

Abstract
In recent years, m6A has emerged as an abundant and dynamically regulated modification throughout the transcriptome. Recent technological advances have enabled the transcriptome-wide identification of m6A residues, which in turn has provided important insights into the biology and regulation of this pervasive regulatory mark. Also central to our current understanding of m6A are the discovery and characterization of m6A readers, writers, and erasers. Over the last few years, studies into the function of these proteins have led to important discoveries about the regulation and function of m6A. However, during this time our understanding of these proteins has also evolved considerably, sometimes leading to the reversal of early conceptions regarding the reading, writing and erasing of m6A. In this review, we summarize recent advances in m6A research, and we highlight how these new findings have reshaped our understanding of how m6A is regulated in the transcriptome.
Keywords: epitranscriptome, m6A, RNA modifications, posttranscriptional control
INTRODUCTION
m6A (N6-methyladenosine) is a highly pervasive modification in mRNA and noncoding RNAs that affects RNA splicing, translation, and stability, as well as the epigenetic effects of certain noncoding RNAs (Dominissini et al. 2012; Meyer et al. 2012, 2015; Patil et al. 2016; X. Wang et al. 2014, 2015; Xiao et al. 2016). m6A was first detected in poly(A) RNA fractions in 1974 (Desrosiers et al. 1974, Perry & Kelley 1974), but the interest in m6A as a mode of mRNA regulation largely subsided by the end of the 1970s due to a lack of methods for detecting m6A sites in mRNAs. At the time, m6A detection was limited to quantifying m6A in RNA hydrolysates. This meant that researchers were unable to identify the individual RNAs that contained m6A and thus could not definitively rule out the possibility that the m6A they detected came from contaminating RNA species that copurified with mRNA in poly(A) fractions. An additional limitation was that m6A behaves like A during reverse transcription and thus was not easily detected in cDNA sequences.
Interest in m6A was revived in 2012, when our group and the Rechavi group described MeRIP-Seq, a next-generation sequencing method to map m6A throughout the transcriptome (Dominissini et al. 2012, Meyer et al. 2012). These mapping approaches revealed that m6A is unequivocally an mRNA modification, and they surprisingly showed that m6A is highly selective: Only certain mRNAs contain m6A, and m6A is often located near stop codons and in 3´UTRs. m6A levels in mRNA were found to be dynamic, with levels varying in development and in response to cellular stresses (Dominissini et al. 2012, Meyer et al. 2012). These studies strongly suggested that m6A is likely to have functional roles that affect mRNA fate and function in cells.
Since that time, MeRIP-Seq has been used in nearly all studies on m6A and has led to the identification of numerous methylated mRNAs that appear to have critical roles in cellular processes such as cancer and cellular differentiation. The m6A mapping concept was quickly applied to other modified nucleotides, resulting in transcriptome-wide maps of pseudouridine and other modified nucleotides (Carlile et al. 2014, Delatte et al. 2016, Lovejoy et al. 2014, Schwartz et al. 2014a).
A major part of the history of m6A was a study that reported, for the first time, that m6A could be demethylated (Jia et al. 2011). This study proposed that FTO, an enzyme previously associated with human obesity, is an m6A demethylase (Jia et al. 2011). This finding suggested the possibility that m6A could be dynamically regulated by its removal, not just its formation. As described in the discussion of m6A erasers below, it is now clear that FTO actually prefers to demethylate a closely related and highly abundant nucleotide, N6,2´-O-dimethyladenosine (m6Am) (Mauer et al. 2017). Nevertheless, the identification of a putative m6A eraser helped to spark enthusiasm in the early days of this field.
The two m6A mapping studies and the FTO study were published just a few months apart and independently of each other. Until that time, m6A had been nearly forgotten as a component of mRNA; however, the appearance of these independent studies in rapid succession helped draw attention to the concept that mRNA function could be regulated by RNA modifications. We coined the term epitranscriptome (Meyer et al. 2012) to refer to these previously hidden posttranscriptional modifications in RNA, such as m6A, and to distinguish them from nucleotides such as inosine that are formed through RNA editing. Since these studies, rapid progress has been made in characterizing other RNA modifications, and it is now clear that, in addition to the information encoded by the nucleotide sequence in mRNAs, there is also regulatory information carried in the epitranscriptomic code.
The effects of m6A are determined by m6A readers, writers, and erasers (Figure 1). The writer complex was established prior to the recent resurgence in interest in m6A, largely due to the pioneering work of Bokar, Rottman, and colleagues, who cloned methyltransferase-like 3 (METTL3) in 1994 (Bokar et al. 1997), and work by Fray and colleagues (Zhong et al. 2008) and Fink and colleagues (Agarwala et al. 2012), who identified the METTL3 adaptor protein WTAP (Wilm’s tumor–associated protein) and showed that WTAP and METTL3 form a complex that is indispensable for m6A formation.
Figure 1.
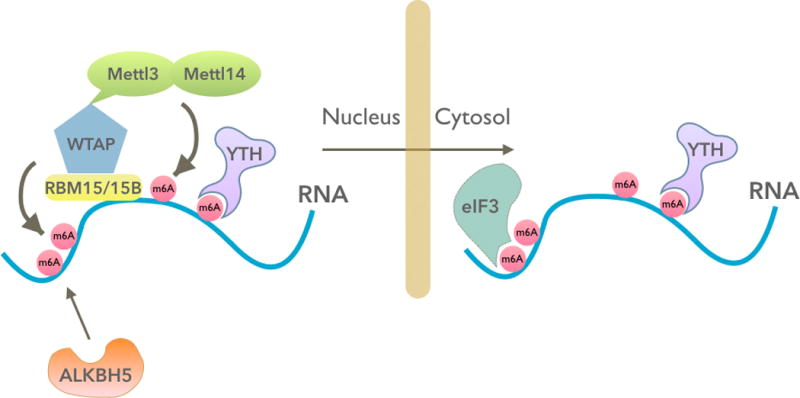
mRNA control by an m6A (N6-methyladenosine) writer complex, an eraser, and readers. The m6A writer complex (RBM15/15B-WTAP-METTL3-METTL14) and the m6A eraser (ALKBH5) are localized primarily in the nucleus. Therefore, any dynamic regulation of m6A in mRNA would occur in the nucleus prior to mRNA export. The m6A “imprint” that is conferred in the nucleus is essentially unchangeable and dictates the mRNA’s fate in the cytoplasm. In the nucleus, m6A can be recognized by m6A readers, the most prominent of which is the YTH protein DC1. In the cytoplasm, m6A can be recognized by the highly similar, and possibly functionally redundant, YTH proteins DF1, DF2, and DF3. Additionally, m6A in 5´UTRs can bind eukaryotic initiation factor 3 (eIF3), which promotes translation independently of the canonical cap-binding protein eIF4E.
More recent publications have added to our understanding of m6A writers and erasers. One study proposed a second m6A writer, METTL14 (Liu et al. 2014). Another study identified α-ketoglutarate-dependent dioxygenase alkB homolog 5 (ALKBH5) as an m6A eraser in mRNA, pointing to the idea that m6A has two erasers, i.e., FTO and ALKBH5 (Zheng et al. 2013). These studies suggested that methylation and demethylation could exhibit the type of complexity seen with epigenetic modifications.
The proposal that there are two distinct methyltransferases that determine m6A dynamics in the transcriptome was a major new concept in the m6A field (Liu et al. 2014). It raised the possibility that differential regulation of METTL3 and METTL14 could result in unique patterns of m6A in the transcriptome. However, recent studies have now overturned this concept. Three studies demonstrated that METTL14 is not enzymatically active and is therefore not a methyltransferase (Sledz & Jinek 2016, P. Wang et al. 2016, X. Wang et al. 2016). METTL14 acts as an adaptor to bind the RNA substrate and to promote the activity of METTL3, the only methyltransferase. Therefore, the specific location and dynamics of m6A in the transcriptome cannot be attributed to differential regulation of METTL3 or METTL14.
The idea that m6A in mRNA is reversible has been an important concept. Conceivably, the activation of m6A demethylases could account for dynamic changes in m6A levels in mRNA. However, despite excitement around the concept, very few studies have explored this concept at the molecular level. The finding that FTO does not target m6A in cells but instead demethylates a related nucleotide, m6Am, suggests that ALKBH5 is the major m6A demethylase in cells (Mauer et al. 2017). However, there is limited evidence so far for direct demethylation of specific m6A sites in mRNA by ALKBH5. ALKBH5 knockout animals appear normal except for defects in spermatogenesis (Zheng et al. 2013), suggesting that m6A demethylation is not required for the diverse signaling and differentiation events required for animal development.
The m6A field has also rapidly evolved with respect to m6A readers. Several m6A readers have been identified, but diverse and often contradictory results have been obtained, especially with the YTHDF class of m6A-binding proteins.
Because many of the foundational studies of m6A and its regulators have been challenged or overturned, it is difficult to keep up with the current concepts of m6A in mRNA. The goal of this review is to provide the current understanding of m6A readers, writers, and erasers. The latest findings give clearer insights into the circumstances and mechanisms by which m6A levels can be regulated and how m6A could exert its effects on mRNAs and lncRNAs.
m6A MAPPING APPROACHES
Transcriptome-Wide m6A Mapping Methods
The first transcriptome-wide identification of m6A sites in individual RNAs was reported in 2012, when two groups independently developed a strategy for global m6A mapping (Dominissini et al. 2012, Meyer et al. 2012). These methods (termed MeRIP-Seq or m6A-seq) relied on highly specific m6A antibodies to immunoprecipitate methylated mRNAs, followed by high-throughput sequencing to determine the identity of the methylated transcripts. This approach revealed thousands of methylated mRNAs throughout the transcriptome. MeRIP-Seq also confirmed earlier studies (Wei & Moss 1977, Wei et al. 1976) indicating a GAC or AAC consensus sequence for m6A. Importantly, MeRIP-Seq also enabled a global view of the distribution of the m6A mark, which revealed that m6A is highly prevalent in coding sequences and 3´UTRs as well as in long exons and is particularly abundant in the vicinity of the stop codon.
Early m6A profiling studies indicated that m6A is highly enriched in long internal exons and near stop codons (Dominissini et al. 2012, Meyer et al. 2012). In principle, only a translating ribosome can detect stop codons, and these are located in the cytoplasm. However, m6A formation is primarily a nuclear event (Sommer et al. 1978). Therefore, the stop codon itself is unlikely to be the cause of the m6A enrichment near stop codons. More recent m6A mapping studies have further refined the localization of m6A to enrichment in the 5´ end of the terminal exon, which often includes the stop codon (Batista et al. 2014, Ke et al. 2015). Although additional studies will be required to determine the mechanism through which terminal exons are preferentially methylated, this finding highlights the importance of m6A mapping methods for expanding our understanding of the biology of m6A.
Single-Nucleotide-Resolution Mapping
The initial MeRIP-Seq technology has now been replaced by a newer single-nucleotide-resolution m6A mapping technique termed miCLIP (m6A individual-nucleotide-resolution crosslinking and immunoprecipitation) (Linder et al. 2015). MeRIP-Seq/m6A-seq has low resolution because it relies solely on immunoprecipitation of fragmented RNA (Figure 2). The resolution of the technique is due to the size of the RNA fragments used as input and typically results in broad peaks that map to genomic regions between 100 and 200 nt in length (Meyer & Jaffrey 2014). However, the key advance in miCLIP was the finding that, when certain m6A antibodies are crosslinked to m6A-containing RNA, the antibody-RNA adduct causes reverse transcriptase–induced mutations at the +1 position in the resulting cDNA (Linder et al. 2015). The resulting crosslinking-induced mutation sites can then be detected by sequencing and used to identify individual m6A residues (Figure 2). miCLIP has been an important technical advance in m6A detection methodology because it maps individual m6A sites in cellular mRNAs with high specificity and sensitivity. The knowledge of individual m6A sites derived from the miCLIP data sets in mouse and human methylomes has enabled investigators to perform site-specific mutations to reveal functions of specific m6A residues.
Figure 2.
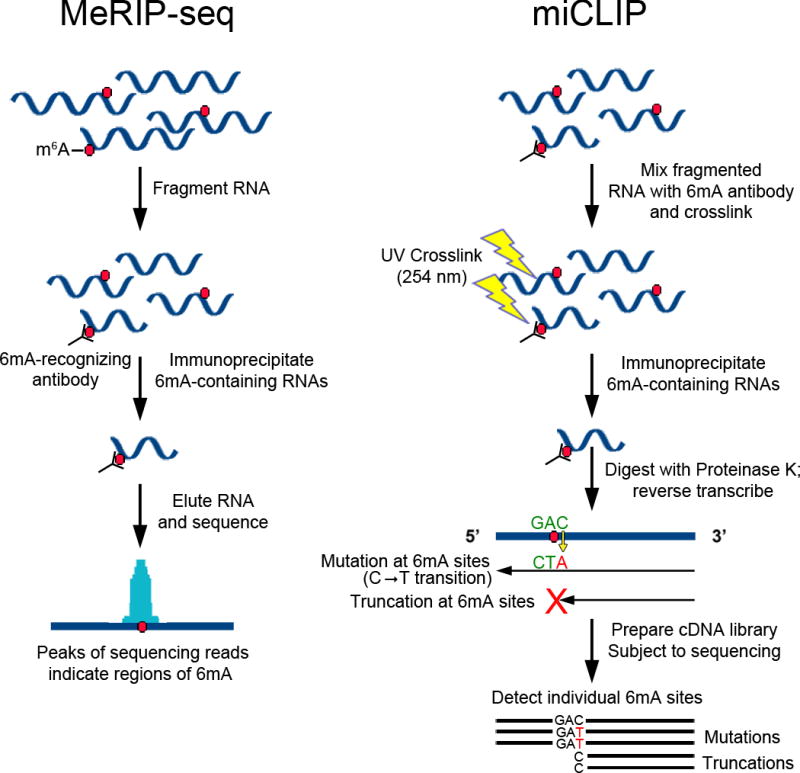
Methods for transcriptome-wide mapping of adenosine methylation. Shown here are schematics outlining the general procedures for MeRIP-Seq (a) and miCLIP (b). In both methods, cellular RNA is fragmented and incubated with 6mA (6-methyladenine)-recognizing antibodies. (a) In MeRIP-Seq, methylated RNAs are directly immunoprecipitated using antibody-binding magnetic beads. Methylated RNA is then eluted by either proteinase K digestion or the addition of free m6A (N6-methyladenosine), and RNA fragments are subjected to high-throughput sequencing. The resulting sequencing reads accumulate around sites of 6mA and enable prediction of an ~100–200-nt-wide region of one or more m6A residues. Enriched reads near the 5´ end of RNAs can be due to either m6A or m6Am (N6,2´-O-dimethyladenosine). (b) In miCLIP, 6mA antibodies are crosslinked to methylated RNA prior to immunoprecipitation. Elution with proteinase K leaves a small crosslinked adduct on the methylated RNA, which causes either truncations or mutations immediately adjacent to methylation sites during reverse transcription. The resulting cDNA is then amplified and subjected to next-generation sequencing, and individual m6A or m6Am sites are identified by detecting sites of mutation or truncation in the sequencing reads.
Quantifying the dynamics of m6A epitranscriptomes
A major challenge is to quantify the dynamic changes in m6A levels at specific sites. For example, many studies have sought to determine how m6A changes after a specific treatment. Typically, MeRIP–qRT-PCR is used. In this method, m6A RNAs are immunoprecipitated in control and experimental conditions, and specific RNAs are quantified by qRT-PCR (Meyer et al. 2012). However, the signal may reflect m6A at physiologically insignificant levels. For example, a fivefold increase in signal may not be meaningful if an mRNA changes from 1% to 5% stoichiometry. Quantitative methods can be used to quantify m6A stoichiometry in single RNAs (Liu et al. 2013), but these methods are laborious and may not be possible for mRNAs that are not highly abundant. Transcriptome-wide mapping approaches such as MeRIP-Seq and miCLIP can be used to generate estimates of m6A abundance, but they do not provide absolute quantitative information on m6A stoichiometry. Thus, methods for precise global measurements of m6A stoichiometry transcriptome wide remain a challenge.
Distinguishing between m6A and m6Am
A major problem with MeRIP-Seq is that it cannot readily distinguish between m6A and m6Am. Both of these nucleotides contain 6mA (6-methyladenine) (Figure 3), and therefore both react with the m6A-specific antibodies. m6Am was discovered in the early 1970s to be a dimethylated adenosine residue that is exclusively found at the first encoded nucleotide of a large fraction of cellular mRNAs, immediately adjacent to the 7-methylguanosine cap structure (Wei et al. 1975). The first nucleotide of every mRNA is typically methylated at the 2´ position of the sugar to form a 2´-O-methylated residue. However, if this base is A, then an additional methylation at the N6 position can occur, forming the dimethylated nucleotide m6Am. Notably, m6Am was shown to be remarkably abundant, with between 30% and 40% of all mRNAs containing this nucleotide following the m7G cap (Wei et al. 1975).
Figure 3.
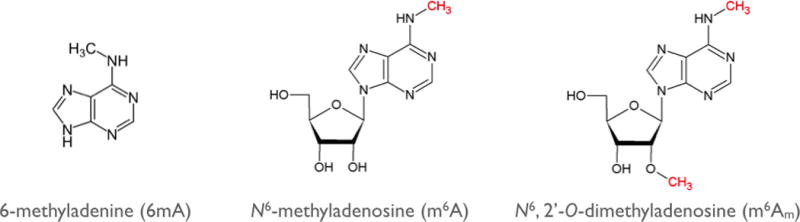
m6A and m6Am are the two 6mA (6-methyladenine)-containing nucleotides in mRNA. 6mA is found in two distinct epitranscriptomic modifications: m6A (N6-methyladenosine) and m6Am (N6,2´-O-dimethyladenosine). The methyl groups that are added enzymatically to alter the function of the nucleotide are shown in red. m6A is found in 5´UTRs, coding sequences, and 3´UTRs. m6Am is found in one place in mRNAs, the first encoded nucleotide, which is adjacent to the m7G (N7-methylguanosine) cap.
Importantly, high-resolution mapping methods such as miCLIP enable m6A to be distinguished from m6Am (Linder et al. 2015). Transcriptome-wide profiling with miCLIP provided the first comprehensive analysis of m6Am residues transcriptome wide and helped expand the repertoire of adenosine methylation events in cellular mRNAs (Linder et al. 2015).
In light of recent discoveries showing that m6Am has marked effects on mRNA stability and is selectively targeted for demethylation by FTO (Mauer et al. 2017), it is critical to have methods for distinguishing between the highly similar m6A and m6Am modifications when global analyses are performed. Similarly, antibodies should be referred to as 6mA-specific antibodies because immunoblotting, immunostaining, and pulldown experiments do not distinguish between m6A and m6Am. On the basis of our understanding of m6Am in the transcriptome, any of these antibody-based analysis methods that have not already taken into account the potential detection of m6Am should be reevaluated in light of the fact that the signal could be due to m6Am rather than m6A. The similarity between m6A and m6Am also underscores the need for using single-nucleotide-resolution m6A mapping strategies rather than MeRIP-Seq in global mapping studies so that m6A residues can be distinguished from m6Am residues.
m6A READERS
The major mechanism by which m6A exerts its effects is by recruiting m6A-binding proteins. As described below, the current data indicate that m6A can be recognized by proteins that contain a YTH (YT521B homology) domain, or alternatively by eukaryotic initiation factor 3 (eIF3).
YTH Domain Proteins
Rechavi and colleagues initially identified YTHDF2 (DF2) and YTHDF3 (DF3) as m6A-binding proteins in an m6A RNA pulldown experiment (Dominissini et al. 2012). These proteins were the most robust interactors among several recovered proteins. These proteins are named for their YTH domain, which is their only recognizable domain. YTH domains were initially identified by Stamm and colleagues using computational homology searches to pinpoint domains similar to those in the YT521B splicing factor [now known as YTHDC1 (DC1)] (Stoilov et al. 2002). This domain was named YTH for YT521B homology. Following the initial identification of DF2 and DF3 as m6A-binding proteins, multiple groups verified these results using gel shift assays and crystallography (Luo & Tong 2014, Theler et al. 2014, Xu et al. 2014). These results demonstrated that the YTH domain selectively binds m6A in RNA and led to numerous efforts to define the function of these YTH domain proteins.
Sequence features
Mammalian genomes contain five YTH domain–containing proteins: DC1, YTHDC2 (DC2), YTHDF1 (DF1), DF2, and DF3 (Table 1). On the basis of sequence, these proteins can be divided into three major classes: DC1; DC2; and the DF family, which comprises three nearly identical paralogs (DF1, DF2, and DF3) (Figure 4). We recently mapped the binding sites of each of the endogenously expressed proteins in the transcriptome by using the iCLIP (individual-nucleotide-resolution UV crosslinking and immunoprecipitation) method (Konig et al. 2010, Patil et al. 2016). This approach demonstrated that the three DF proteins primarily bind all m6A sites in mRNA, whereas the nuclear-enriched DC1 is bound to some m6A sites in mRNAs as well as in nuclear noncoding RNAs. The DC1-binding sites seen in mRNA may reflect binding to m6A in nascent mRNAs prior to their export to the cytosol. DC2 binds to a smaller number of select m6A sites, especially in noncoding RNA. Notably, this mapping demonstrated that all five YTH proteins are physiological m6A readers (Patil et al. 2016).
Table 1.
Documented and putative m6A readers, writers, and erasers in different organisms.
| Organism | DF-like | DC1-like | DC2-like | METTL3 | WTAP | METTL14 | VIRMA (KIAA1429) | FTO | ALKBH5 |
|---|---|---|---|---|---|---|---|---|---|
| Homo sapiens | YTHDF1 | YTHDC1 | YTHDC2 | METTL3 | WTAP | METTL14 | VIRMA | FTO | ALKBH5 |
| YTHDF2 | |||||||||
| YTHDF3 | |||||||||
| Mus musculus | YTHDF1 | YTHDC1 | YTHDC2 | Mettl3 | Wtap | Mettl14 | Virma | Fto | Alkbh5 |
| YTHDF2 | |||||||||
| YTHDF3 | |||||||||
| Xenopus laevis | YTHDF1 | YTHDC1 | Predicted YTHDC2 (XP_018119820) | mettl3 | wtap | mettl14 | virma | fto | alkbh5 |
| YTHDF2 | |||||||||
| YTHDF3 | |||||||||
| Danio rerio | YTHDF1 | YTHDC1 | Predicted YTHDC2 (XP_017211699) | mettl3 | wtap | mettl14 | si:ch211-79l20.4 (orthologous region) | fto | alkbh5 |
| YTHDF2 | |||||||||
| YTHDF3 | |||||||||
| Drosophila melanogaster | CG6422 | YT521-B | Ime4 | Fl(2)D | Kar4 | Virilizer | |||
| Saccharomyces cerevisiae | MRB1 | Ime4 | mum2 | Kar4 | |||||
| Schizosaccharomyces pombe | Mmi1* | Ime4 | |||||||
| Arabidopsis thaliana | >10 proteins that contain a YTH domain | MTA | FIP37 | ||||||
does not bind m6A (Wang et al. 2016)
Figure 4.
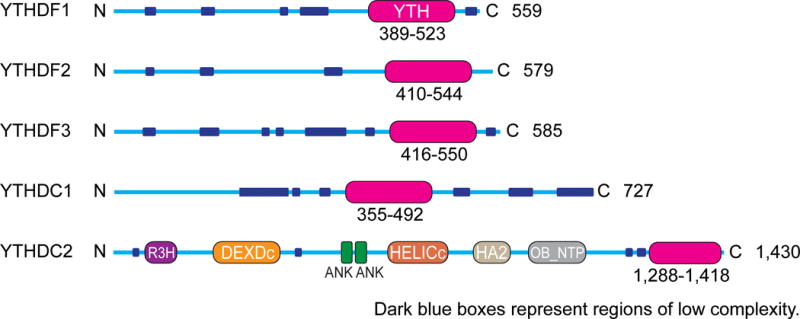
Schematic representation of the domain structures of the five human YTH proteins: DF1, DF2, DF3, DC1, and DC2. The YTH domain (magenta) is at the C-terminal region in DF1, DF2, DF3, and DC2 and is located internally in DC1. DC1 has a different domain organization than DC2. DF1, DF2, and DF3 show highly similar sequence and domain structure. The low-complexity (dark blue) and Glu-rich (red) regions are indicated, as are the R3H, DEXDc, ankyrin repeat (ANK), HELICc, HA2, and OB-fold (OB_NTP) domains for DC2. The length of the protein is indicated at the right of each protein schematic.
The DF family members (DF1, DF2, and DF3) are highly similar to each other, are predominantly cytoplasmic, and contain two domains: a C-terminally located YTH domain and the remaining majority of the protein, which is a large, low-complexity domain enriched largely in Q, N, and P residues (Figure 4). The presence of such a large, low-complexity domain raises the possibility that DF protein function is linked to liquid-liquid phase separations and protein droplets, as is commonly observed for proteins with these domains, especially proteins that also have RNA-binding domains (Aguzzi & Altmeyer 2016). Indeed, each of the DF proteins is found in proteomics analyses of protein droplet structures, including stress granules (Jain et al. 2016) and artificially generated RNA granules (Kato et al. 2012).
In contrast to the DF proteins, DC1 is a predominantly nuclear protein with a YTH domain and multiple other functional domains, including multiple putative nuclear localization elements, and an SH2 domain. DC1 appears to be the major reader of nuclear m6A.
DC2 is a nucleocytoplasmic protein with poorly defined function. It contains a DEAD-box RNA helicase domain, raising the possibility that the helicase is targeted to specific RNAs by m6A. DC2 binds predominantly to noncoding RNAs and to intronic and intergenic regions (Patil et al. 2016), so any functional roles of this protein may not be mediated by its direct binding to m6A residues in mature mRNAs.
Structural mechanism of m6A recognition
The structure of m6A mRNA complexed with a YTH domain was solved by several groups (Luo & Tong 2014, Theler et al. 2014, Xu et al. 2014). The YTH domain folds into a distinct module comprising approximately ~145 amino acids, with a notable tryptophan cage comprising two or three tryptophan residues that form a pyramid-shaped cage around the methyl group of m6A. The YTH domain makes additional interactions with the nucleotides preceding and following m6A, explaining the sequence preference for the DRACH (where D = G, A, or U; R = G or A; and H = C, A, or U) motif (Luo & Tong 2014, Theler et al. 2014, Xu et al. 2014). Notably, the affinity for YTH domain binding to m6A is unusually weak, with most reports showing affinities of ~1–2 μM (Luo & Tong 2014; Theler et al. 2014; Xu et al. 2014, 2015) and ~20–50-fold-weaker binding for the nonmethylated RNA. The weak binding affinity suggests that the YTH domain likely does not form a stable complex with m6A and would need additional interactors to remain bound.
m6A is found in a slightly degenerate context in RNA: DRACH (Linder et al. 2015). There is no evidence that any specific DRACH sequence is strongly preferred by any YTH domain in any YTH protein. One study suggested that the DC1 domain, unlike other YTH domains, prefers a G preceding the m6A (Xu et al. 2015); however, iCLIP analysis of the endogenous YTH proteins showed that all YTH domains bind m6A, with either G or A preceding the m6A (Patil et al. 2016). Thus, primary sequence alone does not appear to dictate differential YTH protein binding to specific mRNAs.
Function of DF family proteins
There have been conflicting studies related to the function of the DF proteins. Multiple groups have performed highly similar assays but have obtained different results, making it difficult to fully understand the function of these proteins. The first study to explore the function of a DF protein examined DF2 (X. Wang et al. 2014), the DF family member that is the most abundant in nearly all cell types. This study proposed that DF2 mediates the well-documented instability of m6A-containing mRNAs, which was first shown using radiolabeled tracer studies in 1978 (Sommer et al. 1978). More recent studies similarly showed that m6A-containing mRNAs have reduced half-lives compared to mRNAs lacking m6A (Batista et al. 2014, Dominissini et al. 2012, Geula et al. 2015). Analysis of DF2-depleted cells showed increased half-lives of mRNAs that contained PAR-CLIP-mapped DF2-binding sites (X. Wang et al. 2014). This effect was proposed to be due to an interaction of DF2 with P-bodies (X. Wang et al. 2014). However, this interaction was infrequent, indicating that DF2 may interact only transiently with P-bodies to deliver mRNAs for decay. Notably, ribosome profiling analysis showed a minimal effect of DF2 on translation efficiency (X. Wang et al. 2014).
In contrast to DF2, DF1 was shown to promote translation (Wang et al. 2015). Unlike DF2, DF1 did not show a substantial effect on mRNA stability on the basis of analysis of DF1-depleted cells. Wang et al. (2015) found that DF1 interacted with eIF3 and other translation initiation factors, which they suggested allowed DF1 to directly influence translation. The authors also proposed an interesting model in which mRNAs are first translated due to binding DF1 and then degraded after being transferred to DF2. This model was generated on the basis of an analysis of metabolic labeling to monitor the fate of mRNAs (Wang et al. 2015).
One of the biggest questions raised by these two studies was how two very similar proteins could have different effects on their target mRNAs and bind different interacting proteins. An additional question was how DF1 could promote translation by binding to initiation factors. DF1-binding sites are enriched near stop codons and 3´UTRs. In contrast, initiation factors are typically recruited to the 5´UTR to facilitate loading of the ribosome to the start codon. Although factors that bind at or near the poly(A) tail enhance translation through an mRNA looping mechanism that places the 3´ end of the mRNA in proximity to the 5´UTR, the majority of 3´UTR m6A sites (and thus DF1-binding sites) are located closer to the coding sequence. Thus, the DF1 findings suggest a potentially novel mode of translation initiation in which initiation factors are initially recruited to stop codon–adjacent regions.
Do DF proteins bind different mRNAs and different m6A sites? The initial DF1 study suggested that DF1 and DF2 share approximately 50% of their target RNAs, with the remaining transcripts unique to each DF protein (Wang et al. 2015). However, more recent CLIP-based transcriptome-wide mapping of all YTH proteins showed that DF1, DF2, and DF3 show essentially identical binding at each m6A site in mRNAs (Patil et al. 2016). This latter finding is consistent with the high sequence identity of the YTH domains shared by these three proteins. One difference is that Patil et al. (2016) examined endogenous YTH proteins rather than heterologously expressed proteins as were used in previous studies. An additional difference is that the earlier studies used a bioinformatic approach in which sites were called on the basis of specific statistical criteria and subthreshold sites were not called. This approach results in stochastic calling efficiencies for sites that are near the threshold. Moreover, these studies compared DF-binding sites independently of whether an m6A site had been called there. However, Patil et al. (2016) focused on m6A sites rather than DF-binding sites. At each m6A site, read densities for each DF protein were compared, revealing highly similar patterns of binding between all DF proteins. On the basis of these new findings, it appears that DF proteins exhibit essentially identical RNA-binding preferences at m6A sites.
DF1 was initially shown to affect translation without affecting mRNA stability (Wang et al. 2015), although other studies have found that DF1 triggers mRNA degradation. Tethering experiments initially showed that only DF2 induces mRNA degradation when tethered to a reporter RNA (X. Wang et al. 2014). However, tethering experiments performed by others showed that all three DF proteins triggered mRNA degradation and deadenylation (Du et al. 2016). Another tethering experiment showed that all DF proteins caused increased mRNA stability, with a corresponding increase in protein expression (Kennedy et al. 2016). Studies of viruses, including Zika virus (Gokhale et al. 2016, Lichinchi et al. 2016) and HIV (Tirumuru et al. 2016), showed that DF proteins promoted degradation of virus-encoded m6A transcripts, consistent with the idea that all these proteins promote mRNA degradation. The common theme in these more recent studies is that all three DF proteins share a similar function, rather than distinct functions. Nevertheless, the highly divergent results seen by different groups raise questions about the functions of these individual proteins that remain to be resolved.
Interestingly, Drosophila contains a single DF-like YTH protein, CG6422, rather than separate homologs for DF1, DF2, and DF3 (Kam et al. 2017) (Table 1). This is more consistent with the model that the DF proteins are functionally redundant in mammals, with a single DF-like protein sufficient to mediate their function in lower organisms.
DC1 and DC2
DC1 was originally characterized as a nucleus-enriched splicing regulator before it was known to interact with m6A. Studies using DC1 overexpression demonstrated that it regulated diverse types of alternative splicing events in endogenous transcripts (Zhang et al. 2010). This effect required the YTH domain. Thus, once the YTH domain was implicated as an m6A-binding module, DC1 was examined for its role in m6A-regulated splicing (Xiao et al. 2016). DC1 depletion caused splicing abnormalities, which could be rescued only by a DC1 protein that contains its YTH domain. Thus, DC1 mediates, at least in part, splicing events regulated by m6A.
Another study suggested that m6A-regulated splicing is caused, in part, by m6A-induced unfolding of mRNA and subsequently enhanced accessibility to HNRNPC, a regulator of splicing (Liu et al. 2015). This mechanism was proposed to contribute to less than 20% of m6A-regulated splicing events. It remains unclear how many m6A-dependent splicing events can be attributed uniquely to DC1 or HNRNPC and how many cannot be explained by either DC1 or HNRNPC.
DC1 also mediates the epigenetic effects of XIST, a noncoding RNA that is critical for silencing genes on one X chromosome in female cells (Lee 2009). XIST contains at least 76 m6A sites, and m6A is required for the gene silencing effects of XIST (Patil et al. 2016). By performing a systematic analysis of the transcriptome-wide binding properties of each YTH protein, we found that only DC1 prominently bound to m6A sites in XIST. Furthermore, depletion of DC1 prevented XIST from inducing gene repression on the X chromosome (Patil et al. 2016). Notably, m6A acts to recruit DC1 to XIST, which triggers transcriptional repression. Depletion of m6A normally prevents XIST function; however, artificial tethering of DC1 to XIST was sufficient to restore XIST function in m6A-deficient cells (Patil et al. 2016). Notably, the DC1 protein interaction network includes multiple epigenetic regulators (Patil et al. 2016). It remains to be determined whether these interactions enable DC1 to mediate the epigenetic silencing effects of XIST.
At present, the function of DC2 is poorly understood. Prior to our finding that DC2 binds m6A (Patil et al. 2016), DC2 was described as a cellular factor that was necessary for HCV genome replication (Morohashi et al. 2011). Recent studies suggest that DC2 enhances the translation of HIF1α mRNA via its helicase function (Tanabe et al. 2016). It is not known whether these effects require its YTH domain or involve m6A recognition in HIF1α mRNA.
eIF3
We identified eIF3 as part of studies attempting to understand the translation-promoting effects of m6A in in vitro reconstituted translation systems (Meyer et al. 2015). In these assays, template RNAs are added to purified translation initiation factors, resulting in ribosome binding to RNA, followed by scanning and then formation of a translation preinitiation complex at the start codon. This method, termed toeprinting, has been widely used to study the initiation factor requirements for viral internal ribosome entry sites (IRES) (de Breyne et al. 2008, Jackson et al. 2010).
Toeprinting showed that a single m6A in the 5´UTR of transcripts results in a marked change in the initiation factors required for translation initiation. Typically, eIF4 proteins, especially the cap-binding protein eIF4E, are required for translation initiation (Jackson et al. 2010). However, m6A-containing mRNAs do not require eIF4E and other components of the eIF4 complex, such as eIF4A and eIF4G, to recruit eIF3 to mRNA. Translation of m6A-containing mRNAs can be achieved with eIF3 and the other initiation factors. This was confirmed using cellular lysates lacking eIF4E (Meyer et al. 2015).
The initial evidence that eIF3 was an m6A reader came from the finding that eIF3 preferentially crosslinks to m6A-containing mRNA over nonmethylated RNA (Meyer et al. 2015). The physiological significance of eIF3 as an m6A-binding protein complex was supported by the finding that ~35% of eIF3 CLIP sites mapped throughout the transcriptome overlap with m6A sites. Thus, m6A is a major mechanism by which eIF3 is recruited to mRNAs.
We also found that mRNAs that contain 5´UTR m6A show enhanced translation in cells. In cells depleted of m6A by METTL3 knockdown, only mRNAs containing 5´UTR m6A showed reduced translation, whereas mRNAs that contained m6A elsewhere, including near stop codons or in the 3´UTR, did not show reduced translation (Meyer et al. 2015). These data highlight the selective link between 5´UTR m6A and translation.
These findings revealed a novel function for m6A in promoting a unique mode of translation initiation. 5´UTR m6A enables mRNAs to switch from canonical eIF4E-dependent translation initiation to eIF4E-independent translation initiation. Because eIF4E activity is impaired in diverse stress and disease states (Banko et al. 2006, Richter & Sonenberg 2005), these studies point to m6A as a potential mechanism by which selective, disease-specific translation could be achieved.
Importantly, the translation effects are due to 5´UTR m6A, and not m6Am. Both of these nucleotides are found in the 5´UTR of mRNAs, so it is possible to mistakenly annotate an mRNA with an m6A when it instead has an m6Am, especially if low-resolution approaches such as MeRIP-Seq/m6A-seq are used. However, the studies on 5´UTR m6A mapped these m6A residues by using the single-nucleotide-resolution m6A mapping approach miCLIP, which can readily discriminate between m6A and m6Am. Additionally, METTL3 knockdown selectively reduces the translation of mRNAs with an annotated 5´UTR m6A. Notably, METTL3 forms only m6A, not m6Am, which is formed by an unknown enzyme (Keith et al. 1978). Thus, 5´UTR m6A is selectively linked to eIF3-dependent, eIF4E-independent translation.
Notably, eIF3 is linked to two modes of m6A-induced translation. In one case, m6A directly binds and recruits eIF3 to the 5´UTR (Meyer et al. 2015). In another case, DF1 binds m6A residues near the stop codon and potentially delivers eIF3 to the 5´UTR through an undefined mechanism (Wang et al. 2015). The direct 5´UTR m6A-eIF3 interaction is similar to two other mechanisms by which eIF3 is recruited to 5´UTRs: certain viral IRES sequences (Sun et al. 2013) and recently defined eIF3-binding specific stem loop structures (Lee et al. 2015). Thus, eIF3 recruitment to the 5´UTR appears to be a general mechanism of translational enhancement. In contrast to viral IRESs, which present large surfaces to bind eIF3 (Jackson et al. 2010), m6A is small and appears to be recognized by a multisubunit interface of eIF3 involving the ΔeIF3a/eIF3c, ΔeIF3c, and eIF3d/eIF3I subunits (Meyer et al. 2015). Similarly, structured motifs in the c-jun 5´UTR also bind to eIF3 via a multisubunit interface comprising eIF3a, -b, -d, and -g (Lee et al. 2015). The direct recruitment of eIF3 by m6A contrasts with the indirect mechanism of recruitment mediated by DF1 (Wang et al. 2015). Surprisingly, we found no substantial change in translation efficiency of mRNAs that contain m6A near the stop codon after METTL3 knockdown (Meyer et al. 2015). These are the major locations of DF1 binding. The reason for this discrepancy is unknown but could be due to any of a number of potential factors, as mentioned above.
Another Protein: HNRNPA2B1
Another potential m6A-binding protein is HNRNPA2B1. This protein was originally identified as a nuclear m6A-binding protein that affects microRNA biogenesis (Alarcon et al. 2015a,b). Although HNRNPA2B1 lacks a YTH domain, the authors of this study suggested that HNRNPA2B1 could be an m6A reader. However, they also mentioned that m6A could instead help unfold RNA and improve the accessibility of HNRNPA2B1 to its binding sites. This concept of m6A-induced RNA unfolding was proposed by Kierzek & Kierzek (2003) and was characterized in detail by Kool and colleagues (Roost et al. 2015). Subsequent transcriptome-wide structure mapping demonstrated that m6A is found in regions with reduced structure (Spitale et al. 2015), supporting the overall idea that m6A can create greater accessibility to a region of RNA by impairing local RNA folding.
The studies of HNRNPA2B1 highlight the difficulty in distinguishing between whether a protein directly binds m6A or simply shows improved binding to RNA due to local unfolding induced by m6A. The binding properties of HNRNPA2B1 suggest that HNRNPA2B1 does not directly bind m6A, unlike the other nuclear reader DC1. HNRNPA2B1-binding sites in the transcriptome show poor overlap with m6A, with only a small subset of m6A sites showing prominent HNRNPA2B1 CLIP reads (Alarcon et al. 2015a). This finding is more compatible with the model in which HNRNPA2B1 binds to a nonmethylated consensus site and some sites become more accessible after a nearby adenosine becomes methylated. HNRNPA2B1 contrasts with DC1, which is bound at essentially all m6A sites in nuclear RNAs (Patil et al. 2016). Further structural studies will be needed to definitively determine whether HNRNPA2B1 contains a domain that directly contacts and recognizes m6A.
Interestingly, a recent analysis reexamined the role of HNRNPA2B1 in processing pre-microRNAs (Martinez et al. 2016). This study failed to find a clear effect of HNRNPA2B1 depletion on mature microRNA expression levels, in contrast to earlier studies (Alarcon et al. 2015a,b). This newer study challenges the idea that HNRNPA2B1 has a general role in microRNA processing. Further work will be needed to address this discrepancy.
m6A WRITERS
Early Studies: Discovery and Cloning of METTL3
Initial investigations into the identity of the protein(s) responsible for m6A formation were facilitated by the development of an in vitro methylation assay in the late 1980s (Narayan & Rottman 1988). In this assay, synthetic mRNAs are mixed with HeLa cell nuclear extracts, and incorporation of [3H]-methyl groups into RNA was used to monitor m6A formation. Using this strategy, researchers showed that in vitro methylation of synthetic mRNAs recapitulates the pattern of m6A observed in vivo and confirmed that a previously reported m6A consensus sequence (GGACU) is also methylated with this system (Harper et al. 1990).
Using this in vitro methylation assay, Rottman and colleagues tested the methylation activity of various nuclear fractions separated by column chromatography and isolated a 70-kDa fraction that had S-adenosylmethionine (SAM)-binding activity (Bokar et al. 1994). Subsequent purification and cloning identified the sequence for this m6A methyltransferase (Bokar et al. 1997), originally termed MT-A70 but now referred to as METTL3. METTL3 is part of a larger family of putative SAM-dependent methyltransferases that is highly conserved in mammals (Schapira 2015). Genetic deletion of METTL3 in plants, yeast, and mammalian cells leads to complete or near-complete loss of m6A in polyadenylated RNA (Agarwala et al. 2012, Geula et al. 2015, Zhong et al. 2008). Thus, METTL3 appears to be the major m6A-forming enzyme in polyadenylated mRNA. Importantly, however, METTL3 does not mediate m6A formation in rRNA or the small nuclear RNA (snRNA) U6 (Shimba et al. 1995).
WTAP: The Key Adaptor of METTL3
The second major component of the m6A methylation complex is WTAP, which was initially identified by Fray and colleagues (Zhong et al. 2008). Fray and colleagues characterized the Arabidopsis homolog of METTL3, MTA, after it was identified in a large-scale screen of plant embryo defective mutants. As part of their analysis, they performed a yeast two-hybrid screen for MTA-binding proteins, which resulted in the identification of FIP37, the plant homolog of WTAP. Their detailed characterization of this interaction confirmed that FIP37 binds MTA and that FIP37 and MTA colocalize in the nucleus (Zhong et al. 2008).
The functional importance of WTAP was revealed by Fink and colleagues (Agarwala et al. 2012). They similarly found that the yeast METTL3 homolog, Ime4, was bound to the yeast WTAP homolog, mum2. Notably, these authors showed that mum2 was required for m6A formation in yeast. Thus, their data showed that the WTAP homolog is a binding partner of the methyltransferase and is essential for RNA methylation. Shortly after these seminal discoveries, the WTAP-METTL3 interaction was confirmed in diverse mammalian cells (Liu et al. 2014, Ping et al. 2014, Schwartz et al. 2014b).
A major function of WTAP is to localize METTL3 and its binding partner, METTL14 (see below), to nuclear speckles (Ping et al. 2014). WTAP depletion causes loss of METTL3 and METTL14 localization from these speckles and loss of m6A formation in mRNA. Thus, WTAP maintains METTL3 in speckles to efficiently methylate mRNA.
KIAA1429/Virilizer
KIAA1429 (also known as vir-like m6A methyltransferase associated, or VIRMA), the mammalian homolog of the Drosophila Virilizer protein, is also associated with the methylation complex. Several lines of evidence hinted that this interaction might be important. A proteomics analysis of WTAP revealed KIAA1429 as one of the top interactors (Horiuchi et al. 2013). Subsequent proteomics studies similarly observed this interaction (Schwartz et al. 2014b). Depletion of KIAA1429 leads to substantial loss of m6A, indicating the necessity of KIAA1429 for the function of the methyltransferase complex (Schwartz et al. 2014b). The Drosophila homologs of KIAA1429 and WTAP [Virilizer and Fl(2)d, respectively] also interact in flies (Ortega et al. 2003), as do FL(2)d and Ime4 (Lence et al. 2016). These proteins influence m6A levels in Drosophila S2R+ cells (Lence et al. 2016), thus suggesting a conserved role in regulating methylation. In flies, Virilizer and Fl(2)d are involved in splicing of Sxl transcripts in females to control sex determination (Granadino et al. 1996, Hilfiker et al. 1995), and this process is dependent on m6A formation catalyzed by Virilizer, Fl(2)d, and IME4, the Drosophila homologs of METTL3 (Haussmann et al. 2016, Kan et al. 2017, Lence et al. 2016). It is currently not known how KIAA1429 influences the methylation complex and why it is important for m6A formation.
RBM15/15B: Mediators of Methylation Specificity
A major mystery in m6A biology has been the mechanism by which specific adenosines are targeted for methylation. The preferred consensus sequence for m6A methylation is DRACH, which is observed with a relatively high frequency and uniform distribution along the length of a given mRNA (Linder et al. 2015). However, m6A residues are enriched only in certain mRNAs and within distinct regions of a transcript (Meyer & Jaffrey 2014). Therefore, only certain DRACH m6A consensus sites are selected for methylation, and this selection occurs through an unknown mechanism.
Recent studies have begun to shed light on how specific m6A consensus sites may be targeted for methylation. We recently showed that two proteins—RBM15 and its paralog, RBM15B—are additional components of the methyltransferase complex (Patil et al. 2016). These proteins were initially identified as interactors of WTAP through proteomics studies (Horiuchi et al. 2013), which led us to speculate that they might also contribute to the methyltransferase complex. Indeed, coimmunoprecipitation analyses revealed that RBM15 and RBM15B interact with METTL3 in a WTAP-dependent manner (Patil et al. 2016). Importantly, knockdown of RBM15 and RBM15B caused significant reductions of m6A in mRNA (Patil et al. 2016), indicating that these proteins are indeed functional components of the methyltransferase complex. These findings were further supported by studies in Drosophila, which showed that the homolog of RBM15, Spenito (Nito), is also necessary for m6A formation in flies (Lence et al. 2016).
Interestingly, iCLIP studies show that RBM15 and RBM15B bind to U-rich regions in mRNAs immediately adjacent to m6A sites (Patil et al. 2016). This finding suggests that, instead of binding directly to DRACH motifs, RBM15 and RBM15B bind uridine-enriched regions and then recruit WTAP/METTL3 complexes to methylate nearby DRACH motifs.
One RNA that is targeted by this mechanism is the lncRNA XIST, which mediates X-inactivation and gene silencing during development. RBM15/15B-assisted XIST methylation is necessary for XIST-mediated silencing, thus revealing the first functional role for transcript-specific methylation directed by RBM15 and RBM15B (Patil et al. 2016).
Additional adaptors may exist. Not all methylated DRACH sites are found near U-rich regions; thus, RBM15/15B-mediated methylation is likely a mechanism for m6A formation in only a subset of cellular RNAs. Methylation complexes directed to specific mRNAs may provide a mechanism to shape m6A levels in individual mRNAs.
METTL14: An RNA Adaptor Needed for METTL3 Activity
Proteomics analyses of METTL3 adaptors revealed METTL14 (Liu et al. 2014, Ping et al. 2014, Schwartz et al. 2014b, Y. Wang et al. 2014). All the studies showed that METTL14 was required for m6A formation in cells. However, He and colleagues showed that purified METTL14 synthesizes m6A, thus identifying METTL14 as the second methyltransferase enzyme (Liu et al. 2014). Indeed, METTL14 has homology to methyltransferases; however, it has a degenerate SAM-binding domain. This study fundamentally transformed the field by raising the idea that distinct m6A writers could mediate formation of different m6A sites in the transcriptome.
However, three separate crystallization studies overturned the concept that METTL14 is a methyltransferase enzyme. Crystal structures of the METTL3-METTL14 complex demonstrated that METTL3 is the only protein to bind SAM and identified residues in the catalytic motif of METTL3 that are critical for methylation (Sledz & Jinek 2016, P. Wang et al. 2016, X. Wang et al. 2016). These studies showed that METTL14 lacks a SAM-binding domain. Additionally, these researchers showed that purified METTL14 does not have methyltransferase activity, in contrast to the earlier finding that reported methyltransferase activity from purified METTL14. Although it is now clear that METTL14 does not transfer a methyl group to RNA, METTL14 plays a critical role by binding substrate RNA by forming interactions with METTL3 that enhance its enzymatic activity and by positioning the methyl group for transfer to adenosine (Sledz & Jinek 2016, P. Wang et al. 2016, X. Wang et al. 2016).
What accounts for the discrepancy between the original data showing METTL14 methyltransferase activity and the subsequent studies? He and colleagues expressed METTL14 in insect cells prior to purification for enzyme assays (Liu et al. 2014). Nam and colleagues showed that METTL14 purified in this manner copurifies complexed with insect METTL3 (P. Wang et al. 2016). As a result, the human METTL14–insect METTL3 heterodimer likely explains the activity attributed to purified METTL14 in the earlier study. Thus, METTL14 is not a methyltransferase, but an adaptor required for METTL3 activity.
Notably, the crystal structure data have provided new insights into cancer-associated mutations in METTL14 (P. Wang et al. 2016). R298P, which is detected in uterine/endometrial cancers, is a mutation in the RNA-binding domain, and the mutated form of METTL14 shows partially reduced methyltransferase activity, most likely due to inefficient RNA binding (P. Wang et al. 2016).
Moving forward, it will be important to understand how the known components of the methyltransferase complex interact with each other and with target RNAs to achieve methylation. Early studies showed that methylation is achieved within 10 min of RNA synthesis (Sommer et al. 1978). These data, combined with the fact that the methyltransferase machinery is predominantly located in nuclear speckles (Bokar et al. 1997, Ping et al. 2014) (sites where transcription and RNA processing can occur), suggest that adenosine methylation is a cotranscriptional process. Recent studies have further supported this idea by showing that METTL3 interacts with chromatin and with the transcription machinery and that m6A formation is regulated by the rate of transcription (Knuckles et al. 2017, Slobodin et al. 2017). Further studies investigating potential interactions between the methyltransferase complex and the transcriptional machinery as well as other mRNA processing factors will likely lead to a better understanding of what drives methylation specificity.
METTL16: An m6A Methyltransferase that Targets U6 snRNA and MAT2A mRNA
Recent studies have revealed that m6A in at least one mammalian mRNA can be formed by methyltransferase-like 16 (METTL16) (Pendleton et al. 2017). METTL16 was found to be an m6A-forming enzyme in mRNA on the basis of studies of SAM-regulated intron retention in the methionine adenosyltransferase 2A (MAT2A) gene, which encodes the major SAM-forming enzyme in most cells. Conrad and colleagues found a hairpin structure in MAT2A pre-mRNA that was required for splicing this intron (Pendleton et al. 2017). They noticed that the hairpin sequence resembled the hairpin in the U6 snRNA that contains m6A and that the hairpins in MAT2A indeed contained m6A at the same site as found in U6 (Pendleton et al. 2017). Because m6A-forming enzymes require SAM, these researchers reasoned that this methylation event may be involved in the SAM-dependent splicing of this intron. After finding that METTL3 failed to methylate these hairpins, they focused on METTL16 because the Schizosaccharomyces pombe homolog forms m6A in U6 RNA. Conrad and colleagues showed that human METTL16 synthesizes m6A in both human U6 and the U6-like hairpins in MAT2A mRNA (Pendleton et al. 2017). Notably, the m6A is found in a C-m6A-G context in MAT2A mRNA, indicating that not all m6A residues in mRNA are in a DRACH sequence context. Although MAT2A is the only mRNA target of METTL16 to date, further investigation may reveal other mRNAs, especially those involved in SAM metabolism, as targets for methylation by METTL16.
m6A ERASERS
An important discovery in the m6A field was the identification of two different enzymes that can demethylate m6A: FTO and ALKBH5 (Jia et al. 2011, Zheng et al. 2013). However, recent studies show that FTO preferentially targets m6Am, a highly prevalent nucleotide in mRNA (Mauer et al. 2017). ALKBH5 was not found to have m6Am demethylase activity (Mauer et al. 2017), although the degree to which ALKBH5 targets specific m6A sites remains unclear. Animals deficient in ALKBH5 have spermatogenesis defects but are otherwise largely normal, demonstrating that m6A removal by this protein is not essential for development or viability. As described below, further studies will be needed to determine whether m6A demethylation has a role in regulating m6A in mRNA as opposed to other types of m6A-containing RNA.
FTO
Although FTO was the first enzyme linked to m6A demethylation, this view of FTO has markedly changed with the recent finding that m6A is not the preferred target of FTO in vivo (Mauer et al. 2017). The initial link between FTO and RNA came from He and colleagues, who showed that FTO demethylates 3-methyluridine in RNA (Jia et al. 2008). This modified nucleotide is not prevalent in mRNA, and this group subsequently showed that FTO could demethylate m6A in mRNA in in vitro reactions (Jia et al. 2011). Subtle changes in m6A in polyA mRNA were also seen in cells that either overexpressed or were depleted of FTO (Jia et al. 2011). These studies provided the initial support for the idea that m6A could be reversed by FTO and generated excitement because it suggested that m6A could be formed and removed in a dynamic, signal-dependent manner.
We and others examined the Fto knockout mouse to understand how Fto affects m6A (Hess et al. 2013). These mice are largely normal except for alterations in dopaminergic neurotransmission, lean body mass, and alterations in metabolic rate (Fischer et al. 2009, Hess et al. 2013). However, instead of detecting global increases in m6A, m6A mapping using MeRIP-Seq showed changes in just a subset of m6A peaks throughout the transcriptome (Hess et al. 2013).
The selective alteration in just a few m6A residues in Fto knockout mice was surprising because all m6A residues are in the same general consensus site. Therefore, each m6A should be equally susceptible to Fto. However, a detailed reanalysis of m6A peak increases in the Fto knockout compared to wild-type mice showed that m6A peaks that showed elevation in Fto knockouts were highly enriched in the 5´UTR (Mauer et al. 2017). The 5´UTR is the site of m6Am, which is highly similar to m6A except that it also contains a 2´-O-methyl modification on the ribose moiety of the nucleotide (Wei et al. 1975).
The previous studies were clear that FTO could demethylate the N6 position of adenine (Jia et al. 2011), but they did not test the other major and highly prevalent cellular nucleotide that contains methylated N6 adenine, i.e., m6Am. We found that FTO showed nearly 100-times-greater catalytic activity against m6Am when presented in its natural context adjacent to the N7-methylguanosine (m7G) cap (Mauer et al. 2017). Notably, quantitative measurements of m6Am in FTO knockout cells showed significant increases in m6Am, with no detectable increase in m6A. In contrast, expression of a cytoplasmically localized FTO caused a reduction in m6Am, with no effect on m6A. These experiments revealed that m6Am is a reversible cap-associated epitranscriptomic mark (Mauer et al. 2017). Additional studies showed that m6Am renders mRNAs less susceptible to decapping, thereby contributing to the markedly elevated mRNA stability associated with mRNAs containing m6Am. Thus, FTO is the eraser for m6Am, and this role has functional consequences for mRNA stability.
Notably, several findings hinted that m6A might not be the correct substrate. First, FTO did not show a preference for m6A in its physiological consensus site (Jia et al. 2011). A lack of sequence selectivity is not consistent with most modifying enzymes. In contrast, FTO has strong preference for the structural context of m6Am, with a requirement for the m7G and triphosphate linker to facilitate its demethylation of m6Am (Mauer et al. 2017). Another hint about FTO was its low catalytic rate toward m6A relative to other enzymes in this class. In contrast, the catalytic rate of FTO for m6Am is much higher (Mauer et al. 2017).
What could explain the small but detectable increase in m6A seen after FTO depletion? In some studies, no increases in m6A levels in poly(A) mRNA were detected in FTO knockdown or knockout tissue (Mauer et al. 2017). However, in a study of FTO in acute myeloid leukemia, an ~20% increase in m6A was seen upon FTO depletion (Li et al. 2017). However, the RNA-Seq gene expression data sets of the FTO-depleted acute myeloid leukemia cells showed a similar 25% increase in METTL14 mRNA. Thus, increases in m6A seen in some cells after FTO depletion may be caused by increased levels of m6A-forming enzymes.
A major reason why the discovery that FTO demethylates m6A generated excitement is that FTO mutations were linked to obesity (Fawcett & Barroso 2010). Thus, improper control of m6A levels could be a major source of human disease. However, this view was overturned by several studies that showed that obesity-associated FTO mutations do not affect the FTO protein; instead, such mutations affect the expression of neighboring genes (Claussnitzer et al. 2015, Smemo et al. 2014, Stratigopoulos et al. 2016). The mutations are located primarily in intron 1 of FTO, and chromosome interaction analysis showed that this region is an enhancer that controls the expression of neighboring genes Irx3 and RPGRIP1L. Despite extensive studies, there is no direct evidence that the obesity-associated FTO mutations affect FTO splicing or FTO expression. Thus, human obesity is linked to mutations in the FTO gene, but not to alterations in FTO protein levels or RNA methylation.
ALKBH5
ALKBH5 was initially discovered in a biochemical screen of demethylase proteins for enzymes that exhibit m6A demethylase activity (Zheng et al. 2013). ALKBH5 knockdown resulted in subtle increases and ALKBH5 overexpression caused subtle decreases in m6A in cellular poly(A) mRNA, supporting the idea that ALKBH5 might regulate m6A in mRNA (Zheng et al. 2013). Whereas FTO demethylates m6Am, ALKBH5 has no activity toward m6Am and appears selective for m6A (Mauer et al. 2017).
The localization of ALKBH5 in the nucleus (Zheng et al. 2013) makes it unlikely that it demethylates m6A in mature mRNA in the cytoplasm. If ALKBH5 demethylates mRNA, this demethylation can occur only during mRNA biogenesis in the nucleus. ALKBH5 could also demethylate m6A in nucleus-enriched, m6A-containing ncRNAs, such as MALAT1, Neat1, small nucleolar RNAs, snRNAs, and others (Linder et al. 2015).
Few studies have shown specific mRNA m6A sites that are increased in ALKBH5-depleted cells. In one case, NANOG mRNA was shown to have decreased m6A levels in breast cancer cells exhibiting increased ALKBH5 (Zhang et al. 2016a). Subsequent studies showed that these cells also have decreased methyltransferase activity, and thus whether the reduced m6A levels in NANOG mRNA are due to increased ALKBH5 or decreased methyltransferase activity is unclear (Zhang et al. 2016b).
In a study of glioma stem cells that exhibit elevated ALKBH5 expression, the numbers of m6A peaks and m6A-containing mRNAs were quantified after ALKBH5 knockdown (Zhang et al. 2017). Notably, ALKBH5 knockdown was associated with a reduction in both the number of mapped m6A sites and the number of m6A-containing mRNAs, which is contrary to the expectation that ALKBH5 demethylates m6A. However, Zhang et al. (2017) identified novel m6A peaks in the ALKBH5-depleted cells by comparing m6A peaks in the control and knockdown cells. The significance of this development is unclear because MeRIP-Seq/m6A-seq is relatively noisy. Therefore, there will be numerous peak differences between any two data sets. It will be important to analyze replicate data sets and to account for peak-calling variability so as to determine whether the observed differences are larger than what would be expected by chance.
Nevertheless, Zhang et al. (2017) identified a specific m6A in FOXMI mRNA that appears to be regulated by ALKBH5. These authors showed that demethylation of this m6A was dependent on the formation of a RNA-RNA hybrid between FOXM1 mRNA and an antisense transcript, AS-FOXM1. The authors speculated that this unique RNA-RNA hybrid enabled the recruitment and demethylation mediated by ALKBH5. Although biochemical measurements of demethylation rates of m6A in single-stranded RNA and in double-stranded RNA were not presented, the data raise the exciting possibility that ALKBH5 selectively demethylates a subset of m6A residues that are located in highly unique sequence and structural contexts, rather than serving as a general m6A demethylase. Notably, previous characterization of the enzymatic catalytic rates of ALKBH5 (Zheng et al. 2013) revealed unusually slow kinetics toward m6A in RNA. However, these m6A-containing RNAs were linear substrates. Conceivably, m6A in specific structural contexts such as that found in the FOXM1 RNA-RNA hybrid may result in improved kinetics, supporting the idea that these specialized m6A sites are the endogenous targets of ALKBH5.
The phenotype of ALKBH5 knockout mice suggests that m6A demethylation has very subtle roles in signaling. ALKBH5 knockout mice are largely normal except for defects in spermatogenesis (Zheng et al. 2013). Thus, none of the diverse signaling pathways necessary for development require m6A demethylation, unless there exists an as-yet-undiscovered m6A demethylase. Importantly, ALKBH5 may have more subtle phenotypes that may be revealed following further analysis of the ALKBH5 knockout mouse.
CONCLUSIONS AND FUTURE DIRECTIONS
The first studies of m6A in the 1970s predicted that m6A was likely to be a widespread modification with the potential to mediate selective control of gene expression (Desrosiers et al. 1974). Now, more than 40 years later, the development of methods for recognizing m6A transcriptome wide has elevated our understanding of this modification and has revealed that m6A is indeed capable of conferring selective regulation to subsets of the transcriptome.
The ability to map m6A at single-nucleotide resolution has been a significant technological advance that will now enable more targeted studies of m6A function through site-specific mutations of adenosine residues that are methylated. Mutagenesis is particularly important to avoid potential pleiotropic effects of depletion of m6A readers, writers, and erasers and to definitively establish the biological function of specific m6A residues in individual mRNAs.
Improvements in our ability to detect m6A have also helped shape our views of this modification and of other mRNA methylation events. Early profiling studies reported m6A antibody-based enrichment of RNA fragments mapping near the transcription start site of mRNAs (Meyer & Jaffrey 2014), but due to the low resolution of these techniques, it was difficult to determine whether this enrichment reflected m6A or m6Am residues. More recent single-nucleotide-resolution m6A mapping approaches revealed the prevalence of m6Am and enabled researchers to better distinguish these two modifications in global profiling studies (Linder et al. 2015, Schwartz et al. 2014b). The mapping of m6Am throughout the transcriptome, in turn, prompted functional studies that led to the unexpected discovery that m6Am is the preferred substrate for FTO (Mauer et al. 2017). This recent discovery not only adds to the repertoire of functional dynamic mRNA modifications but also indicates that previous studies relying on FTO depletion to investigate m6A function must be reinterpreted as they reveal roles of m6Am, not m6A.
Another major question relates to whether m6A demethylation is a physiological mechanism for global regulation of m6A levels in the transcriptome. The targets of ALKBH5, the putative m6A eraser in mRNA, are poorly defined, with current evidence indicating that ALKBH5 depletion has minimal effects on the levels of most m6A residues. Definitive identification of additional m6A sites that are targeted by ALKBH5 throughout the transcriptome will be critical to determining the degree to which demethylation regulates m6A levels in cells. Moreover, if ALKBH5 is shown to target specific m6A sites in mRNAs, it will be important to establish a mechanism by which ALKBH5 achieves this type of selectivity.
As our understanding of m6A processing has transformed, so too has our understanding of m6A function. Advances have been facilitated in large part by the discovery of m6A readers. Among the best-studied m6A-binding proteins are those in the YTH family, whose documented m6A-dependent functions include the regulation of mRNA stability (DF1, DF2, DF3), translation (DF1, DF3), splicing (DC1), and lncRNA-mediated gene silencing (DC1). The discrepancies among studies on the function of DF family proteins need to be resolved. However, these studies have revealed that m6A plays diverse functional roles within the cell and that recognition by m6A readers plays a critical role in determining the effects of m6A within a given mRNA.
Studies from our group have shown that methylation of cellular 5´UTRs is triggered by diverse stress-response pathways that are often activated in human disease (Meyer et al. 2015, Zhou et al. 2015). We find that these 5´UTR m6A residues promote the translation of stress-response proteins and suggest the existence of a so-called m6A stress response, in which m6A is a potential mechanism for fine-tuning the production of critical proteins during disease states. Additional physiological roles for m6A-mediated gene regulation that have been uncovered include regulation of circadian rhythms (Fustin et al. 2013), stem cell differentiation (Batista et al. 2014, Geula et al. 2015, Y. Wang et al. 2014), and noncoding RNA function (Patil et al. 2016). Additionally, several studies have uncovered links between m6A regulation and disease states: Multiple reports implicate various m6A regulatory proteins in cancer (Cui et al. 2017, Jaffrey & Kharas 2017, McGuinness & McGuinness 2014, Zhang et al. 2017), and several recent studies link m6A to viral RNA stability (Gokhale & Horner 2017). Such studies have been critical for our understanding of the broader roles of RNA methylation in physiological processes and human disease.
Although we have made tremendous progress in our understanding of the function and regulation of m6A since its discovery more than 40 years ago, much work lies ahead to gain a complete picture of this mark and how it controls gene expression. Through continued efforts to improve m6A detection methods, to identify additional readers, writers, and erasers, and to uncover functional roles for m6A, we will undoubtedly expand our understanding of the biology of m6A and its contribution to human health and disease.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Literature Cited
- Agarwala SD, Blitzblau HG, Hochwagen A, Fink GR. RNA methylation by the MIS complex regulates a cell fate decision in yeast. PLOS Genet. 2012;8:e1002732. doi: 10.1371/journal.pgen.1002732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguzzi A, Altmeyer M. Phase separation: linking cellular compartmentalization to disease. Trends Cell Biol. 2016;26:547–58. doi: 10.1016/j.tcb.2016.03.004. [DOI] [PubMed] [Google Scholar]
- Alarcon CR, Goodarzi H, Lee H, Liu X, Tavazoie S, Tavazoie SF. HNRNPA2B1 is a mediator of m6A-dependent nuclear RNA processing events. Cell. 2015a;162:1299–308. doi: 10.1016/j.cell.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-Methyladenosine marks primary microRNAs for processing. Nature. 2015b;519:482–85. doi: 10.1038/nature14281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banko JL, Hou L, Poulin F, Sonenberg N, Klann E. Regulation of eukaryotic initiation factor 4E by converging signaling pathways during metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2006;26:2167–73. doi: 10.1523/JNEUROSCI.5196-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista PJ, Molinie B, Wang J, Qu K, Zhang J, et al. m6A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15:707–19. doi: 10.1016/j.stem.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokar JA, Rath-Shambaugh ME, Ludwiczak R, Narayan P, Rottman F. Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex. J Biol Chem. 1994;269:17697–704. [PubMed] [Google Scholar]
- Bokar JA, Shambaugh ME, Polayes D, Matera AG, Rottman FM. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA. 1997;3:1233–47. [PMC free article] [PubMed] [Google Scholar]
- Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM, Gilbert WV. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014;515:143–46. doi: 10.1038/nature13802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claussnitzer M, Dankel SN, Kim KH, Quon G, Meuleman W, et al. FTO obesity variant circuitry and adipocyte browning in humans. N Engl J Med. 2015;373:895–907. doi: 10.1056/NEJMoa1502214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Shi H, Ye P, Li L, Qu Q, et al. m6A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18:2622–34. doi: 10.1016/j.celrep.2017.02.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Breyne S, Yu Y, Pestova TV, Hellen CU. Factor requirements for translation initiation on the Simian picornavirus internal ribosomal entry site. RNA. 2008;14:367–80. doi: 10.1261/rna.696508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delatte B, Wang F, Ngoc LV, Collignon E, Bonvin E, et al. Transcriptome-wide distribution and function of RNA hydroxymethylcytosine. Science. 2016;351:282–85. doi: 10.1126/science.aac5253. [DOI] [PubMed] [Google Scholar]
- Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. PNAS. 1974;71:3971–75. doi: 10.1073/pnas.71.10.3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–6. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- Du H, Zhao Y, He J, Zhang Y, Xi H, et al. YTHDF2 destabilizes m6A-containing RNA through direct recruitment of the CCR4-NOT deadenylase complex. Nat Commun. 2016;7:12626. doi: 10.1038/ncomms12626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett KA, Barroso I. The genetics of obesity: FTO leads the way. Trends Genet. 2010;26:266–74. doi: 10.1016/j.tig.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer J, Koch L, Emmerling C, Vierkotten J, Peters T, et al. Inactivation of the Fto gene protects from obesity. Nature. 2009;458:894–98. doi: 10.1038/nature07848. [DOI] [PubMed] [Google Scholar]
- Fustin J-M, Doi M, Yamaguchi Y, Hida H, Nishimura S, et al. RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell. 2013;155:793–806. doi: 10.1016/j.cell.2013.10.026. [DOI] [PubMed] [Google Scholar]
- Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, et al. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science. 2015;347:1002–6. doi: 10.1126/science.1261417. [DOI] [PubMed] [Google Scholar]
- Gokhale NS, Horner SM. RNA modifications go viral. PLOS Pathog. 2017;13:e1006188. doi: 10.1371/journal.ppat.1006188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhale NS, McIntyre AB, McFadden MJ, Roder AE, Kennedy EM, et al. N6-Methyladenosine in Flaviviridae viral RNA genomes regulates infection. Cell Host Microbe. 2016;20:654–65. doi: 10.1016/j.chom.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granadino B, Penalva LO, Sanchez L. The gene fl(2)d is needed for the sex-specific splicing of transformer pre-mRNA but not for double-sex pre-mRNA in Drosophila melanogaster. Mol Gen Genet. 1996;253:26–31. doi: 10.1007/s004380050292. [DOI] [PubMed] [Google Scholar]
- Harper JE, Miceli SM, Roberts RJ, Manley JL. Sequence specificity of the human mRNA N6-adenosine methylase in vitro. Nucleic Acids Res. 1990;18:5735–41. doi: 10.1093/nar/18.19.5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussmann IU, Bodi Z, Sanchez-Moran E, Mongan NP, Archer N, et al. m6A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature. 2016;540:301–4. doi: 10.1038/nature20577. [DOI] [PubMed] [Google Scholar]
- Hess ME, Hess S, Meyer KD, Verhagen LA, Koch L, et al. The fat mass and obesity associated gene (Fto) regulates activity of the dopaminergic midbrain circuitry. Nat Neurosci. 2013;16:1042–48. doi: 10.1038/nn.3449. [DOI] [PubMed] [Google Scholar]
- Hilfiker A, Amrein H, Dubendorfer A, Schneiter R, Nothiger R. The gene virilizer is required for female-specific splicing controlled by Sxl, the master gene for sexual development in Drosophila. Development. 1995;121:4017–26. doi: 10.1242/dev.121.12.4017. [DOI] [PubMed] [Google Scholar]
- Horiuchi K, Kawamura T, Iwanari H, Ohashi R, Naito M, et al. Identification of Wilms’ tumor 1–associating protein complex and its role in alternative splicing and the cell cycle. J Biol Chem. 2013;288:33292–302. doi: 10.1074/jbc.M113.500397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010;11:113–27. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffrey SR, Kharas MG. Emerging links between m6A and misregulated mRNA methylation in cancer. Genome Med. 2017;9:2. doi: 10.1186/s13073-016-0395-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, Wheeler JR, Walters RW, Agrawal A, Barsic A, Parker R. ATPase-modulated stress granules contain a diverse proteome and substructure. Cell. 2016;164:487–98. doi: 10.1016/j.cell.2015.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–87. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Yang CG, Yang S, Jian X, Yi C, et al. Oxidative demethylation of 3-methylthymine and 3-methyluracil in single-stranded DNA and RNA by mouse and human FTO. FEBS Lett. 2008;582:3313–19. doi: 10.1016/j.febslet.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan L, Grozhik AV, Vedanayagam J, Patil DP, Pang N, et al. The m6A pathway facilitates sex determination in Drosophila. Nat Commun. 2017 doi: 10.1038/ncomms15737. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Han TW, Xie S, Shi K, Du X, et al. Cell-free formation of RNA granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–67. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke S, Alemu EA, Mertens C, Gantman EC, Fak JJ, et al. A majority of m6A residues are in the last exons, allowing the potential for 3´ UTR regulation. Genes Dev. 2015;29:2037–53. doi: 10.1101/gad.269415.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith JM, Ensinger MJ, Mose B. HeLa cell RNA (2´-O-methyladenosine-N6-)-methyltransferase specific for the capped 5´-end of messenger RNA. J Biol Chem. 1978;253:5033–39. [PubMed] [Google Scholar]
- Kennedy EM, Bogerd HP, Kornepati AV, Kang D, Ghoshal D, et al. Posttranscriptional m6A editing of HIV-1 mRNAs enhances viral gene expression. Cell Host Microbe. 2016;19:675–85. doi: 10.1016/j.chom.2016.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kierzek E, Kierzek R. The thermodynamic stability of RNA duplexes and hairpins containing N6-alkyladenosines and 2-methylthio-N6-alkyladenosines. Nucleic Acids Res. 2003;31:4472–80. doi: 10.1093/nar/gkg633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuckles P, Carl SH, Musheev M, Niehrs C, Wenger A, Buhler M. RNA fate determination through cotranscriptional adenosine methylation and microprocessor binding. Nat Struct Mol Biol. 2017 doi: 10.1038/nsmb.3419. In press; https://doi.org/10.1038/nsmb.3419. [DOI] [PubMed]
- Konig J, Zarnack K, Rot G, Curk T, Kayikci M, et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. 2010;17:909–15. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AS, Kranzusch PJ, Cate JH. eIF3 targets cell-proliferation messenger RNAs for translational activation or repression. Nature. 2015;522:111–14. doi: 10.1038/nature14267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT. Lessons from X-chromosome inactivation: long ncRNA as guides and tethers to the epigenome. Genes Dev. 2009;23:1831–42. doi: 10.1101/gad.1811209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lence T, Akhtar J, Bayer M, Schmid K, Spindler L, et al. m6A modulates neuronal functions and sex determination in Drosophila. Nature. 2016;540:242–47. doi: 10.1038/nature20568. [DOI] [PubMed] [Google Scholar]
- Li Z, Weng H, Su R, Weng X, Zuo Z, et al. FTO plays an oncogenic role in acute myeloid leukemia as a N6-methyladenosine RNA demethylase. Cancer Cell. 2017;31:127–41. doi: 10.1016/j.ccell.2016.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichinchi G, Zhao BS, Wu Y, Lu Z, Qin Y, et al. Dynamics of human and viral RNA methylation during Zika virus infection. Cell Host Microbe. 2016;20:666–73. doi: 10.1016/j.chom.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12:767–72. doi: 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N6-Methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–64. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, Parisien M, Dai Q, Zheng G, He C, Pan T. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA. 2013;19:1848–56. doi: 10.1261/rna.041178.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovejoy AF, Riordan DP, Brown PO. Transcriptome-wide mapping of pseudouridines: Pseudouridine synthases modify specific mRNAs in S. cerevisiae. PLOS ONE. 2014;9:e110799. doi: 10.1371/journal.pone.0110799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S, Tong L. Molecular basis for the recognition of methylated adenines in RNA by the eukaryotic YTH domain. PNAS. 2014;111:13834–39. doi: 10.1073/pnas.1412742111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FJ, Pratt GA, van Nostrand EL, Batra R, Huelga SC, et al. Protein-RNA networks regulated by normal and ALS-associated mutant HNRNPA2B1 in the nervous system. Neuron. 2016;92:780–95. doi: 10.1016/j.neuron.2016.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, et al. Reversible methylation of m6Am in the 5´ cap controls mRNA stability. Nature. 2017;541:371–75. doi: 10.1038/nature21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuinness D, McGuinness D. m6A RNA methylation: the implications for health and disease. J Cancer Sci Clin Oncol. 2014;1:1–7. [Google Scholar]
- Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol. 2014;15:313–26. doi: 10.1038/nrm3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, et al. 5´ UTR m6A promotes cap-independent translation. Cell. 2015;163:999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3´ UTRs and near stop codons. Cell. 2012;149:1635–46. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morohashi K, Sahara H, Watashi K, Iwabata K, Sunoki T, et al. Cyclosporin A associated helicase-like protein facilitates the association of hepatitis C virus RNA polymerase with its cellular cyclophilin B. PLOS ONE. 2011;6:e18285. doi: 10.1371/journal.pone.0018285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan P, Rottman FM. An in vitro system for accurate methylation of internal adenosine residues in messenger RNA. Science. 1988;242:1159–62. doi: 10.1126/science.3187541. [DOI] [PubMed] [Google Scholar]
- Ortega A, Niksic M, Bachi A, Wilm M, Sanchez L, et al. Biochemical function of female-lethal (2)D/Wilms’ tumor suppressor-1-associated proteins in alternative pre-mRNA splicing. J Biol Chem. 2003;278:3040–47. doi: 10.1074/jbc.M210737200. [DOI] [PubMed] [Google Scholar]
- Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, et al. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537:369–73. doi: 10.1038/nature19342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendleton KE, Chen B, Liu K, Hunter OV, Xie Y, et al. The U6 snRNA m6A methyltransferase METTL16 regulates SAM synthetase intron retention. Cell. 2017;169:824–35.e14. doi: 10.1016/j.cell.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RP, Kelley DE. Existence of methylated messenger RNA in mouse L cells. Cell. 1974;1:37–42. [Google Scholar]
- Ping XL, Sun BF, Wang L, Xiao W, Yang X, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24:177–89. doi: 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Sonenberg N. Regulation of cap-dependent translation by eIF4E inhibitory proteins. Nature. 2005;433:477–80. doi: 10.1038/nature03205. [DOI] [PubMed] [Google Scholar]
- Roost C, Lynch SR, Batista PJ, Qu K, Chang HY, Kool ET. Structure and thermodynamics of N6-methyladenosine in RNA: a spring-loaded base modification. J Am Chem Soc. 2015;137:2107–15. doi: 10.1021/ja513080v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira M. Structural chemistry of human RNA methyltransferases. ACS Chem Biol. 2015;11:575–82. doi: 10.1021/acschembio.5b00781. [DOI] [PubMed] [Google Scholar]
- Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell. 2014a;159:148–62. doi: 10.1016/j.cell.2014.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5´ sites. Cell Rep. 2014b;8:284–96. doi: 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimba S, Bokar JA, Rottman F, Reddy R. Accurate and efficient N-6-adenosine methylation in spliceosomal U6 small nuclear RNA by HeLa cell extract in vitro. Nucleic Acids Res. 1995;23:2421–26. doi: 10.1093/nar/23.13.2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sledz P, Jinek M. Structural insights into the molecular mechanism of the m6A writer complex. eLife. 2016;5:e18434. doi: 10.7554/eLife.18434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slobodin B, Han R, Calderone V, Vrielink JA, Loayza-Puch F, et al. Transcription impacts the efficiency of mRNA translation via co-transcriptional N6-adenosine methylation. Cell. 2017;169:326–37.e12. doi: 10.1016/j.cell.2017.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507:371–75. doi: 10.1038/nature13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer S, Lavi U, Darnell JE., Jr The absolute frequency of labeled N-6-methyladenosine in HeLa cell messenger RNA decreases with label time. J Mol Biol. 1978;124:487–99. doi: 10.1016/0022-2836(78)90183-3. [DOI] [PubMed] [Google Scholar]
- Spitale RC, Flynn RA, Zhang QC, Crisalli P, Lee B, et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature. 2015;519:486–90. doi: 10.1038/nature14263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoilov P, Rafalska I, Stamm S. YTH: a new domain in nuclear proteins. Trends Biochem Sci. 2002;27:495–97. doi: 10.1016/s0968-0004(02)02189-8. [DOI] [PubMed] [Google Scholar]
- Stratigopoulos G, Burnett LC, Rausch R, Gill R, Penn DB, et al. Hypomorphism of Fto and Rpgrip1l causes obesity in mice. J Clin Investig. 2016;126:1897–910. doi: 10.1172/JCI85526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Querol-Audi J, Mortimer SA, Arias-Palomo E, Doudna JA, et al. Two RNA-binding motifs in eIF3 direct HCV IRES-dependent translation. Nucleic Acids Res. 2013;41:7512–21. doi: 10.1093/nar/gkt510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe A, Tanikawa K, Tsunetomi M, Takai K, Ikeda H, et al. RNA helicase YTHDC2 promotes cancer metastasis via the enhancement of the efficiency by which HIF-1α mRNA is translated. Cancer Lett. 2016;376:34–42. doi: 10.1016/j.canlet.2016.02.022. [DOI] [PubMed] [Google Scholar]
- Theler D, Dominguez C, Blatter M, Boudet J, Allain FH. Solution structure of the YTH domain in complex with N6-methyladenosine RNA: a reader of methylated RNA. Nucleic Acids Res. 2014;42:13911–19. doi: 10.1093/nar/gku1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirumuru N, Zhao BS, Lu W, Lu Z, He C, Wu L. N6-Methyladenosine of HIV-1 RNA regulates viral infection and HIV-1 Gag protein expression. eLife. 2016;5:e15528. doi: 10.7554/eLife.15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Zhu Y, Bao H, Jiang Y, Xu C, et al. A novel RNA-binding mode of the YTH domain reveals the mechanism for recognition of determinant of selective removal by Mmi1. Nucleic Acids Res. 2016;44:969–82. doi: 10.1093/nar/gkv1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell. 2016;63:306–17. doi: 10.1016/j.molcel.2016.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Feng J, Xue Y, Guan Z, Zhang D, et al. Structural basis of N6-adenosine methylation by the METTL3-METTL14 complex. Nature. 2016;534:575–78. doi: 10.1038/nature18298. [DOI] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, et al. N6-Methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–20. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, et al. N6-Methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161:1388–99. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-Methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol. 2014;16:191–98. doi: 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C, Gershowitz A, Moss B. N6, O2´-dimethyladenosine a novel methylated ribonucleoside next to the 5´ terminal of animal cell and virus mRNAs. Nature. 1975;257:251–53. doi: 10.1038/257251a0. [DOI] [PubMed] [Google Scholar]
- Wei CM, Gershowitz A, Moss B. 5´-Terminal and internal methylated nucleotide sequences in HeLa cell mRNA. Biochemistry. 1976;15:397–401. doi: 10.1021/bi00647a024. [DOI] [PubMed] [Google Scholar]
- Wei CM, Moss B. Nucleotide sequences at the N6-methyladenosine sites of HeLa cell messenger ribonucleic acid. Biochemistry. 1977;16:1672–76. doi: 10.1021/bi00627a023. [DOI] [PubMed] [Google Scholar]
- Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, et al. Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61:507–19. doi: 10.1016/j.molcel.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Xu C, Liu K, Ahmed H, Loppnau P, Schapira M, Min J. Structural basis for the discriminative recognition of N6-methyladenosine RNA by the human YT521-B homology domain family of proteins. J Biol Chem. 2015;290:24902–13. doi: 10.1074/jbc.M115.680389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Wang X, Liu K, Roundtree IA, Tempel W, et al. Structural basis for selective binding of m6A RNA by the YTHDC1 YTH domain. Nat Chem Biol. 2014;10:927–29. doi: 10.1038/nchembio.1654. [DOI] [PubMed] [Google Scholar]
- Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. PNAS. 2016a;113:E2047–56. doi: 10.1073/pnas.1602883113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Zhi WI, Lu H, Samanta D, Chen I, et al. Hypoxia-inducible factors regulate pluripotency factor expression by ZNF217- and ALKBH5-mediated modulation of RNA methylation in breast cancer cells. Oncotarget. 2016b;7:64527–42. doi: 10.18632/oncotarget.11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, et al. m6A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31:591–606.e6. doi: 10.1016/j.ccell.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Theler D, Kaminska KH, Hiller M, de la Grange P, et al. The YTH domain is a novel RNA binding domain. J Biol Chem. 2010;285:14701–10. doi: 10.1074/jbc.M110.104711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S, Li H, Bodi Z, Button J, Vespa L, et al. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell. 2008;20:1278–88. doi: 10.1105/tpc.108.058883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature. 2015;526:591–94. doi: 10.1038/nature15377. [DOI] [PMC free article] [PubMed] [Google Scholar]