renal and intestinal epithelia. (A,B) OD-MDCK cells expressing EGFP-OCLNWT (WT), EGFP-OCLNDM (DM) and EGFP vector (Vec) were grown to confluence, and cell migration assay was performed by scrape wounding. Phase-contrast images were captured at various time points (A); the purple lines indicate the origin of migration. Area of migration was measured using ImageJ and presented in arbitrary units (B). Values are means±s.e.m. (n=5; each value is an average of five images from the same monolayer). Asterisks indicate the values that are significantly (P<0.05) different from corresponding values for Vec cells, and the hash signs indicate the values that are different from corresponding values for EGFP-OCLNWT cells. (C,D) IEC-6 cells expressing EGFP-OCLNWT (WT), EGFP-OCLNDM (DM) and EGFP vector (Vec) were grown to confluence, and cell migration assay was performed by scratch wounding. Phase-contrast images were captured at 12 h or 24 h (C). The wound area was measured using ImageJ and presented in arbitrary units (D). Values are means±s.e.m. (n=5; each value is an average of five images from the same monolayer). Asterisks indicate the values that are significantly (P<0.05) different from corresponding values for Vec cells, and the hash signs indicate the values that are different from corresponding values for EGFP-OCLNWT cells. (E,F) OD-MDCK cells (E) and IEC-6 cells (F) expressing EGFP vector (Vec), EGFP-OCLNWT (WT) or EGFP-OCLNDM (DM) (105 cells /well), and MDCK cells (E) were seeded at low density on to transwells. Cells that migrated to the bottom surface of transwells were counted (E). Similar to MDCK cells, IEC-6 cells transfected with EGFP vector (Vec), EGFP-OCLNWT (WT) or EGFP-OCLNDM (DM) were seeded on to transwells to measure transmigration (F). Values are means±s.e.m., and the values within the bars are the number of samples per group. Asterisks indicate significant differences (P<0.05) between the groups.
DISCUSSION
Besides forming a seal that maintains tissue compartmentation, TJs serve to regulate the paracellular transport across the epithelium under various physiological and pathophysiological conditions. The TJ protein complex is dynamic in nature (Shen, 2012), although the precise function of this property is poorly understood. The importance of phosphorylation in the assembly/disassembly of TJs (Rao, 2009) and the identification of phosphorylation sites in the C-terminal domain of occludin (Elias et al., 2009; Kale et al., 2003; Suzuki et al., 2009) led to the recognition of a highly conserved phosphorylation hotspot in occludin – the ORM. We hypothesized that ORM plays a regulatory role in TJ dynamics. Although deletion of occludin prevents neither in vitro nor in vivo TJ assembly (Saitou et al., 1998, 2000), the results of our current study provide evidence for a role of occludin and ORM in the regulation of the dynamic property of TJs and AJs.
Interaction with ZO-1 is crucial for its assembly into the TJ. Our results indicate that ORM is not required for ZO-1 binding and, therefore, ORM deletion does not prevent TJ assembly or barrier function. On the contrary, assembly of OCLNDM at the junctions is significantly greater than that of OCLNWT. On days 3–4 after seeding, OCLNDM and Vec cell monolayers maintained low TER compared with OCLNWT and MDCK cell monolayers, but the inulin permeability in OCLNDM and Vec cell monolayers was as low as that in OCLNWT and MDCK monolayers. This raised the question whether low resistance on days 3–4 after seeding is caused by higher expression of pore-forming claudins. A previous study showed that occludin regulates the localization of Cldn-2, a cation-selective pore-forming claudin, in Caco-2 cell monolayers through a mechanism that depends on phosphorylation of S408 (Raleigh et al., 2011). The present study shows that the levels and junctional distribution of Cldn-2 were significantly higher in Vec and OCLNDM cell monolayers. Occludin and ORM may indirectly regulate the expression and distribution of Cldn-2. However, on post seeding days 1–2, OCLNDM and Vec cell monolayers maintained low TER and high inulin permeability compared with those in OCLNWT and MDCK cell monolayers. Therefore, in addition to the difference in Cldn-2 levels, the TJ barrier properties or TJ assembly kinetics may also be altered in OCLNDM and Vec cell monolayers. To draw a meaningful conclusion regarding the interaction between occludin and Cldn-2, detailed future studies in cell lines and mouse tissues are necessary. A previous study has shown that TER of OD-MDCK cells is significantly lower than that in control cell monolayers during post-seeding assembly or Ca2+-mediated reassembly of TJs (Yu et al., 2005). This study has also shown that the FITC-dextran permeabilities of 6-day-old MDCK and OD-MDCK cell monolayers were not different, although the permeability characteristics were different for small molecular weight solutes.
The cytoplasmic TJ plaque is closely associated with the perijunctional actomyosin ring through its interaction with ZO-1, and, therefore, indirectly with occludin; this interaction is essential for the assembly and maintenance of TJs (Fanning et al., 1998). Our data show that lack of ORM enhances localization of occludin in the actin-rich detergent-insoluble fraction of the cell, suggesting an enhanced interaction of occludin with the actin cytoskeleton in the absence of ORM. However, it should be noted that the detergent-insoluble fraction may also contain other components, such as lipid rafts. FRAP analysis demonstrated that TJ proteins are highly dynamic at steady state and each TJ protein has a distinct dynamic behavior, with occludin having the highest mobile fraction (Shen et al., 2008). Our data show that ORM deletion reduces the mobile fraction of occludin, indicating that it is more static at the intercellular junctions in the absence of ORM. We speculate that the increased association of OCLNDM with the actin cytoskeleton determines the reduced mobile fraction or vice versa.
Many previous studies have associated hyperphosphorylated occludin with TJs, mostly assessed by the association of hyperphosphorylated occludin with the detergent-insoluble fraction. The results of the present study show that non-phosphorylated occludin also associates with the detergent-insoluble fraction and TJs. Although hyperphosphorylated occludin has been shown to be associated with TJs and the detergent-insoluble fraction in several studies, it is unclear whether occludin is phosphorylated before or after its assembly into TJs. A previous study has indicated that lack of occludin phosphorylation delays, but does not prevent, assembly of occludin into TJs (Suzuki et al., 2009). Our present study demonstrates that phosphorylation of occludin is not a prerequisite for its assembly into TJs, and that absence of phosphorylation confers resistance to disassembly of occludin from TJs. Therefore, irrespective of whether the phosphorylation occurs prior to or after assembly, it is required for optimal disassembly of occludin from the TJ. Interestingly, our study also shows that non-phosphorylated occludin confers resistance to disassembly of an entire TJ. Therefore, occludin may not be needed for TJ assembly but, once it is assembled into a TJ, it regulates the TJ dynamics by a phosphorylation-dependent mechanism.
The Ca2+-switch assay is a widely used technique to study the de novo assembly of TJs. We used this method to investigate the role of ORM in TJ disruption and assembly. Live cell imaging showed that incubation with LCM induced redistribution of EGFP-OCLNWT from the intercellular junctions into the intracellular compartment. Intriguingly, LCM failed to induce redistribution of EGFP-OCLNDM from the intercellular junctions even after overnight incubation, indicating that ORM is required for Ca2+-depletion-mediated redistribution of junctional occludin. Immunofluorescence staining of fixed cell monolayers indicated that the lack of occludin or ORM blocks the redistribution of both occludin and ZO-1. Furthermore, absence of occludin or ORM prevents Ca2+-depletion-mediated decrease in electrical resistance and inulin permeability. These observations demonstrate that ORM in occludin is required for Ca2+-depletion-mediated disruption of TJs and barrier dysfunction.
Absence of occludin or ORM also blocked the Ca2+-depletion-mediated redistribution of E-cadherin and β-catenin from the junctions, indicating that the absence of ORM interferes with the Ca2+-depletion-mediated disruption of AJs. The mechanism involved in Ca2+-depletion-mediated TJ disassembly remains to be elucidated. It could be assumed that the extracellular Ca2+ depletion leads to AJ disruption – as E-cadherin requires Ca2+ for its trans homophilic interactions – in turn, disrupting the TJs. Another mechanism that is likely to be involved is that the depletion of extracellular Ca2+ leads to a decrease in intracellular Ca2+, which activates a signaling cascade that mediates the disassembly of AJCs. Furthermore, the absence of ORM blocks Ca2+-depletion-mediated disruption of actin cytoskeleton and microtubules. Therefore, it is likely that Ca2+ depletion triggers a common signaling pathway that leads to disruption of TJs, AJs and the cytoskeleton, and that ORM is required for activation of this signaling cascade. The lack of differences in detergent insoluble to soluble ratios of actin distribution suggests that actin polymerization rates in OCLNWT and OCLNDN cell monolayers are the same. However, a resistance to latrunculin-mediated barrier disruption in EGFP-OCLNDM and Vec cell monolayers suggested that the rate for F-actin disassembly is reduced by ORM deletion.
To determine whether ORM is involved in TJ disruption by other stressors, we evaluated its influence on TJ disruption in response to osmotic stress or hydrogen peroxide. Osmotic stress and oxidative stress are known to induce TJ disruption by Src-mediated Tyr phosphorylation and PP2A-induced Thr dephosphorylation of occludin (Basuroy et al., 2003; Rao, 2008; Samak et al., 2015, 2011; Sheth et al., 2003). Our current data show that the absence of ORM attenuates both osmotic stress-mediated and hydrogen peroxide-mediated disruption of TJs and AJs. These observations confirm that the role of ORM is not restricted to Ca2+-depletion-mediated TJ disruption. Rather, it is a general phenomenon affecting TJ and AJ disruption by several factors. This view is further supported by the previous observation that OD-MDCK cell monolayers are resistant to methyl-β-cyclodextrin-induced barrier disruption, whereas native MDCK cell monolayers are not (Yu et al., 2005).
Resistance to AJC disruption due to multiple stressors, such as Ca2+ depletion, osmotic stress, hydrogen peroxide and latrunculin A, in occludin-deficient Vec cell monolayers suggests that occludin has a role in promoting AJC disruption. This is an intriguing observation. Although it appears that occludin negatively affects the AJC, the ability to disassemble is likely an important characteristic feature of the epithelial TJs, AJs and the cytoskeleton. It is already established that the ability of the actin cytoskeleton to disassemble is an essential feature of actin dynamics as actin depolymerization is needed for cell migration and other cellular functions. Our data raise the question whether the presence of occludin in TJs enhances the flexibility of the actin cytoskeleton to dissociate when needed for certain cellular functions. Our data demonstrate that the phosphorylation hotspot, i.e. ORM, in the C-terminal domain of occludin is necessary for this function of occludin.
Our previous studies have demonstrated that occludin is phosphorylated during the assembly and disassembly of TJs (Rao, 2009; Rao et al., 2002; Seth et al., 2007). Phosphorylation of occludin on T403 and T404 promotes TJ assembly (Suzuki et al., 2009), whereas phosphorylation of Y398 and Y402 prevents TJ interaction with ZO-1 (Kale et al., 2003) and is involved in the disruption of TJs (Elias et al., 2009). As these phosphorylation sites are located in the ORM sequence, we conducted studies to determine whether the response to ORM deletion could be mimicked by point mutations to these residues. Attenuation of occludin mobility, Ca2+-depletion-mediated disruption of the AJC and barrier dysfunction by T403A/T404A double mutations are similar to the effects of ORM deletion, demonstrating that the phosphorylation of ORM determines the dynamic property of occludin and, therefore, AJC dynamics. This observation is in contrast to our previous observation that T403/T404 phosphorylation promotes TJ assembly. The likely explanation for this discrepancy is that, in our previous study, we expressed T403/404 mutants in native MDCK cells with unaltered endogenous WT occludin. Therefore, T403/404 mutants were likely to oligomerize with the endogenous WT occludin. Although the absence of T403/404 phosphorylation delayed the assembly of occludin into TJs, eventually it did assemble. In the present study, we expressed occludin mutants in occludin-deficient cells. Occludin phosphorylation dynamics can promote TJ assembly and, once the TJ is formed, the phosphorylation dynamics are required to recognize the signaling elements needed to disrupt the AJC.
Interestingly, LCM-mediated barrier dysfunction and occludin/ZO-1 redistribution was partially blocked by Y398A/Y402A or Y398D/Y402D mutations. The likely explanation is that these tyrosine residues are transiently phosphorylated during LCM-induced TJ disruption and, therefore, both phosphorylation and dephosphorylation of these residues are necessary for TJ disruption. The importance of Tyr phosphorylation in LCM-mediated TJ disruption and barrier dysfunction was confirmed by the observation that genistein, a tyrosine kinase inhibitor, prevents the Ca2+-depletion-mediated loss of TJ integrity. FRAP analysis showed that mutation of T403/404 and Y398/402 alters the mobile fraction of occludin in parallel to its effect on Ca2+-depletion-mediated TJ disruption. We speculate that the interaction of signaling elements with ORM by a phosphorylation-dependent mechanism may be involved in the regulation of the dynamic property of the AJC in different epithelial tissues.
It is unclear whether the reduction in mobile fraction of occludin is related to the resistance of EGFP-OCLNDM cell monolayers to TJ disruption. The effect of ORM deletion on the resistance to AJC disruption upon Ca2+ depletion is greater than that observed on the mobile fraction of occludin. It is possible that these two responses are independent of each other. However, FRAP is likely to be a result of multiple pools of mobile fraction. The lack of ORM might affect only one pool of mobile fraction that is relevant to TJ disruption. The logical explanation for AJC disruption by Ca2+ depletion is loss of cadherin-dependent adhesion that requires extracellular Ca2+ for homophilic interactions between extracellular domains of E-cadherin. The results of the present study unexpectedly showed that the non-phosphorylated occludin mutant confers cell monolayers with the resistance to Ca2+-mediated disruption of AJs and the actin cytoskeleton. This observation suggests that the process of Ca2+-depletion-mediated disruption of TJs, AJs and the actin cytoskeleton involves modification of a common intracellular mechanism. Previous studies have demonstrated the role of Rho-associated kinase II (ROCKII) (Samarin et al., 2007) and c-Jun N-terminal kinase (JNK) (Naydenov et al., 2009) in AJC disruption by Ca2+ depletion. Our present observation confirms that AJ disruption is not the primary mechanism involved in Ca2+-depletion-mediated TJ disruption. Our previous studies have shown that dephosphorylation of TJ and AJ proteins occurs during Ca2+-depletion-mediated disruption of TJs and AJs (Seth et al., 2007). Data presented in this article (Fig. 3K-M) show that Ca2+ depletion mediates dephosphorylation of E-cadherin, β-catenin and ZO-1 on Thr residues in MDCK and OCLNWT cell monolayers, whereas the Ca2+-depletion-mediated Thr dephosphorylation of E-cadherin, β-catenin and ZO-1 is absent in OCLNDM cell monolayers. This suggests that ORM forms a platform to recruit signaling molecules such as protein kinases and phosphatases involved in the regulation of the phosphorylation status of TJ and AJ proteins.
The studies described here were conducted in cell lines derived from MDCK cells, a renal tubular epithelial cell line. To determine whether the ORM-mediated regulation of AJC dynamics is observed in another epithelium, we performed similar studies in the intestinal epithelium using IEC-6 cells that lack occludin and OCLN−/− mice. Similar to the observation made in MDCK cells, FRAP analysis in IEC-6 cells expressing OCLNWT or OCLNDM revealed that the mobile fraction of occludin is reduced in the absence of ORM. ORM deletion also attenuated Ca2+-depletion-mediated barrier dysfunction and disruption of TJs and AJs, suggesting that ORM is also required for occludin and TJ dynamics in the intestinal epithelium as well. Resistance to EGTA-mediated inulin absorption and redistribution of AJC proteins from the intestinal epithelial junctions of OCLN−/− mice compared with WT mouse intestines further supports the role of occludin in the regulation of TJ and AJ dynamics. Therefore, the role of occludin and ORM appears to be not only universal to different types of stress, but also a common property of different epithelia.
Although the precise function of occludin is unclear at this time, there are pieces of evidence that indicate its potential role in several cellular functions, one of which is regulation of cell migration. A previous study has shown that occludin is involved in directional cell migration (Du et al., 2010). During directional cell migration, occludin appears to be localized to the leading edge, and knockdown of occludin attenuates cell migration. Phosphorylation of occludin on Y473 allowed recruitment of p85α to the leading edge. Knockdown of occludin blocked activation of PI 3-kinase, leading to disorganization of the actin cytoskeleton and prevention of lamellipodia extension. Our data show that occludin deficiency or the absence of ORM attenuates cell migration in MDCK and IEC-6 cells; cell migration was rescued by OCLNWT but not by EGFP-OCLNDM. In addition to its suggested role at the leading edge, the reduced mobile fraction of occludin and, in turn, reduced TJ fluidity in the absence of ORM might be responsible for the obstruction of cell migration. These data suggest that interaction of signaling elements with ORM is involved in the mechanism of cell migration. ORM and TJ dynamics may be important in collective epithelial migration rather than migration of an individual cell. Collective cell migration is implicated in physiological conditions, such as crypt-to-villus migration of intestinal epithelial cells, and pathophysiological conditions, such as wound healing (Friedl and Gilmour, 2009).
To determine whether the effect of ORM is specific for collective cell migration, we evaluated single-cell migration by transmigration assay. Surprisingly, our preliminary data show that occludin deficiency and ORM deletion increases single-cell migration in both MDCK and IEC-6 cells, which is in contrast to their effect on collective cell migration. Single-cell migration may be relevant to tumor metastasis, and our data open a new avenue for future investigations to understand the differences in the mechanisms involved in collective cell migration and single-cell migration in reference to their regulation by occludin.
In summary, this study demonstrates that ORM, a highly conserved phosphorylation hotspot in the C-terminal domain of occludin, determines the dynamic property of TJs in renal and intestinal epithelial monolayers. ORM confers TJs with a dynamic property under multiple conditions of stress in renal and intestinal epithelial tissues. The results of this study also indicate that ORM may directly or indirectly influence the integrity of AJs and the cytoskeleton. Therefore, the TJ dynamics regulated by ORM might play an important role in collective cell migration in epithelial tissues.
MATERIALS AND METHODS
Plasmids, gene knockdown and mutations
For occludin gene silencing in canine MDCK cells, vector-based short hairpin RNAs (shRNAs) were designed using the Dharmacon website (siDesign® Center, http://www.dharmacon.com/DesignCenter/) and cloned into pRNAtin-U6 vector (GenScript), as described previously (Suzuki et al., 2008). Two targeting sequences were chosen against the nucleotide sequence of the canine occludin gene (Target 1: 5′-TATGTCAGACCTTATAACG-3′; Target 2: 5′-TATGCTACCACCCATTAAG-3′). The sequences were further verified by a BLAST search against the canine genome database to confirm the uniqueness of these sequences. To construct the shRNA vectors, two pairs of oligonucleotides containing the antisense sequence, a hairpin loop region (TTGATATCCG) and the sense sequence with cohesive BamHI and HindIII sites were synthesized (Sigma Genosys, St Louis, MO). Primer sequences are provided in Table S1. For a control, a mutant form of shRNA was designed by replacing adenine and guanine nucleotides with cytosine and adenine, respectively.
The construct pEGFP-occludin (human) was used for ORM deletion by inverse PCR using primers flanking the ORM region. The primers were as follows: sense primer 5′-TGGATCAGGGAATATCCACCTATC-3′ and antisense primer 5′-TGCTCTTCCCTTTGCAGGTGCTCT-3′. The DPn1-treated PCR product was ligated by incubation with T4 polynucleotide kinase and T4 DNA ligase. The mutation was confirmed by restriction digestion, PCR amplification and sequencing. This resulted in an ORM deletion mutant that is missing 28 amino acids in its C-terminal domain (Δ388-415: GRSKRTEQDHYETDYTTGGESCDELEED). Double point mutations of T403A/T404A, T403D/T404D, Y398A/Y402A and Y398D/Y402D were introduced in wild-type pEGFP-occludin, as described previously (Elias et al., 2009; Suzuki et al., 2009), by using QuikChange site-directed mutagenesis kit (Stratagene).
Cell culture and transfection
Stable clones of OD-MDCK cells were generated by shRNA transfection of MDCK.2 cells (ATCC, CRL-2936), and stable knockdown clones were isolated by dilution cloning. The pEGFP-OCLNWT or pEGFP-OCLNDM constructs or empty vector was transfected into stable OD-MDCK cells. Cells grown in six-well cluster plates to 70% confluence were transfected in opti-MEM (Invitrogen) containing 1 µg plasmid DNA and 3 µl lipofectamine 2000 (Invitrogen) and incubated at 37°C for 6 h. Fetal bovine serum (FBS) was then added to the medium to make a final concentration of 10% serum and the cells were incubated at 37°C. After 24 h, the cell monolayers were trypsinized and seeded onto 100 mm Petri dishes for selection. Stable clones expressing EGFP (Vector), EGFP-OCLNWT or EGFP-OCLNDM were isolated by geneticin (G418) selection and dilution cloning. At least 12 clones from each group were screened for EGFP fluorescence by live-cell imaging and western blot analysis. Point mutant occludin construct pEGFP-OCLNT403/404A or pEGFP-OCLNT403/404D or pEGFP-OCLNY398/402A or pEGFP-OCLNY398/402D was transfected into OD-MDCK clones as described above. IEC-6 cells expressing EGFP-OCLNWT, EGFP-OCLNDM or EGFP were generated by transfection with FuGENE HD (Promega).
Detergent-insoluble fraction
Cell monolayers in 60 mm culture plates were incubated with 500 µl lysis buffer-CS [Tris buffer containing 10 µl/ml protease inhibitor cocktail, 1 mM vanadate and 1 mM phenylmethane sulfonyl fluoride (PMSF)] on ice for 15 min. Cell lysates were centrifuged at 15,600 g for 4 min at 4°C to sediment the high-density actin cytoskeleton. The supernatant was used as the triton-soluble fraction. The pellet, which is known to contain membrane-associated TJ components, was suspended in 200 μl of lysis buffer-CS and sonicated to homogenize the actin cytoskeleton. This was used as the triton-insoluble fraction. Protein contents in both fractions were measured by the BCA assay (Pierce Biotechnology, Rockford, IL). The protein fractions were mixed with an equal volume of 2× Laemmli's sample buffer and heated at 100°C for 10 min.
Immunoprecipitation
EGFP was immunoprecipitated from total protein extracts under native conditions as previously described (Suzuki et al., 2009). Confluent monolayers (60 mm culture plates) were washed with ice-cold PBS, and proteins were extracted in 500 µl ice-cold lysis buffer (10 mM Tris-Cl pH 7.5 containing 150 mM NaCl, 0.5 mM EDTA, 0.5% NP40, 1 mM PMSF, 1× Protease inhibitor cocktail). The lysates were centrifuged at 15,600 g for 4 min at 4°C. The supernatant was separated, and the pellet was suspended in 200 µl lysis buffer and sonicated for 10 s to induce fragmentation of F-actin filaments and release actin-bound protein complexes in the supernatant fraction. This was centrifuged at 17,135 g for 10 min and the supernatants were pooled. The protein concentration was quantified by the BCA method. The total lysates (1.0 mg protein/ml) were incubated with prewashed 25 µl GFP-Trap_A beads at 4°C for 1 h. Immune complexes were isolated by spinning at 4500 g for 2 min at 4°C, after which they were washed with Tris buffer, mixed with an equal volume of Laemmli's sample buffer (2× concentrated), heated at 100°C for 10 min and centrifuged at 17,135 g for 10 min.
For p-Thr analysis of TJ and AJ proteins, proteins from MDCK, Vec, OCLNWT and OCLNDM cell monolayers were placed under denaturing conditions using lysis buffer-D (0.3% SDS w/v, 10 mM Tris-HCl, pH 7.4, containing 10 µg/ml leupeptin, 10 µg/ml pepstatin A, 10 µg/ml aprotinin, 10 µg/ml bestatin and 0.1 mM PMSF) and pre-heated to 100°C. After repeated pipetting to homogenize samples, they were heated at 100°C for 10 min. Protein extracts (400 μg) were incubated with anti-p-Thr antibodies overnight as described above. Protein complexes were immunoprecipitated with Protein A/G-Sepharose beads and extracted in Laemmli sample buffer for immunoblot analysis for ZO-1, E-cadherin and β-catenin.
Immunoblot analysis
EGFP immunoprecipitates and protein extracts were separated by SDS-polyacrylamide gel (7%) electrophoresis and transferred to PVDF membranes. Proteins on the membrane were probed with specific primary antibodies in combination with HRP-conjugated anti-mouse IgG or HRP-conjugated anti-rabbit IgG antibodies. The blots were developed by an enhanced chemiluminescence (ECL) method (Amersham, Arlington Heights, IL). The bands were quantified by densitometric analysis using ImageJ software (National Institutes of Health, Bethesda, MD).
Analysis of barrier function and junction integrity
Barrier function of epithelial monolayers was evaluated by measuring TER and the flux of FITC-inulin as described before (Suzuki et al., 2008). TJ and AJ integrity was assessed by immunofluorescence staining of fixed cell monolayers for GFP-occludin, ZO-1, E-cadherin and β-catenin as described before (Suzuki et al., 2009).
FRAP analysis
By using a FRAP module in a Zeiss 710 confocal microscope and Zen software, FRAP analyses were performed in cell lines expressing EGFP-OCLNWT and its mutants. Three to four regions of interest (ROIs), provided in the module, were defined at the intercellular junctions of the monolayer. A high-intensity laser was used at 488 nm to bleach the ROIs, with a specific number of iterations. The module was set to capture at least three images before the bleach and time-lapse images up to ∼25 min after the bleach. All the time-lapse images were collected at the same focal plane. The time-series images were processed by using ImageJ software and confirmed by manual calculation. Fluorescence intensity was measured at different times after bleaching to calculate percentage recovery. Percentage recovery at 25 min was considered to be the percentage mobile fraction.
Ca2+-switch assay
OD-MDCK and IEC-6 cells expressing EGFP-OCLNWT or its mutants were grown to confluence in transwells (12 or 6.5 mm) or on 60 mm culture plates and incubated in LCM (low-Ca2+ medium; DMEM containing only 10 µM CaCl2). After 16 h in LCM, the medium was switched with NCM (normal-Ca2+ medium). Barrier function was monitored at different time points by measuring TER and inulin flux. TJ integrity was monitored by live-cell imaging, in the case of culture plates, or by fixing cell monolayers, in the case of transwells. In another model, cell monolayers were treated with 4 mM EGTA to induce Ca2+ depletion.
Cell migration assay
For collective cell migration, OD-MDCK and IEC-6 cells expressing EGFP-OCLNWT, EGFP-OCLNDM and EGFP were grown to confluence in six-well cluster plates. The cell monolayers were scraped with a razor blade in MDCK cells, whereas a pipette tip was used for scratch wounding in IEC-6 cells. Phase-contrast images of the wounded area were captured at different time points and processed using ImageJ software. The average rate of cell migration in each monolayer was calculated from at least five spots.
For single-cell migration, EGFP-OCLNWT, EGFP-OCLNDM, Vector and MDCK cells (1×105 cells/100 µl) in DMEM containing 0.1% BSA without serum were seeded on top of an 8 µm filter membrane in a 12-well transwell insert. DMEM containing 10% FBS was added to the bottom chamber of transwells. Cells were allowed to incubate for 6 h at 37°C and 5% CO2. Migrated cells were fixed with 90% ethanol for 10 min and air dried. Cells were stained with 2% Crystal Violet at room temperature for 10 min. Cells were photographed, photographs converted to 16-bit images, and cells counted using ImageJ software.
Osmotic stress and oxidative stress
Cell monolayers in transwell inserts were treated with DMEM containing mannitol (0.3 M) to induce osmotic stress or incubated with hydrogen peroxide (100 µM) to induce oxidative stress, as previously described (Elias et al., 2009; Samak et al., 2011). Barrier function and TJ integrity were monitored at various time points.
Intestinal loops ex vivo
Adult WT and OCLN−/− mice that had ad libitum access to regular laboratory chow and water were used in this study. OCLN−/− mice backcrossed to C57BL/6 mice were provided by Dr Jerrold Turner (Harvard University, Boston, MA). All experiments were conducted according to an IACUC approved protocol. Mid small intestine was harvested and intestinal sacs (∼2 cm long) were prepared. One end of the segment was ligated with a nylon thread. Saline-containing FITC-inulin (0.5 mg/ml) with or without 1 mM EGTA was injected from the other end immediately before ligating the loop to prepare sacs. The sacs were incubated in DMEM at 37°C for 30 min. After the incubation, contents of the sac were collected, and fluorescence in the luminal flushing was measured. Cryosections of loops were stained for ZO-1, F-actin, E-cadherin, β-catenin and nucleus for immunofluorescence analysis.
Reagents
Dulbecco's modified Eagle's medium (DMEM), FBS and antibiotics were purchased from Cellgro® (Manassas, VA) whereas Ca2+ free DMEM was from Thermo Fisher Scientific (Tustin, CA). Rabbit polyclonal anti-ZO-1, rabbit polyclonal anti-p-Thr, mouse monoclonal anti-occludin, anti-Cldn-2, Alexa Fluor 488-conjugated anti-mouse IgG and phalloidin were purchased from Invitrogen (Carlsbad, CA). Mouse monoclonal anti-EGFP antibody was purchased from Clontech (Mountain View, CA). Hoechst 33342 was obtained from Life Technologies (Carlsbad, CA). Mouse monoclonal anti-E-cadherin and rabbit polyclonal anti-β-catenin were purchased from BD Biosciences (San Jose, CA). Latrunculin-A, genistein, Cy3-conjugated anti-rabbit IgG, HRP-conjugated anti-mouse IgG, HRP-conjugated anti-rabbit IgG and mouse monoclonal anti-β-actin antibodies were obtained from Sigma (St Louis, MO). For immunofluorescence staining, all antibodies were used at 1:100 dilution and Hoechst dye was used at 1:1000 dilution. For immunoblot analysis, all primary antibodies were used at 1:1000 dilution and secondary antibodies were used at 1:10,000 dilution. GFP-Trap A was purchased from Chromotek (Hauppauge, NY). Restriction enzymes T4 DNA ligase, polynucleotide kinase and DPn1 were obtained from New England Biolabs (Beverly, MA). Full details of all antibodies used are provided in Table S2.
Supplementary Material
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: B.M., R.K.R.; Methodology: B.M., H.M., R.G., A.S.M., S.A., P.K.S., K.D., T.S.; Software: B.M.; Validation: B.M.; Formal analysis: B.M.; Investigation: B.M., A.S.M., S.A., K.D.; Resources: H.M.; Data curation: B.M., A.S.M.; Writing - original draft: B.M.; Writing - review & editing: R.K.R.; Supervision: R.K.R.; Project administration: R.K.R.; Funding acquisition: R.K.R.
Funding
Bhargavi Manda is a recipient of J. Paul Quigley memorial scholarship and Dorothy K. and Daniel L. Gerwin graduate scholarship award from the University of Tennessee Health Science Center. This study was funded by National Institutes of Health grants DK55532 and AA12307. Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://jcs.biologists.org/lookup/doi/10.1242/jcs.206789.supplemental
References
- Anderson J. M. and Van Itallie C. M. (1995). Tight junctions and the molecular basis for regulation of paracellular permeability. 269, G467-G475. 10.1152/ajpgi.1995.269.4.G467 [DOI] [PubMed] [Google Scholar]
- Anderson J. M. and Van Itallie C. M. (2009). Physiology and function of the tight junction. 1, a002584 10.1101/cshperspect.a002584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basuroy S., Sheth P., Kuppuswamy D., Balasubramanian S., Ray R. M. and Rao R. K. (2003). Expression of kinase-inactive c-Src delays oxidative stress-induced disassembly and accelerates calcium-mediated reassembly of tight junctions in the Caco-2 cell monolayer. 278, 11916-11924. 10.1074/jbc.M211710200 [DOI] [PubMed] [Google Scholar]
- Beeman N., Webb P. G. and Baumgartner H. K. (2012). Occludin is required for apoptosis when claudin-claudin interactions are disrupted. 3, e273 10.1038/cddis.2012.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruewer M., Samarin S. and Nusrat A. (2006). Inflammatory bowel disease and the apical junctional complex. 1072, 242-252. 10.1196/annals.1326.017 [DOI] [PubMed] [Google Scholar]
- Cummins P. M. (2012). Occludin: one protein, many forms. 32, 242-250. 10.1128/MCB.06029-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörfel M. J. and Huber O. (2012). Modulation of tight junction structure and function by kinases and phosphatases targeting occludin. 2012, 807356 10.1155/2012/807356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dörfel M. J., Westphal J. K., Bellmann C., Krug S. M., Cording J., Mittag S., Tauber R., Fromm M., Blasig I. E. and Huber O. (2013). CK2-dependent phosphorylation of occludin regulates the interaction with ZO-proteins and tight junction integrity. 11, 40 10.1186/1478-811X-11-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du D., Xu F., Yu L., Zhang C., Lu X., Yuan H., Huang Q., Zhang F., Bao H., Jia L. et al. (2010). The tight junction protein, occludin, regulates the directional migration of epithelial cells. 18, 52-63. 10.1016/j.devcel.2009.12.008 [DOI] [PubMed] [Google Scholar]
- Elias B. C., Suzuki T., Seth A., Giorgianni F., Kale G., Shen L., Turner J. R., Naren A., Desiderio D. M. and Rao R. (2009). Phosphorylation of Tyr-398 and Tyr-402 in occludin prevents its interaction with ZO-1 and destabilizes its assembly at the tight junctions. 284, 1559-1569. 10.1074/jbc.M804783200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanning A. S., Jameson B. J., Jesaitis L. A. and Anderson J. M. (1998). The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. 273, 29745-29753. 10.1074/jbc.273.45.29745 [DOI] [PubMed] [Google Scholar]
- Friedl P. and Gilmour D. (2009). Collective cell migration in morphogenesis, regeneration and cancer. 10, 445-457. 10.1038/nrm2720 [DOI] [PubMed] [Google Scholar]
- Furuse M., Hirase T., Itoh M., Nagafuchi A., Yonemura S., Tsukita S. and Tsukita S. (1993). Occludin: a novel integral membrane protein localizing at tight junctions. 123, 1777-1788. 10.1083/jcb.123.6.1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse M., Itoh M., Hirase T., Nagafuchi A., Yonemura S., Tsukita S. and Tsukita S. (1994). Direct association of occludin with ZO-1 and its possible involvement in the localization of occludin at tight junctions. 127, 1617-1626. 10.1083/jcb.127.6.1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S., Suzuki T., Seth A., Samak G. and Rao R. (2011). Protein kinase Czeta phosphorylates occludin and promotes assembly of epithelial tight junctions. 437, 289-299. 10.1042/BJ20110587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang A.-S. (2014). The apical junctional complex in respiratory diseases. 50, 1-5. 10.4068/cmj.2014.50.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kale G., Naren A. P., Sheth P. and Rao R. K. (2003). Tyrosine phosphorylation of occludin attenuates its interactions with ZO-1, ZO-2, and ZO-3. 302, 324-329. 10.1016/S0006-291X(03)00167-0 [DOI] [PubMed] [Google Scholar]
- Matter K. and Balda M. S. (1999). Occludin and the functions of tight junctions. 186, 117-146. 10.1016/S0074-7696(08)61052-9 [DOI] [PubMed] [Google Scholar]
- Naydenov N. G., Hopkins A. M. and Ivanov A. I. (2009). c-Jun N-terminal kinase mediates disassembly of apical junctions in model intestinal epithelia. 8, 2110-2121. 10.4161/cc.8.13.8928 [DOI] [PubMed] [Google Scholar]
- Rachow S., Zorn-Kruppa M., Ohnemus U., Kirschner N., Vidal-y-Sy S., von den Driesch P., Börnchen C., Eberle J., Mildner M., Vettorazzi E. et al. (2013). Occludin is involved in adhesion, apoptosis, differentiation and Ca2+-homeostasis of human keratinocytes: implications for tumorigenesis. 8, e55116 10.1371/journal.pone.0055116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raleigh D. R., Boe D. M., Yu D., Weber C. R., Marchiando A. M., Bradford E. M., Wang Y., Wu L., Schneeberger E. E., Shen L. et al. (2011). Occludin S408 phosphorylation regulates tight junction protein interactions and barrier function. 193, 565-582. 10.1083/jcb.201010065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R. (2008). Oxidative stress-induced disruption of epithelial and endothelial tight junctions. 13, 7210-7226. 10.2741/3223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R. (2009). Occludin phosphorylation in regulation of epithelial tight junctions. 1165, 62-68. 10.1111/j.1749-6632.2009.04054.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R. K., Baker R. D., Baker S. S., Gupta A. and Holycross M. (1997). Oxidant-induced disruption of intestinal epithelial barrier function: role of protein tyrosine phosphorylation. 273, G812-G823. 10.1152/ajpgi.1997.273.4.G812 [DOI] [PubMed] [Google Scholar]
- Rao R. K., Basuroy S., Rao V. U., Karnaky K. J. Jr and Gupta A. (2002). Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. 368, 471-481. 10.1042/bj20011804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou M., Fujimoto K., Doi Y., Itoh M., Fujimoto T., Furuse M., Takano H., Noda T. and Tsukita S. (1998). Occludin-deficient embryonic stem cells can differentiate into polarized epithelial cells bearing tight junctions. 141, 397-408. 10.1083/jcb.141.2.397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitou M., Furuse M., Sasaki H., Schulzke J.-D., Fromm M., Takano H., Noda T. and Tsukita S. (2000). Complex phenotype of mice lacking occludin, a component of tight junction strands. 11, 4131-4142. 10.1091/mbc.11.12.4131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samak G., Narayanan D., Jaggar J. H. and Rao R. (2011). CaV1.3 channels and intracellular calcium mediate osmotic stress-induced N-terminal c-Jun kinase activation and disruption of tight junctions in Caco-2 CELL MONOLAYERS. 286, 30232-30243. 10.1074/jbc.M111.240358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samak G., Chaudhry K. K., Gangwar R., Narayanan D., Jaggar J. H. and Rao R. (2015). Calcium/Ask1/MKK7/JNK2/c-Src signalling cascade mediates disruption of intestinal epithelial tight junctions by dextran sulfate sodium. 465, 503-515. 10.1042/BJ20140450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samarin S. N., Ivanov A. I., Flatau G., Parkos C. A. and Nusrat A. (2007). Rho/Rho-associated kinase-II signaling mediates disassembly of epithelial apical junctions. 18, 3429-3439. 10.1091/mbc.E07-04-0315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlüter M. A. and Margolis B. (2012). Apicobasal polarity in the kidney. 318, 1033-1039. 10.1016/j.yexcr.2012.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth A., Sheth P., Elias B. C. and Rao R. (2007). Protein phosphatases 2A and 1 interact with occludin and negatively regulate the assembly of tight junctions in the CACO-2 cell monolayer. 282, 11487-11498. 10.1074/jbc.M610597200 [DOI] [PubMed] [Google Scholar]
- Shen L. (2012). Tight junctions on the move: molecular mechanisms for epithelial barrier regulation. 1258, 9-18. 10.1111/j.1749-6632.2012.06613.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L., Weber C. R. and Turner J. R. (2008). The tight junction protein complex undergoes rapid and continuous molecular remodeling at steady state. 181, 683-695. 10.1083/jcb.200711165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L., Weber C. R., Raleigh D. R., Yu D. and Turner J. R. (2011). Tight junction pore and leak pathways: a dynamic duo. 73, 283-309. 10.1146/annurev-physiol-012110-142150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheth B., Fontaine J.-J., Ponza E., McCallum A., Page A., Citi S., Louvard D., Zahraoui A. and Fleming T. P. (2000). Differentiation of the epithelial apical junctional complex during mouse preimplantation development: a role for rab13 in the early maturation of the tight junction. 97, 93-104. 10.1016/S0925-4773(00)00416-0 [DOI] [PubMed] [Google Scholar]
- Sheth P., Basuroy S., Li C., Naren A. P. and Rao R. K. (2003). Role of phosphatidylinositol 3-kinase in oxidative stress-induced disruption of tight junctions. 278, 49239-49245. 10.1074/jbc.M305654200 [DOI] [PubMed] [Google Scholar]
- Shin K., Fogg V. C. and Margolis B. (2006). Tight junctions and cell polarity. 22, 207-235. 10.1146/annurev.cellbio.22.010305.104219 [DOI] [PubMed] [Google Scholar]
- Suzuki T., Seth A. and Rao R. (2008). Role of phospholipase Cgamma-induced activation of protein kinase Cepsilon (PKCepsilon) and PKCbetaI in epidermal growth factor-mediated protection of tight junctions from acetaldehyde in Caco-2 cell monolayers. 283, 3574-3583. 10.1074/jbc.M709141200 [DOI] [PubMed] [Google Scholar]
- Suzuki T., Elias B. C., Seth A., Shen L., Turner J. R., Giorgianni F., Desiderio D., Guntaka R. and Rao R. (2009). PKC eta regulates occludin phosphorylation and epithelial tight junction integrity. 106, 61-66. 10.1073/pnas.0802741106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Itallie C. M. and Anderson J. M. (1997). Occludin confers adhesiveness when expressed in fibroblasts. 110, 1113-1121. [DOI] [PubMed] [Google Scholar]
- Vogelmann R. and Nelson W. J. (2005). Fractionation of the epithelial apical junctional complex: reassessment of protein distributions in different substructures. 16, 701-716. 10.1091/mbc.E04-09-0827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelmann R., Amieva M. R., Falkow S. and Nelson W. J. (2004). Breaking into the epithelial apical-junctional complex--news from pathogen hackers. 16, 86-93. 10.1016/j.ceb.2003.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A. S. L., McCarthy K. M., Francis S. A., McCormack J. M., Lai J., Rogers R. A., Lynch R. D. and Schneeberger E. E. (2005). Knockdown of occludin expression leads to diverse phenotypic alterations in epithelial cells. 288, C1231-C1241. 10.1152/ajpcell.00581.2004 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.