

Abstract
Cardiovascular disease and susceptibility to infection are leading causes of morbidity and mortality for individuals with spinal cord injury (SCI). A major contributor to these is autonomic dysreflexia (AD), an amplified reaction of the autonomic nervous system (hallmarked by severe hypertension) in response to sensory stimuli below the injury. Maladaptive plasticity of the spinal sympathetic reflex circuit below the SCI results in AD intensification over time. Mechanisms underlying this maladaptive plasticity are poorly understood, restricting the identification of treatments. Thus, no preventative treatments are currently available. Neuroinflammation has been implicated in other pathologies associated with hyperexcitable neural circuits. Specifically, the soluble form of TNFα (sTNFα) is known to play a role in neuroplasticity. We hypothesize that persistent expression of sTNFα in spinal cord underlies AD exacerbation. To test this, we intrathecally administered XPro1595, a biologic that renders sTNFα nonfunctional, after complete, high-level SCI in female rats. This dramatically attenuated the intensification of colorectal distension-induced and naturally occurring AD events. This improvement is mediated via decreased sprouting of nociceptive primary afferents and activation of the spinal sympathetic reflex circuit. We also examined peripheral vascular function using ex vivo pressurized arterial preparations and immune function via flow cytometric analysis of splenocytes. Diminishing AD via pharmacological inhibition of sTNFα mitigated ensuing vascular hypersensitivity and immune dysfunction. This is the first demonstration that neuroinflammation-induced sTNFα is critical for altering the spinal sympathetic reflex circuit, elucidating a novel mechanism for AD. Importantly, we identify the first potential pharmacological, prophylactic treatment for this life-threatening syndrome.
SIGNIFICANCE STATEMENT Autonomic dysreflexia (AD), a disorder that develops after spinal cord injury (SCI) and is hallmarked by sudden, extreme hypertension, contributes to cardiovascular disease and susceptibility to infection, respectively, two leading causes of mortality and morbidity in SCI patients. We demonstrate that neuroinflammation-induced expression of soluble TNFα plays a critical role in AD, elucidating a novel underlying mechanism. We found that intrathecal administration after SCI of a biologic that inhibits soluble TNFα signaling dramatically attenuates AD and significantly reduces AD-associated peripheral vascular and immune dysfunction. We identified mechanisms behind diminished plasticity of neuronal populations within the spinal sympathetic reflex circuit. This study is the first to pinpoint a potential pharmacological, prophylactic strategy to attenuate AD and ensuing cardiovascular and immune dysfunction.
Keywords: autonomic dysreflexia; plasticity; soluble TNFα, spinal cord injury
Introduction
Autonomic dysreflexia (AD), a life-threatening disorder that manifests in up to 90% of people who have sustained a severe, high-level spinal cord injury (SCI) (Curt et al., 1997), is hallmarked by extreme, sudden hypertension and reflexive bradycardia that can lead to strokes and even death. Episodes are triggered by an amplified reaction of the spinal sympathetic reflex circuit in response to sensory stimuli below the injury (e.g., bladder expansion, rectal distension). Chronic, frequent AD bouts have secondary consequences; they instigate detrimental changes to peripheral vasculature, such as increased contractility in response to adrenergic input (Alan et al., 2010; West et al., 2013a) and fewer lymphocytes (Zhang et al., 2013; Ueno et al., 2016) that promote cardiovascular disease and susceptibility to infection, respectively, two leading causes of mortality and morbidity for those living with chronic SCI (Garshick et al., 2005; Meisel et al., 2005; Myers et al., 2007; Brommer et al., 2016). Unfortunately, the only treatments currently available are palliative, aimed at merely managing the hypertensive symptoms after an AD episode has already been initiated until the triggering stimulus is identified and addressed. There are no prophylactic treatments that limit the onset of AD.
One mechanism for AD after SCI is the interruption of descending, modulatory input to sympathetic preganglionic neurons (SPNs) that reside within the spinal cord [thoracic (T) level 1, lumbar (L) level 2] and regulate sympathetic output to splanchnic circulation. Critically, AD worsens over time with events becoming more frequent and extreme (Krassioukov and Weaver, 1995; Mayorov et al., 2001; Rabchevsky et al., 2012; Zhang et al., 2013; West et al., 2015). This secondary, intensifying phase of AD has been attributed to plasticity caudal to the injury (e.g., sprouting of nociceptive primary afferents) (Krenz et al., 1999; Cameron et al., 2006; Hou et al., 2009), which results in hyperexcitable spinal sympathetic circuits. Thus, a sensory stimulus below the injury leads to activation of more interneurons (Krassioukov et al., 2002; Landrum et al., 2002; Hou et al., 2008), including those that project to SPNs, triggering an exaggerated sympathetic discharge that causes hypertension (see Fig. 12). What prompts this maladaptive plasticity is still obscure. A better understanding of mechanisms behind it could elucidate potential targets for prophylactic treatments to attenuate the onset or severity of AD after SCI and avert ensuing cardiovascular and immune dysfunction.
Figure 12.
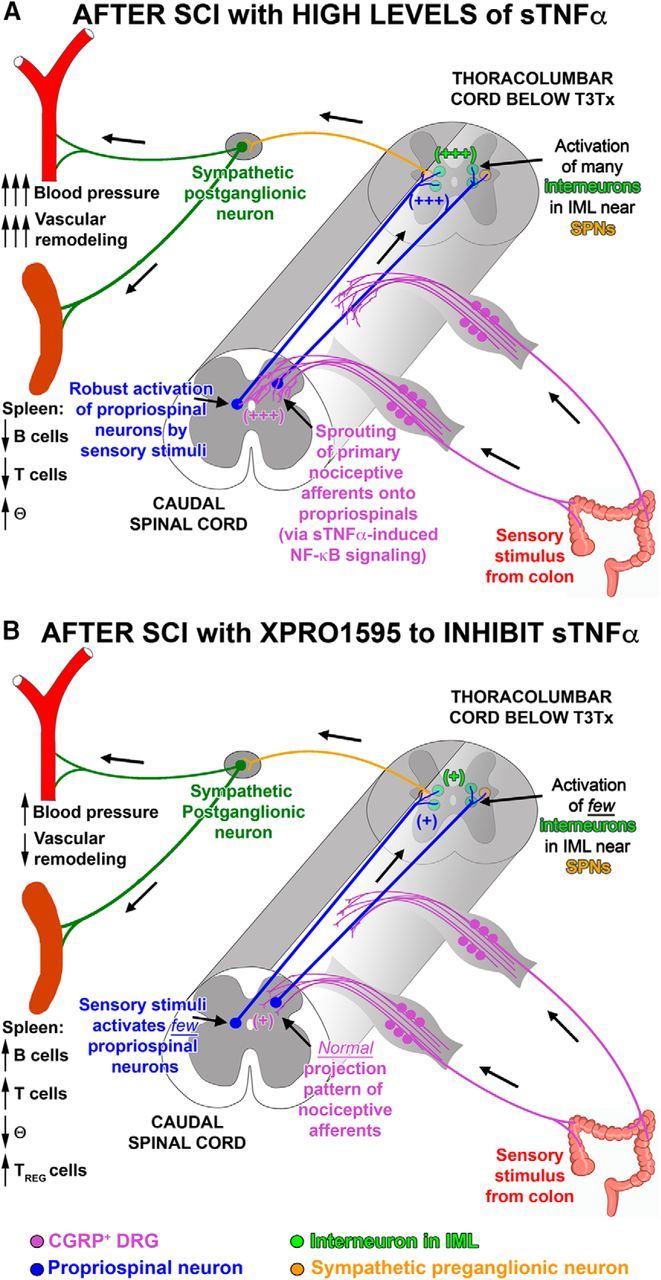
Summary of how SCI-induced sTNFα affects the sympathetic spinal reflex circuit and peripheral immune and vascular function. CGRP, calcitonin gene-related peptide; IML, intermediolateral cell column; SCI, spinal cord injury; SPN, sympathetic preganglionic neuron.
Aberrant plasticity and hyperexcitable circuits are not only associated with AD; they are correlated with other conditions, including epilepsy and neuropathic pain. Interestingly, an activated immune system is thought to underlie these pathologies (Marchi et al., 2014; Walters, 2014). Whether inflammation instigates plasticity and subsequent AD exacerbation, however, has not been investigated.
The “master regulator” cytokine TNFα has been implicated in initiating inflammation in a variety of contexts. TNFα-converting enzyme cleaves the transmembrane form of TNFα into the soluble form (sTNFα). sTNFα homotrimerizes and binds preferentially to TNF receptor 1 (TNFR1) (Grell et al., 1995; Probert, 2015). TNFR1 signaling is necessary for mounting a full inflammatory response (Loetscher et al., 1993). After SCI, there is substantial upregulation of TNFα within the spinal cord, both at the epicenter (Bethea et al., 1999) and below the injury (Detloff et al., 2008). Intriguingly, increased TNFα is associated with various forms of plasticity that can increase neuronal excitability (Beattie et al., 2002; Stellwagen et al., 2005; Stück et al., 2012).
We hypothesized that sTNFα signaling plays a crucial role in injury-induced plasticity caudal to a severe (i.e., complete), high-level SCI that mediates the intensification of AD over time. We previously used XPro1595, a biologic that forms nonfunctional heterotrimers with sTNFα (Steed et al., 2003), to modulate a neuroimmune response (Brambilla et al., 2011). We surmised that inhibiting sTNFα signaling caudal to a SCI via intrathecal delivery of XPro1595 after injury would diminish AD. Last, we examined whether reducing AD via blocking sTNFα signaling centrally improved vascular and immune function peripherally.
Materials and Methods
Animal use.
Adult, female Wistar rats (225–250 g; Charles River) were used for all experiments. Animals were housed, given unlimited access to food and water, and used in accordance with Drexel University Institutional Animal Care and Use Committee and National Institutes of Health guidelines for experimentation with laboratory animals. Animals were allowed to acclimate for a least 1 week after arrival before any procedure was done before being randomly assigned to treatment groups. After all surgical procedures, animals were given ampicillin (200 mg/kg), slow-release buprenorphine (ZooPharm; 0.05 mg/kg), and lactated Ringer's solution perioperatively and placed on a thermal barrier to recover. They were returned to their cages once they became alert and responsive. All animals that received spinal transections had their bladders manually expressed at least twice a day for the duration of the study.
Radiotelemeter implantation into the descending aorta.
As indicated in the experimental timeline in Figure 1, radiotelemeter pressure transducers (either model HD-S10 or modified HD-S11; Data Sciences International) were transplanted into the descending aorta of naive animals at least 1 week before spinal transection (Tx) to give the animals ample recovery time after the implantation surgery (Rabchevsky et al., 2012). As we did previously (Hou et al., 2013b), naive rats were deeply anesthetized with isoflurane and the femoral artery was exposed. After securing the vessel distally and temporarily blocking blood flow proximally, the artery was punctured, the tip of the catheter was inserted and advanced rostrally into the abdominal aorta. After the catheter was secured in the vessel, the transmitter body was placed into a subcutaneous pocket along the flank. The surrounding tissue at the end of transmitter was sutured to prevent the telemeter from shifting position. The skin was closed with wound clips.
Figure 1.

Overview of the experimental timeline.
Preparation of osmotic minipumps to deliver XPro1595.
Osmotic minipumps (Alzet, no. 2002) were used to intrathecally deliver the sTNFα biologic XPro1595 or saline vehicle continuously for 28 d. The amount of XPro1595 used here (60 μg per day) successfully conferred neuroprotection at a lesion epicenter of a rodent contusive SCI model (Novrup et al., 2014). The minipumps were prepared 1 d before spinal Tx and implantation so that either compound would be delivered to the spinal cord starting immediately upon placement in vivo. Either XPro1595 (10 mg/ml; generously provided by David E. Szymkowski of Xencor) or saline was loaded into the osmotic minipumps. An intrathecal catheter (ReCathCo) was attached to the minipump. The loaded pumps were incubated in sterile saline at 37°C overnight and were removed from the warmed saline solution just before implantation.
Thoracic spinal transection and osmotic minipump implantation.
Animals were deeply anesthetized using isoflurane. Similar to what we did previously (Xu et al., 2014; Wu et al., 2015), the spinal cord at thoracic level 3 (T3) was exposed by dorsal laminectomy of the T2 and the T5 vertebral processes (overlying T3 and T6 spinal cord levels, respectively). After a small incision was made in the dura overlying T3 using a microknife, a ∼2-mm-long segment of the spinal cord was removed by vacuum aspiration using a pulled glass micropipette. The lesion cavity was filled with gelfoam saturated with saline to achieve hemostasis. The gelfoam was removed, and the cavity was visually examined to ensure that the Tx was complete. The dura was sutured shut with 9-0 sutures.
Immediately after Tx, a small incision was made in the dura above T6 spinal cord. The catheter attached to an osmotic minipump was carefully threaded subdurally until the end lay just caudal to the Tx site (Fig. 1). The catheter was held in place via suturing to both the dura and the neighboring muscle. The minipump was placed in a subcutaneous pocket and was secured to the underlying muscle. Both laminectomy sites were covered with a SILASTIC membrane (BioBrane; UDL Laboratories, Rockford, IL) and the overlying musculature was sutured with Vicryl 5-0 sutures. Wound clips were used to close the skin incision.
Blood pressure (BP) recordings to assess naturally occurring AD.
To monitor heart rate (HR) and mean arterial pressure (MAP), radiotelemeters implanted in the saline- or XPro1595-treated animals were turned on and cages containing individual animals were placed on the telemetry receivers (RC-1; Data Sciences International). We obtained baseline recordings of all animals after telemeter implantation and before SCI to determine whether HR and MAP values were within a normal range (to ensure that the catheters were not occluded and were providing accurate measurements). At 2, 3, and 4 weeks after injury, MAP and HR were monitored continuously in these animals while they moved freely in their cages using Dataquest A.R.T. acquisition software (Data Sciences International) for 24 h (MAP and HR values sampled every 2 s). Naturally occurring AD events were detected via a two-pass method. First, the 24 h data from each individual animal at each time point was imported into MATLAB (The MathWorks). Using a modified version of established algorithms (Rabchevsky et al., 2012; Zhang et al., 2013; West et al., 2015), time-matched BP and HR were compared simultaneously. Rolling MAP and HR baselines were established by continuously averaging a 6 min period. AD events were defined as when MAP was at least 20 mmHg greater than baseline for at least 30 s and was accompanied by bradycardia of at least 20 beats per minute. Afterward, all “detected” events went through a second pass and were visually verified by a blinded observer according to the criterion described above. Any false-positives were disregarded. Additionally, any events that occurred within 2 min of each other were considered to be 1 event. Last, any detected events within 15 min of manual bladder expression were not included in additional comparative analyses. Once an event was verified, the average MAP during the event, the change in MAP from baseline, the HR during the event, and the duration of the bout were calculated.
Colorectal distension to induce AD.
At 2, 3, and 4 weeks after Tx, BP and HR were monitored before, during, and after colorectal distension (CRD) in unanesthetized rats, a well-established means that reliably induces an AD episode (Mayorov et al., 2001; Cameron et al., 2006). CRD expands the colon just as large fecal boluses would. Similar to what we did previously (Hou et al., 2013b), a silicone balloon-tipped catheter (2-way pediatric Foley catheter, 10 French, 3 cc, Coloplast) was gently inserted 2 cm inside the rectums of all T3Tx rats treated with saline or XPro1595. The external portion of the catheter was secured to the tails with tape. The animals were allowed to acclimate to the presence of the catheter for at least 30 min. To initiate spinal viscero-sympathetic reflexes and induce AD, the balloon catheter was gradually inflated (over 10 s) with 2.0 ml of air and the distension was sustained for 1 min. Two or three trials were conducted per animal per time point, with an intertrial interval of at least 20 min. For each animal, the difference between the baseline MAP and the CRD-induced MAP was calculated for each trial and averaged per animal per time point.
Intermittent colorectal distension to induce c-Fos expression.
To determine the extent of neuronal activation in response to a visceral sensory stimulus (e.g., CRD, indicated by induction of c-Fos; see Histology), 4 weeks after SCI, some T3Tx-saline or T3Tx-XPro1595 animals were given intermittent CRD over an extended period of time (Landrum et al., 2002; Hou et al., 2008; Ueno et al., 2016). Briefly, after the balloon catheter was inserted into the rectum, as described above, the balloon was inflated with 2.0 ml of air for 30 s and then deflated for 60 s. This 90 s cycle was repeated for 45 min. At 1.5 h after the last round of CRD, animals were given an overdose of Euthasol and perfused with 0.9% saline followed by 4% PFA.
Ex vivo mesenteric artery reactivity studies.
At 4 weeks after injury, T3Tx-saline (n = 7) or T3Tx-XPro1595 (n = 6) animals were given an overdose of Euthasol. Naive, uninjured animals (n = 5) were used as an additional control group. A midline incision was made first through the skin and then through the abdominal muscle to access the intestine. The entire intestine was excised and placed in chilled physiological saline solution (PSS), which was composed of the following (in mm): 140 NaCl, 5 KC1, 1.2 MgSO4, 2.0 CaC12, 10 NaAcetate, 10 HEPES, 1.2 Na2H2PO4, 5 glucose, and pH adjusted to 7.4 with NaOH. As done previously (Osei-Owusu et al., 2012, 2014; Jie et al., 2016), sections of isolated vessels 2–3 mm in length were excised and immediately placed in an organ chamber for mounting on glass pipettes. Excised pieces of small mesenteric arteries were transferred and cannulated under no-flow conditions at both ends with glass pipettes and secured with nylon ligature. The lumen of the vessel was then filled with PSS. The vessel was observed with a video camera system (MTI CCD-72) to track and record changes in vessel diameter in response to vasoactive agents. The baseline internal diameter of the vessel was measured online at 60 mmHg and 37°C with IonOptix vessel dimension acquisition and analysis software as previously described (Sweazea and Walker, 2012). After vessels were equilibrated for 30 min, the bath solution was replaced with warmed PSS containing increasing concentrations of either the vasoconstrictor phenylephrine (PE) or the vasodilator acetylcholine (ACh). After application of the highest concentration of PE or ACh, the bath solution was replaced with PSS to allow for complete relaxation. The changes in vasoconstriction were measured as a percentage change from the baseline lumen diameter. The pEC50 was calculated using SigmaPlot 12.0.
Flow cytometry.
At 4 weeks after SCI, animals were overdosed with Euthasol and spleens were harvested. Spleens were dissociated through a 40 μm cell strainer and collected in 10 ml MACS buffer (PBS, 0.5% BSA, 2 mm EDTA). Splenocytes were centrifuged (300 × g, 5 min) and washed once with 10 ml MACS buffer. Cells were incubated in 3 ml ACK Lysing Buffer (Thermo Fisher Scientific) for 3 min to deplete red blood cells, washed again with 10 ml MACS buffer, and then stained for different immune cell markers using fluorescence-labeled antibodies (Miltenyi Biotec). For staining of cytoplasmic proteins, the FoxP3 Staining Buffer Set was used according to manufacturer's instructions (Miltenyi Biotec catalog #130-093-142). Data were acquired using a FACS Canto (BD Bioscience) and analyzed with FlowJo (TreeStar).
Preparation of tissue samples for biochemical analysis.
Naive, T3Tx-saline, or T3Tx-XPro1595 rats at 3 d, 1 week, 2 weeks, 3 weeks, and 4 weeks after SCI were killed with Euthasol overdose, and fresh spinal cord tissue was immediately extracted, blocked into lumbar and thoracic segments below the Tx, flash-frozen in liquid nitrogen, and stored at −80°C until further processing. Protein extraction was conducted as previously described (Bethea et al., 1999; Bracchi-Ricard et al., 2013). Briefly, samples were homogenized in RIPA buffer (0.01 m sodium phosphate, pH 7.2, 0.15 m NaCl, 1% NP40, 1% sodium deoxycholate, 0.1% SDS, 2 mm EDTA) supplemented with Roche complete protease inhibitor mixture, mixed end-over-end at 4°C for 30 min, and centrifuged at 14,000 rpm for 10 min at 4°C. The supernatants were transferred to fresh tubes and stored at −80°C. Protein quantification of each sample was performed using DC Protein Assay (Bio-Rad).
ELISA to determine TNFα levels in lumbar spinal cord after a T3Tx.
TNFα (Abcam catalog #AB100785) or nerve growth factor (NGF) ELISA kits (Millipore catalog #CYT304) were used to measure expression levels of TNFα or NGF in samples of lumbar spinal cord (as prepared above) from naive, uninjured rats (n = 4) or rats at 3 d, 1, 2, 3, and 4 weeks after a T3Tx (for the TNFα ELISA; n = 4 per time point) or from naive (n = 4), T3Tx-saline (n = 5), and T3Tx-XPro1595 (n = 5) rats 4 weeks after T3Tx (for the NGF ELISA) per the manufacturer's instructions.
Western blotting.
Equal amounts of proteins were resolved by SDS-PAGE on 10% or 15% gels, transferred to nitrocellulose membranes, and blocked in 5% nonfat milk in 0.1 m TBS-T for 1 h at room temperature. Following blocking, membranes were probed overnight at 4°C with an antibody recognizing either TNFR1 (Santa Cruz Biotechnology catalog #sc-8436, RRID:AB_628377), the phosphorylated form of the p65 subunit of the transcription factor NF-κB (pNF-κB p65; Cell Signaling Technology catalog #3031, RRID:AB_330559), or the NGF receptor TrkA (Cell Signaling Technology catalog #2505, RRID:AB_561332). Proteins were visualized with a chemiluminescent kit (ECL; GE Healthcare). After extensive washes in TBS-T, membranes were incubated with appropriate HRP-conjugated secondary antibodies for 30 min at room temperature. Detection was performed with Super-Signal West Pico chemiluminescent substrate (Thermo Fisher Scientific). Quantification was performed using Quantity One software from Bio-Rad. Blots were also probed for β-actin (Santa Cruz Biotechnology catalog #sc-47778, RRID:AB_626632) to allow for normalization.
Histology.
Naive animals and T3Tx-saline and -XPro1595 animals 4 weeks after SCI (n = 4 or 5 per group) were killed with Euthasol and perfused with 0.9% saline followed by 4% PFA. The spinal cords were dissected out, postfixed in the same fixative overnight at 4°C, and then cryoprotected in 30% sucrose for at least 48 h before cutting 30 μm transverse sections of lumbar spinal cord or 30 μm longitudinal sections of T6-T10 cord on a cryostat. Sections were blocked in 5% normal goat serum, 10% BSA, 0.1% Triton X-100 in PBS for 1 h. After blocking, thoracic sections were incubated with anti-c-Fos (Santa Cruz Biotechnology catalog #sc-52, RRID:AB_2106783) to visualize neurons activated by CRD and anti-ChAT (Millipore catalog #AB144, RRID:AB_90650) to visualize SPNs in the intermediolateral cell column (IML). Lower lumbar sections (L6) were incubated with anti-c-Fos to visualize neurons activated by CRD, anti-CGRP (Peninsula Laboratories catalog #T-5053.0050, RRID:AB_1113068) to visualize nociceptive primary afferents axons, anti-Iba1 (Wako catalog #019-19741, RRID:AB_839504) to visualize microglia, and anti-GFAP (Dako catalog #Z0334, RRID:AB_10013382) to visualize astrocytes. After overnight incubation with primary antibodies at 4°C, sections were washed in PBS, incubated with appropriate AlexaFluor-conjugated secondary antibodies (Invitrogen) for 2 h at room temperature, washed again in PBS, mounted onto slides, and coverslipped with FluorSave (EMD Chemical). Stained sections were analyzed on Olympus BX51 and Leica DM5500B epifluorescent microscopes and a Leica TCS SP2 confocal microscope equipped with a Leica DMRE microscope.
Quantification of histology.
Images of 4 equivalent sections per animal per were captured using Olympus BX51 and Leica DM5500B epifluorescent microscopes. For each of the antibodies listed above, sections were stained at the same time and images were taken with the same exposure time. Using ImageJ, an intensity threshold was applied to each image to include the positive labeling while minimizing the inclusion of nonspecific, background staining in sections containing ChAT+ SPNs near the central canal (for thoracic sections) or the dorsal horn and/or around the central canal (for lower lumbar sections). The same threshold values were used for each antibody. To assess the extent of immunoreactivity for c-Fos, the number of c-Fos+ nuclei in identically sized regions of dorsal horn and around the central canal (in lumbar sections) or in intermediate gray adjacent to the IML were counted. To assess the extent of immunoreactivity for Iba1 and GFAP, the thresholded pixel area in identically sized regions of dorsal horn was determined for each section. To determine CGRP immunoreactivity, thresholded pixel areas were determined in equally sized regions encompassing laminae I-IV or lamina VII/X (i.e., dorsal gray commissure around the central canal).
In vitro analysis of CGRP+ DRG neurite outgrowth.
Single-cell suspensions of DRG neurons were prepared as described previously (Tom et al., 2004). DRGs were harvested from adult Wistar rats (225–250 g, Charles River). After trimming the roots, the DRGs were incubated with collagenase (2000 U/ml) and neutral protease (25 U/ml; Worthington Biochemical) in HBSS (Invitrogen) at 37°C for 30 min. DRGs were rinsed several times with HBSS and then gently triturated in culture media that consisted of Neurobasal-A, B-27, GlutaMax, and penicillin/streptomycin (Invitrogen). After two rounds of low-speed spins (2000 rpm × 2 min), the pellet containing the dissociated DRG neurons was resuspended in culture media containing 20 μm of the antimitotic agent 5-fluoro-2′-deoxyuridine (Sigma-Aldrich) and plated onto glass coverslips coated with poly-L-lysine (0.1 mg/ml; Sigma-Aldrich) at a density of 2000 neurons/ml. In addition to a media control group, some cultures were treated with recombinant human TNFα (50 ng/ml; Millipore), the cell-permeable NF-êB inhibitor SN50 (10 μm; Millipore), or a combination of both. Two days later, the cultures were fixed with 4% PFA in 0.1 m PBS and then processed for immunocytochemistry. The coverslips were rinsed in fresh PBS, incubated in blocking solution (5% normal goat serum, 0.1% BSA, 0.1% Triton X-100 in PBS) for 1 h at room temperature and then in anti-CGRP diluted in blocking solution overnight at 4°C. The next day, the coverslips were rinsed in PBS and then incubated in the appropriate AlexaFluor-conjugated secondary antibody (Invitrogen) for 2 h at room temperature. The coverslips were rinsed in PBS, mounted onto glass slides using FluorSave (EMD Biosciences), and examined using an Olympus BX51 fluorescence microscope.
To quantify the CGRP+ neurites in the cultures, CGRP+ neurons (30 per condition) whose axons did not overlap with any other neuron were imaged. Using ImageJ, each CGRP+ neurite was traced using the freehand tool and the travel distance was measured. The longest neurite for each neuron was recorded, and the total CGRP+ neurite length per neuron was summed.
Experimental design and statistical analysis.
Animals were randomly assigned to treatment groups. Sample sizes were determined based upon our previous published studies using similar techniques. All analyses were performed blinded to treatment group. To determine differences between two groups, a Student's t test was performed. A two-way ANOVA and post hoc Fisher's LSD tests were performed to ascertain differences between three groups over time (i.e., hemodynamic recording analyses). A one-way ANOVA and post hoc Fisher's LSD tests were performed to assess for differences between three or more groups in which time was not a consideration (i.e., histological and biochemical analyses). A p value <0.05 was considered significant. All statistical tests were performed using GraphPad Prism 7.
Results
Sustained elevation of TNFα and TNFR1 below a complete, high-thoracic SCI
We assessed whether there was a persistent neuroimmune response in lumbar spinal cord, far from a high-thoracic, complete SCI site, where aberrant plasticity associated with AD has been observed (Krenz et al., 1999; Marsh et al., 2002; Cameron et al., 2006; Hou et al., 2009). We transected (Tx) the spinal cord of adult Wistar rats at thoracic level 3 (T3), which reliably results in AD by 2 weeks after injury (Krassioukov and Weaver, 1995; Krenz et al., 1999; Cameron et al., 2006). We examined whether sTNFα is elevated in lumbar cord at several time points (3 d, 4 weeks) after T3Tx using ELISA (Fig. 2a). Although there was some sTNFα in naive lumbar tissue, expression was significantly higher at 1 week and 4 weeks after T3Tx (Fig. 2a; F(5,25) = 2.426, p = 0.0635, post hoc, vs 1 week p = 0.0478; vs 4 weeks p = 0.0094). Moreover, levels of TNFR1, the primary receptor for sTNFα, were significantly higher below a T3Tx than in naive tissue at all time points (Fig. 2b,c; F(5,12) = 82.02, p < 0.0001, post hoc, vs 3 d p = 0.0004; vs 1 week p < 0.0001; vs 2 weeks p = 0.0075; vs 3 weeks p < 0.0001; vs 4 weeks p < 0.0001). Additionally, there was a marked additional spike in TNFR1 levels between 2 weeks and 3 weeks after T3Tx that remained at 4 weeks (F(5,12) = 82.02, p < 0.0001, post hoc, 2 weeks vs 3 weeks p < 0.0001; 2 weeks vs 4 weeks p < 0.0001).
Figure 2.

Intrathecal administration of XPro1595 diminishes signaling downstream of persistent sTNFα/TNFR1 expression below a high-thoracic SCI. a, Levels of TNFα protein in lumbar spinal cord were assessed via ELISA in naive rats and injured rats at different after T3Tx time points (3 d, 4 weeks; n = 4 or 5 per group). Even 1 month after injury, sTNFα levels well below a T3Tx were significantly higher than in naive lumbar spinal cord. b–e, An expression time course of TNFR1 or the phosphorylated (i.e. activated) p65 subunit of NF-κB (pNF-κB p65), a downstream effector of TNFR1 activation, in lumbar spinal cord of uninjured rats or T3Tx rats treated with saline, was determined via Western blot (n = 3 per group). Levels of TNFR1 (b, c) and pNF-κB p65 (d, e) well below a T3Tx were significantly higher than that observed in naive controls, regardless of time point, after SCI. Additionally, there was a significant spike in both TNFR1 and pNF-kB p65 levels between 2 and 3 weeks after T3Tx. f–i, Expression levels of TNFR1 and pNF-κB p65 in lumbar spinal cord of naive rats or T3Tx rats treated with saline or the sTNFα biologic XPro1595 4 weeks after SCI were determined via Western blot (n = 4 or 5 per group). Levels of TNFR1 well below a T3Tx were significantly higher than that observed in naive controls, regardless of whether saline or XPro1595 was infused (f, g). However, despite similarities in TNFR1 expression, inhibiting sTNFα signaling with XPro1595 significantly diminished levels of pNF-κB p65, compared with T3Tx-saline animals. Levels in T3Tx-XPro1595 animals were indistinguishable from that observed in naive animals (h, i). Data are mean ± SEM. p values were determined by a Student's t test or one-way ANOVA and post hoc Fisher's LSD tests: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, #p < 0.0001 versus 2 weeks. %p < 0.001 versus 2 weeks. @p < 0.01.
We examined whether this increase in sTNFα and TNFR1 levels augmented activation (i.e., phosphorylation) of the p65 subunit of nuclear factor-κB (pNF-κB p65), a transcription-regulating protein complex that is downstream of sTNFα binding to and activating TNFR1 (Baud and Karin, 2001). Because sTNFα and TNFR1 levels are elevated after SCI (Fig. 2a–c), it was not surprising to see that pNF-κB p65 levels were also significantly higher after T3Tx at all time points examined (Fig. 2d,e; F(5,12) = 20.6, p < 0.0001, post hoc, vs 3 d p = 0.0004; vs 1 week p = 0.0002; vs 2 weeks p = 0.0048; vs 3 weeks p < 0.0001; vs 4 weeks p < 0.0001). Similar to what we saw with TNFR1 levels, we saw an additional surge in pNF-κB p65 levels after 2 weeks (F(5,12) = 20.6, p < 0.0001, post hoc, 2 weeks vs 3 weeks p = 0.0001; 2 weeks vs 4 weeks p = 0.0013). These data indicate that SCI induces a long-lasting increase in both the proinflammatory cytokine sTNFα and its receptor that results in increased downstream, NF-κB signaling.
XPro1595 decreases NF-κB activation downstream of sTNFα-TNFR1 signaling
To ensure that intrathecal administration of XPro1595 after SCI sufficiently dampened sTNFα signaling caudal to a complete SCI, we examined whether 4 weeks of continuous XPro1595 affected levels of TNFR1 or pNF-κB p65. Although XPro1595 did not affect the injury-induced increase in TNFR1 levels at 4 weeks after SCI (Fig. 2f,g; F(2,10) = 12.895, p = 0.0017, post hoc, vs naive p = 0.0024), it did return pNF-κB p65 levels to those seen in uninjured cord (Fig. 2h,i), indicating that XPro1595 significantly stifled the effect of elevated sTNFα/TNFR1 well below an SCI (F(2,10) = 69.07, p < 0.0001, post hoc, vs T3Tx-saline p < 0.0001).
Microglia and astrocytes are persistently reactive remote to an SCI
To determine whether prolonged sTNFα/TNFR1 signaling was associated with increased neuroinflammation well below an SCI, we stained sections of lumbar spinal cord from naive, uninjured animals and T3Tx animals treated with either saline or XPro1595 4 weeks after SCI for Iba-1 to identify microglia and for GFAP to identify astrocytes. Immunostaining across each section appeared uniform, so we focused our analysis on dorsal horn, an area in which plasticity has been implicated in AD (Krenz et al., 1999; Cameron et al., 2006). Iba-1+ microglia in naive cord were highly ramified (Fig. 3a,a′), indicative of a resting phenotype. There was significantly more Iba-1+ immunoreactivity in lumbar cord below a T3Tx (Fig. 3d; F(2,8) = 13.07, p = 0.0030, post hoc, p = 0.0012) and the Iba-1+ microglia had short processes, characteristic of a reactive phenotype (Fig. 3b,b′). We observed significantly less Iba-1 immunoreactivity in the T3Tx-XPro1595 animals than the T3Tx-saline ones (Fig. 3d; F(2,8) = 13.07, p = 0.0030, post hoc, p = 0.0156), although more than the uninjured animals (F(2,8) = 13.07, p = 0.0030, post hoc, p = 0.0443). Furthermore, the Iba-1+ microglia in the XPro1595-administered animals appeared to have more ramified processes than in saline-treated animals (Fig. 3c,c′).
Figure 3.

Inhibiting sTNFα/TNFR1 dampens microglial reactivity but does not reduce astrocytic gliosis. a–c, Representative confocal images of immunostaining of Iba-1+ microglia in lumbar spinal cord from naive, uninjured animals or T3Tx animals 4 weeks after SCI that were treated with either saline or XPro1595 (n = 3 per group). In naive animals, Iba-1+ microglia took on a highly ramified morphology typical of resting microglia (a, a′). There was significantly more Iba-1+ immunoreactivity in tissue from T3Tx-saline animals (d). The Iba-1+ microglia in these animals had short processes and took on the phenotype of more reactive cells (b, b′). Iba-1+ immunoreactivity in T3Tx-XPro1595 animals were less than their saline-treated counterparts but also were greater than naive levels (c, c′, d). e–g, Representative confocal images of immunostaining of GFAP+ astrocytes in lumbar spinal cord from naive, uninjured animals or T3Tx animals 4 weeks after SCI that were treated with either saline or XPro1595 (n = 4 per group). Higher magnification images of the boxed regions in e–g are shown in e′–g′. Astrocytes in naive animals show morphology typical of a resting astrocyte (e). T3Tx resulted in significantly more GFAP+ immunoreactivity, regardless of treatment (f–h). The GFAP+ astrocytes in both T3Tx-saline and -XPro1595 animals appeared hypertrophic and took on a phenotype typical of more reactive astrocytes (f, g). There was no difference in GFAP+ immunoreactivity between treatment groups (h). Data are mean ± SEM. *p < 0.05 (one-way ANOVA and post hoc Fisher's LSD test). **p < 0.01 (one-way ANOVA and post hoc Fisher's LSD test). ****p < 0.0001 (one-way ANOVA and post hoc Fisher's LSD test). Scale bars: a–c, 37.5 μm; e–g, 25 μm.
In similarly located sections, there was significantly more GFAP+ immunoreactivity in animals with T3Tx, regardless of treatment, than in uninjured animals, suggesting that astrocytes remain reactive long after an SCI (Fig. 3e–h; F(2,13) = 30.92, p < 0.0001, post hoc, vs T3Tx-saline p < 0.0001; vs T3Tx-XPro1595 p < 0.0001). We did not observe any difference in GFAP+ immunoreactivity between T3Tx-saline and -XPro1595 rats, suggesting that inhibiting sTNFα does not grossly attenuate astrocytic gliosis in lumbar cord. These data indicate that inhibiting sTNFα with XPro1595 attenuates activation of microglia but not astrocytes in tissue well below an SCI.
Intrathecal administration of XPro1595 after SCI diminishes the severity of AD
To determine whether sTNFα plays a role in the progressive exacerbation of AD over time (Krassioukov and Weaver, 1995; Rabchevsky et al., 2012; Zhang et al., 2013; West et al., 2015), we implanted a radiotelemetry pressure transducer into the descending aortas of adult rats before T3Tx and intrathecal treatment with saline or XPro1595 (Fig. 1), allowing for longitudinal monitoring of BP and HR in the same animals at various time points. Baseline MAP (Fig. 4a) and HR (Fig. 4b) were similar between the T3Tx-saline and T3Tx-XPro1595 animals at all time points, indicating that chronic XPro1595 infusion did not affect basal hemodynamics.
Figure 4.

XPro1595 does not affect basal hemodynamics. T3Tx animals treated with XPro1595 or saline (n = 8 or 9 per group) were implanted with radiotelemetry pressure transducers into the descending aorta to allow for recording BP and HR in animals at various time points. Baseline MAP (a) and HR (b) of T3Tx-saline and T3Tx-XPro1595 were similar at all time points, indicating that XPro1595 did not grossly affect hemodynamics.
CRD is a well-established experimental means to induce an AD episode (Krassioukov and Weaver, 1995; Krenz et al., 1999; Cameron et al., 2006; Hou et al., 2013b) that mimics constipation. We examined hemodynamic parameters before, during, and after subjecting the T3Tx-saline and -XPro1595 animals to CRD. CRD quickly elicited AD events in both T3Tx-saline and T3Tx-XPro1595 animals (Fig. 5a,b). However, in the T3Tx-saline animals, there was a strong trend for an increase in the magnitude of CRD-induced hypertension between 2 and 4 weeks (p = 0.0524). Moreover, the MAP spikes were significantly reduced in the T3Tx-XPro1595 animals compared with those in the T3Tx-saline rats at 2, 3, and 4 weeks after injury (Fig. 5c; F(1,48) = 49.14, p < 0.0001; post hoc, at 2 weeks p = 0.0025, at 3 weeks p = 0.0002, at 4 weeks p < 0.0001). Moreover, XPro1595 treatment markedly reduced the time to return to baseline MAP after the CRD was relieved at all time points (Fig. 5d; F(1,45) = 33.08, p < 0.0001, post hoc, at 2 weeks p = 0.0054, at 3 weeks p = 0.0167, at 4 weeks p < 0.0001). We did not observe significant differences in reflexive bradycardia, although there was a strong trend toward a significant difference between groups at 4 weeks (Fig. 5e).
Figure 5.

XPro1595 treatment mitigates colorectal distension-induced AD. At 2, 3, and 4 weeks after T3Tx, HR and BP were determined in rats before, during, and after CRD, a well-established experimental means to elicit an AD episode that mimics constipation, a common trigger in SCI patients. a, b, Representative beat-to-beat arterial pressure traces from T3Tx-saline and T3Tx-XPro1595 animals 4 weeks after SCI. Red line indicates the 1 min CRD interval. White line indicates MAP. T3Tx-saline animals had stereotypical sharp spikes in MAP in response to CRD. T3Tx-XPro1595 animals exhibited a dramatically smaller increase in MAP. c, Quantification of the increase in MAP above baseline in response to CRD. The MAP increase in XPro1595-treated animals was significantly smaller than in saline-treated animals at every time point. Furthermore, there was a strong trend that the BP spike in the saline-treated animals progressively increased from 2 to 4 weeks (p = 0.054), further suggesting that AD worsens over time. Conversely, the MAP increase in the T3Tx-XPro1595 animals was stable at all 3 time points. d, The time that it took to return to baseline MAP after 1 min of CRD was significantly longer in T3Tx-saline animals than T3Tx-XPro1595 animals. e, We observed no significant differences in bradycardia (bpm; beats per minute) in response to CRD between groups, although there was a strong trend at 4 weeks (p = 0.051). Data are mean ± SEM. *p < 0.05 (two-way ANOVA and post hoc Fisher's LSD test). **p < 0.01 (two-way ANOVA and post hoc Fisher's LSD test). ***p < 0.001 (two-way ANOVA and post hoc Fisher's LSD test). ****p < 0.0001 (two-way ANOVA and post hoc Fisher's LSD test).
Although CRD is a well-controlled means to trigger an AD episode, patients experience multiple, “naturally occurring” episodes daily that are triggered by “everyday” stimuli, such as blocked catheters, pressure sores, impacted fecal matter, etc. Similarly, rodents also experience such naturally occurring episodes (Rabchevsky et al., 2012; Zhang et al., 2013; West et al., 2015). To further assess the degree to which XPro1595 diminishes AD, at 2, 3, and 4 weeks after T3Tx, the animals' BP and HR were recorded over a continuous 24 h period while they moved freely in their cages. Using a combination of parameters from established algorithms (Rabchevsky et al., 2012; Zhang et al., 2013; West et al., 2015), we determined the number of naturally occurring AD bouts during the recording interval (Fig. 6a,b). Both T3Tx-saline and T3Tx-XPro1595 animals had similar numbers of events at 2 weeks (Fig. 6c). However, AD in the T3Tx-saline animals intensified over time, the number of detected events (Fig. 6c; F(1,47) = 8.162, p = 0.0064, post hoc, p = 0.0263) and the peak MAP during each event significantly increased between 2 and 4 weeks (Fig. 6d; F(1,340) = 41.6, p < 0.0001, post hoc, p = 0.0171). In contrast, the AD events in T3Tx-XPro1595 animals were remarkably consistent over time and did not increase in frequency (Fig. 6c) or magnitude (Fig. 6d). Moreover, XPro1595-treated animals had lower peak BP per event at all time points (Fig. 6d; F(1,340) = 41.6, p < 0.0001, post hoc, at 2 weeks p < 0.0001, at 3 weeks p = 0.0105, at 4 weeks p < 0.0001) and significantly shorter events at 3 weeks (Fig. 6e; F(1,119) = 7.07, p = 0.0089, post hoc, p = 0.0299). Collectively, these data indicate that inhibiting spinal sTNFα below SCI attenuates the severity and progressive exacerbation of AD.
Figure 6.

Inhibiting sTNFα signaling decreases the frequency and severity of naturally occurring AD episodes after complete T3 SCI. At 2, 3, and 4 weeks after injury, BP and HR were continuously recorded over 24 h in T3Tx animals treated with XPro1595 or saline (n = 8–9 per group) while they moved freely in their cages. a, b, Representative MAP traces from a T3Tx-saline or a T3Tx-XPro1595 animal over a 24 h period 4 weeks after injury. Red asterisks indicate naturally occurring (i.e., not experimentally induced) AD events detected using MATLAB followed by manual verification. c, Quantification of the frequency of naturally occurring events at 2, 3, and 4 weeks after T3Tx. At 2 weeks, animals from both groups experienced similar numbers of AD episodes. The number of events did not change over time in the XPro1595-treated animals. However, the number of daily events the saline-treated animals experienced significantly increased by 4 weeks. Furthermore, these animals had sharply more episodes than their XPro1595-treated counterparts. d, T3Tx-XPro1595 animals had significantly lower peak MAP during each detected event at all time points assessed. Furthermore, while the highest MAP in the XPro1595-treated animals across weeks were similar, the peak MAP in the saline-treated animals significantly increased over time. e, The time that MAP remained >20 mmHg above baseline was considered the duration of each naturally occurring AD event. XPro1595-treated T3Tx rats had significantly shorter events than saline-treated rats at 3 weeks. Data are mean ± SEM. *p < 0.05 (two-way ANOVA and post hoc Fisher's LSD test). ****p < 0.0001 (two-way ANOVA and post hoc Fisher's LSD test).
Inhibiting sTNFα within the spinal cord decreases peripheral vascular dysfunction after complete high-thoracic SCI
Because AD is associated with detrimental changes to the peripheral vasculature that contribute to cardiovascular disease (e.g., hyper-responsiveness to vasopressors) (Alan et al., 2010; Rummery et al., 2010; West et al., 2016), we assessed whether intrathecal delivery of XPro1595 that diminishes AD also attenuates pathological remodeling of arteries in the splanchnic vascular bed. We isolated small mesenteric arteries from T3Tx-saline and -XPro1595 animals 4 weeks after SCI for comparison with arteries from naive, uninjured animals. Mesenteric arteries from T3Tx-saline rats had smaller lumen diameters (Fig. 7a; F(2,13) = 3.892, p = 0.0473, post hoc, p = 0.0382) and thinner walls (Fig. 7b; F(2,14) = 4.595, p = 0.0292, post hoc, p = 0.0244) than naive arteries. We did not see significant differences between arteries from naive animals that do not experience AD (Krassioukov and Weaver, 1995) and T3Tx-XPro1595 animals that experienced less frequent and less intense AD (Figs. 5, 6).
Figure 7.

Intrathecally administered XPro1595 diminishes peripheral vascular dysfunction after high-level SCI. Mesenteric arteries were harvested from naive animals (n = 5) or T3Tx-saline (n = 7) and T3Tx-XPro1595 rats (n = 6) 4 weeks after SCI for ex vivo analysis. a, b, The lumen diameters and wall thicknesses of isolated small mesenteric arteries were determined. Arteries from T3Tx-saline animals had a significantly narrower lumen and a thinner wall than those from naive animals. Arteries from T3Tx-XPro1595 animals were not statistically different from naive animals. c–e, We assessed mesenteric artery reactivity to increasing concentrations of the vasopressor PE. There was a leftward shift of the PE concentration-response curve for arteries from T3Tx-saline animals (c). Moreover, the concentration of PE required for arteries to reach 50% constriction (pEC50) was lowest in T3Tx-saline animals (which experienced the most severe AD) (d), indicating that arteries from T3Tx-saline animals were hyper-responsive to PE. XPro1595 decreased percentage maximum constriction in response to PE (e). f, g, We also examined the response of the isolated arteries to the endothelium-dependent vasodilator ACh. We did not observe any group differences in ACh-induced vasodilation at any concentration. The concentration-response curves (f), pEC50 (g), and percentage maximal dilation (h) of the arteries from the four groups were similar, indicating that the enhanced constriction (c, d) was not due to diminished vasodilation. Data are mean ± SEM. p values were determined by one-way ANOVA and post hoc Fisher's LSD tests: ∧T3Tx-saline versus Naive (p < 0.01); T3Tx-saline versus T3Tx-XPro1595 (p < 0.05). #T3Tx-saline versus T3Tx-XPro1595 (p < 0.05). *p < 0.05.
We also assessed vascular reactivity of the isolated arteries to increasing concentrations of the vasoconstrictor PE or the vasodilator ACh using an ex vivo pressurized vessel preparation. The PE concentration-response curve of T3Tx-saline animals was shifted leftward relative to curves generated with arteries from naive and T3Tx-XPro1595 animals (Fig. 7c). Arteries from T3Tx-saline rats showed more robust constriction at 10−7 m (Fig. 7c) than either naive, uninjured animals (F(2,14) = 4.408, p = 0.0327, post hoc, p = 0.0142) or T3Tx-XPro1595 animals (F(2,14) = 4.408, p = 0.0327, post hoc, p = 0.0449). Arteries from T3Tx-saline animals also had the lowest PE pEC50 (i.e., the concentration to achieve 50% constriction) compared with the other groups (Fig. 7d; F(2,14) = 3.528, p = 0.0574, post hoc, vs naive p = 0.0476, vs T3Tx-XPro1595 p = 0.0320). Maximal constriction to PE in T3Tx-XPro1595 animals was similar to naive animals but was significantly lower than T3Tx-saline animals (Fig. 7e; F(2,15) = 3.718, p = 0.0488, post hoc, p = 0.0226). Arteries from all groups showed similar endothelium-dependent vasodilation; we saw no significant differences in the ACh concentration-response curves (Fig. 7f,g) or maximal vasodilatory response (Fig. 7h), indicating that the increased constriction to PE was not a secondary consequence of impaired endothelium-dependent vasodilatation. Collectively, these data demonstrate that inhibiting sTNFα locally in the spinal cord mitigates peripheral vascular dysfunction after high-thoracic SCI.
Inhibiting sTNFα within the spinal cord decreases peripheral immune dysfunction after complete high thoracic SCI
We wanted to determine whether XPro1595 treatment also attenuates peripheral immune dysfunction resultant from AD (Zhang et al., 2013; Ueno et al., 2016). Splenic leukocytes were harvested from uninjured animals and T3Tx-saline and -XPro1595 animals 4 weeks after SCI and characterized via flow cytometry. T3Tx-saline animals had significantly fewer CD45R+ B cells (Fig. 8a; F(2,15) = 15.31, p = 0.0002, post hoc, vs naive p < 0.0001, vs T3Tx-XPro1595 p = 0.0012) and CD8+ T cells (Fig. 8b; F(2,15) = 4.837, p = 0.0239, post hoc, vs naive p = 0.0094, vs T3Tx-XPro1595 p = 0.0388). T3Tx-saline animals also had the most CD11b/c+ monocytes/macrophages (Fig. 8e; F(2,15) = 7.398, p = 0.0058; post hoc, vs naive p = 0.0028, vs T3Tx-XPro1595 p = 0.0085) and CD68+ monocytes/macrophages (Fig. 8f; F(2,15) = 7.398, p = 0.0058; post hoc, vs naive p = 0.0028, vs T3Tx-XPro1595 p = 0.0085), suggestive of compromised immune function. T3Tx-XPro1595 had normal (i.e., similar to naive) levels of CD45R+ B cells, CD8+ T cells, and CD11b/c+ monocytes/macrophages (Fig. 8a,b,e). Levels of CD68+ macrophages were in between those seen in uninjured and T3Tx-saline animals (Fig. 8f). Although all groups had similar numbers of CD4+ T cells (Fig. 8c), interestingly, T3Tx-XPro1595 animals had significantly more CD4+CD25+FoxP3+ regulatory T cells (TReg) than either T3Tx-saline or naive animals (Fig. 8d; F(2,15) = 6.213, p = 0.0108, post hoc, vs naive p = 0.0323, vs T3Tx-saline p = 0.0036). These data indicate that inhibiting central sTNFα prevents AD-mediated compromised immune function.
Figure 8.

Intrathecally administering XPro1595 averts peripheral immune dysfunction after T3 SCI. Spleens from naive, uninjured animals and T3Tx-saline and -XPro1595 animals 4 weeks after SCI were harvested and processed for flow cytometry (n = 6 per group). a, b, T3Tx-saline animals had a smaller proportion of total leukocytes that were CD45R+ B cells (a) or CD8+ T cells (b) than naive and T3Tx-XPro1595 animals. There was no significant difference in the percentage of cells containing these markers between naive and T3Tx-XPro1595 animals. c, The proportion of leukocytes that were CD4+ T cells was similar across groups. d, T3Tx-XPro1595 animals had a greater percentage of CD4+ T cells that were also CD25+ FoxP3+ regulatory T cells (TReg) than both uninjured and T3Tx-saline animals. e, T3Tx-saline animals had a higher percentage of total leukocytes that were CD11b/c+ than both uninjured and T3Tx-XPro1595 animals. f, Moreover, T3Tx-saline animals had a greater percentage that were CD68+ monocytes/macrophages than uninjured animals. T3Tx-XPro1595 animals had levels of CD68+ monocytes/macrophages that were in between and statistically similar to those seen in naive and T3Tx-saline animals. Data are mean ± SEM. *p < 0.05 (one-way ANOVA and post hoc Fisher's LSD test). **p < 0.01 (one-way ANOVA and post hoc Fisher's LSD test). ****p < 0.0001 (one-way ANOVA and post hoc Fisher's LSD test).
Inhibiting sTNFα decreases neuronal activation below T3Tx in response to colorectal distension
SCI induces changes in the spinal sympathetic reflex circuit that results in sensory stimuli below the level of injury (e.g., CRD) activating more sympathetically correlated interneurons caudal to T3Tx than in a naive setting (Krassioukov et al., 2002; Landrum et al., 2002; Hou et al., 2008; Ueno et al., 2016). We examined the degree of c-Fos induction, a marker of neuronal activation, after an extended period of intermittent CRD (Landrum et al., 2002; Hou et al., 2008; Ueno et al., 2016) in T3Tx-saline or T3Tx-XPro1595 animals 4 weeks after SCI. We focused on L6 spinal cord because that is the spinal entrance point of primary afferents carrying visceral sensory information (e.g., CRD) (Cameron et al., 2006). CRD in T3Tx-saline animals robustly induced c-Fos expression in interneurons in deeper layers of L6 dorsal horn (Fig. 9a,c) and laminae VII/X (Fig. 9d,f). CRD in T3Tx-XPro1595 animals induced detectable c-Fos in significantly fewer interneurons in both the dorsal horn region (Fig. 9b,c; t(18) = 9.291, p < 0.0001) and laminae VII/X (Fig. 9d,f; t(19) = 5.995, p < 0.0001). This is particularly notable because these deeper laminae contain sympathetically correlated interneurons that receive sensory input and project to the IML in thoracic spinal cord (Clarke et al., 1998; Matsushita, 1998; Tang et al., 2004; Hofstetter et al., 2005).
Figure 9.

Inhibiting sTNFα signaling decreases CRD-elicited activation of interneurons caudal to T3 SCI. a–i, Activated c-Fos+ (green) interneurons after CRD were immunohistochemically assessed in T3Tx-saline or T3Tx-XPro1595 animals 4 weeks after SCI (n = 3 or 4/group). CRD in T3Tx-saline animals induced c-Fos in many interneurons within L6 dorsal horn (a, c) and dorsal gray commissure in laminae VII/X (d, f), which contains neurons that relay visceral sensory input (e.g., CRD) to SPNs in thoracic cord. There were fewer c-Fos+ neurons in these regions in T3Tx-XPro1595 animals (b, c, e, f). We also observed fewer c-Fos+, ChAT− interneurons in close proximity to ChAT+ (red) SPNs in the T9 IML in T3Tx-XPro1595 animals than T3Tx-saline animals. This indicates that CRD activated fewer sympathetically correlated interneurons near SPNs (g–i). Data are mean ± SEM. *p < 0.05 (Student's t test). ****p < 0.0001 (Student's t test). Scale bar, 50 μm. cc, Central canal.
Interneurons near SPNs within the IML also play a crucial role in relaying sensory information to the SPNs and eliciting an AD episode (Ueno et al., 2016). CRD in T3Tx-saline animals induced c-Fos in many ChAT+ SPNs (Fig. 9g) within the IML and neighboring ChAT− interneurons (Fig. 9g,i) in T9 cord. There were significantly fewer c-Fos+/ChAT− interneurons after CRD in XPro1595 animals (Fig. 9h,i; t(11) = 2.743, p = 0.0191). Collectively, these data indicate that inhibiting sTNFα after T3Tx dampens CRD-triggered activation of interneurons within the spinal sympathetic reflex circuit.
sTNFα signaling promotes arborization of CGRP+ primary afferents after SCI and CGRP+ neurite growth in vitro
As mentioned, sympathetically correlated interneurons in lumbosacral cord (e.g., those examined in Fig. 9) receive input from primary afferents, including calcitonin gene-related peptide (CGRP)+ fibers that carry visceral nociceptive information (e.g., CRD). After T3Tx, the heightened response of these interneurons to sensory stimuli caudal to the injury (Landrum et al., 2002; Hou et al., 2008) (corroborated in Fig. 9) is thought to result from sprouting of CGRP+ fibers onto these interneurons (Krenz and Weaver, 1998; Jacob et al., 2001; Weaver et al., 2001; Cameron et al., 2006) that then relay this activation to the SPNs in the IML. Moreover, the degree of CGRP+ sprouting positively correlates with the magnitude of AD episodes, inhibiting injury-induced sprouting of CGRP+ fibers attenuates AD (Krenz et al., 1999; Weaver et al., 2001; Marsh et al., 2002; Cameron et al., 2006).
We examined CGRP+ immunoreactivity in lower lumbar spinal cord from naive or T3Tx-saline and T3Tx-XPro animals 4 weeks after SCI. Normally, CGRP+ afferents are restricted to the superficial Rexed laminae (I-II) of dorsal horn, although a few fibers are located in deeper dorsal horn laminae (Fig. 10a,a′) and in laminae VII/X around the central canal (Fig. 10e,e′). As mentioned above, neurons in the dorsal gray commissure in these latter laminae have been postulated to be a hub for nociceptive and visceral sensory information that is then relayed rostrally (Matsushita, 1998), including to the SPNs (Hou et al., 2008). There was significantly more CGRP+ immunoreactivity in the T3Tx-saline animals, both in the dorsal horn, particularly in the deeper layers (Fig. 10b,b′,d; F(2,24) = 7.505, p = 0.0029, post hoc, p = 0.0190), and around the central canal (Fig. 10f,f′,h; F(2,25) = 5.661, p = 0.0094, post hoc, p = 0.0066). Inhibiting sTNFα with XPro1595 attenuated this arborization. There was less CGRP+ staining in the dorsal horn (Fig. 10c,c′; F(2,24) = 7.505, p = 0.0029, post hoc, p = 0.0012) and the dorsal gray commissure in laminae VII/X (Fig. 10g,g′; F(2,25) = 5.661, p = 0.0094, post hoc, p = 0.0211) in T3Tx-XPro1595 animals than in T3Tx-saline animals and was similar to naive animals (Fig. 10d,h).
Figure 10.

Inhibiting sTNFα signaling decreases CGRP+ afferent sprouting caudal to T3 SCI. a–h, Transverse sections of L6 cord from naive or T3Tx-saline and T3Tx-XPro1595 animals 4 weeks after SCI immunostained for CGRP (n = 4 or 5/group). In naive animals, CGRP+ staining was primarily in superficial layers, although some was in the deeper layers of dorsal horn (a, a′). We observed few CGRP+ fibers around laminae VII/X (i.e., dorsal gray commissure) (e, e′). There was increased CGRP immunoreactivity in the dorsal horn of T3Tx-saline animals (b, d), particularly in the deeper layers (b′), and around the central canal (f, f′, h). In T3Tx-XPro1595 animals, we observed normal CGRP+ fiber density in the dorsal horn (c, c′, d) and laminae VII/X (g, g′, h). i, ELISA analysis of NGF expression in lumbar cord from naive (n = 6) or T3Tx-saline (n = 4) and T3Tx-XPro1595 (n = 5) animals 4 weeks after SCI. T3Tx increased NGF expression, even with XPro1595 treatment. j, k, TrkA protein levels in lumbar cord of T3Tx-saline animals 4 weeks after SCI were lower than in naive animals. Inhibiting sTNFα signaling further diminished TrkA expression. Data are mean ± SEM. *p < 0.05 (one-way ANOVA and post hoc Fisher's LSD test). **p < 0.01 (one-way ANOVA and post hoc Fisher's LSD test). ***p < 0.001 (one-way ANOVA and post hoc Fisher's LSD test). k, The last sample in the blot was an outlier, as determined by the Grubbs' method with α = 0.05, and was excluded from the bar graph in j. Scale bars, 100 μm. cc, Central canal.
One established mechanism for CGRP+ fiber growth is NGF/TrkA signaling. CGRP+ fibers are virtually the only spinal axon population to express TrkA and respond to NGF (Averill et al., 1995; Michael et al., 1997; Abbott et al., 2006; Molofsky et al., 2014). After T3Tx, NGF levels in lumbar cord increase (Fig. 10i; F(2,12) = 8.55, p = 0.0049, post hoc, p = 0.0304) and TrkA levels decrease (Fig. 10j,k; F(2,9) = 19.42, p = 0.0005, post hoc, p = 0.0037). XPro1595 treatment did not affect the injury-triggered induction of NGF (Fig. 10i; F(2,12) = 8.55, p = 0.0049, post hoc, p = 0.0016), but it did further decrease TrkA expression levels (Fig. 10j,k; F(2,9) = 19.42, p = 0.0005, post hoc, vs Naive p = 0.0002, vs T3Tx-saline p = 0.0485). These data suggest that inhibiting sTNFα signaling diminishes afferent sprouting via decreasing TrkA-mediated sensitivity to NGF. As NGF is expressed primarily in non-neuronal cells after SCI (Krenz and Weaver, 2000; Brown et al., 2004) and TrkA is exclusively expressed in neuronal processes, predominantly CGRP+ afferents (Averill et al., 1995; Michael et al., 1997; Molofsky et al., 2014), these data also suggest that sTNFα triggers afferent sprouting by acting upon the neurons directly.
To further explore this possibility, we turned to a dissociated adult DRG neuronal culture system. Notably, these adult neurons can be reliably maintained in serum-free media in the absence of any neurotrophic factors (Tom et al., 2004). Treating adult DRG neurons with sTNFα in vitro enhances growth of CGRP+ neurites, both total growth (Fig. 11b,e; F(3,94) = 10.14, p < 0.0001, post hoc, p < 0.0001) and the length of the longest neurite (Fig. 11b,f; F(3,94) = 13.15, p < 0.0001, post hoc, p < 0.0001), compared with media controls (Fig. 11a). Adding SN50, a cell-permeable compound that specifically inhibits NF-κB activation (Lin et al., 1995) that occurs downstream of sTNFα/TNFR1 signaling, abolished the growth-enhancing effects of sTNFα (Fig. 11d–f; total growth: F(3,94) = 10.14, p < 0.0001, post hoc, p = 0.0015; longest neurite: F(3,94) = 13.15, p < 0.0001, post hoc, p = 0.0213). SN50 alone had no effect on growth (Fig. 11c) and CGRP+ neurite outgrowth was similar to that in control cultures (Fig. 11a). In all CGRP+ axons, regardless of treatment, CGRP+ staining extended the length of βIII-tubulin+ staining, suggesting that there was no significant difference in the transport of CGRP down the axon (data not shown). These data, combined with those in Figure 10, indicate that injury-induced sTNFα signaling exacerbates AD by triggering CGRP+ sprouting through a variety of mechanisms.
Figure 11.

sTNFα promotes growth of CGRP+ neurites in an NF-κB -dependent manner. Dissociated adult DRGs plated on poly-L-lysine were incubated with just serum-free media or serum-free media containing sTNFα (50 ng/ml), the cell-permeable NF-κB inhibitor SN50 (10 μm), or both sTNFα and SN50 for 48 h. Cultures were stained to visualize CGRP+ neurons and their neurites. Compared with the growth seen in the control cultures (a), adding sTNFα to the culture media increases both the total CGRP+ growth per neuron (b, e) as well as the length of the longest neurite (f). While the presence of the NF-κB inhibitor SN50 alone did not negatively affect growth compared with control cultures (c, e, f), its presence did nullify the growth-promoting effects of sTNFα (d–f). These data indicate that sTNFα is also capable of directly promoting the growth of CGRP+ nociceptive sensory neurons. Data are mean ± SEM. *p < 0.05 (one-way ANOVA and post hoc Fisher's LSD tests). **p < 0.01 (one-way ANOVA and post hoc Fisher's LSD tests). ****p < 0.0001 (one-way ANOVA and post hoc Fisher's LSD tests). n = 30 neurons per condition. Scale bar, 50 μm.
Discussion
Although the exacerbation of AD after SCI is well documented (Krassioukov and Weaver, 1995; Zhang et al., 2013), the mechanisms underlying AD are not well understood, limiting the development of treatments aimed at managing it. We identified a novel mechanism for AD progression, persistent sTNFα signaling and associated neuroinflammation well below the site of SCI long after the time of injury and found that inhibiting sTNFα is sufficient to attenuate AD intensification over time (Fig. 6). The timing of this exacerbation (between 2 and 3 weeks after SCI) corresponds with a significant, additional surge in TNFR1 and pNF-κB p65 activation (Fig. 2), further supporting that sTNFα/TNFR1 signaling plays a role. To our knowledge, this is the first study to elucidate a potential preventative strategy that mitigates AD and averts ensuing cardiovascular and immune function after SCI. Intrathecal delivery of XPro1595 not only diminished AD but also had profound, beneficial effects on peripheral vasculature and splenic leukocytes (summarized in Fig. 12). Future studies will determine the critical period when injury-induced sTNFα exerts its effects to elucidate the window of XPro1595 administration. While SCI quickly induces sTNFα expression in microglia, astrocytes, neurons, and oligodendrocytes (Pineau and Lacroix, 2007; Kroner et al., 2014), various factors, such as protracted Wallerian degeneration after SCI (Buss and Schwab, 2003), could sustain sTNFα expression that would require prolonged XPro1595 infusion.
Because TNFR1 is ubiquitously expressed (Probert, 2015), decreasing TNFR1 activation could affect multiple neural cell types. One population impacted by sTNFα after T3Tx is CGRP+ DRG neurons that carry nociceptive information from the periphery into the spinal cord. After SCI, sprouting of CGRP+ primary afferents is thought to underlie AD intensification. CGRP+ fibers are virtually the only population in the spinal cord to express TrkA (Averill et al., 1995; Michael et al., 1997; Abbott et al., 2006; Molofsky et al., 2014) and respond to injury-induced, glial (i.e., astrocytes, microglia) upregulation of NGF (Krenz and Weaver, 2000; Brown et al., 2004). Previous studies established a positive correlation between NGF/TrkA-mediated CGRP+ afferent sprouting after SCI and AD severity (Krenz and Weaver, 1998; Krenz et al., 1999; Marsh et al., 2002; Cameron et al., 2006). Interestingly, XPro1595-treated animals had CGRP+ staining patterns that were virtually identical to that in uninjured animals, indicating that sTNFα is a critical trigger for CGRP+ sprouting. Inhibiting sTNFα after T3Tx did not affect NGF levels. While TNFα signaling has been implicated with heightened NGF expression (Hattori et al., 1993, 1996; Kuno et al., 2006), other factors that are likely present in tissue below an SCI (e.g., IL-1β) are also capable of driving NGF expression (Yoshida and Gage, 1992). XPro1595 did, however, decrease TrkA levels (Fig. 10), suggesting that it diminished CGRP+ sprouting by decreasing available TrkA that binds NGF.
Additionally, sTNFα stimulated CGRP+ neurite growth in the absence of exogenous NGF (Fig. 11). Although we posit that NF-κB activation in DRGs mediates this growth because the cultures are enriched for neurons, some satellite cells, which also express TNFR1 (Ohtori et al., 2004; Dubový et al., 2006), are present (Steinmetz et al., 2005). Cell-specific deletion of TNFR1 or NF-κB is needed to more definitively determine the role of signaling in neurons versus glia.
Where the sprouted CGRP+ fibers were located is noteworthy. In the T3Tx-saline animals, there was abundant CGRP+ arborization in lumbosacral dorsal horn and laminae VII/X (i.e., dorsal gray commissure), which contain sympathetically correlated interneurons (Matsushita, 1998; Landrum et al., 2002; Hou et al., 2008) (Fig. 9). Furthermore, CRD in T3Tx-XPro1595 animals activated fewer interneurons within lumbosacral cord and near thoracic SPNs than in T3Tx-saline animals (Fig. 9). Thus, XPro1595 attenuated the intensification of AD, at least in part, by decreasing CGRP+ primary afferent plasticity onto interneurons within the dorsal horn and the dorsal gray commissure that, in turn, relay the nociceptive information to SPNs to trigger a sympathetic response.
There are likely other mechanisms, as sTNFα affects a variety of spinal cell populations other than the few examined here. TNFα signaling can increase membrane levels of excitatory glutamatergic receptors and decrease levels of inhibitory GABAergic receptors (Beattie et al., 2002; Stellwagen et al., 2005; Stück et al., 2012; Patel et al., 2017). Moreover, sTNFα/TNFR1 signaling can increase synaptic strength in multiple species (Beattie et al., 2002; Stellwagen et al., 2005; Stellwagen and Malenka, 2006; Kawasaki et al., 2008; Weinberg et al., 2013; Patel et al., 2017), suggesting that it is a fundamental mechanism for this process. It will be important to understand whether and how sTNFα mediates plasticity of propriospinal input to SPNs (Weaver et al., 1997; Llewellyn-Smith et al., 2006; Hou et al., 2008) or of SPNs themselves. Additionally, astrocytic or microglial sTNFα/TNFR1 signaling may alter synaptic function (Korn et al., 2005; Domercq et al., 2006; Tilleux and Hermans, 2008; Gruber-Schoffnegger et al., 2013; Liu et al., 2017). Administering XPro1595 diminished microglial reactivity well below an SCI (Fig. 3). As sTNFα is a “master cytokine” that regulates the expression of itself and other proinflammatory cytokines, decreasing sTNFα signaling may affect the expression of other cytokines (e.g., IL-1β) that also have been implicated in shaping neural plasticity and neuronal excitability (Kawasaki et al., 2008; Clark et al., 2015; Semple et al., 2017). Future studies will determine whether these are other mechanisms by which sTNFα contributes to AD.
Inhibiting sTNFα activity specifically within the cord normalized mesenteric arterial responses to the vasopressor PE (Fig. 7), the first demonstration of such an improvement. This more standard response to vasoconstrictors is likely one reason behind AD events being less extreme in the XPro1595-treated animals. Given the unlikelihood that centrally administered XPro1595 directly affects peripheral vasculature, these data suggest that pathological remodeling of vasculature after high-level SCI is, at least partly, mediated by sTNFα-induced plasticity within the spinal cord. How an exaggerated sympathetic reflex circuit following high-level SCI underlies changes to resistance arteries is unclear. Our data support the postulation that AD-associated hemodynamic oscillations are a cause (Alan et al., 2010; West et al., 2013b). Although T3Tx-XPro1595 animals still experience AD (likely because descending, modulatory input to the SPNs remains absent), their episodes are not nearly as numerous, intense, or enduring as in T3Tx-saline animals (Figs. 4, 5). Repeating, rapid, and severe BP spikes may alter myogenic constriction, which is intrinsic to vascular smooth muscle cells and triggered by pressure-induced stretches of the cell membrane (Davis and Hill, 1999). Additionally, alteration to sympathetic activity may lead to sensitization of adrenergic receptors via modifying downstream G-protein signaling (Jie et al., 2016). A better understanding of maladaptive remodeling within the vasculature will inform improved treatments to normalize BP during an AD episode.
Pharmacologically inhibiting spinal sTNFα signaling also prevented the recurrent AD-dependent decrease in B cells and T cells that is associated with chronic immune suppression after SCI (Zhang et al., 2013; Ueno et al., 2016) (Fig. 8). This corroborates data in which pharmacogenetically silencing glutamatergic interneurons near SPNs to diminish AD also lessens immune suppression (Ueno et al., 2016). Interestingly, XPro1595 animals had more TReg cells, which modulate leukocyte function and are critical for normal immune function (Sakaguchi et al., 2010), than their saline counterparts. It will be important to ascertain whether T3Tx-XPro1595 animals having more B and T cells improves their ability to mount a robust immune response to a pathogen and fight off infection.
Although inhibiting sTNFα decreased the magnitude of the hypertension during an AD episode, there was no change in AD-induced bradycardia, similar to what was observed when Sema3a-mediated inhibition of CGRP+ sprouting diminished hypertension but not bradycardia during AD (Cameron et al., 2006). It is not apparent why this is. There was a strong trend toward significance at 4 weeks, suggesting that interanimal variability in the parasympathetic response is one reason. It is also possible that a consistent degree of reflexive bradycardia is triggered once the BP rises above a threshold.
We chose to test efficacy of the biologic in a well-established, high-thoracic, complete SCI model that reliably results in AD (Krassioukov and Weaver, 1995; Cameron et al., 2006; Rabchevsky et al., 2012; Hou et al., 2013b) for several reasons, XPro1595 modulates the immune response at an incomplete lesion epicenter and confers neuroprotection (Novrup et al., 2014); sparing of descending vasomotor projections decreases AD severity (Karlsson, 1999; Furlan et al., 2003; Gris et al., 2004; Krassioukov, 2006; Hou et al., 2013a). Removing tissue sparing as a confound allowed us to directly assess the role of sTNFα signaling in maladaptive neuroplasticity within spinal cord caudal to an injury. However, it will be interesting to use XPro1595 in a severe, yet incomplete, SCI model (Gris et al., 2004; Squair et al., 2017) to assess efficacy.
Although we focus on the role of sTNFα/TNFR1 signaling in the development of AD here, future studies will determine the role of transmembrane form of TNFα/TNFR2 signaling (Grell et al., 1995, 1998; Probert, 2015). Activation of TNFR1 versus TNFR2 triggers different signaling cascades with divergent consequences (Faustman and Davis, 2010). Interestingly, TNFR1 and TNFR2 are differentially regulated in rodent epileptic models; expression of TNFR1 increases while TNFR2 decreases (Weinberg et al., 2013; Patel et al., 2017). Moreover, administration of a TNFR1/2 agonist attenuates seizure sensitivity (Weinberg et al., 2013). It will be interesting to determine whether TNFR2 activation mitigates AD.
In conclusion, this study is the first to determine that persistent neuroinflammation plays a role in the development of AD after SCI. Furthermore, we identify a potential pharmacological, prophylactic strategy to attenuate life-threatening AD and, importantly, improve cardiovascular and immune function that greatly impact quality of life for SCI patients.
Footnotes
This work was supported by National Institutes of Health Grant R01 NS085426 to V.J.T., Department of Defense Congressionally Directed Medical Research Program Grant W81XWH-14-1-0605 to V.J.T., National Institutes of Health Grant R01 NS1069080 to V.J.T. and J.R.B., Craig H. Neilsen Foundation Grant 382566 to V.J.T., National Institutes of Health Grant R01 NS051709 to J.R.B., National Institutes of Health Grant R01 HL139754 to P.O.-O., American Heart Association Scientist Development Grant 16SDG27260276 to P.O.-O., Margaret Q. Landenberger Foundation Grant 002745 to P.O.-O., and Drexel University Dean's Fellowship for Excellence in Collaborative or Themed Research Training to E.M. We thank Dr. David E. Szymkowski (Xencor) for supplying XPro1595; Dr. Alexander G. Rabchevsky for providing the MATLAB algorithm that served as the basis for what we used to analyze the naturally occurring events; Dr. Vladimir Zhukarev for assistance with the confocal microscope; and the Drexel University Spinal Cord Research Center for use of its core facilities.
The authors declare no competing financial interests.
References
- Abbott NJ, Rönnbäck L, Hansson E (2006) Astrocyte-endothelial interactions at the blood–brain barrier. Nat Rev Neurosci 7:41–53. 10.1038/nrn1824 [DOI] [PubMed] [Google Scholar]
- Alan N, Ramer LM, Inskip JA, Golbidi S, Ramer MS, Laher I, Krassioukov AV (2010) Recurrent autonomic dysreflexia exacerbates vascular dysfunction after spinal cord injury. Spine J 10:1108–1117. 10.1016/j.spinee.2010.09.018 [DOI] [PubMed] [Google Scholar]
- Averill S, McMahon SB, Clary DO, Reichardt LF, Priestley JV (1995) Immunocytochemical localization of trkA receptors in chemically identified subgroups of adult rat sensory neurons. Eur J Neurosci 7:1484–1494. 10.1111/j.1460-9568.1995.tb01143.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud V, Karin M (2001) Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol 11:372–377. 10.1016/S0962-8924(01)02064-5 [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC (2002) Control of synaptic strength by glial TNFalpha. Science 295:2282–2285. 10.1126/science.1067859 [DOI] [PubMed] [Google Scholar]
- Bethea JR, Nagashima H, Acosta MC, Briceno C, Gomez F, Marcillo AE, Loor K, Green J, Dietrich WD (1999) Systemically administered interleukin-10 reduces tumor necrosis factor-alpha production and significantly improves functional recovery following traumatic spinal cord injury in rats. J Neurotrauma 16:851–863. 10.1089/neu.1999.16.851 [DOI] [PubMed] [Google Scholar]
- Bracchi-Ricard V, Lambertsen KL, Ricard J, Nathanson L, Karmally S, Johnstone J, Ellman DG, Frydel B, McTigue DM, Bethea JR (2013) Inhibition of astroglial NF-kappaB enhances oligodendrogenesis following spinal cord injury. J Neuroinflammation 10:92. 10.1186/1742-2094-10-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambilla R, Ashbaugh JJ, Magliozzi R, Dellarole A, Karmally S, Szymkowski DE, Bethea JR (2011) Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain 134:2736–2754. 10.1093/brain/awr199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brommer B, Engel O, Kopp MA, Watzlawick R, Muller S, Prüss H, Chen Y, DeVivo MJ, Finkenstaedt FW, Dirnagl U, Liebscher T, Meisel A, Schwab JM (2016) Spinal cord injury-induced immune deficiency syndrome enhances infection susceptibility dependent on lesion level. Brain 139:692–707. 10.1093/brain/awv375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Ricci MJ, Weaver LC (2004) NGF message and protein distribution in the injured rat spinal cord. Exp Neurol 188:115–127. 10.1016/j.expneurol.2004.03.017 [DOI] [PubMed] [Google Scholar]
- Buss A, Schwab ME (2003) Sequential loss of myelin proteins during wallerian degeneration in the rat spinal cord. Glia 42:424–432. 10.1002/glia.10220 [DOI] [PubMed] [Google Scholar]
- Cameron AA, Smith GM, Randall DC, Brown DR, Rabchevsky AG (2006) Genetic manipulation of intraspinal plasticity after spinal cord injury alters the severity of autonomic dysreflexia. J Neurosci 26:2923–2932. 10.1523/JNEUROSCI.4390-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AK, Gruber-Schoffnegger D, Drdla-Schutting R, Gerhold KJ, Malcangio M, Sandkühler J (2015) Selective activation of microglia facilitates synaptic strength. J Neurosci 35:4552–4570. 10.1523/JNEUROSCI.2061-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke HA, Dekaban GA, Weaver LC (1998) Identification of lamina V and VII interneurons presynaptic to adrenal sympathetic preganglionic neurons in rats using a recombinant herpes simplex virus type 1. Neuroscience 85:863–872. 10.1016/S0306-4522(97)00658-1 [DOI] [PubMed] [Google Scholar]
- Curt A, Nitsche B, Rodic B, Schurch B, Dietz V (1997) Assessment of autonomic dysreflexia in patients with spinal cord injury. J Neurol Neurosurg Psychiatry 62:473–477. 10.1136/jnnp.62.5.473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MJ, Hill MA (1999) Signaling mechanisms underlying the vascular myogenic response. Physiol Rev 79:387–423. 10.1152/physrev.1999.79.2.387 [DOI] [PubMed] [Google Scholar]
- Detloff MR, Fisher LC, McGaughy V, Longbrake EE, Popovich PG, Basso DM (2008) Remote activation of microglia and pro-inflammatory cytokines predict the onset and severity of below-level neuropathic pain after spinal cord injury in rats. Exp Neurol 212:337–347. 10.1016/j.expneurol.2008.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domercq M, Brambilla L, Pilati E, Marchaland J, Volterra A, Bezzi P (2006) P2Y1 receptor-evoked glutamate exocytosis from astrocytes: control by tumor necrosis factor-alpha and prostaglandins. J Biol Chem 281:30684–30696. 10.1074/jbc.M606429200 [DOI] [PubMed] [Google Scholar]
- Dubový P, Jancálek R, Klusáková I, Svízenská I, Pejchalová K (2006) Intra- and extraneuronal changes of immunofluorescence staining for TNF-alpha and TNFR1 in the dorsal root ganglia of rat peripheral neuropathic pain models. Cell Mol Neurobiol 26:1205–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faustman D, Davis M (2010) TNF receptor 2 pathway: drug target for autoimmune diseases. Nat Rev Drug Discov 9:482–493. 10.1038/nrd3030 [DOI] [PubMed] [Google Scholar]
- Furlan JC, Fehlings MG, Shannon P, Norenberg MD, Krassioukov AV (2003) Descending vasomotor pathways in humans: correlation between axonal preservation and cardiovascular dysfunction after spinal cord injury. J Neurotrauma 20:1351–1363. 10.1089/089771503322686148 [DOI] [PubMed] [Google Scholar]
- Garshick E, Kelley A, Cohen SA, Garrison A, Tun CG, Gagnon D, Brown R (2005) A prospective assessment of mortality in chronic spinal cord injury. Spinal Cord 43:408–416. 10.1038/sj.sc.3101729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grell M, Douni E, Wajant H, Löhden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, Scheurich P (1995) The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 83:793–802. 10.1016/0092-8674(95)90192-2 [DOI] [PubMed] [Google Scholar]
- Grell M, Wajant H, Zimmermann G, Scheurich P (1998) The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc Natl Acad Sci U S A 95:570–575. 10.1073/pnas.95.2.570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gris D, Marsh DR, Oatway MA, Chen Y, Hamilton EF, Dekaban GA, Weaver LC (2004) Transient blockade of the CD11d/CD18 integrin reduces secondary damage after spinal cord injury, improving sensory, autonomic, and motor function. J Neurosci 24:4043–4051. 10.1523/JNEUROSCI.5343-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber-Schoffnegger D, Drdla-Schutting R, Hönigsperger C, Wunderbaldinger G, Gassner M, Sandkühler J (2013) Induction of thermal hyperalgesia and synaptic long-term potentiation in the spinal cord lamina I by TNF-alpha and IL-1beta is mediated by glial cells. J Neurosci 33:6540–6551. 10.1523/JNEUROSCI.5087-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori A, Tanaka E, Murase K, Ishida N, Chatani Y, Tsujimoto M, Hayashi K, Kohno M (1993) Tumor necrosis factor stimulates the synthesis and secretion of biologically active nerve growth factor in non-neuronal cells. J Biol Chem 268:2577–2582. [PubMed] [Google Scholar]
- Hattori A, Hayashi K, Kohno M (1996) Tumor necrosis factor (TNF) stimulates the production of nerve growth factor in fibroblasts via the 55-kDa type 1 TNF receptor. FEBS Lett 379:157–160. 10.1016/0014-5793(95)01502-7 [DOI] [PubMed] [Google Scholar]
- Hofstetter CP, Card JP, Olson L (2005) A spinal cord pathway connecting primary afferents to the segmental sympathetic outflow system. Exp Neurol 194:128–138. 10.1016/j.expneurol.2005.01.028 [DOI] [PubMed] [Google Scholar]
- Hou S, Duale H, Cameron AA, Abshire SM, Lyttle TS, Rabchevsky AG (2008) Plasticity of lumbosacral propriospinal neurons is associated with the development of autonomic dysreflexia after thoracic spinal cord transection. J Comp Neurol 509:382–399. 10.1002/cne.21771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou S, Duale H, Rabchevsky AG (2009) Intraspinal sprouting of unmyelinated pelvic afferents after complete spinal cord injury is correlated with autonomic dysreflexia induced by visceral pain. Neuroscience 159:369–379. 10.1016/j.neuroscience.2008.12.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou S, Lu P, Blesch A (2013a) Characterization of supraspinal vasomotor pathways and autonomic dysreflexia after spinal cord injury in F344 rats. Auton Neurosci 176:54–63. 10.1016/j.autneu.2013.02.001 [DOI] [PubMed] [Google Scholar]
- Hou S, Tom VJ, Graham L, Lu P, Blesch A (2013b) Partial restoration of cardiovascular function by embryonic neural stem cell grafts after complete spinal cord transection. J Neurosci 33:17138–17149. 10.1523/JNEUROSCI.2851-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob JE, Pniak A, Weaver LC, Brown A (2001) Autonomic dysreflexia in a mouse model of spinal cord injury. Neuroscience 108:687–693. 10.1016/S0306-4522(01)00436-5 [DOI] [PubMed] [Google Scholar]
- Jie L, Owens EA, Plante LA, Fang Z, Rensing DT, Moeller KD, Osei-Owusu P (2016) RGS2 squelches vascular Gi/o and Gq signaling to modulate myogenic tone and promote uterine blood flow. Physiol Rep 4:e12692. 10.14814/phy2.12692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson AK. (1999) Autonomic dysreflexia. Spinal Cord 37:383–391. 10.1038/sj.sc.3100867 [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Zhang L, Cheng JK, Ji RR (2008) Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J Neurosci 28:5189–5194. 10.1523/JNEUROSCI.3338-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Magnus T, Jung S (2005) Autoantigen specific T cells inhibit glutamate uptake in astrocytes by decreasing expression of astrocytic glutamate transporter GLAST: a mechanism mediated by tumor necrosis factor-alpha. FASEB J 19:1878–1880. 10.1096/fj.05-3748fje [DOI] [PubMed] [Google Scholar]
- Krassioukov A. (2006) Which pathways must be spared in the injured human spinal cord to retain cardiovascular control? Prog Brain Res 152:39–47. 10.1016/S0079-6123(05)52003-X [DOI] [PubMed] [Google Scholar]
- Krassioukov AV, Weaver LC (1995) Episodic hypertension due to autonomic dysreflexia in acute and chronic spinal cord-injured rats. J Physiol 268:H2077–H2083. 10.1152/ajpheart.1995.268.5.H2077 [DOI] [PubMed] [Google Scholar]
- Krassioukov AV, Johns DG, Schramm LP (2002) Sensitivity of sympathetically correlated spinal interneurons, renal sympathetic nerve activity, and arterial pressure to somatic and visceral stimuli after chronic spinal injury. J Neurotrauma 19:1521–1529. 10.1089/089771502762300193 [DOI] [PubMed] [Google Scholar]
- Krenz NR, Weaver LC (1998) Sprouting of primary afferent fibers after spinal cord transection in the rat. Neuroscience 85:443–458. 10.1016/S0306-4522(97)00622-2 [DOI] [PubMed] [Google Scholar]
- Krenz NR, Weaver LC (2000) Nerve growth factor in glia and inflammatory cells of the injured rat spinal cord. J Neurochem 74:730–739. 10.1046/j.1471-4159.2000.740730.x [DOI] [PubMed] [Google Scholar]
- Krenz NR, Meakin SO, Krassioukov AV, Weaver LC (1999) Neutralizing intraspinal nerve growth factor blocks autonomic dysreflexia caused by spinal cord injury. J Neurosci 19:7405–7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroner A, Greenhalgh AD, Zarruk JG, Passos Dos Santos R, Gaestel M, David S (2014) TNF and increased intracellular iron alter macrophage polarization to a detrimental M1 phenotype in the injured spinal cord. Neuron 83:1098–1116. 10.1016/j.neuron.2014.07.027 [DOI] [PubMed] [Google Scholar]
- Kuno R, Yoshida Y, Nitta A, Nabeshima T, Wang J, Sonobe Y, Kawanokuchi J, Takeuchi H, Mizuno T, Suzumura A (2006) The role of TNF-alpha and its receptors in the production of NGF and GDNF by astrocytes. Brain Res 1116:12–18. 10.1016/j.brainres.2006.07.120 [DOI] [PubMed] [Google Scholar]
- Landrum LM, Jones SL, Blair RW (2002) The expression of fos-labeled spinal neurons in response to colorectal distension is enhanced after chronic spinal cord transection in the rat. Neuroscience 110:569–578. 10.1016/S0306-4522(01)00548-6 [DOI] [PubMed] [Google Scholar]
- Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J (1995) Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem 270:14255–14258. 10.1074/jbc.270.24.14255 [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhou LJ, Wang J, Li D, Ren WJ, Peng J, Wei X, Xu T, Xin WJ, Pang RP, Li YY, Qin ZH, Murugan M, Mattson MP, Wu LJ, Liu XG (2017) TNF-alpha differentially regulates synaptic plasticity in the hippocampus and spinal cord by microglia-dependent mechanisms after peripheral nerve injury. J Neurosci 37:871–881. 10.1523/JNEUROSCI.2235-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llewellyn-Smith IJ, Weaver LC, Keast JR (2006) Effects of spinal cord injury on synaptic inputs to sympathetic preganglionic neurons. Prog Brain Res 152:11–26. 10.1016/S0079-6123(05)52001-6 [DOI] [PubMed] [Google Scholar]
- Loetscher H, Stueber D, Banner D, Mackay F, Lesslauer W (1993) Human tumor necrosis factor alpha (TNF alpha) mutants with exclusive specificity for the 55-kDa or 75-kDa TNF receptors. J Biol Chem 268:26350–26357. [PubMed] [Google Scholar]
- Marchi N, Granata T, Janigro D (2014) Inflammatory pathways of seizure disorders. Trends Neurosci 37:55–65. 10.1016/j.tins.2013.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh DR, Wong ST, Meakin SO, MacDonald JI, Hamilton EF, Weaver LC (2002) Neutralizing intraspinal nerve growth factor with a trkA-IgG fusion protein blocks the development of autonomic dysreflexia in a clip-compression model of spinal cord injury. J Neurotrauma 19:1531–1541. 10.1089/089771502762300201 [DOI] [PubMed] [Google Scholar]
- Matsushita M. (1998) Ascending propriospinal afferents to area X (substantia grisea centralis) of the spinal cord in the rat. Exp Brain Res 119:356–366. 10.1007/s002210050351 [DOI] [PubMed] [Google Scholar]
- Mayorov DN, Adams MA, Krassioukov AV (2001) Telemetric blood pressure monitoring in conscious rats before and after compression injury of spinal cord. J Neurotrauma 18:727–736. 10.1089/089771501750357663 [DOI] [PubMed] [Google Scholar]
- Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U (2005) Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci 6:775–786. 10.1038/nrn1765 [DOI] [PubMed] [Google Scholar]
- Michael GJ, Kaya E, Averill S, Rattray M, Clary DO, Priestley JV (1997) TrkA immunoreactive neurones in the rat spinal cord. J Comp Neurol 385:441–455. 10.1002/(SICI)1096-9861(19970901)385:3%3C441::AID-CNE8%3E3.0.CO;2-3 [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Kelley KW, Tsai HH, Redmond SA, Chang SM, Madireddy L, Chan JR, Baranzini SE, Ullian EM, Rowitch DH (2014) Astrocyte-encoded positional cues maintain sensorimotor circuit integrity. Nature 509:189–194. 10.1038/nature13161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers J, Lee M, Kiratli J (2007) Cardiovascular disease in spinal cord injury: an overview of prevalence, risk, evaluation, and management. Am J Phys Med Rehabil 86:142–152. 10.1097/PHM.0b013e31802f0247 [DOI] [PubMed] [Google Scholar]
- Novrup HG, Bracchi-Ricard V, Ellman DG, Ricard J, Jain A, Runko E, Lyck L, Yli-Karjanmaa M, Szymkowski DE, Pearse DD, Lambertsen KL, Bethea JR (2014) Central but not systemic administration of XPro1595 is therapeutic following moderate spinal cord injury in mice. J Neuroinflammation 11:159. 10.1186/s12974-014-0159-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtori S, Takahashi K, Moriya H, Myers RR (2004) TNF-alpha and TNF-alpha receptor type 1 upregulation in glia and neurons after peripheral nerve injury: studies in murine DRG and spinal cord. Spine (Phila Pa 1976) 29:1082–1088. 10.1097/00007632-200405150-00006 [DOI] [PubMed] [Google Scholar]
- Osei-Owusu P, Sabharwal R, Kaltenbronn KM, Rhee MH, Chapleau MW, Dietrich HH, Blumer KJ (2012) Regulator of G protein signaling 2 deficiency causes endothelial dysfunction and impaired endothelium-derived hyperpolarizing factor-mediated relaxation by dysregulating Gi/o signaling. J Biol Chem 287:12541–12549. 10.1074/jbc.M111.332130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osei-Owusu P, Knutsen RH, Kozel BA, Dietrich HH, Blumer KJ, Mecham RP (2014) Altered reactivity of resistance vasculature contributes to hypertension in elastin insufficiency. Am J Physiol Heart Circ Physiol 306:H654–H666. 10.1152/ajpheart.00601.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel DC, Wallis G, Dahle EJ, McElroy PB, Thomson KE, Tesi RJ, Szymkowski DE, West PJ, Smeal RM, Patel M, Fujinami RS, White HS, Wilcox KS (2017) Hippocampal TNFalpha signaling contributes to seizure generation in an infection-induced mouse model of limbic epilepsy. eNeuro 4:ENEURO.0105–17.2017. 10.1523/ENEURO.0105-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineau I, Lacroix S (2007) Proinflammatory cytokine synthesis in the injured mouse spinal cord: multiphasic expression pattern and identification of the cell types involved. J Comp Neurol 500:267–285. 10.1002/cne.21149 [DOI] [PubMed] [Google Scholar]
- Probert L. (2015) TNF and its receptors in the CNS: the essential, the desirable and the deleterious effects. Neuroscience 302:2–22. 10.1016/j.neuroscience.2015.06.038 [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG, Patel SP, Lyttle TS, Eldahan KC, O'Dell CR, Zhang Y, Popovich PG, Kitzman PH, Donohue KD (2012) Effects of gabapentin on muscle spasticity and both induced as well as spontaneous autonomic dysreflexia after complete spinal cord injury. Front Physiol 3:329. 10.3389/fphys.2012.00329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rummery NM, Tripovic D, McLachlan EM, Brock JA (2010) Sympathetic vasoconstriction is potentiated in arteries caudal but not rostral to a spinal cord transection in rats. J Neurotrauma 27:2077–2089. 10.1089/neu.2010.1468 [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Miyara M, Costantino CM, Hafler DA (2010) FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 10:490–500. 10.1038/nri2785 [DOI] [PubMed] [Google Scholar]
- Semple BD, O'Brien TJ, Gimlin K, Wright DK, Kim SE, Casillas-Espinosa PM, Webster KM, Petrou S, Noble-Haeusslein LJ (2017) Interleukin-1 receptor in seizure susceptibility after traumatic injury to the pediatric brain. J Neurosci 37:7864–7877. 10.1523/JNEUROSCI.0982-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squair JW, West CR, Popok D, Assinck P, Liu J, Tetzlaff W, Krassioukov AV (2017) High thoracic contusion model for the investigation of cardiovascular function after spinal cord injury. J Neurotrauma 34:671–684. 10.1089/neu.2016.4518 [DOI] [PubMed] [Google Scholar]
- Steed PM, Tansey MG, Zalevsky J, Zhukovsky EA, Desjarlais JR, Szymkowski DE, Abbott C, Carmichael D, Chan C, Cherry L, Cheung P, Chirino AJ, Chung HH, Doberstein SK, Eivazi A, Filikov AV, Gao SX, Hubert RS, Hwang M, Hyun L, et al. (2003) Inactivation of TNF signaling by rationally designed dominant-negative TNF variants. Science 301:1895–1898. 10.1126/science.1081297 [DOI] [PubMed] [Google Scholar]
- Steinmetz MP, Horn KP, Tom VJ, Miller JH, Busch SA, Nair D, Silver DJ, Silver J (2005) Chronic enhancement of the intrinsic growth capacity of sensory neurons combined with the degradation of inhibitory proteoglycans allows functional regeneration of sensory axons through the dorsal root entry zone in the mammalian spinal cord. J Neurosci 25:8066–8076. 10.1523/JNEUROSCI.2111-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC (2006) Synaptic scaling mediated by glial TNF-alpha. Nature 440:1054–1059. 10.1038/nature04671 [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Beattie EC, Seo JY, Malenka RC (2005) Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J Neurosci 25:3219–3228. 10.1523/JNEUROSCI.4486-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stück ED, Christensen RN, Huie JR, Tovar CA, Miller BA, Nout YS, Bresnahan JC, Beattie MS, Ferguson AR (2012) Tumor necrosis factor alpha mediates GABA(A) receptor trafficking to the plasma membrane of spinal cord neurons in vivo. Neural Plast 2012:261345. 10.1155/2012/261345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweazea KL, Walker BR (2012) Impaired myogenic tone in mesenteric arteries from overweight rats. Nutr Metab (Lond) 9:18. 10.1186/1743-7075-9-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Neckel ND, Schramm LP (2004) Spinal interneurons infected by renal injection of pseudorabies virus in the rat. Brain Res 1004:1–7. 10.1016/j.brainres.2004.01.016 [DOI] [PubMed] [Google Scholar]
- Tilleux S, Hermans E (2008) Down-regulation of astrocytic GLAST by microglia-related inflammation is abrogated in dibutyryl cAMP-differentiated cultures. J Neurochem 105:2224–2236. 10.1111/j.1471-4159.2008.05305.x [DOI] [PubMed] [Google Scholar]
- Tom VJ, Steinmetz MP, Miller JH, Doller CM, Silver J (2004) Studies on the development and behavior of the dystrophic growth cone, the hallmark of regeneration failure, in an in vitro model of the glial scar and after spinal cord injury. J Neurosci 24:6531–6539. 10.1523/JNEUROSCI.0994-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno M, Ueno-Nakamura Y, Niehaus J, Popovich PG, Yoshida Y (2016) Silencing spinal interneurons inhibits immune suppressive autonomic reflexes caused by spinal cord injury. Nat Neurosci 19:784–787. 10.1038/nn.4289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters ET. (2014) Neuroinflammatory contributions to pain after SCI: roles for central glial mechanisms and nociceptor-mediated host defense. Exp Neurol 258C:48–61. 10.1016/j.expneurol.2014.02.001 [DOI] [PubMed] [Google Scholar]
- Weaver LC, Cassam AK, Krassioukov AV, Llewellyn-Smith IJ (1997) Changes in immunoreactivity for growth associated protein-43 suggest reorganization of synapses on spinal sympathetic neurons after cord transection. Neuroscience 81:535–551. 10.1016/S0306-4522(97)00151-6 [DOI] [PubMed] [Google Scholar]
- Weaver LC, Verghese P, Bruce JC, Fehlings MG, Krenz NR, Marsh DR (2001) Autonomic dysreflexia and primary afferent sprouting after clip-compression injury of the rat spinal cord. J Neurotrauma 18:1107–1119. 10.1089/08977150152693782 [DOI] [PubMed] [Google Scholar]
- Weinberg MS, Blake BL, McCown TJ (2013) Opposing actions of hippocampus TNFalpha receptors on limbic seizure susceptibility. Exp Neurol 247:429–437. 10.1016/j.expneurol.2013.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West CR, Bellantoni A, Krassioukov AV (2013a) Cardiovascular function in individuals with incomplete spinal cord injury: a systematic review. Top Spinal Cord Inj Rehabil 19:267–278. 10.1310/sci1904-267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West CR, Alyahya A, Laher I, Krassioukov A (2013b) Peripheral vascular function in spinal cord injury: a systematic review. Spinal Cord 51:10–19. 10.1038/sc.2012.136 [DOI] [PubMed] [Google Scholar]
- West CR, Popok D, Crawford MA, Krassioukov AV (2015) Characterizing the temporal development of cardiovascular dysfunction in response to spinal cord injury. J Neurotrauma 32:922–930. 10.1089/neu.2014.3722 [DOI] [PubMed] [Google Scholar]
- West CR, Crawford MA, Laher I, Ramer MS, Krassioukov AV (2016) Passive hind-limb cycling reduces the severity of autonomic dysreflexia after experimental spinal cord injury. Neurorehabil Neural Repair 30:317–327. 10.1177/1545968315593807 [DOI] [PubMed] [Google Scholar]
- Wu D, Klaw MC, Connors T, Kholodilov N, Burke RE, Tom VJ (2015) Expressing constitutively active Rheb in adult neurons after a complete spinal cord injury enhances axonal regeneration beyond a chondroitinase-treated glial scar. J Neurosci 35:11068–11080. 10.1523/JNEUROSCI.0719-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Klaw MC, Lemay MA, Baas PW, Tom VJ (2014) Pharmacologically inhibiting kinesin-5 activity with monastrol promotes axonal regeneration following spinal cord injury. Exp Neurol 263C:172–176. 10.1016/j.expneurol.2014.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Gage FH (1992) Cooperative regulation of nerve growth factor synthesis and secretion in fibroblasts and astrocytes by fibroblast growth factor and other cytokines. Brain Res 569:14–25. 10.1016/0006-8993(92)90364-F [DOI] [PubMed] [Google Scholar]
- Zhang Y, Guan Z, Reader B, Shawler T, Mandrekar-Colucci S, Huang K, Weil Z, Bratasz A, Wells J, Powell ND, Sheridan JF, Whitacre CC, Rabchevsky AG, Nash MS, Popovich PG (2013) Autonomic dysreflexia causes chronic immune suppression after spinal cord injury. J Neurosci 33:12970–12981. 10.1523/JNEUROSCI.1974-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]