

Abstract
Objectives
Diagnosing biliary atresia (BA) quickly is critical, because earlier treatment correlates with delayed or reduced need for liver transplantation. However, diagnosing BA quickly is also difficult, with infants usually treated after 60 days of life. In this study, we aim to accelerate BA diagnosis and treatment, by better understanding factors influencing the diagnostic timeline.
Methods
Infants born between 2007–2014 and diagnosed with BA at our institution were included (n=65). Two periods were examined retrospectively: P1, the time from birth to specialist referral, and P2, the time from specialist referral to treatment. How sociodemographic factors associate with P1 and P2 were analyzed with Kaplan-Meier curves and Cox proportional hazard models. In addition, to better characterize P2, laboratory results and early tissue histology were studied.
Results
P1 associated with race/ethnicity, with shorter times in non-Hispanic white infants compared to non-Hispanic black and Hispanic infants (p=0.007 and p=0.004, respectively). P2 associated with referral age, with shorter times in infants referred after 30, 45, or 60 days of life (p<0.001, p<0.001, and p=0.001, respectively). One potential reason for longer P2 in infants referred ≤30 days is that aminotransferase levels were normal or near-normal. However, despite reassuring laboratory values, tissue histology in early cases showed key features of BA.
Conclusions
Our findings suggest two opportunities to accelerate BA diagnosis and treatment. First, to achieve prompt referrals for all races/ethnicities, universal screening strategies should be considered. Second, to ensure efficient evaluations independent of age, algorithms designed to detect early features of BA can be developed.
Introduction
Biliary atresia (BA), the most common indication for pediatric liver transplantation, is a serious liver disease of infancy progressing from cholestasis to cirrhosis to end-stage liver disease in the first year of life.1 One intervention—the Kasai portoenterostomy (KP)—has been proven to slow BA’s rapid course. Importantly, the age at which the KP is performed is critical.2–8 Infants receiving the KP before 30-45 days of life (DoL) have the best chances of delaying or avoiding need for liver transplant.3,4 In contrast, infants receiving the KP after 90-120 DoL are less likely to benefit.9 Hence, an important goal in pediatric hepatology is diagnosing BA quickly to optimize timing of the KP.
Unfortunately, diagnosing BA is difficult, with the average age of KP in the United States greater than 60 DoL.9,10 Reasons for later diagnoses have focused on the challenges primary care providers (PCPs) face in identifying and referring infants.11 For example, many PCPs schedule newborn visits in the first two weeks and then again at two months of life, in accord with current recommendations. As a result, PCPs may not see infants within this interval, when concerning symptoms such as pale stools typically develop.12 In addition, PCPs may not always suspect BA initially in infants with persistent jaundice. This is because BA is rare (incidence 1:10,000-18,000 births) compared to benign physiological jaundice affecting as many as 15% of all healthy newborns.13
The objective of this study is to identify opportunities to accelerate BA treatments. To accomplish this, we sought to identify additional factors influencing the timing of the BA diagnosis. We focus our analysis on two sequential periods: P1, the time in which PCPs recognize and refer infants, and P2, the time in which specialists such as pediatric gastroenterologists and surgeons evaluate infants and perform the KP.
Methods
Patient selection
This study was approved by the institutional review board at Baylor College of Medicine. All infants born between 2007-2014 and diagnosed with BA at Texas Children’s Hospital (TCH), a large pediatric specialty center in Houston, Texas, were eligible. In subjects receiving a KP, the BA diagnosis was established by intraoperative cholangiogram at the time of KP and/or pathological examination of bile duct remnants after KP. In subjects not receiving a KP, the diagnosis of BA was inferred based on the clinical picture, liver biopsy at presentation, absence of alternative diagnoses explaining persistent cholestasis, and pathological examination of the explant after transplant. The decision to not perform a KP was made on a case-by-case basis, and was influenced by the subject’s age, fibrosis stage on liver biopsy, and/or signs of portal hypertension already present at the time of presentation. Patients diagnosed with BA elsewhere and referred to TCH for further care were not eligible for the study.
Data Acquisition
The BA diagnostic timeline was divided into two sequential periods: P1, the time from birth to specialist referral, and P2, the time from first specialist visit to KP (Figure, Supplemental Digital Content 1). To calculate these intervals and analyze associations between sociodemographic variables, the following were collected retrospectively from each subject’s electronic medical record: birth date, date of first specialist appointment at TCH, occurrence of prior specialist care before referral, date of KP, gender, race, ethnicity, preferred language, insurance type, zip code (to determine distance from TCH), birth era, and birth season. Race and ethnicity were combined and categorized as non-Hispanic white, non-Hispanic black, non-Hispanic Asian, or Hispanic. Distance was designated as ≤50 miles and >50 miles from TCH, to distinguish those who could and could not travel easily between home and hospital during the evaluation. Birth era was either 2007-2010 or 2011-2014, to help detect any changes in P1 or P2 as advances in the field were made. Birth season was divided into Winter (January-March), Spring (April-June), Summer (July-September), or Fall (October-December).
To correlate initial laboratory results at TCH with age, values and the dates they were drawn were obtained retrospectively from each subject’s electronic medical record. Aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (AlkP), and gamma glutamyltransferase (GGT) values were measured using standard techniques. Unconjugated bilirubin (Bu) and conjugated bilirubin (Bc) were measured using direct spectrophotometry with the VITROS analyzer (Ortho Clinical Diagnsotics, Raritan, NJ). Total and direct bilirubin were not measured. Reference intervals for AST, ALT, Bc, AlkP, and GGT were derived by the TCH laboratory with the middle 95% of values from age-matched populations.14 Reference intervals for Bc ratios were borrowed from the existing literature, with a Bc ratio >0.20 signifying liver disease and a Bc ratio ≤0.20 marking red blood cell turnover and Bu processing.15
To analyze histopathology in early BA cases, liver and biliary remnant tissue were obtained from the TCH Department of Pathology archives. Samples were stained with the following: hematoxyline and eosin (H&E), to study morphology and identify bile plugs; trichrome, to measure the amount of fibrosis; anti-Cytokeratin 7 (CK7) antibodies, to outline bile duct/ductule morphology and number; and anti-CD56 antibody, to detect reactive bile duct/ductules.16–18 Liver samples were evaluated by an experienced pathologist (MJF) based on common findings in BA as outlined by ChiLDReN.19 Bile duct proliferation was categorized as none, focal, or generalized. Portal fibrosis was graded as absent/some tracts, most tracts, focal bridging, marked bridging, or cirrhosis. Bile plugs were rated as absent, canalicular only, bile duct only, or bile duct and canalicular.
Statistical Analysis
For the time-to-event analyses, two outcomes were studied: (1) first specialist appointment, where P1 represented time-to-event and was calculated as ([date of first specialist appointment at TCH] ─ [birth date]); and (2) KP, where P2 represented time-to-event and was calculated as ([date of KP] ─ [date of first specialist appointment at TCH]). Kaplan-Meier curves and the log-rank test were used to characterize P1 or P2 relative to subject attributes, while univariate Cox proportional hazards models were used to determine the direction of associations between P1 or P2 and subject attributes. We tested the proportional hazard assumption for our variables of interest by evaluating the Schoenfeld residuals after fitting each model, and also by using the time varying covariates option in Stata. In these analyses, the proportional hazards assumption was not violated. Multivariable analyses were not performed because of limited sample sizes.
For analysis of laboratory values, Bc ratios were calculated as ([Bc]/[Bc+Bu]) based on the equation suggested by Moyer and colleagues.15 Spearman’s rank correlation coefficients (rs) with one-tailed p-values were used to describe correlations between test results and age, based on assumptions that laboratory data is non-parametric, right-skewed (away from the zero value), and expected to increase with age if a correlation exists. All calculations were performed using Microsoft Excel (Seattle, WA), GraphPad Prism (La Jolla, CA), and Stata 14.0 (College Station, TX) software.
Results
There were 65 subjects born between 2007-2014 and diagnosed with BA at TCH (Table, Supplemental Digital Content 2). This represents the majority of predicted cases in the region, assuming an incidence of 1:10,000-18,000 births and reported birth rate of 90,000/year.20 More subjects were female than male, as has been reported previously in some BA studies. In addition, more subjects were Hispanic than non-Hispanic white, non-Hispanic black, or non-Hispanic Asian, reflecting the racial/ethnic composition of children in the study city. Most subjects were born to parents who spoke English, had public insurance, and lived within 50 miles of the study hospital. Approximately the same number of subjects were born between 2007-2010 and 2011-2014, and subjects’ birth dates were evenly distributed among the seasons.
We first focused on the beginning of the diagnostic timeline, when PCPs identify infants and refer them to specialists for further evaluation (denoted as P1 in this study). For this analysis, 12 subjects referred directly from the nursery before the first PCP visit and nine subjects referred to specialists at other institutions before evaluation at our center were excluded (Figure, Supplemental Digital Content 1). The mean P1 time was 72.9±39.9 days for the remaining 44 subjects (Table, Supplemental Digital Content 2). Race/ethnicity was associated with age at referral, with non-Hispanic white subjects having significantly shorter P1 times than Hispanic and non-Hispanic black subjects (p<0.01) (Figure 1). These associations were confirmed with Cox analyses, with non-Hispanic black (HR 0.23, 95% CI 0.08-0.67, p=0.007) and Hispanic (HR 0.30, 95% CI 0.13-0.68, p=0.004) subjects having a lower likelihood of early referral (Table, Supplemental Digital Content 3). No significant differences in P1 were found among other attributes such as gender, preferred language, insurance type, distance from our center, birth era, or birth season (Table, Supplemental Digital Content 3; Figure, Supplemental Digital Content 4).
Figure 1. Later referrals for non-Hispanic black and Hispanic subjects.
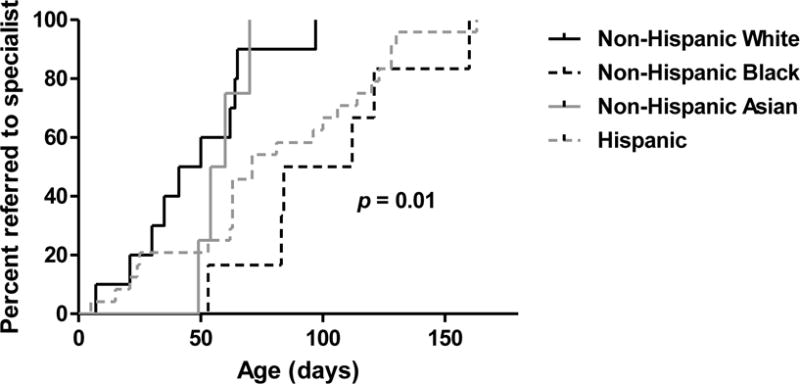
Kaplan-Meier curves for time to referral based on race/ethnicity. Differences among groups were calculated using the log-rank test.
We next focused on the second part of the diagnostic timeline, when specialists such as pediatric gastroenterologists and surgeons evaluate and perform the KP (denoted as P2 in this study). For this analysis, 16 subjects referred too late for the KP and one subject with congenital heart disease precluding further operations were excluded (Figure, Supplemental Digital Content 1). The mean P2 time was 15.6±23.0 days for the remaining 48 subjects (Table, Supplemental Digital Content 2). Shorter P2 times were strongly associated with older ages at referral. Figure 2a is a scatter plot demonstrating an inverse relationship between evaluation time and age, and Figures 2b-d show significant differences in P2 times for subjects referred before or after 30, 45, and 60 DoL, respectively. These associations were confirmed with Cox analyses, with subjects presenting after 30 DoL (HR 15.47, 95% CI 4.43-53.99, p<0.001), 45 DoL (HR 6.94, 95% CI 2.72-17.71, p<0.001) and 60 DoL (HR 3.12, 95% CI 1.55-6.25, p=0.001) having a higher likelihood of faster specialist evaluation and treatment (Table, Supplemental Digital Content 5). Shorter P2 times were also associated with non-summer birth months and living beyond 50 miles of the study hospital (Table, Supplemental Digital Content 5; Figure, Supplemental Digital Content 6).
Figure 2. Evaluation times vary inversely with age at referral.
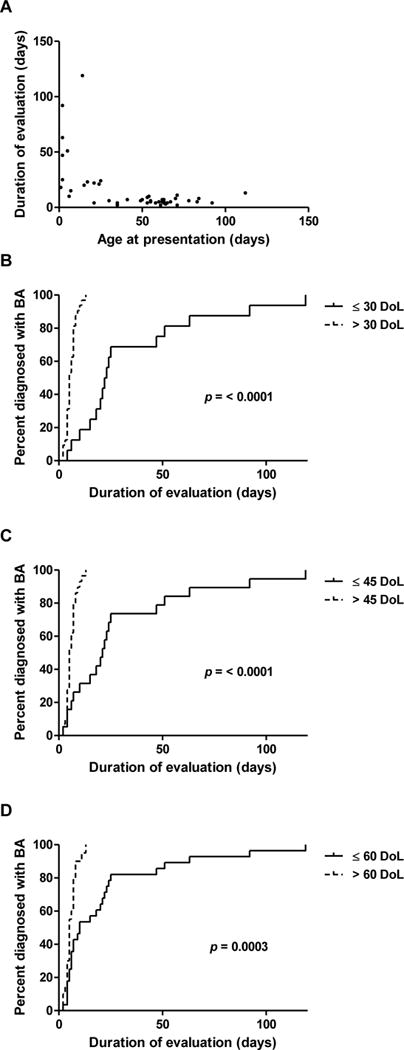
(A) Scatter plot of P2 times and age at presentation demonstrating an inverse relationship. (B-D) Kaplan-Meier curves for time to KP after specialist referral, based on age at presentation. Differences between groups were calculated using the log-rank test.
We hypothesized that one reason for longer P2 times in younger infants is that laboratory results may be equivocal in early stages of disease. To test this, initial AST, ALT, Bc, AlkP, GGT, and Bc ratios were plotted versus age for all 65 subjects. In accord with our hypothesis, initial AST and ALT were within laboratory reference intervals in infants less than 30 DoL, a result which could be falsely reassuring (Figure 3). Initial AST and ALT were higher in older infants, with a positive correlation with age (rs=0.77, 95% CI 0.64-0.85, p<0.0001 and rs=0.79, 95% CI 0.67-0.87, p<0.0001, respectively). In contrast, initial Bc was elevated in all ages. Initial AlkP and GGT were less predictable, and were normal or elevated at different ages (Figure, Supplemental Digital Content 7). Finally, initial Bc ratios were elevated at all ages except when calculated in the immediate post-natal period, consistent with a previous report.21
Figure 3. Normal/near-normal transaminases, but elevated Bc, in first 30 DoL.
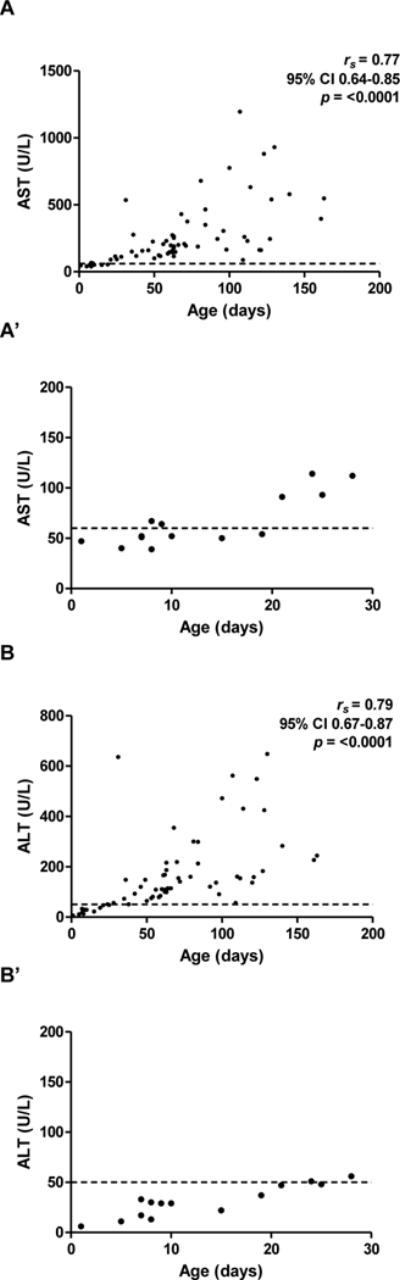
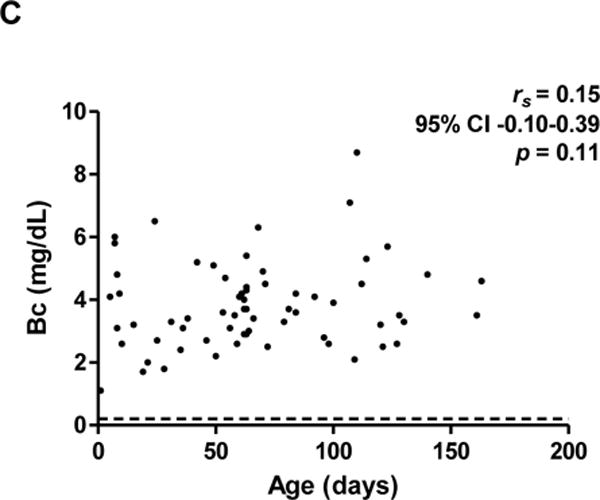
Initial AST (A) and ALT (B) levels, with insets (A′) and (B′) highlighting first 30 DoL. (C) Initial Bc levels. Dashed lines denote upper limits of normal (reference intervals: AST 20-60 U/L; ALT 6-50 U/L; Bc 0-0.2 mg/dL).
Another potential reason for longer P2 times in younger infants is that specialists may wait to allow time for histological changes to occur and disease to become more apparent. To investigate this concept further, samples from the six subjects undergoing liver biopsy before 30 DoL were studied (Table, Supplemental Digital Content 8). Contrary to our expectations, liver tissue from all six cases showed signs of extrahepatic obstruction, including bile duct proliferation (highlighted best with cytokeratin and CD56 immunohistochemistry), portal fibrosis, and bile duct plugging (Figure 4a-d). In addition, despite pigmented stools in five of six cases, biliary remnants from all six cases had areas devoid of patent ducts (Figure 4e-f). These results suggest that even at early ages there can be complete obliteration of the biliary tract.
Figure 4. Abnormal early histology in liver and biliary remnants.
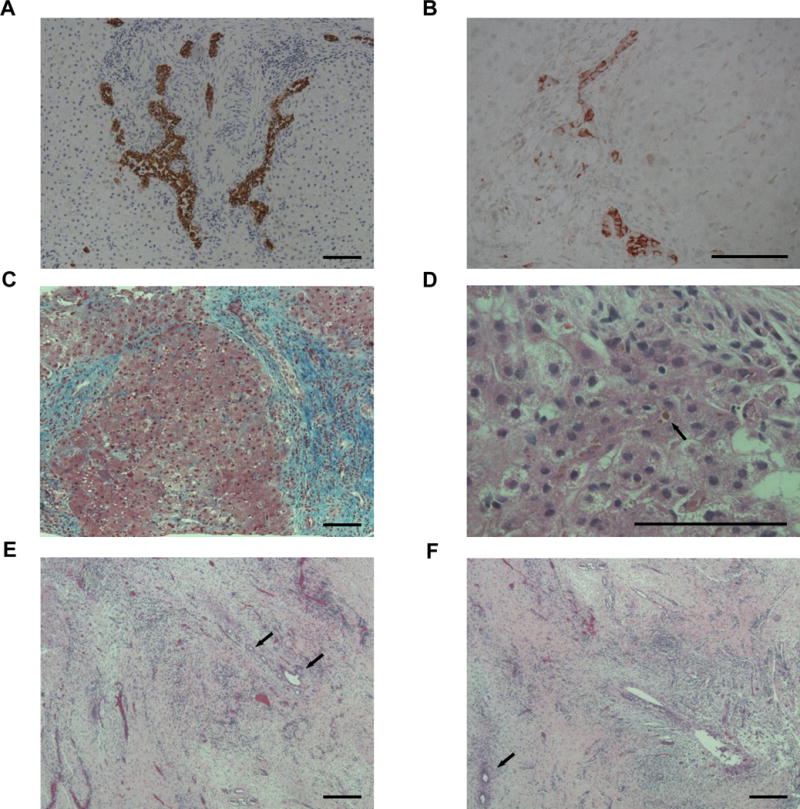
(A-D) Liver. (A) Duct proliferation (brown) at DoL 15 (case 1, 100X, anti-CK7). (B) Reactive ducts/ductules (red) at DoL 30 (case 6, 200X, anti-CD56). (C) Bridging fibrosis (blue) at DoL 21 (case 4, 100X, trichrome). (D) Canalicular bile plugging (arrow) at DoL 24 (case 5, 400X, H&E). (E-F) Biliary remnants. Areas of left (E) and right (F) hepatic duct tissue devoid of large, epithelial-lined ducts at DoL 16. Some small duct branches with epithelial cells loss, distorted lumens, and circumferential fibrosis are seen (arrows) (case 2, 100X, H&E). Bar=100 uM.
Discussion
We analyzed the BA diagnostic timeline to identify factors which correlate with how quickly BA is diagnosed. For the first period (P1), non-Hispanic white infants were referred sooner than non-Hispanic black and Hispanic infants. PCPs could be detecting jaundice more readily in light-skinned infants, which could explain later referrals in non-Hispanic black infants and some Hispanic infants. Another possibility is that parents from certain cultures, races, or ethnicities may be more likely to access the health care system and/or attend scheduled well-child visits. Based on our findings, such a culture effect would be independent of language spoken, insurance status, or travel distance from a specialty center. A third explanation is that PCPs may have implicit biases leading to uneven treatment across populations. Implicit biases have been recognized in other pediatric disease, but their role in BA has not been explored.22
For the second period (P2), infants have longer evaluation times when referred earlier and vice versa. This may be because specialists could be less suspicious of BA in younger infants who have normal or near normal aminotransferase levels. Alternatively, specialists may purposely postpone evaluations in early referrals to allow time for the disease to progress. Such an approach avoids “over-testing” infants whose cholestasis would otherwise self-resolve. It also theoretically minimizes the potential of false-negative biopsy results, which are assumed to occur in early stages of disease.23 Shorter P2 times were also found in infants born in non-summer months and living further from the treatment center. The former could reflect reduced availability of pediatric gastroenterologists, interventional radiologists, pathologists, and/or pediatric surgeons during summer vacation months, whereas the latter suggests that diagnostic tests are consolidated for those who cannot easily travel back and forth between home and hospital.
One important question and potential limitation is whether our results are applicable to other regions. Our study population may be atypical, with high percentages of Hispanic infants (54%) and of infants identified too late to receive the KP (17/65 or 26%). However, a number of previously published findings suggest our observations may be similar in other parts of the United States. First, older KP ages for Hispanic and/or African American infants have been reported in single- and multiple-hospital studies.9,24,25 Second, large variations have been described in the time North American specialists take to evaluate infants, though the reasons for these variations were not explored.3,9 Third, guidelines for specialists caution against liver biopsies in younger infants, to avoid false-negative results (see below).23 Hence, specialists following these guidelines would be expected to delay evaluations in younger infants. Future prospective studies in different regions can further examine the applicability of our findings.
Assuming our results are applicable to other regions, specific policy solutions could accelerate the diagnosis and treatment of BA. For P1, screening programs could be applied universally independent of race/ethnicity. Two such programs were discussed in a recent American Academy of Pediatrics technical report.26 The most studied approach is the stool color card program.27–30 This program relies on PCPs and/or parents identifying infants who may have BA based on the pale-colored stools which develop over time. A second approach still under investigation is newborn direct/conjugated bilirubin screening.31–34 This approach capitalizes on elevated direct/conjugated bilirubin levels shortly after birth in infants with BA.21
For P2, efforts to help specialists appreciate the urgency of evaluations—even in infants less than 30 DoL—should be prioritized. To help accomplish this, specialists need diagnostic algorithms which account for findings in very young infants. Current algorithms have been derived from older infants who already have markedly elevated AST and ALT levels.35,36 In young infants, however, AST and ALT levels should be normal or close to normal, and, perhaps counter-intuitively, do not exclude BA. Current algorithms also require infants to have high Bc levels, such as above 2.5 mg/dL in one study.36 While young infants with BA will have Bc levels above laboratory reference intervals, their levels may not always exceed those found in older infants.21,37 Similarly, some algorithms use Bc ratios greater than 0.2 to indicate cholestasis.15 Most measurements from younger infants fit this rule, except in some circumstances in the few days after birth.
Our findings suggest that liver biopsies could be helpful in diagnostic algorithms to detect early BA. The utility of such early biopsies has been controversial. On one hand, current guidelines caution against early biopsies, because of potentially false-negative results if sampling occurs before disease has had time to progress.23 On the other hand, both a meta-analysis and large consortium study found no correlation with young age and false-negative biopsy results. 38,39 Interestingly, reports which caution against early biopsies usually cite a case series which featured an infant with BA who had a normal biopsy at three weeks of life.40 However, the same infant had normal biopsies at five and nine weeks of life as well, raising the possibility that the false-negative results may have been related to other factors rather than age at sampling.
Finally, the issue of “over-testing” young infants who may or may not have BA deserves special mention. Waiting potentially protects infants from the risks of diagnostic tests, such as pain from blood draws, radiation exposure from imaging, and/or side-effects of anesthesia. However, waiting also potentially exposes infants to risks of progressing liver disease. We found complete obliteration of bile ducts in the earliest ages sampled, suggesting that waiting would lead to worsening bile back-up and resulting liver injury. Furthermore, the high-grade fibrosis found in some early cases argues strongly against delaying evaluations simply based on age. This interpretation agrees with data from centers around the world, which correlate an earlier KP with the best chance of delaying or preventing need for liver transplantation.2 For example, a KP performed before 30 DoL in a Canadian study correlated with the highest 10-year liver transplant-free survival rates.3 Similarly, a KP performed before 45 DoL in a French study was associated with 12.1% higher transplant-free survival rates after 15 years, and a KP performed before 30-45 DoL correlated with reduced need for liver transplant in Swiss and United States studies.4–6
Conclusion
By examining factors influencing the time-to-diagnosis in BA, this study identifies opportunities to accelerate the timeline and potentially improve outcomes. In the period from birth to specialist referral, infants of certain races/ethnicities are referred sooner. To ensure even referral times for all races/ethnicities, PCPs could be aided by universal screening programs such as the stool color card and/or newborn direct/conjugated bilirubin measurements. In the period from specialist referral to KP, infants referred later receive faster evaluations. To ensure even evaluation times independent of referral ages, specialists could follow diagnostic algorithms designed for younger infants. Such algorithms would recognize normal aminotransferase in cases ≤30 DoL, and not wait for disease progression if liver biopsy is used as a diagnostic test.
Supplementary Material
Figure, Supplemental Digital Content 1 Schematic of biliary atresia (BA) diagnostic timeline and patient flow diagram. (A) P1 and P2 comprise the time from birth to specialist referral to Kasai Portoenterostomy (KP). (B) P1 subjects were referred by PCPs directly to our institution for specialist care. (C) P2 subjects received an evaluation and KP at our institution.
Table, Supplemental Digital Content 2 Subject Characteristics
Table, Supplemental Digital Content 3 Predictors of Earlier Specialist Referral using Cox Proportional Hazard Models (n=44)
Figure, Supplemental Digital Content 4 Kaplan-Meier curves for time to specialist referral. Attributes compared were gender (A), preferred language (B), insurance type (C), location of residence (D), birth era (E), and birth season (F). Differences among attributes were calculated using the log-rank test.
Table, Supplemental Digital Content 5 Predictors of Earlier KP after Specialist Referral using Cox Proportional Hazard Models (n=48)
Figure, Supplemental Digital Content 6 Kaplan-Meier curves for time to KP after specialist referral. Attributes compared were gender (A), race/ethnicity (B), preferred language (C), insurance type (D), location of residence (E), birth era (F), and birth season (G). Differences among attributes were calculated using the log-rank test.
Figure, Supplemental Digital Content 7 Initial AlkP (A), GGT (B), and Bc ratio (C). Dashed lines denote upper limits of normal (reference intervals: AlkP 77-265 U/L [0-7 DoL], 91-375 U/L [8-30 DoL], 60-360 U/L [31-150 DoL], 55-325 U/L [151-240 DoL]; GGT 10-160 U/L [0-90 DoL], 11-82 U/L [>90 DoL]; Bc ratio 0.00-0.20).
Table, Supplemental Digital Content 8 Histological Features of Early BA (n=6)
What is Known
Infants with biliary atresia (BA) must be identified and treated quickly to delay or avoid need for liver transplant.
Unfortunately, early diagnoses before 30-45 days of life are uncommon, for reasons which have not been fully explored.
What is New
We identify factors which correlate with the length of time from (i) birth to specialist referral and (ii) specialist referral to diagnosis and treatment.
Policy makers can use our findings to design programs which accelerate the BA diagnostic timeline.
Acknowledgments
Source of Funding: Funding for this study came from the Cade R. Alpard Foundation for Pediatric Liver Disease, the American Association for the Study of Liver Diseases Jan Albrecht Clinical and Translational Research Award, the Baylor College of Medicine Junior Faculty Seed Award, and NIH 1 K23 DK109207.
Abbreviations
- BA
biliary atresia
- KP
Kasai portoenterostomy
- DoL
day of life
- PCP
primary care physician
- TCH
Texas Children’s Hospital
- AST
aspartate aminotransferase
- ALT
alanine aminotransferase
- AlkP
alkaline phosphatase
- GGT
gamma glutamyltransferase
- Bu
unconjugated bilirubin
- Bc
conjugated bilirubin
- H&E
hematoxyline and eosin
- CK7
cytokeratin 7
Footnotes
Conflicts of Interest: The authors have no conflicts of interest or financial relationships relevant to this article to disclose.
Contributor’s Statement:
Dr. Harpavat conceptualized and designed the study, acquired the data, analyzed and interpreted the data, drafted the initial manuscript, reviewed and revised the manuscript, and approved the final manuscript as submitted.
Dr. Lupo designed the study, analyzed and interpreted the data, reviewed and revised the manuscript, and approved the final manuscript as submitted.
Ms. Liwanag acquired the data, analyzed, reviewed and revised the manuscript, and approved the final manuscript as submitted.
Drs. Hollier and Brandt analyzed and interpreted the data, reviewed and revised the manuscript, and approved the final manuscript as submitted.
Dr. Finegold acquired the data, analyzed and interpreted the data, reviewed and revised the manuscript, and approved the final manuscript as submitted.
Dr. Shneider conceptualized and designed the study, analyzed and interpreted the data, reviewed and revised the manuscript, and approved the final manuscript as submitted.
References
- 1.Sokol RJ, Shepherd RW, Superina R, Bezerra JA, Robuck P, Hoofnagle JH. Screening and outcomes in biliary atresia: summary of a National Institutes of Health workshop. Hepatology. 2007;46(2):566–581. doi: 10.1002/hep.21790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jimenez-Rivera C, Jolin-Dahel KS, Fortinsky KJ, Gozdyra P, Benchimol EI. International incidence and outcomes of biliary atresia. J Pediatr Gastroenterol Nutr. 2013;56(4):344–354. doi: 10.1097/MPG.0b013e318282a913. [DOI] [PubMed] [Google Scholar]
- 3.Schreiber RA, Barker CC, Roberts EA, et al. Biliary atresia: the Canadian experience. J Pediatr. 2007;151(6):659–65, 665.e1. doi: 10.1016/j.jpeds.2007.05.051. [DOI] [PubMed] [Google Scholar]
- 4.Serinet M-O, Wildhaber BE, Broué P, et al. Impact of age at Kasai operation on its results in late childhood and adolescence: a rational basis for biliary atresia screening. Pediatrics. 2009;123(5):1280–1286. doi: 10.1542/peds.2008-1949. [DOI] [PubMed] [Google Scholar]
- 5.Wildhaber BE, Majno P, Mayr J, et al. Biliary atresia: Swiss national study, 1994-2004. J Pediatr Gastroenterol Nutr. 2008;46(3):299–307. doi: 10.1097/MPG.0b013e3181633562. [DOI] [PubMed] [Google Scholar]
- 6.Karrer FM, Lilly JR, Stewart BA, Hall RJ. Biliary atresia registry, 1976 to 1989. J Pediatr Surg. 1990;25(10):1076–80. doi: 10.1016/0022-3468(90)90222-u. discussion 1081. http://www.ncbi.nlm.nih.gov/pubmed/2262862. Accessed May 31, 2015. [DOI] [PubMed] [Google Scholar]
- 7.de Carvalho E, dos Santos JL, da Silveira TR, et al. Biliary atresia: the Brazilian experience. J Pediatr (Rio J) 2010;86(6):473–479. doi: 10.2223/JPED.2054. [DOI] [PubMed] [Google Scholar]
- 8.Tiao M-M, Tsai S-S, Kuo H-W, Chen C-L, Yang C-Y. Epidemiological features of biliary atresia in Taiwan, a national study 1996-2003. J Gastroenterol Hepatol. 2008;23(1):62–66. doi: 10.1111/j.1440-1746.2007.05114.x. [DOI] [PubMed] [Google Scholar]
- 9.Shneider BL, Brown MB, Haber B, et al. A multicenter study of the outcome of biliary atresia in the United States, 1997 to 2000. J Pediatr. 2006;148(4):467–474. doi: 10.1016/j.jpeds.2005.12.054. [DOI] [PubMed] [Google Scholar]
- 10.Superina R, Magee JC, Brandt ML, et al. The anatomic pattern of biliary atresia identified at time of Kasai hepatoportoenterostomy and early postoperative clearance of jaundice are significant predictors of transplant-free survival. Ann Surg. 2011;254(4):577–585. doi: 10.1097/SLA.0b013e3182300950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Palermo JJ, Joerger S, Turmelle Y, Putnam P, Garbutt J. Neonatal cholestasis: opportunities to increase early detection. Acad Pediatr. 2012;12(4):283–287. doi: 10.1016/j.acap.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sokol RJ. Biliary atresia screening: why, when, and how? Pediatrics. 2009;123(5):e951–2. doi: 10.1542/peds.2008-3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Logan S, Stanton A. Screening for biliary atresia. Lancet. 1993;342(8866):256. doi: 10.1016/0140-6736(93)91814-3. http://www.ncbi.nlm.nih.gov/pubmed/8101299. Accessed May 28, 2015. [DOI] [PubMed] [Google Scholar]
- 14.Clinical and Laboratory Standards Institute. EP28-A3c: Defining, Establishing, and Verifying Reference Intervals in the Clinical Laboratory; Approved Guideline. (Third) 2010 Oct [Google Scholar]
- 15.Moyer V, Freese DK, Whitington PF, et al. Guideline for the evaluation of cholestatic jaundice in infants: recommendations of the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2004;39(2):115–128. doi: 10.1097/00005176-200408000-00001. http://www.ncbi.nlm.nih.gov/pubmed/15269615. Accessed January 19, 2015. [DOI] [PubMed] [Google Scholar]
- 16.Torbenson M, Wang J, Abraham S, Maitra A, Boitnott J. Bile ducts and ductules are positive for CD56 (N-CAM) in most cases of extrahepatic biliary atresia. Am J Surg Pathol. 2003;27(11):1454–1457. doi: 10.1097/00000478-200311000-00008. [DOI] [PubMed] [Google Scholar]
- 17.Sira MM, El-Guindi MA-S, Saber MA, Ehsan NA, Rizk MS. Differential hepatic expression of CD56 can discriminate biliary atresia from other neonatal cholestatic disorders. Eur J Gastroenterol Hepatol. 2012;24(10):1227–1233. doi: 10.1097/MEG.0b013e328356aee4. [DOI] [PubMed] [Google Scholar]
- 18.Zhang R-Z, Yu J-K, Peng J, et al. Role of CD56-expressing immature biliary epithelial cells in biliary atresia. World J Gastroenterol. 2016;22(8):2545. doi: 10.3748/wjg.v22.i8.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Russo P, Magee JC, Boitnott J, et al. Design and validation of the biliary atresia research consortium histologic assessment system for cholestasis in infancy. Clin Gastroenterol Hepatol. 2011;9(4):357–362.e2. doi: 10.1016/j.cgh.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Texas Department of State Health Services. No Title. Texas Health Data Births to Texas Residents. http://soupfin.tdh.state.tx.us/birth.htm. Accessed June 1, 2016.
- 21.Harpavat S, Finegold MJ, Karpen SJ. Patients with biliary atresia have elevated direct/conjugated bilirubin levels shortly after birth. Pediatrics. 2011;128(6):e1428–33. doi: 10.1542/peds.2011-1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sabin JA, Greenwald AG. The influence of implicit bias on treatment recommendations for 4 common pediatric conditions: pain, urinary tract infection, attention deficit hyperactivity disorder, and asthma. Am J Public Health. 2012;102(5):988–995. doi: 10.2105/AJPH.2011.300621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fawaz R, Baumann U, Ekong U, et al. Guideline for the Evaluation of Cholestatic Jaundice in Infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition (NASPGHAN) and the European Society for Pediatric Gastroenterology, Hepatology. J Pediatr Gastroenterol Nutr. 2016 doi: 10.1097/MPG.0000000000001334. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 24.Lee H, Lewis J, Schoen BT, Brand T, Ricketts RR. Kasai portoenterostomy: Differences related to race. J Pediatr Surg. 2001;36(8):1196–1198. doi: 10.1053/jpsu.2001.25761. [DOI] [PubMed] [Google Scholar]
- 25.Raval MV, Dzakovic A, Bentrem DJ, Reynolds M, Superina R. Trends in age for hepatoportoenterostomy in the United States. Surgery. 2010;148(4):785–91-2. doi: 10.1016/j.surg.2010.07.028. [DOI] [PubMed] [Google Scholar]
- 26.Wang KS. Newborn Screening for Biliary Atresia. Pediatrics. 2015;136(6):e1663–9. doi: 10.1542/peds.2015-3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsiao C-H, Chang M-H, Chen H-L, et al. Universal screening for biliary atresia using an infant stool color card in Taiwan. Hepatology. 2008;47(4):1233–1240. doi: 10.1002/hep.22182. [DOI] [PubMed] [Google Scholar]
- 28.Lien T-H, Chang M-H, Wu J-F, et al. Effects of the infant stool color card screening program on 5-year outcome of biliary atresia in Taiwan. Hepatology. 2011;53(1):202–208. doi: 10.1002/hep.24023. [DOI] [PubMed] [Google Scholar]
- 29.Tseng J-J, Lai M-S, Lin M-C, Fu Y-C. Stool color card screening for biliary atresia. Pediatrics. 2011;128(5):e1209–15. doi: 10.1542/peds.2010-3495. [DOI] [PubMed] [Google Scholar]
- 30.Gu Y-H, Yokoyama K, Mizuta K, et al. Stool color card screening for early detection of biliary atresia and long-term native liver survival: a 19-year cohort study in Japan. J Pediatr. 2015;166(4):897–902.e1. doi: 10.1016/j.jpeds.2014.12.063. [DOI] [PubMed] [Google Scholar]
- 31.Keffler S, Kelly DA, Powell JE, Green A. Population screening for neonatal liver disease: a feasibility study. J Pediatr Gastroenterol Nutr. 1998;27(3):306–311. doi: 10.1097/00005176-199809000-00007. http://www.ncbi.nlm.nih.gov/pubmed/9740202. Accessed May 12, 2015. [DOI] [PubMed] [Google Scholar]
- 32.Powell JE, Keffler S, Kelly DA, Green A. Population screening for neonatal liver disease: potential for a community-based programme. J Med Screen. 2003;10(3):112–116. doi: 10.1258/096914103769010996. [DOI] [PubMed] [Google Scholar]
- 33.Harpavat S, Ramraj R, Finegold MJ, et al. Newborn Direct/Conjugated Bilirubin Measurements As A Potential Screen for Biliary Atresia. J Pediatr Gastroenterol Nutr. 2016;62(6):799–803. doi: 10.1097/MPG.0000000000001097. [DOI] [PubMed] [Google Scholar]
- 34.Harpavat S, Garcia-Prats JA, Shneider BL. Newborn Bilirubin Screening for Biliary Atresia. N Engl J Med. 2016;375(6):605–606. doi: 10.1056/NEJMc1601230. [DOI] [PubMed] [Google Scholar]
- 35.El-Guindi MA-S, Sira MM, Sira AM, et al. Design and validation of a diagnostic score for biliary atresia. J Hepatol. 2014;61(1):116–123. doi: 10.1016/j.jhep.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 36.Jancelewicz T, Barmherzig R, Chung CT-S, et al. A screening algorithm for the efficient exclusion of biliary atresia in infants with cholestatic jaundice. J Pediatr Surg. 2015;50(3):363–370. doi: 10.1016/j.jpedsurg.2014.08.014. [DOI] [PubMed] [Google Scholar]
- 37.Terui K, Higashimoto Y, Saito E, et al. Diagnosis of biliary atresia can not be excluded by declining trend of serum direct bilirubin. Pediatr Rep. 2013;5:73–75. doi: 10.4081/pr.2013.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee JYJ, Sullivan K, El Demellawy D, Nasr A. The value of preoperative liver biopsy in the diagnosis of extrahepatic biliary atresia: A systematic review and meta-analysis. J Pediatr Surg. 2016;51(5):753–761. doi: 10.1016/j.jpedsurg.2016.02.016. [DOI] [PubMed] [Google Scholar]
- 39.Russo P, Magee JC, Anders RA, et al. Key Histopathologic Features of Liver Biopsies That Distinguish Biliary Atresia From Other Causes of Infantile Cholestasis and Their Correlation With Outcome. Am J Surg Pathol. 2016;40(12):1601–1615. doi: 10.1097/PAS.0000000000000755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Azar G, Beneck D, Lane B, Markowitz J, Daum F, Kahn E. Atypical morphologic presentation of biliary atresia and value of serial liver biopsies. J Pediatr Gastroenterol Nutr. 2002;34(2):212–215. doi: 10.1097/00005176-200202000-00020. http://www.ncbi.nlm.nih.gov/pubmed/11840042. Accessed August 13, 2016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure, Supplemental Digital Content 1 Schematic of biliary atresia (BA) diagnostic timeline and patient flow diagram. (A) P1 and P2 comprise the time from birth to specialist referral to Kasai Portoenterostomy (KP). (B) P1 subjects were referred by PCPs directly to our institution for specialist care. (C) P2 subjects received an evaluation and KP at our institution.
Table, Supplemental Digital Content 2 Subject Characteristics
Table, Supplemental Digital Content 3 Predictors of Earlier Specialist Referral using Cox Proportional Hazard Models (n=44)
Figure, Supplemental Digital Content 4 Kaplan-Meier curves for time to specialist referral. Attributes compared were gender (A), preferred language (B), insurance type (C), location of residence (D), birth era (E), and birth season (F). Differences among attributes were calculated using the log-rank test.
Table, Supplemental Digital Content 5 Predictors of Earlier KP after Specialist Referral using Cox Proportional Hazard Models (n=48)
Figure, Supplemental Digital Content 6 Kaplan-Meier curves for time to KP after specialist referral. Attributes compared were gender (A), race/ethnicity (B), preferred language (C), insurance type (D), location of residence (E), birth era (F), and birth season (G). Differences among attributes were calculated using the log-rank test.
Figure, Supplemental Digital Content 7 Initial AlkP (A), GGT (B), and Bc ratio (C). Dashed lines denote upper limits of normal (reference intervals: AlkP 77-265 U/L [0-7 DoL], 91-375 U/L [8-30 DoL], 60-360 U/L [31-150 DoL], 55-325 U/L [151-240 DoL]; GGT 10-160 U/L [0-90 DoL], 11-82 U/L [>90 DoL]; Bc ratio 0.00-0.20).
Table, Supplemental Digital Content 8 Histological Features of Early BA (n=6)