

Abstract
Chronic Obstructive Pulmonary Disease (COPD) is predicted to become the third leading cause of death and disability worldwide by 2030; with cigarette smoking (active or passive) being the chief cause of its occurrence. Cigarette smoke exposure has been found to result in excessive inflammation and tissue injury, which might lead to COPD, although the exact pathophysiology of the disease remains elusive. While previous studies have demonstrated the role of membrane-bound Toll-like receptors (TLRs) in cigarette smoke (CS)-induced inflammation, scant information is available about the role of cytosolic NOD-like receptors (NLR) in regulating CS-mediated inflammatory responses. Thus, we investigated the role of NLRP10 and NLRP12 in regulating inflammatory responses in human alveolar type II epithelial cells (A549) and human monocytic cells (THP-1) in response to a challenge with cigarette smoke extract (CSE). We observed a CSE-mediated increase in caspase-1 activity, production of IL-1β and IL-18, and NLRP10/NLRP12 expression in A549 and THP-1 cells. Interestingly, immunofluorescence imaging results demonstrated an increase in the colocalization of NLRP10/NLRP12 proteins to the cell membrane of CSE-challenged THP-1 cells. We also observed an increase in the expression of lipid raft proteins (caveolin-1, caveolin-2, and flotillin-1) and an induction of lipid raft assembly following CSE-exposure in A549 cells. Lipid rafts are cholesterol-rich membrane microdomains well known to act as harbors for signaling molecules. Here we demonstrate membrane recruitment of NLRP10 and NLRP12 in lipid raft entities as well as the interaction of NLRP12 with the lipid raft protein caveolin-1 in CSE-challenged A549 cells. Furthermore, enrichment of lipid raft entities with poly-unsaturated fatty acids (PUFA) rescued A549 cells from CSE-induced inflammation. Interestingly, we also observed that PUFA rescued filipin (chemical agent used for disrupting lipid rafts)-mediated exacerbated responses in CSE-challenged cells. Overall, our results demonstrate an important role of lipid rafts in NLRP10/NLRP12-mediated signalling in CSE-challenged A549 cells.
Keywords: Cigarette Smoke Extract (CSE), NLRP10, NLRP12, Lipid Rafts, COPD
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is a leading cause of morbidity and mortality with a global disease burden of 251 million cases reported in 2016 (WHO 2017). Cigarette smoking is the chief etiologic factor for COPD, which causes functional and structural alterations in the airways and alveoli thus resulting in progressive, irreversible damage to the lungs (WHO 2016). Mounting evidence demonstrates an important role of smoking-dependent inflammatory responses in the development of COPD (Shaykhiev and Crystal 2013). However, the precise mechanisms underlying the onset and progression of COPD remains elusive (Lewis et al. 2017).
Pattern-recognition receptors (PRRs) such as membrane bound Toll-like receptors (TLRs) and cytosolic nucleotide-binding oligomerization domain (NOD)-like receptors (NLR), work as surveillance components of innate immunity (Fukata et al. 2009). While the role of TLRs in the pathogenesis of COPD has been well documented(Zuo et al. 2015), the importance of NLR proteins in the development and advancement of COPD or smoke-mediated inflammation has not been extensively explored. NLRs are a heterogeneous class of cytosolic proteins characterized by a tripartite structure comprised of a central nucleotide-binding domain termed the NACHT domain (or NOD domain), N-terminal effector domains including PYRIN, caspase recruitment domain (CARD), or baculovirus inhibitor of apoptosis protein repeat (BIR) domains for binding downstream signalling molecules, and a stretch of carboxyl-terminal leucine-rich repeat (LRR) motifs(Shaw et al. 2008). Upon activation, either due to infection or tissue damage, some NLR family members can form inflammasome complex which regulates caspase-1 dependent maturation and release of pro-inflammatory cytokines IL-1β and IL-18. Evidence suggests the involvement of NLR/inflammasome-mediated signalling in eliciting inflammatory responses in tissues obtained from chronic smokers or patients with COPD (Faner et al. 2016; Qian et al. 2015). However, not all the NLR proteins have been studied with regards to cigarette smoke exposure. Considering this fact, we investigated the role of NLRP10 and NLRP12 in eliciting CSE-mediated inflammatory responses. NLRP10 (also known as NALP10, NOD8, PAN5, Pynod, and CLR11.1) is the only member of the NLR family that lacks an LRR domain, which is essential for detection of pathogen associated molecular patterns (PAMPs) or danger associated molecular patterns (DAMPs)(Su et al. 2013). Therefore, it is speculated that NLRP10 might play a regulatory role in inflammatory signalling through its association with other NLR proteins. An earlier report by Wang et al (2002) suggested a role of NLRP12(also known as NALP12, PAN6,PYPAF7,RNO and CLR19.3)as a positive regulator of caspase-1 activation, IL-1β production, and NF-κB activation(Wang et al. 2002). Another study demonstrated NLRP12 to be an important modulator of inflammasome-mediated production of IL-1β and IL-18 in Yersinia pestis infection using C57Bl/6 mice as a study model (Vladimer et al. 2012). Likewise, in this study we observed a CSE-mediated increase in the expression of both NLRP10 and NLRP12 in A549 cells, which prompted us to explore the role of these NLR family members in response to CSE-challenge.
Despite being cytosolic receptors, some NLRs have been shown to be recruited to the plasma membrane to activate downstream signalling (Eitel et al. 2008; Kufer et al. 2008; Lautz et al. 2012). Membrane microdomains, referred to as lipid rafts, serve as platforms for localization of different receptors and other proteins to induce cell signalling in response to external stimulus(Barnich et al. 2005; Bodas et al. 2015; Shaw 2006). Lipid rafts have been studied in relation to several lung pathologies and are known to play an important role in mediating cellular activation and inflammation. Dudez and co-workers found that cystic fibrosis transmembrane receptors (CFTRs) harboured in lipid raft domains mediate inflammatory responses in Madin-Darby canine kidney type I cells (Dudez et al. 2008). Another study demonstrated a direct correlation between lipid raft CFTR expression and advancement of emphysema through increased ceramide gathering in cigarette smoke-exposed C57Bl/6 mice (Bodas et al. 2011). A significant accumulation of ceramide in lipid-rafts within the lungs of COPD patients has been reported to occur with increasing severity of emphysema (Bodas et al. 2015). Collectively, these reports indicate that lipid rafts may play a role in regulating intracellular signalling during COPD.
To understand the roles of lipid rafts, NLRP10, and NLRP12 in CSE-induced inflammation, we exposed human alveolar type II epithelial (A549) cells and human monocytic (THP-1) cells to CSE. Our results demonstrated increased expression and co-localization of NLRP10 and NLRP12 proteins in A549 and THP-1 cells. We further observed membrane recruitment of NLRP10 and NLRP12 in lipid raft entities and proximity of NLRP12 and caveolin-1 (lipid raft protein) following CSE challenge in A549 cells. To study the role of lipid rafts in CSE-mediated inflammation, we either enriched cell media with polyunsaturated fatty acids (PUFA) to form larger rafts or used the chemical agent filipin to disrupt lipid rafts. Interestingly, enrichment of membrane microdomains with PUFA significantly diminished CSE-induced inflammation and caused decreased recruitment of NLRP10 and NLRP12 in the membrane while depletion of membrane cholesterol (a major component of lipid rafts) by filipin resulted in exacerbated response in terms of IL-1β and CCL2 (MCP-1)production. Furthermore, PUFA enrichment was able to inhibit filipin-mediated hyper inflammation in CSE-challenged cells. Overall, our findings suggest an important role of lipid rafts in NLRP10 and NLRP12-mediated signalling during CSE-exposure. These findings provide novel insight into the molecular mechanisms associated with CSE-induced inflammatory responses and may help facilitate the design of improved therapeutic strategies targeting the membrane raft assembly to manage the onset and progression of COPD.
MATERIALS AND METHODS
Chemicals
For A549 cell culture, F-12K medium containing L-Glutamine (Corning Inc., Corning, NY) supplemented with Fetal Bovine serum (FBS) (Corning Inc., Corning, NY) and Penicillin-Streptomycin (GE Healthcare, Logan, UT) was used. For THP-1 culture, RPMI 1640 medium (GE Healthcare, Logan, UT) supplemented with FBS and Penicillin-Streptomycin was employed. To detect cytokine/chemokine levels in cell culture media, we used ELISA kits from R&D Systems (Minneapolis, MN) or Affymetrix (eBioscience, Santa Clara, CA). Antibodies against NLRP10, NLRP12, Caveolin-1, Caveolin-2, and Flotillin-1, used for immunoblotting were purchased from Santa Cruz Biotechnology (Dallas, TX) or Cell Signaling (Danvers, MA). While, F(ab′)2-Goat anti -Rabbit IgG (H+L) Alexa Fluor® 488 conjugate (Cat# A11070) and Goat Anti-Mouse IgG (H+L) Alexa Fluor® 594 conjugate (Cat# A11058) for immunofluorescence and Vybrant® lipid raft labelling kit (Cat#V34405) were procured from Life Technologies (Carlsbad, CA). HRP conjugated anti-mouse IgG (H+L) and anti-Rabbit IgG (H+L) were obtained from Thermo Fisher (Waltham, MA). Enhanced chemiluminescence kit was purchased from LI-COR Biosciences (Lincoln, CE). Electrophoresis reagents were procured from Amresco (Solon, OH). Duolink In Situ Red Starter Kit (Cat# DUO92105) for Proximity Ligation assay and Caveolae/Rafts Isolation Kit (Cat#CS0750) were purchased from Sigma Chemicals (St. Louis, MO).
Cell culture and treatments
Human adenocarcinoma (A549) cells (Cat No# ATCC®CCL-185) and human monocytic cells (THP-1) (Cat No# ATCC® TIB-202) were obtained from American Type Culture Collection (ATCC, Rockville, MD) and maintained in F-12K and RPMI 1640 medium respectively, supplemented with 10% FBS and 1%v/v Penicillin-Streptomycin. Of note, A549 are widely used as a model system for human type II alveolar epithelial cells (AECII) as the antigen presentation in resting and stimulated conditions and metabolic and biochemical properties of both these cells lines have been found to be similar through various characterization studies (Balis et al. 1984; Corbiere et al. 2011; Foster et al. 1998; Hawdon et al. 2010; Mortaz et al. 2017; Smith 1977). Cells were seeded in 6-well culture plates (0.35×106/well) or poly-L-lysine coated 8-well glass slides (3.5×104/well) and incubated overnight at 37°C in 5% CO2 incubator. Cells were challenged with commercially available cigarette smoke extract (CSE; Murty Pharmaceuticals Inc., Lexington, KY) at 0.25μg/ml and 1μg/ml concentration for 6h. According to the manufacturer, CSE was prepared by smoking University of Kentucky’s 1R3F standard research cigarettes on a Phipps-Bird 20-channel smoking machine and stored in DMSO at −20°C (Huang et al. 2015; Kispert et al. 2015; Pierson et al. 2013; Wang et al. 2014). The stock concentration of CSE was 40mg/ml of total particulate matter containing 6% nicotine. Cells treated with dimethyl sulfoxide (DMSO) (Cat# D8418) (Sigma-Aldrich Inc., St. Louis, MO) served as negative control for our experiments.
Lipid raft enrichment studies were performed by maintaining the cells in culture media containing low endotoxin FBS, supplemented with 15.77ng/ml (approximately 50μM, calculated from the average molecular weight of EPA and DHA) of polyunsaturated fatty acid from animal source (PUFA) (Cat# 47015) or 25μM Docosahexaenoic acid (DHA) (Cat# D2534) (Sigma-Aldrich, Inc., St. Louis, MO) for 24h followed by 6h challenge with CSE or DMSO. The PUFA used for this study is a mixture of: Methyl myristate; Methyl palmitate; Methyl palmitoleate; Methyl stearate; Methyl oleate; cis-11-Octadecenoic methyl ester; Methyl linoleate; Methyl linolenate; Methyl γ-linolenate; Methyl cis-11-eicosenoate; Methyl arachidonate; Methyl all-cis-5,8,11,14,17-eicosapentaenoate; cis-7,10,13,16-Docosatetraenoic acid methyl ester; cis-4,7,10,13,16,19-Docosahexaenoic acid methyl.
We also used saturated fatty acid (arachidic acid) at 25μM concentration to determine its effect on CSE induced inflammation. Enrichment with PUFA or DHA was carried out in complete media as described by Schumann and co-workers(Schumann et al. 2011). Cell viability between FBS or BSA (Fatty Acid Free) complexes of PUFA in FBS-containing and FBS-free media was compared. Lipid raft disruption was achieved by pre-treatment of A549 cells with 5μg/ml Filipin (Cat# F9675) (from Streptomyces filipinensis; Sigma Aldrich, St Louis, MO) for 2h in PUFA enriched or control group of cells followed by a 6h challenge with CSE or DMSO. Culture media was collected and stored at −80°C for determination of cytokines/chemokines, while cells were collected for western blotting and mRNA expression analysis.
ELISA
Following treatment of cells, culture media was collected to determine IL-1β (R&D Systems, Minneapolis, MN), IL-18, CCL-2 and CXCL-8 (IL-8)(eBioscience, Santa Clara, CA) production by ELISA following the manufacturer’s protocol. Colorimetric detection was performed at 450nm.
mRNA expression
Cell pellets were stored in TRIzol® Reagent (Sigma-Aldrich, St. Louis, MO) at −80°C for mRNA expression analysis. To determine the expression of our genes of interest, mRNA was isolated from the cells and used to prepare cDNA for RT-PCR analysis. We used Direct-zoL™ RNA Kits (Cat# R2072)(Zymo Research, Irwin, CA) for RNA isolation as per manufacturer’s protocol. Using iScript cDNA Synthesis kit (Cat# 1708841)(BIO-RAD Laboratories, Hercules, CA), cDNA was prepared from 1μg of isolated total RNA, and used to determine the expression of IL-1β, IL-18, CCL-2, and CXCL-8 using gene specific human forward and reverse primers: IL-1β (Fw- 5′-CAA AGG CGG CCA GGA TAT AA-3′, Rv- 5′-CTA GGG ATT GAG TCC ACA TTC AG-3′), IL-18 (Fw- 5′-CCA AGG AAA TCG GCC TCT ATT-3′, Rv- 5′-CAT ACC TCT AGG CTG GCT ATC T-3′), CCL-2 (Fw- 5′-GGC TGA GAC TAA CCC AGA AAC-3′, Rv- 5′-GAA TGA AGG TGG CTG CTA TGA-3′), and CXCL-8 (Fw- 5′-CTT GGC AGC CTT CCT GAT TT-3′, Rv- 5′-GGG TGG AAA GGT TTG GAG TAT G-3′), and, for transcriptional analysis, while Actin (Fw- 5′-CCC TAG TTC CAG TTC CAA GAT G-3′, Rv- 5′-GGA TTT GGA GGC AGG ATG AA-3′) was used as the internal control in q-RT PCR reaction. Relative mRNA expression of each gene was determined using 7500 Fast Real-Time PCR System (Applied Biosystems, CA).
Caspase-1 activity
Caspase-1 activity was determined using caspase-1 fluorometric kit (Cat# K110) (Bio Vision, Milpitas, CA) or Caspase-Glo® 1 homogeneous luminescent assay (Cat# G9951)(Promega Corporation, Madison, WI) kit as per manufacturer’s protocol. Change in fluorescence or luminescence (depending on the kit) was plotted as relative fluorescence units (RFU) or relative luminescence units (RLU) respectively.
Immunoblotting
For western blotting experiments, cells were lysed using Urea/CHAPS/Tris buffer (8M urea, 40mM Tris, 40mg CHAPS,) containing a protease inhibitor cocktail (Cat# P8340) and a phosphatase inhibitor cocktail 3 (Cat# P0044) (Sigma-Aldrich, St. Louis, MO). The total protein content was determined using the Bradford protein assay kit (Thermo Fisher, Waltham, MA). A total of 25μg protein from each sample was mixed with 4X Laemmeli Buffer (Bio-Rad, Hercules, CA) and resolved by SDS-PAGE and electro-blotted on PVDF membrane (Bio-Rad, Hercules, CA). Blots were incubated overnight using anti-NLRP10 (Cat#sc-50608), anti-NLRP12 (Cat#sc-390666) (Santa Cruz Biotechnology, Dallas, TX), anti-Caveolin-1 (Cat#3267) (Cell Signaling, Danvers, MA), anti-Flotillin-1 (Cat#sc-74566) (Santa Cruz Biotechnology, Dallas, TX), anti-Caveolin-2 (Cat#8522) and anti-GAPDH (Cat#5174) (Cell Signaling, Danvers, MA). Immunostaining was performed using appropriate secondary antibody (Cell Signaling, Danvers, MA) at the dilution of 1:2000. The blots were visualized on ChemiDoc Touch Imaging System (Bio-Rad, Hercules, CA) by enhanced chemiluminescence using WesternSure PREMIUM Chemiluminescent Substrate (LI-COR, Lincoln, CE).
In silico Protein-Protein Interaction prediction
To determine potential interactions between lipid rafts, NLRP10 and NLRP12, we conducted in-silico molecular docking studies. The 3D-NMR structures were obtained from European Protein Data Bank (PDBe) (http://www.ebi.ac.uk/pdbe/). For the proteins whose complete structure is unknown, ab initio modelling was used for the in-silico structure prediction. Protein sequences of Caveolin-1, Caveolin-2, Flotillin-1 and Flotillin-2 were submitted to LOMETS (Local Meta-Threading-server) (http://zhanglab.ccmb.med.umich.edu/LOMETS/) for protein structure prediction (Wu and Zhang 2007). Of note, protein sequences for the PYD domain of NLRP10 (3D NMR solved structure PDBe ID 2m5v) and NLRP12 (3D NMR solved structure PDBe ID2l6a) were used for this study; as the crystal structure of this domain has been solved and the domain has been identified as putative protein-protein interaction domain. The predicted as well as 3D-NMR structure were then used for docking through ZDOCK (http://zdock.umassmed.edu/) an automated web-based docking tool. PyMol, a molecular visualization software was used for analysis of our docking results (Pierce et al. 2014; PyMOL).
Immunofluorescence
Immunofluorescence staining was conducted using 8-chamber glass slides to perform localization studies. Slides were coated for 3 hours with poly L-lysine (Sigma Aldrich, USA). Cells were seeded at the cell density of 3.5×104/well and incubated overnight in CO2 incubator at 37°C. Thereafter, cells were challenged with CSE (0.25μg/ml) or DMSO for 6 hours. Lipid Rafts labelling of treated cells was performed using Vybrant® lipid raft labelling kit (Cat# V34405; Life Technologies, Carlsbad, CA) following the manufacturer’s protocol. Cells were then fixed at room temperature with 2% paraformaldehyde for 20 minutes and permeabilized using permeabilization buffer. Cells were incubated in goat serum which served as blocking medium, followed by overnight incubation with anti-NLRP10 and anti-NLRP12 antibodies (Santa Cruz Biotechnology, Dallas, TX). After immunostaining with primary antibody, cells were washed and subsequently incubated at room temperature in dark humid chamber with appropriate fluorochrome-conjugated secondary antibody at 1:3000 dilution. The immune stained slides were mounted using VECTASHIELD antifade mounting medium (Vector Laboratories, Burlingame, CA). We also conducted experiments using cell-based cholesterol labelling kit (Abcam, Cat#ab133116) to identify the distribution/level of cholesterol within the cell following vendor protocol. Leica and Olympus FluoView® FV10i Confocal Microscope were used for the analysis.
Proximity Ligation Assay (PLA)
To determine the possible interaction between NLRP10 and NLRP12 following CSE challenge we performed Proximity Ligation Assay (PLA) using Duolink In Situ Red Starter Kit (Sigma Aldrich, St Louis, MO) as per the manufacturer’s protocol. Briefly, 8-chamber glass slides were coated with poly L-lysine for 3 hours. Cells were then seeded at a density of 3.5×104 per well and incubated overnight at 37°C in CO2 incubator. Cells were challenged with 0.25μg/ml of CSE or DMSO for 6h and then fixed using 2% paraformaldehyde for 20 minutes at room temperature. After cell permeabilization and blocking, the cells were incubated overnight with primary antibody against NLRP10 (Cat#sc-50609) and NLRP12 (Cat# sc-30650) (Santa Cruz Biotechnology, Dallas, TX) at 1:300 dilution at room temperature. Thereafter the cells were incubated for 1h with PLA probe (anti-mouse PLUS and anti-goat MINUS) after which the ligation mixture was added and kept for1h incubation. This was followed by addition of an amplification mixture to each chamber and the slides were then incubated in the dark for 1.5h at 37°C. Cells were then washed thrice and dried before mounting with antifade mounting medium. The images were acquired using Olympus FluoView FV10i Confocal Microscope.
Co-Immunoprecipitation
To determine the interaction between NLRP12 and NLRP10 or lipid raft proteins, co-immunoprecipitation studies were performed using Pierce Co-Immunoprecipitation Kit (Cat# 26149; Thermo Fisher, Waltham, MA) following the manufacturer’s protocol with some modifications. Approximately 5×106, A549 cells were seeded in a T-75 flask and allowed to grow overnight at 37°C in 5% CO2 incubator. Cells were treated with CSE (0.25μg/ml &1μg/ml) or DMSO (1μg/ml) for 6h and lysed in IP lysis buffer. Lysate was centrifuged at 13000g for 10 minutes at 4°C and supernatant was collected. Protein concentration in each sample was estimated with the help of Pierce BCA Protein Assay Kit (Thermo Fisher, Waltham, MA). Total 500μg of protein per sample in 200μl volume was taken for immunoprecipitation. Samples were pre-cleared using 80μl of control agarose resin slurry. Equal volume of pre-cleared protein lysate was transferred to fresh micro centrifuge tube and 2μg of anti-NLRP12 antibody (Cat# sc-30650) (Santa Cruz Biotechnology, Dallas, TX) was added followed by slow end to end mixing on a rotator at 4°Covernight. Amino link plus coupling resin was equilibrated at room temperature and washed in IP lysis buffer three times. Lysate (with antibody) was transferred to amino link plus coupling beads and incubated for slow end to end mixing on rotator at 4°C for 4 hours’ duration. Following incubation, beads were separated by centrifugation and washed three times. For preparing immunoblotting samples, 1× loading dye was directly added to the beads and samples were heated at 95°C for 6-8 min. Samples were probed with antibodies targeting anticipated binding partners of NLRP12.
Membrane Extraction
Approximately 3×106, A549 cells were seeded in T-75 flask and incubated overnight at 37°C in CO2 incubator. Thereafter, cells were enriched with PUFA or DHA as discussed earlier. Following 24h of membrane enrichment, cells were treated with 0.25μg/ml of CSE for 6h. Membrane and cytosol proteins were extracted using Mem-PERTM Plus Membrane Protein Extraction Kit (Cat#89842) (Thermo Fisher Scientific, Meridian Road, IL) as per manufacturer’s protocol. In short, cells were collected after 6h of CSE treatment, washed with Cell Wash Solution and pelleted at 300g for 5 min. Cells were then permeabilized at 4°C for 10min with constant mixing. Samples were centrifuged at 16000g for 15min and supernatant containing cytosolic proteins was collected. Cell pellet was resuspended in membrane solubilisation buffer and incubated at 4°C for 30min with constant mixing. Following centrifugation at 16000g for 15min, supernatant containing solubilized membrane and membrane-associated proteins was transferred to a fresh tube. Immunoblotting of membrane samples was performed using equal amount of proteins and the expression of NLRP10 and NLRP12 was determined.
Gene knockdown
NLRP10 and NLRP12 knockdown experiments were performed using Nlrp10-siRNA (Cat# sc61140) and Nlrp12-siRNA (Cat# sc45388) (Santa Cruz Biotechnology, Dallas, TX), while Control siRNA was used as negative control. TransIT-X2system (Cat# MIR6000) (Mirus Bio LLC Madison, WI) was used to deliver siRNA into the cells following manufacturer’s protocol. Around 0.3×106 A549 cells were seeded in 6-well plate and incubated overnight at 37°C in CO2 incubator. Following incubation culture media was replaced with fresh media. To perform transfection, TransIT-X2system was mixed with siRNA for specific gene or control-siRNA in culture media. The mix was added to cells in complete culture media and incubated for 48h in CO2 incubator. After incubation, cells were treated with 0.25μg/ml, 1μg/ml of CSE or DMSO. Gene knockdown was confirmed using immunoblotting assay for NLRP10 and NLRP12 protein expression.
Cell viability MTS assay
Viability of cells was determined using Cell Titer 96 Aqueous One Solution Cell Proliferation Assay kit (Cat# G3580) (Promega Corporation, Madison, WI). Cells (A549; 3.5×103 cells/100μ1/well in 96 well plate) were cultured in PUFA (employed FBS- or BSA-complexes as delivery vehicle) or DHA enriched media for 24h followed by addition of 20μl of MTS [3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] per well. Absorbance was measured at 490nm after 1h incubation at 37°C in CO2 incubator. Standard curve was plotted (cell number vs. absorbance) to determine the viability of cells in various culture conditions.
Isolation of Lipid rafts
Lipid rafts were isolated from A549 cells using Caveolae/Rafts Isolation Kit (Sigma-Aldrich, Cat#CS0750) as per the manufacturer’s protocol. Approximately 3×106 A549 cells were seeded in T-75 flask and incubated overnight at 37°C in CO2 incubator. Thereafter, cells were enriched with PUFA or DHA. Following 24h of membrane enrichment, cells were lysed in 1% Triton X-100 containing lysis buffer and equal amount of proteins were used for lipid rafts isolation. Concentration gradient ranging between 0-35percent was prepared using OptiPrep density gradient medium to isolate lipid rafts. Samples spun at 200,000g for 4 hours using table-top ultra-centrifuge (Hitachi, CS150NX). After ultra-centrifugation process, fractions were collected, and dot blot was performed using anti-caveolin-1 (lipid raft marker) to determine localization of membrane rafts in specific fractions. The fractions found positive for lipid rafts were analysed for presence of different lipid components using lipidomic analysis.
Extraction of sphingolipids and lipidomic analysis
For lipidomic analysis, internal standards procured from Avanti Polar Lipids (Alabaster, AL) were added to the samples in 10 μl ethanol: methanol: water (7:2:1) cocktail at 250 pmol concentration each. Samples were disrupted using ultra-sonicator (30 secs at room temperature), followed by overnight incubation at 48°C. Further preparatory steps included addition of KOH (1M) solubilized in methanol, followed by a short sonication step, and thereafter incubation for 2 h at 37°C in a shaking water bath to cleave potentially interfering glycerolipids. Samples were neutralized followed by centrifugation to collect the supernatant. The extracts were Speed Vac dried and reconstituted in starting mobile phase solvent for LC-MS/MS (Liquid Chromatography with tandem Mass Spectrometry) analysis. Samples were further sonicated for 15 seconds and centrifuged for 5 min to collect the clear supernatant which was then transferred to the auto injector vial for analysis. LC-MS/MS analyses were performed for sphingoid bases, sphingoid base-1-phosphates, and complex sphingolipids. Separation of compounds was carried out by reverse phase LC using a Supelco 2.1 (i.d.) × 50 mm Ascentis Express C18 column (Sigma, St. Louis, MO) and a binary solvent system at 35°C. Prior to the injection of each sample, column was equilibrated for 0.5 min with a solvent mixture of 95% mobile phase.
Statistical Significance
Data was analysed by ordinary one-way ANOVA and expressed as mean ± SE. Experiments were conducted in triplicates and were repeated 3-4 times. All statistical calculations were performed using GraphPad Prizm 6.0. Differences were considered statistically significant at * p <0.05 when compared with controls.
RESULTS
CSE induces Caspase-1 activity and release of pro-inflammatory cytokines and chemokines in A549 and THP-1 cells
Production of pro-inflammatory cytokine/chemokine is the key indicator of inflammation and induction of an immune response in mammals (Jun-Ming Zhang 2007). To determine the production of key cytokines/chemokines at transcriptional and translational levels following CSE-exposure; we conducted mRNA expression analysis and ELISA. Increased production of IL-1β, IL-18, CCL-2, and CXCL-8 was observed by ELISA in CSE-challenged A549 cells (Figure 1B). The mRNA results further confirmed CSE-mediated increase in the production of IL-1β, CCL-2 and CXCL-8 by A549 cells (Figure 1C). To confirm if the observed changes are cell-specific we conducted similar studies with THP-1 cells and found increased production of pro-inflammatory cytokines. i.e., IL-1β, CCL-2 and CXCL-8 (Figure 1A). We also estimated Caspase-1 activity (essential for processing pro-forms of IL-1β and IL-18 into mature forms) in both THP-1 and A549 cells following CSE-exposure. We observed a significant increase in the Caspase-1 activity in CSE-challenged vs. control group (Figure 1A-B).
Figure 1. CSE-exposure induces cytokine/chemokine production and Caspase-1 activity in A549 and THP-1 cells.
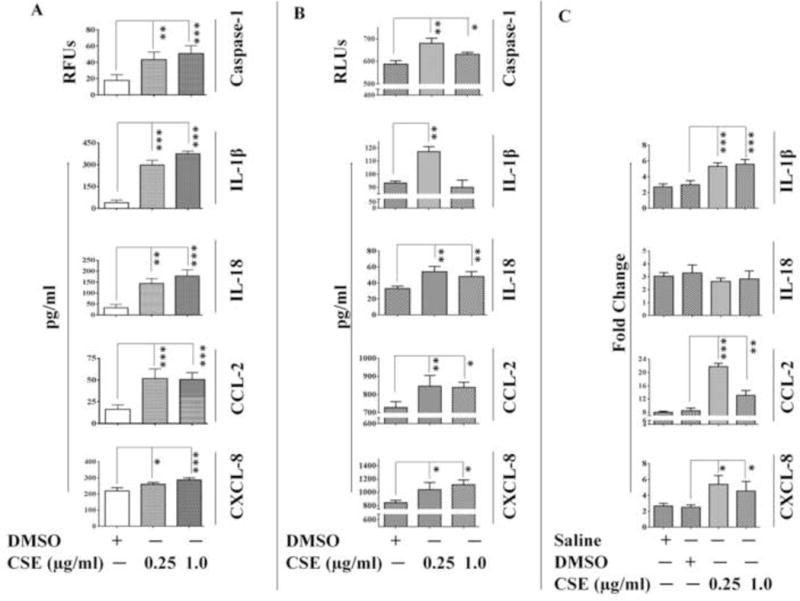
Caspase-1 activity and cytokine/chemokine levels in (A) THP-1 cells and (B) A549 cells as determined by ELISA; and (C) mRNA level expression of cytokine/chemokines in A549 cells following 6-hr CSE-challenge.(n=3-4/group). Results are a representation of three independent experiments. Error bars represent SEM, * (p<0.05), ** (p<=0.01) and *** (p<=0.001), as per one-way ANOVA for multiple comparisons.
Increase in the expression and membrane recruitment of NLRP10 and NLRP12 during CSE challenge
We next sought to evaluate the expression of NLRP10 and NLRP12 in CSE-challenged A549cells. Immunoblotting results demonstrated increased expression of NLRP10 and NLRP12 proteins in CSE-challenged A549 cells as compared to DMSO controls (Figure 2A). Previous reports have suggested that the membrane recruitment of some NLR proteins are essential to activate downstream signalling (Eitel et al. 2008; Kufer et al. 2008; Lecine et al. 2007). Therefore, we also assessed the expression of lipid raft proteins in our study model and observed increased expression of Caveolin-1, Caveolin-2 and Flotillin-1 following a 6h CSE-challenge in A549 cells (Figure 2A). Lipid Raft labelling experiments demonstrated increased formation of lipid raft entities in CSE challenged A549 cells as indicated by increased staining of GM-1 (monosialotetrahexosylganglioside); widely used as a lipid raft marker (Figure 2B). We further conducted immunofluorescence analysis to target NLRP10 and NLRP12 and determine their cellular distribution following CSE-challenge. Our results point towards membrane recruitment of NLRP10 and NLRP12 (which are cytosolic proteins in unstimulated conditions) in CSE-exposed A549 cells (Figure 2B). To further substantiate our findings, we performed immunoblotting on the membrane fraction obtained from CSE-challenged A549 cells. We found increased recruitment of NLRP10 and NLRP12 proteins in the membrane fractions obtained from CSE-exposed cells as compared to the controls (DMSO) (Supplementary Figure 1).
Figure 2. CSE-challenge promotes the expression of lipid raft proteins and membrane localization of NLRP10/12 in A549 cells.
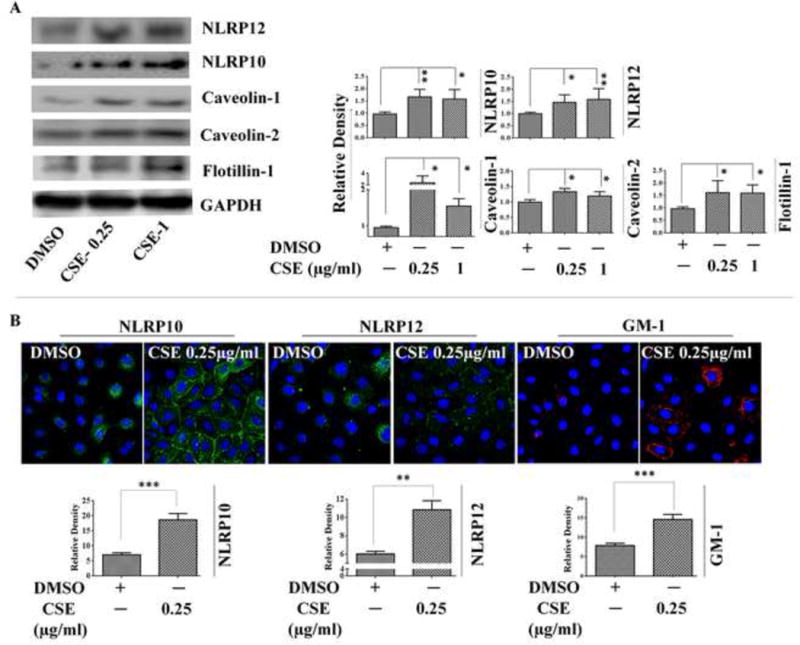
(A) Western Blot images and Densitometry results for the expression of NLRP10/12 and lipid raft proteins (Caveolin-1, Caveolin-2 and Flotillin-1) following 6-hr CSE-challenge in A549 cells; and (B) Immunofluorescence results demonstrating cellular localization of NLRP10, NLRP12; and GM-1 (lipid raft marker) following 6-hr CSE-challenge in A549 cells. (n=3-4/group). Results are a representation of 4 independent experiments. Error bars represent SEM, * (p<0.05) and ** (p<=0.01), as per oneway ANOVA for multiple comparisons. Here, CSE-0.25: CSE (0.25 μg/ml) and CSE-1: CSE (1 μg/ml)
NLRP10 and NLRP12 localize in lipid raft entities following CSE-exposure
Recruitment of NLRP10 and NLRP12 proteins in the cell membranes along with increased expression of lipid raft proteins and their assembly in response to CSE-exposure, prompted us to investigate possible interaction between NLRP10, NLRP12 and lipid raft associated proteins. To explore this possibility, we first used in silico protein-protein docking server ZDOCK to predict the interactions of NLR proteins (NLRP10 and NLRP12) with lipid raft proteins (Caveolin-1, Caveolin-2, Foltillin-1 and Flotillin-2). ZDOCK accomplishes high analytical accuracy on protein-protein interactions and shows precision around more than 70% in top 1000 predictions. ZDOCK data simulated interactions between NLRP10/NLRP12 and lipid raft proteins. We did not observe hydrophilic interaction but found hydrophobic interaction between NLRP10 and Caveolin-1 using in-silico experiments (Table 1). Interactions between NLRP10 and Caveolin-2, Flotillin-1, and Flotillin-2 were also predicted by ZDOCK (Table 1)(Figure 3A). Similarly, in silico studies predicted the possibility of NLRP12 interaction with Caveolin-1, Caveolin-2, Flotillin-1 and Flotillin-2 (Figure 3B). To validate the in-silico predictions we employed immunofluorescence imaging and PLA. Immunofluorescence data demonstrated significant translocation of NLRP10 and NLRP12 in the membrane rafts following a 6-hr CSE-challenge in A549 cells (Figure 4A-B). PLA findings further proved close proximity between NLRP12 and lipid raft marker, GM-1 in CSE-challenged A549 cells as compared to the controls (Figure 4C). These observations were further confirmed by co-immunoprecipitation studies using anti-NLRP12. Here we observed concentration (CSE)-dependent increase in the interaction between NLRP12 and caveolin-1 in CSE-challenged A549 cells (Figure 4D). On the other hand, our results from the proximity ligation assay and co-immunoprecipitation studies did not confirm the physical interaction of NLRP10 with lipid raft (proteins) in CSE-challenged cells (data not presented), which point towards the possibility of involvement of other binding partners for NLRP10 that help in modulating CS-mediated responses in vitro.
Table 1.
List of interacting amino acids from in silico results.
| Interacting amino acids | ||
|---|---|---|
| S. No. | NLRP12 amino acid | Caveolin-1 Amino acids |
| 1 | Arg98 | Thr78 |
| 2 | Thr100 | Glu76 |
| 3 | Val97 | Asn173 |
| 4 | Asp95 | Arg171 |
| 5 | Glu94 | Ser168 |
| 6 | Leu96 | Gly 164, Arg171 |
| 7 | Glu102 | Asn169 |
| 8 | Glu89 | Ser88 |
| 9 | Leu87, Arg90 | Glu177 |
| 10 | Gln92, Trp88 | Gln 175 |
| 11 | Glu89, Arg90 | Ile178 |
| Interacting amino acids | ||
| S. No. | NLRP10 amino acid | Caveolin-1 Amino acids |
| 1 | None | None |
| Interacting amino acids | ||
| S. No. | NLRP10 amino acid | Caveolin-2 Amino acids |
| 1 | Gly1 | Val56, Phe104 |
| 2 | Met5 | Asp55 |
| 3 | Ala8 | Phe118 |
| 4 | Lys10 | Ser160, Leu115 |
| 5 | Asn102 | Lys7 |
| 6 | Met83 | Asn6 |
| 7 | Arg101 | Asp138 |
| 8 | Tyr100 | Val139, Asp138 |
| 9 | Leu97 | Val136 |
| 10 | Leu92 | Trp133 |
| 11 | Asn87 | Leu25, Tyr27 |
| 12 | Ser20 | Leu123 |
| 13 | Trp17 | Val125 |
| 14 | Asp90 | Phe91, Leu92 |
| Interacting amino acids | ||
| S. No. | NLRP12 amino acid | Caveolin-2 Amino acids |
| 1 | Asn39 | His48 |
| 2 | Asn39, Leu40 | Ile71 |
| 3 | Asn26 | Asp17 |
| 4 | Gly91 | Ser18 |
| 5 | Gly92 | Asp17 |
| 6 | Cys13 | Ala97 |
| 7 | Val25, Phe30 | Thr62 |
| 8 | Thr64 | Leu144, Val148 |
| 9 | Ile63 | Ala105, Thr106 |
| 10 | Arg73 | Leu88 |
| 11 | Asn70 | Thr89 |
| 12 | Asn42 | Phe118 |
| Interacting amino acids | ||
| S. No. | NLRP10 amino acid | FlotiHin-1 Amino acids |
| 1 | Ser 93 | Arg 124 |
| 2 | Ala 2 | Ser 64 |
| 3 | Asp 99 | Gln 125, Leu 151, Thr 150 |
| 4 | Glu 102 | Lys126 |
| 5 | Met 3 | His 155 |
| Interacting amino acids | ||
| S. No. | NLRP12 amino acid | FlotiHin-1 Amino acids |
| 1 | Arg-90 | His-59, Thr-116, |
| 2 | Arg-81 | Asp-159 |
| 3 | Trp-88 | Asp-156 |
| 4 | Leu-62 | Asp-153 |
| 5 | Thr-64 | Asn-49 |
| Interacting amino acids | ||
| S. No. | NLRP10 amino acid | Flotillin-2 Amino acids |
| 1 | Glu 102 | Gln 80 |
| 2 | Ser 93 | Arg 78 |
| 3 | Gly 1 | Tyr 109 |
| 4 | Ala 2 | Lys 111 |
| 5 | His 98 | Ile 105 |
| 6 | Asp 99 | Thr104 |
| Interacting amino acids | ||
| S. No. | NLRP12 amino acid | Flotillin-2 Amino acids |
| 1 | Ala 7 | Gln 80 |
| 2 | Leu 22 | Arg 63 |
| 3 | Glu 94 | G119 |
| 4 | Glu 69 | Arg 78 |
| 5 | Arg 90 | Glu 14, G15 |
| 6 | Lys 52 | Gln 48 |
| 7 | Arg 93 | Glu 72 |
| 8 | Thr 78 | Lys 111 |
| 9 | Leu 61 | Thr 21 |
| 10 | Arg 84 | Asp 9 |
| 11 | Trp 88 | Asp 110 |
| 12 | Tyr 18 | Leu 69 |
| 13 | Arg 5 | Gln 73, Asp 77 |
Figure 3. In silico studies demonstrate possible interaction between lipid raft proteins and NLRP10/12.

Protein-protein docking results showing probable interaction between (A) NLRP10 (Green, top left) and Flotillin-1 (Yellow; top left); NLRP10 (Blue, top right) and Caveolin-1 (Red, top right); NLRP10 (Green, bottom left) and Flotillin-2(Blue, bottom left); and NLRP10 (Green, bottom right) and Caveolin-2 (Cyan, bottom right) and (ii) NLRP12 (Orange, top left) and Flotillin-1 (Yellow, top left); NLRP12 (Orange, top right) and Caveolin-1 (Blue, top right); NLRP12 (Blue, bottom left) and Flotillin-2 (Orange, bottom left) and NLRP12 (Orange, bottom right) and Caveolin-2 (Cyan, bottom right), where N10:NLRP10; N12:NLRP12; Cav1:Caveolin-1; Cav2: Caveolin-2; Flot1:Flotillin-1; Flot2: Flotillin-2.
Figure 4. CSE-challenge induces recruitment of NLRP10 and 12 to the lipid rafts and leads to interaction between NLRP12 and Caveolin-1 in A549 cells.
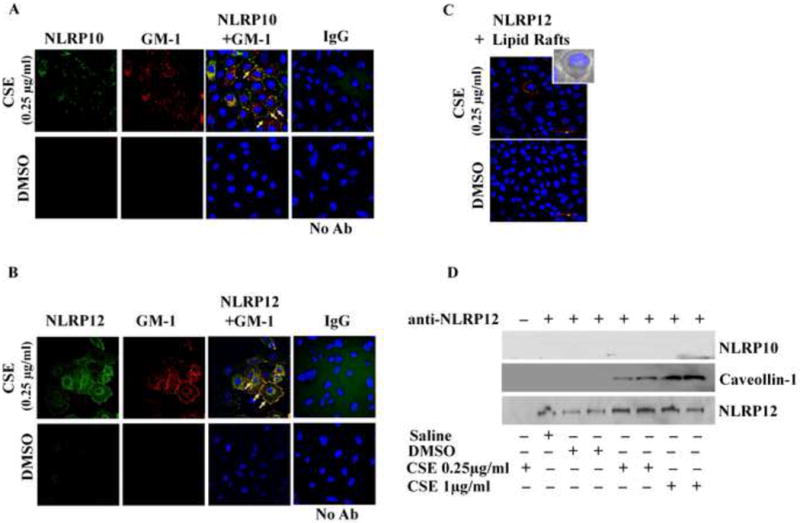
Immunofluorescence results demonstrating recruitment of (A) NLRP10 (Green) and (B) NLRP12 (Green) in the lipid rafts entities (red, shown by labeling GM-1, lipid raft marker), (C) Sub-cellular proximity between NLRP12 and lipid raft protein, and (D) Co-immunoprecipitation of caveolin-1 (lipid raft protein) with the help of anti-NLRP12 antibody following 6-hr CSE challenge in A549 cells.(n=3-4/group). Results are a representation of three independent experiments. Here, No Ab: No Antibody Control and IgG: anti immunoglobulin G control.
Co-localization and molecular proximity between NLRP10 & NLRP12 following CSE challenge
NLRP10 is the only member of the NLR family of proteins that lacks an LRR domain. The LRR domain functions as a protein recognition motif for sensing pathogen associated molecular patterns. We, therefore, speculated that in absence of a recognition signal motif, NLRP10 may have a crucial regulatory role during CSE-induced inflammation by regulating downstream responses possibly in conjugation with NLRP12. To test this possibility, we first performed an in-silico protein docking study as described earlier and found probable interaction sites between NLRP10 and NLRP12 PYD domains (Table 1) (Figure 5A). Using confocal imaging, we observed co-localization of NLRP10 and NLRP12 proteins on CSE-challenge in THP-1 cells (Figure 5B). Furthermore, PLA analyses demonstrated proximity (less than 16nm) between NLRP10 and NLRP12 in CSE exposed A549 cells as compared to DMSO controls (Figure 5C) thus strengthening our hypothesis. To confirm our results, we performed co-immunoprecipitation using anti-NLRP10 and anti-NLRP-12 antibodies. However, we were unable to detect physical interaction between these two NLR family members in CSE-challenged cells under our study conditions (Figure 4D).
Figure 5. CSE-challenge demonstrates cellular co-localization of NLRP10 and NLRP12.
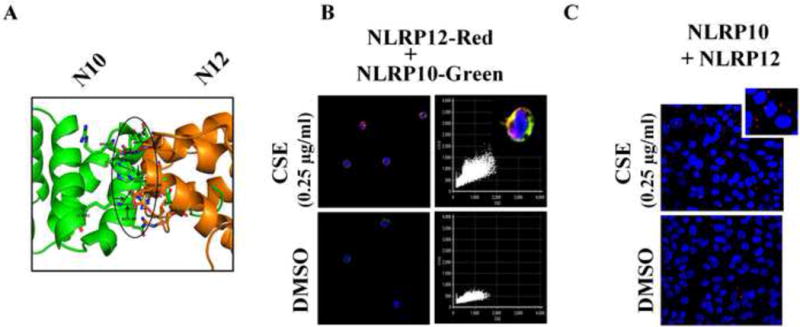
(A) In silico docking results showing probable interactions between NLRP10 (green) and NLRP12 (orange). (B) Immunofluorescence results demonstrating co-localization of NLRP10 (Green) and NLRP12 (red) following 6-hr CSE challenge in THP-1 cells. (n=3-4/group); and (C) Sub-cellular proximity between NLRP10 and NLRP12 as observed by Proximity Ligation Assay following 6-hr CSE challenge in A549 cells. (n=3-4/group). Results are a representation of three independent experiments.
NLRP10 regulates the production of IL-1β during CSE-challenge
To substantiate the role of NLRP10 and NLRP12 in regulating CSE-mediated inflammation we performed siRNA-mediated knockdown experiments using siRNA-Nlrp10 or siRNA-Nlrp12 to transfect A549 cells. NLRP10/12 silencing was confirmed with the help of immunoblotting using anti-NLRP10 and anti-NLRP12 respectively [Figure 6A (i) and B (i)]. On further investigation, we observed a significant reduction in the release of IL-1β in CSE-challenged siRNA-Nlrp10 transfected A549 cells as compared to the controls (Figure 6A (ii)). We also observed NLRP12 mediated reduction in IL-1β following CSE-challenge, however the response was of much lower magnitude [Figure 6B (ii)]. Our results thus point towards an important regulatory role of NLRP10 in CSE-induced inflammation in vitro. However, there exist the possibility of other binding partner(s) of NLRP10 which can mediate CSE-induced inflammation in vitro.
Figure 6. NLRP10 regulates IL-1 β production in CSE-challenged A549 cells.
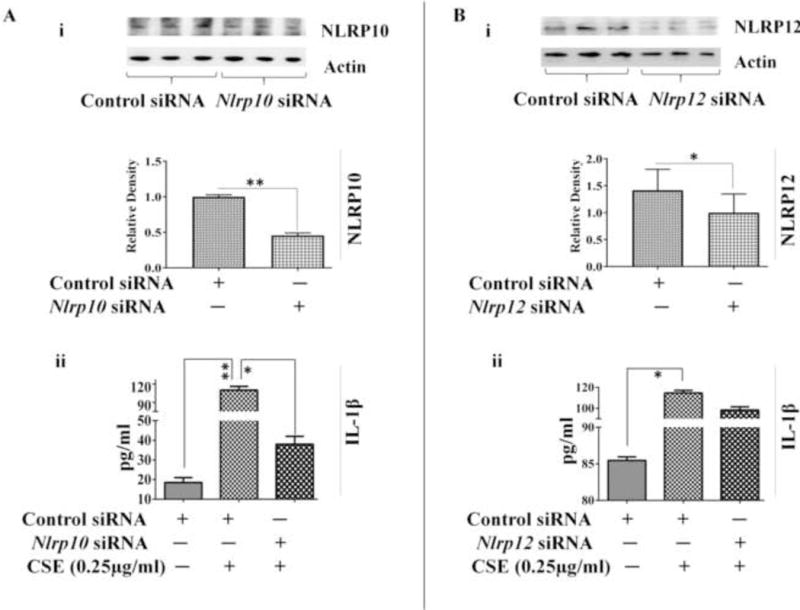
(A) (i) siRNA-mediated silencing of NLRP10 and (ii) IL-1β production in knockdown sample in CSE-challenged A549 cells. (n=3-4/group). (B) (i) siRNA-mediated knockdown of NLRP12 and (ii) IL-1β production in CSE-challenged A549 cells. (n=3-4/group). Results are a representation of three independent experiments. Error bars represent SEM, * (p<0.05) and ** (p<=0.01), as per one-way ANOVA for multiple comparisons.
Enrichment of lipid rafts with PUFA or DHA rescues A549 cells from CSE-induced inflammation
To delineate the role of lipid rafts in CSE-mediated inflammation, we enriched the membrane rafts with PUFA or DHA and challenged them with CSE for 6h to determine inflammatory responses. Both DHA and PUFA have been shown to modulate the membrane microdomains by affecting the membrane characteristics (Stillwell and Wassall 2003; Wassall et al. 2004)thereby leading to alterations in lipid raft-mediated cell signalling (Chapkin et al. 2008; Kim et al. 2008a; Shaikh et al. 2009). We conducted lipidomic analyses of the lipid raft fractions obtained from A549 cells cultured in PUFA- or DHA-enriched media. We observed a significant increase in the levels of DHA, eicosenoic acid (monounsaturated fatty acid) and arachidonic acid (polyunsaturated fatty acid) in the raft fractions isolated from DHA/PUFA enriched cells vs controls, confirming successful incorporation of DHA/PUFA in the membrane lipid rafts of the A549 cells (Figure7B). Furthermore, enrichment with PUFA had no effect on the cell viability. Interestingly, we observed significant reduction in the cell viability on replacing endotoxin free FBS with BSA in our PUFA enrichment media (Figure 7A).
Figure 7. Incorporation of unsaturated fatty acids in the lipid raft entities following enrichment of A549-cells with PUFA/DHA.
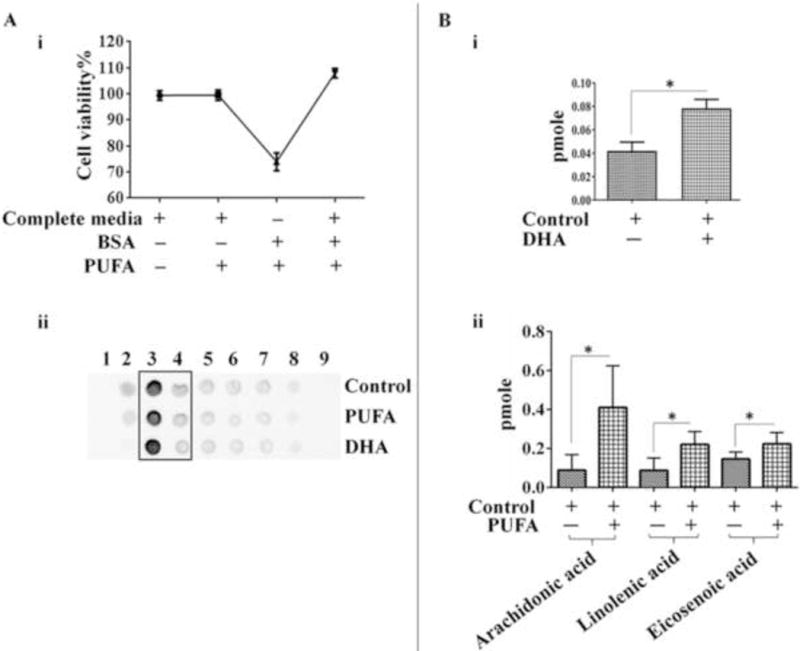
(A) (i) Cell viability on enrichment with PUFA/DHA complexed in (Fatty acid free) BSA in cultures maintained in media with or without FBS (n=3-4/group).(ii) Dot blots of lipid raft fractions from PUFA/DHA-enriched or control A549 cells used for lipidomic analysis.(B) Levels of (i) DHA, or (ii) unsaturated fatty acids detected in lipid raft fractions isolated from PUFA/DHA-enriched or control A549 cells on lipidomic analysis. Equal amount of protein were used for lipid raft purification to normalize the results. (n=3-4/group). Results are a representation of three independent experiments. Error bars represent SEM, * (p<0.05), as per two tailed student t-test.
PUFA or DHA enrichment abrogated CSE-induced transcription and production of IL-1β, CCL-2 and CXCL-8 by A549 cells (Figure 8A-B). Simultaneously, we also observed a reduction in CSE-mediated Caspase-1 activity in PUFA and DHA enriched A549 cells (Figure 8C). Additionally, cholesterol labelling experiments revealed significant reduction in membrane cholesterol in PUFA enriched A549 cells compared to control group (Supplementary Figure 2B). It is important to note here, that the expression of NLRP10 and NLRP12 in the membrane fractions from CSE-challenged PUFA-enriched A549 cells was abrogated as compared to the control group (Supplementary Figure 1) further supporting our hypothesis. Overall our observations suggest that enrichment of lipid rafts with PUFA-could rescue CSE-mediated inflammation and Caspase-1 activation in A549 cells.
Figure 8. PUFA enrichment rescues A549 cells from CSE-induced inflammation.
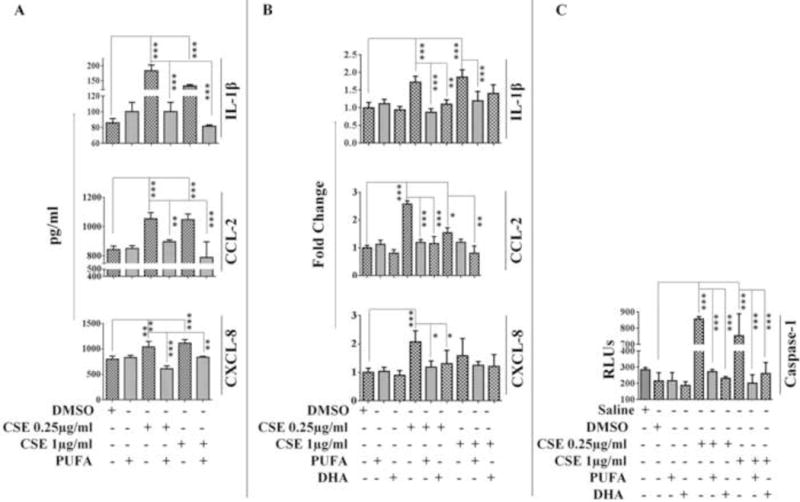
Cytokine/chemokine (IL-1β, CCL-2, and CXCL-2) production at (A) translational and (B) transcriptional levels and (C) Caspase-1 activity in PUFA/DHA-enriched A549 cells challenged with CSE. (n=3-4/group). Results are a representation of three independent experiments. Error bars represent SEM, * (p<0.05), ** (p<=0.01) and *** (p<=0.001), as per one-way ANOVA for multiple comparisons.
PUFA rescues filipin mediated exacerbated responses in CSE-challenged A549 cells
To substantiate our claim in relation to the role of lipid rafts in modulating CSE-mediated inflammation in A549 cells, we disrupted the lipid raft domains by pre-treating cells with filipin prior to CSE-challenge. Pre-treatment with filipin (5 μg/ml) exacerbated CSE-induced mRNA levels of IL-1β and CCL-2. However, no significant variation in CXCL-8 mRNA expression was observed in CSE-exposed cells with/without filipin pre-treatment. We next enriched lipid rafts using PUFA prior to filipin treatment to observe the role of modified rafts in CSE-induced inflammation. Our findings reveal a noticeable rescue of exacerbated responses by PUFA in filipin treated CSE-challengedA549 cells (Figure 9).
Figure 9. Membrane enrichment with PUFA rescues CSE-challenged A549 cells from filipin mediated hyper-responses.
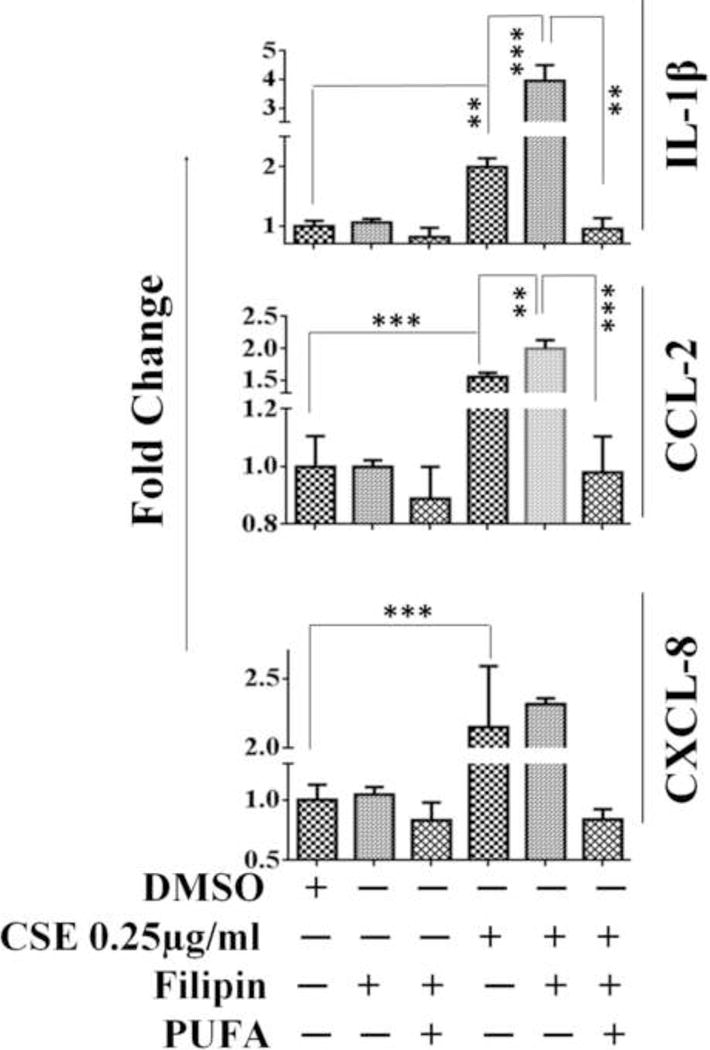
CSE-mediated transcriptional regulation of IL-1β, CCL-2 and CXCL-8 in PUFA-enriched A549 cells pre-treated with filipin. (n=3-4/group). Results are a representation of three independent experiments. Error bars represent SEM, * (p<0.05), ** (p<=0.01), and *** (p<=0.001), as per one-way ANOVA for multiple comparisons.
Enrichment of A549 cells with saturated fatty acids is unable to rescue CSE-induced responses in A549 cells
To test the possibility of rescue from CSE-induced inflammation, we enriched A549 cells with saturated fatty acid (arachidic acid). Our results demonstrate that enrichment of A549 cells with arachidic acid was unable to provide any rescue from CSE induced inflammation. Instead, we observed further increase in CSE-induced IL-1β production following saturated fatty acid enrichment. While no significant change was observed in CCL-2 or CXCL-8production between our study groups (Supplementary Figure 2A).
DISCUSSION
The NLR family of proteins are widely studied for their role in inflammasome formation and signalling. To date, 22 human NLR genes have been identified, amongst which NLRP1, NLRP3, and NLRC4 have been shown to form inflammasome complexes (Proell et al. 2008; Shaw et al. 2010). NLRP10 and NLRP12 are two less well characterized members of this family whose actual function during inflammation remains controversial. Furthermore, NLRP10 is the only member of this family that lacks an LRR domain responsible for identification or detection of PAMPs/DAMPs in humans; thus suggesting the potential for a regulatory role of this protein in cellular signalling (Ng and Xavier 2011).
Earlier studies performed by our group showed increased expression of NLRP10 and NLRP12 in second-hand smoke-exposed C57Bl/6 mice (unpublished work). In the current study, we investigated the role of NLRP10 and NLRP12 in regulating inflammatory responses during CSE exposure in human alveolar type II pulmonary epithelial (A549) and human monocytic (THP-1) cells.
Experiments revealed increased production of inflammatory cytokines/chemokines (IL-1β, IL-18, CCL-2, and CXCL-8) following a 6h CSE challenge in both A549 and THP-1 cells. Increased production of IL-1β has been shown in various inflammatory diseases including COPD, rheumatoid arthritis, multiple sclerosis, inflammatory bowel disease and osteoarthritis(Ren and Torres 2009; Singh et al. 2010). Inflammasome mediated Caspase-1 activation is responsible for the production of IL-1β, IL-18 and other inflammatory mediators (Latz et al. 2013). Our results demonstrate increased activation of Caspase-1 in response to CSE-challenge in both A549 and THP-1 cells (Figure 1A-B). Furthermore, we found increased expression of NLRP10 and NLRP12 proteins in response to 6h CSE-exposure in A549 cells, as compared to controls using immunoblotting and immunofluorescence (Figure 2A-B). In fact, immunofluorescence results further demonstrated membrane recruitment of NLRP10 and NLRP12 proteins in CSE-challenged A549 cells (Figure 2B).
Membrane localization of cytosolic NLR proteins has been reported earlier to regulate inflammatory responses following exposure to various environmental triggers (Eitel et al. 2008; Kufer et al. 2008; Lecine et al. 2007). For instance, NOD2 has been shown to translocate to the membrane thereby promoting the recruitment of RIP2 in MDP (muramyl dipeptide)-treated monocytic cells (Eitel et al. 2008; Lecine et al. 2007). Similarly, NLRP10 has been identified to act as a scaffold for the assembly of the NOD1 signalling complex in a cellular model of Shigella infection (Lautz et al. 2012). To our knowledge, the current study is the first to confirm the membrane localization of NLRP10 and NLRP12 in response to CSE challenge. Moreover, membrane localization of NLRP10 and NLRP12 proteins raises the possibility of their interaction with membrane lipids or proteins. In this respect, cholesterol and sphingolipid-rich detergent-insoluble membrane microdomains, commonly known as lipid rafts, could act as an interaction platform (Shaw 2006). A plethora of information suggests the harbouring and translocation of various signalling molecules to these microdomains and their subsequent interaction with lipid raft proteins (Caveolin-1/2 and Flotillin-1/2) (Anderson et al. 1998; Bagam et al. 2017; Barak and Muchova 2013; Bavari et al. 2002; Lautz et al. 2012). In this study, a significant induction in the expression of lipid raft proteins Caveolin-1/2 and Flotillin-1 following 6hCSE challenge in A549 cells was observed (Figure 2A). In addition, lipid raft assembly (as observed by increased labelling of lipid raft marker, GM-1) was also found to be enhanced in CSE-challenged A549 cells as compared to controls (Figure 2B).
In silico modelling studies of protein-protein interactions provided strong evidence in support of a probable interaction between NLRP10 and NLRP12 and lipid raft proteins (caveolin-1/2 and flotillin-1/2) (Figure 3A-B and 5A). In silico predictions were validated by immunofluorescence studies. Confocal Imaging showed co-localization of NLRP10 or NLRP12 proteins with lipid raft marker (GM-1) (Figure 4A-B). In fact, we detected proximity between NLRP12 and lipid raft protein in the membrane of CSE-challenged A549 cells using the PLA (Figure 4C). Moreover, co-immunoprecipitation studies confirmed the physical interaction of NLRP12 with caveolin-1 in CSE-challenged A549 cells (Figure 4D). However, despite the evidence of a possible interaction between NLRP10 and NLRP12 obtained through in-silico studies (Figure 5A) and their proximity in CSE-challenged A549 membrane fractions (Figure 5C), we were unable to detect physical interaction between NLRP10 and NLRP12 in CSE-challenged A549 cells using co-immunoprecipitation studies (Figure 4D). Taken together these results suggest that though NLRP10 and NLRP12 may not interact with each other directly, their proximity and co-localization in lipid rafts might be critical for cigarette smoke induced inflammation. Also, the formation of a multi-molecular complex involving NLRP10, NLRP12, and possibly other functional partner(s), thereby directing CSE-mediated inflammatory responses, cannot be ruled out.
To further illustrate the roles of NLRP10 and NLRP12 in regulating inflammatory responses during CSE exposure, we employed a knockdown strategy. Knockdown of NLRP10 significantly abrogated CSE-induced IL-1β production by A549 cells however, the similar response by NLRP12 knockdown cells was of much lower magnitude [Figure 6A (ii) and 6B (ii)]. These findings provide evidence that NLRP10 has a critical role in regulating IL-1β-mediated inflammation in response to CSE-challenge in A549 cells.
Since lipid raft-mediated downstream signalling appears to be dependent on the structure and integrity of rafts, we studied the effect of lipid raft disruption (using filipin) and enrichment (using PUFA and DHA) in CSE-mediated inflammatory responses including IL-1β production. At the molecular level, PUFA alters the clustering of lipid rafts, leading to the formation of large rafts which modifies the lateral organization of signalling molecules thereby altering their function (Kim et al. 2008b; Turk and Chapkin 2013). DHA alters membrane fluidity, permeability, phase behaviour, fusion, and resident protein activity (Turk and Chapkin 2013). We observed a PUFA and DHA-mediated decline in Caspase-1 activity and cytokine/chemokine (IL-1β, CCL-2, and CXCL-8) production in CSE-challenged A549 cells at both transcriptional and translational level (Figure 8). Interestingly, immunoblotting of membrane fractions isolated from PUFA or DHA enriched CSE-challenged A549 cells showed reduced recruitment of NLRP10 and NLRP12 compared to CSE-challenged cells maintained in regular growth media (Supplemental File 1). Further enrichment of A549 cells with arachidic acid (saturated fatty acid) was unable to provide any rescue against CSE-induced inflammation, instead we observed exacerbated response in terms of IL-1β production following CSE-challenge (Supplementary Figure 2). Furthermore, enrichment with saturated fatty acid did not show any significant effect on the production of CCL-2 or CXCL-8. To validate our study model we conducted lipidomic analysis of the isolated raft fractions from PUFA/DHA enriched and control set of cells which confirmed increase in DHA, eicosenoic acid (monounsaturated fatty acid) and arachidonic acid (polyunsaturated fatty acid) in the fractions of enriched cells demonstrating successful incorporation of poly-unsaturated fatty acids in the membrane microdomains (Figure 7B). Further, we did not observe any change in the viability of cells cultured in PUFA/DHA enriched media compared to cells maintained in the recommended media (low endotoxin FBS). However, we did observe a significant decline in the viability of cells cultured with PUFA complexed with fatty acid free-BSA (No FBS), supporting the study model used for our findings (complete media with FBS) to determine the effect of PUFA and DHA (Figure 7A)(Schumann et al. 2011).
Filipin has been known to disrupt the lipid raft integrity by binding to and depleting cholesterol (Zaas et al. 2005). Our results show exacerbated response in terms of transcriptional expression of inflammatory mediators (IL-1β and CCL-2) in A549 cells pre-treated with filipin compared to CSE-challenged cells (Figure 9). This further emphasizes the role of lipid rafts in CSE-mediated inflammation. However, the effect of lipid raft disruption was found to be reversed in PUFA-enriched CSE-challenged cells. This is not surprising as previous reports suggest that PUFA perturbs the lipid composition of membrane rafts thereby altering their function (Fan et al. 2004; Ma et al. 2004). Raft cholesterol content was found to be lowered by 46% in the colonocytes of mice fed with n-3 PUFA-enriched diet as compared to n-6 PUFA-enriched diet (Ma et al. 2004). Similarly, the sphingomyelin and sphingolipid content in T-cells was found to be reduced by 30% and 45% respectively in mice fed withn-3 PUFA enriched diet (Fan et al. 2004; Ma et al. 2004). Along similar lines we observed significantly reduced membrane cholesterol in the PUFA enriched CSE-challenged cells compared to CSE-challenged cells maintained in recommended growth media (Supplementary Figure 2B). These findings suggest that reduced cholesterol levels and overall change in the microenvironment of lipid rafts in PUFA enriched cells, renders the rafts less susceptible to filipin mediated disruption and rescues them from CSE-induced inflammation (Figure 8). Furthermore, the alterations in raft components during PUFA enrichment possibly regulates the recruitment of signaling molecules like NLRP10 and NLRP12 as observed in isolated membrane fractions from our study group (Supplementary Figure 1). Our results therefore indicate a potential role of lipid rafts in NLRP10/NLRP12 mediating inflammation in CSE exposed A549 cells.
In conclusion, this is one of the first reports to demonstrate the membrane localization of NLRP10 and NLRP12 proteins in response to CSE-challenge. Our experiments provide evidence of (1) increased expression of lipid raft proteins, NLRP10 and NLRP12proteins, and (2) physical interaction between NLRP12 and caveolin-1 in CSE-challenged A549 cells. Using lipid raft enrichment and disruption, we were able to demonstrate the crucial role played by membrane microdomains in regulating CSE-mediated NLRP10/NLRP12-dependent inflammatory responses (Scheme 1). Future investigations will focus on identifying other interacting partners of NLRP10 and NLRP12, and modulation of these interactions affected by changes in lipid raft assembly. Our findings will advance our understanding about the molecular mechanisms associated with cigarette smoke induced inflammation. Results from these studies will be helpful for the design of improved intervention strategies specifically determining the strategies to alter the composition/assembly of lipid rafts to combat the onset and progression of inflammatory responses due to excessive smoking or smoke induced pulmonary complications.
Scheme 1. Schematic representation of molecular mechanisms during CSE-induced inflammation.
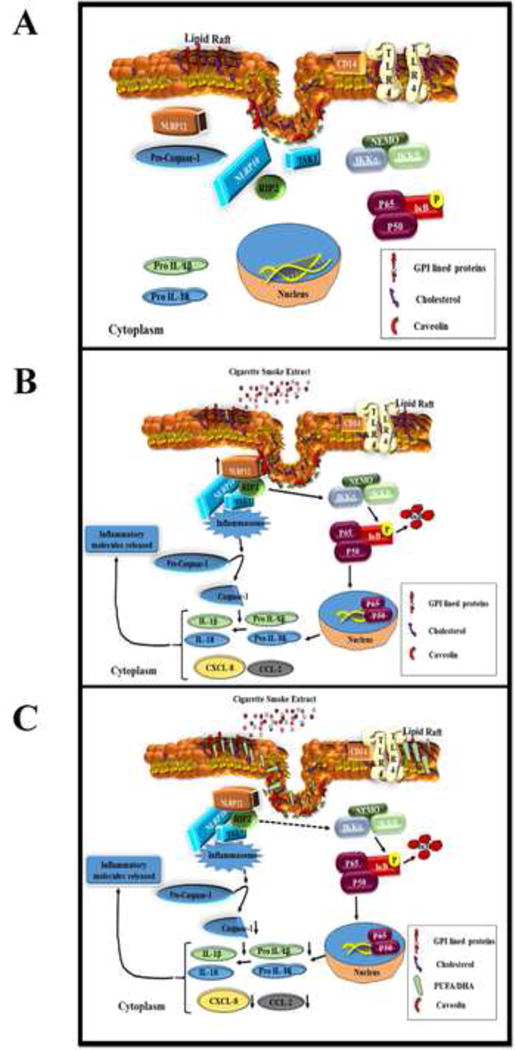
(A) In a resting stage, the NLR family proteins-NLRP10 and NLRP12; are localized in the cytosol. (B) Challenge with CSE induces NLRP10 and NLRP12 expression and membrane recruitment. Both NLRP10 and NLRP12 are co-localized in cholesterol enriched lipid raft domains. The proximity of these two NLR family members and their recruitment to membrane rafts appear to be essential for caspase-1 activation and production of IL-1β. The downstream events of NLR signaling also includes NF-κB activation following CSE-challenge.(C) Enrichment of A549 with PUFA alters the clustering and assembly of lipid raft domains and abrogates recruitment of NLRP10 and NLRP12 in lipid raft entities, caspase-1 activity and IL-1β production.
Supplementary Material
Acknowledgments
This work was supported by Young Clinical Scientist Award from the Flight Attendant Medical Research Institute (FAMRI-123253_YCSA_Faculty); NIH R15 (7 R15 ES023151 02); Southern University Foundation Grant (FY2017-017/FY2018-020); and LBRN Startup Grant (2P20GM103424-14 -Subaward No. 100011) to SB. Authors would like to acknowledge the VCU Lipidomics/Metabolomics Core, the NIH-NCI Cancer Center Support Grant P30 CA016059, as well as a shared resource grant (S10RR031535) from the National Institutes of Health to the VCU Massey Cancer Center. Authors would also like to sincerely thank Ms. Karen McDonough, AgCenter Cell Culture Facility, Louisiana State University, Baton Rouge, Louisiana for maintaining the cell culture for our experiments.
Footnotes
CONFLICT OF INTEREST
Authors declare no conflict of interest.
References
- Anderson HA, Chen Y, Norkin LC. MHC class I molecules are enriched in caveolae but do not enter with simian virus 40. J Gen Virol. 1998;79(Pt 6):1469–1477. doi: 10.1099/0022-1317-79-6-1469. [DOI] [PubMed] [Google Scholar]
- Bagam P, Singh DP, Inda ME, et al. Unraveling the role of membrane microdomains during microbial infections. Cell Biol Toxicol. 2017 doi: 10.1007/s10565-017-9386-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balis JU, Bumgarner SD, Paciga JE, et al. Synthesis of lung surfactant-associated glycoproteins by A549 cells: description of an in vitro model for human type II cell dysfunction. Exp Lung Res. 1984;6(3–4):197–213. doi: 10.3109/01902148409109248. [DOI] [PubMed] [Google Scholar]
- Barak I, Muchova K. The role of lipid domains in bacterial cell processes. Int J Mol Sci. 2013;14(2):4050–4065. doi: 10.3390/ijms14024050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnich N, Aguirre JE, Reinecker HC, et al. Membrane recruitment of NOD2 in intestinal epithelial cells is essential for nuclear factor-{kappa}B activation in muramyl dipeptide recognition. J Cell Biol. 2005;170(1):21–26. doi: 10.1083/jcb.200502153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavari S, Bosio CM, Wiegand E, et al. Lipid raft microdomains: a gateway for compartmentalized trafficking of Ebola and Marburg viruses. J Exp Med. 2002;195(5):593–602. doi: 10.1084/jem.20011500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodas M, Min T, Vij N. Critical role of CFTR-dependent lipid rafts in cigarette smoke-induced lung epithelial injury. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2011;300(6):L811–L820. doi: 10.1152/ajplung.00408.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodas M, Min T, Vij N. Lactosylceramide-accumulation in lipid-rafts mediate aberrant-autophagy, inflammation and apoptosis in cigarette smoke induced emphysema. Apoptosis. 2015;20(5):725–739. doi: 10.1007/s10495-015-1098-0. [DOI] [PubMed] [Google Scholar]
- Chapkin RS, McMurray DN, Davidson LA, et al. Bioactive dietary long-chain fatty acids: emerging mechanisms of action. British Journal of Nutrition. 2008;100(06):1152–1157. doi: 10.1017/S0007114508992576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbiere V, Dirix V, Norrenberg S, et al. Phenotypic characteristics of human type II alveolar epithelial cells suitable for antigen presentation to T lymphocytes. Respir Res. 2011;12:15. doi: 10.1186/1465-9921-12-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudez T, Borot F, Huang S, et al. CFTR in a lipid raft-TNFR1 complex modulates gap junctional intercellular communication and IL-8 secretion. Biochim Biophys Acta. 2008;1783(5):779–788. doi: 10.1016/j.bbamcr.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eitel J, Krull M, Hocke AC, et al. Beta-PIX and Rac1 GTPase mediate trafficking and negative regulation of NOD2. J Immunol. 2008;181(4):2664–2671. doi: 10.4049/jimmunol.181.4.2664. [DOI] [PubMed] [Google Scholar]
- Fan YY, Ly LH, Barhoumi R, et al. Dietary docosahexaenoic acid suppresses T cell protein kinase C theta lipid raft recruitment and IL-2 production. J Immunol. 2004;173(10):6151–6160. doi: 10.4049/jimmunol.173.10.6151. [DOI] [PubMed] [Google Scholar]
- Faner R, Sobradillo P, Noguera A, et al. The inflammasome pathway in stable COPD and acute exacerbations. ERJ Open Res. 2016;2(3) doi: 10.1183/23120541.00002-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KA, Oster CG, Mayer MM, et al. Characterization of the A549 cell line as a type II pulmonary epithelial cell model for drug metabolism. Exp Cell Res. 1998;243(2):359–366. doi: 10.1006/excr.1998.4172. [DOI] [PubMed] [Google Scholar]
- Fukata M, Vamadevan AS, Abreu MT. Toll-like receptors (TLRs) and Nod-like receptors (NLRs) in inflammatory disorders. Semin Immunol. 2009;21(4):242–253. doi: 10.1016/j.smim.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Hawdon NA, Aval PS, Barnes RJ, et al. Cellular responses of A549 alveolar epithelial cells to serially collected Pseudomonas aeruginosa from cystic fibrosis patients at different stages of pulmonary infection. FEMS Immunol Med Microbiol. 2010;59(2):207–220. doi: 10.1111/j.1574-695X.2010.00693.x. [DOI] [PubMed] [Google Scholar]
- Huang C, Wang JJ, Jing G, et al. Erp29 Attenuates Cigarette Smoke Extract-Induced Endoplasmic Reticulum Stress and Mitigates Tight Junction Damage in Retinal Pigment Epithelial Cells. Invest Ophthalmol Vis Sci. 2015;56(11):6196–6207. doi: 10.1167/iovs.15-16795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun-Ming Zhang JA. Cytokines, Inflammation and Pain. Int Anesthesiol Clin. 2007;45(2):27–37. doi: 10.1097/AIA.0b013e318034194e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Fan Y-Y, Barhoumi R, et al. n-3 polyunsaturated fatty acids suppress the localization and activation of signaling proteins at the immunological synapse in murine CD4+ T cells by affecting lipid raft formation. The Journal of Immunology. 2008a;181(9):6236–6243. doi: 10.4049/jimmunol.181.9.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Fan YY, Barhoumi R, et al. n-3 polyunsaturated fatty acids suppress the localization and activation of signaling proteins at the immunological synapse in murine CD4+ T cells by affecting lipid raft formation. J Immunol. 2008b;181(9):6236–6243. doi: 10.4049/jimmunol.181.9.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kispert S, Marentette J, McHowat J. Cigarette smoke induces cell motility via platelet-activating factor accumulation in breast cancer cells: a potential mechanism for metastatic disease. Physiol Rep. 2015;3(3) doi: 10.14814/phy2.12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufer TA, Kremmer E, Adam AC, et al. The pattern-recognition molecule Nod1 is localized at the plasma membrane at sites of bacterial interaction. Cell Microbiol. 2008;10(2):477–486. doi: 10.1111/j.1462-5822.2007.01062.x. [DOI] [PubMed] [Google Scholar]
- Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13(6):397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautz K, Damm A, Menning M, et al. NLRP10 enhances Shigella-induced proinflammatory responses. Cell Microbiol. 2012;14(10):1568–1583. doi: 10.1111/j.1462-5822.2012.01822.x. [DOI] [PubMed] [Google Scholar]
- Lecine P, Esmiol S, Metais JY, et al. The NOD2-RICK complex signals from the plasma membrane. J Biol Chem. 2007;282(20):15197–15207. doi: 10.1074/jbc.M606242200. [DOI] [PubMed] [Google Scholar]
- Lewis JB, Hirschi KM, Arroyo JA, et al. Plausible Roles for RAGE in Conditions Exacerbated by Direct and Indirect (Secondhand) Smoke Exposure. Int J Mol Sci. 2017;18(3) doi: 10.3390/ijms18030652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma DW, Seo J, Davidson LA, et al. n-3 PUFA alter caveolae lipid composition and resident protein localization in mouse colon. FASEB J. 2004;18(9):1040–1042. doi: 10.1096/fj.03-1430fje. [DOI] [PubMed] [Google Scholar]
- Mortaz E, Alipoor SD, Movassaghi M, et al. Water-pipe smoke condensate increases the internalization of Mycobacterium Bovis of type II alveolar epithelial cells (A549) BMC Pulm Med. 2017;17(1):68. doi: 10.1186/s12890-017-0413-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng A, Xavier RJ. Leucine-rich repeat (LRR) proteins: integrators of pattern recognition and signaling in immunity. Autophagy. 2011;7(9):1082–1084. doi: 10.4161/auto.7.9.16464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce BG, Wiehe K, Hwang H, et al. ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics. 2014;30(12):1771–1773. doi: 10.1093/bioinformatics/btu097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierson T, Learmonth-Pierson S, Pinto D, et al. Cigarette smoke extract induces differential expression levels of beta-defensin peptides in human alveolar epithelial cells. Tob Induc Dis. 2013;11(1):10. doi: 10.1186/1617-9625-11-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proell M, Riedl SJ, Fritz JH, et al. The Nod-like receptor (NLR) family: a tale of similarities and differences. PLoS One. 2008;3(4):e2119. doi: 10.1371/journal.pone.0002119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PyMOL The PyMOL Molecular Graphics System. Schrödinger, LLC. Version 1.8
- Qian YJ, Wang X, Gao YF, et al. Cigarette Smoke Modulates NOD1 Signal Pathway and Human beta Defensins Expression in Human Oral Mucosa. Cell Physiol Biochem. 2015;36(2):457–473. doi: 10.1159/000430112. [DOI] [PubMed] [Google Scholar]
- Ren K, Torres R. Role of interleukin-1beta during pain and inflammation. Brain Res Rev. 2009;60(1):57–64. doi: 10.1016/j.brainresrev.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann J, Leichtle A, Thiery J, et al. Fatty acid and peptide profiles in plasma membrane and membrane rafts of PUFA supplemented RAW264.7 macrophages. PLoS One. 2011;6(8):e24066. doi: 10.1371/journal.pone.0024066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh SR, Rockett BD, Salameh M, et al. Docosahexaenoic acid modifies the clustering and size of lipid rafts and the lateral organization and surface expression of MHC class I of EL4 cells. The Journal of nutrition. 2009;139(9):1632–1639. doi: 10.3945/jn.109.108720. [DOI] [PubMed] [Google Scholar]
- Shaw AS. Lipid rafts: now you see them, now you don’t. Nat Immunol. 2006;7(11):1139–1142. doi: 10.1038/ni1405. [DOI] [PubMed] [Google Scholar]
- Shaw MH, Reimer T, Kim Y-G, et al. NOD-like receptors (NLRs): bona fide intracellular microbial sensors. Current opinion in immunology. 2008;20(4):377–382. doi: 10.1016/j.coi.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PJ, Lamkanfi M, Kanneganti TD. NOD-like receptor (NLR) signaling beyond the inflammasome. Eur J Immunol. 2010;40(3):624–627. doi: 10.1002/eji.200940211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaykhiev R, Crystal RG. Innate immunity and chronic obstructive pulmonary disease: a mini-review. Gerontology. 2013;59(6):481–489. doi: 10.1159/000354173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Arora S, Khanna V. Association of severity of COPD with IgE and interleukin-1 beta. Monaldi Arch Chest Dis. 2010;73(2):86–87. doi: 10.4081/monaldi.2010.303. [DOI] [PubMed] [Google Scholar]
- Smith BT. Cell line A549: a model system for the study of alveolar type II cell function. Am Rev Respir Dis. 1977;115(2):285–293. doi: 10.1164/arrd.1977.115.2.285. [DOI] [PubMed] [Google Scholar]
- Stillwell W, Wassall SR. Docosahexaenoic acid: membrane properties of a unique fatty acid. Chemistry and physics of lipids. 2003;126(1):1–27. doi: 10.1016/s0009-3084(03)00101-4. [DOI] [PubMed] [Google Scholar]
- Su MY, Kuo CI, Chang CF, et al. Three-dimensional structure of human NLRP10/PYNOD pyrin domain reveals a homotypic interaction site distinct from its mouse homologue. PLoS One. 2013;8(7):e67843. doi: 10.1371/journal.pone.0067843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk HF, Chapkin RS. Membrane lipid raft organization is uniquely modified by n-3 polyunsaturated fatty acids. Prostaglandins Leukot Essent Fatty Acids. 2013;88(1):43–47. doi: 10.1016/j.plefa.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vladimer GI, Weng D, Paquette SW, et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity. 2012;37(1):96–107. doi: 10.1016/j.immuni.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Kondo N, Cano M, et al. Nrf2 signaling modulates cigarette smoke-induced complement activation in retinal pigmented epithelial cells. Free Radic Biol Med. 2014;70:155–166. doi: 10.1016/j.freeradbiomed.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Manji GA, Grenier JM, et al. PYPAF7, a novel PYRIN-containing Apaf1-like protein that regulates activation of NF-kappa B and caspase-1-dependent cytokine processing. J Biol Chem. 2002;277(33):29874–29880. doi: 10.1074/jbc.M203915200. [DOI] [PubMed] [Google Scholar]
- Wassall SR, Brzustowicz MR, Shaikh SR, et al. Order from disorder, corralling cholesterol with chaotic lipids: The role of polyunsaturated lipids in membrane raft formation. Chemistry and physics of lipids. 2004;132(1):79–88. doi: 10.1016/j.chemphyslip.2004.09.007. [DOI] [PubMed] [Google Scholar]
- WHO. Chronic obstructive pulmonary disease (COPD) 2016 http://www.who.int/mediacentre/factsheets/fs315/en/ Accessed Feb 7 2017.
- WHO. Fact Sheet: Top ten causes of death. 2017 http://www.who.int/mediacentre/factsheets/fs310/en/ Accessed April 5 2017.
- Wu S, Zhang Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 2007;35(10):3375–3382. doi: 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaas DW, Duncan M, Rae Wright J, et al. The role of lipid rafts in the pathogenesis of bacterial infections. Biochim Biophys Acta. 2005;1746(3):305–313. doi: 10.1016/j.bbamcr.2005.10.003. [DOI] [PubMed] [Google Scholar]
- Zuo L, Lucas K, Fortuna CA, et al. Molecular regulation of toll-like receptors in asthma and COPD. Frontiers in physiology. 2015;6:312. doi: 10.3389/fphys.2015.00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.