

Abstract
Non-steroidal anti-inflammatory drugs (NSAIDs) inhibit cyclooxygenase (COX) activity and are commonly used for pain relief and fever reduction. NSAIDs are used following childhood vaccinations and cancer immunotherapies; however, how NSAIDs influence the development of immunity following these therapies is unknown. We hypothesized that NSAIDs would modulate the development of an immune response to Listeria monocytogenes-based immunotherapy. Treatment of mice with the non-specific COX-inhibitor, indomethacin, impaired the generation of cell-mediated immunity. This phenotype was due to inhibition of the inducible COX-2 enzyme as treatment with the COX-2-selective inhibitor Celecoxib similarly inhibited the development of immunity. In contrast, loss of COX-1 activity improved immunity to L. monocytogenes. Impairments in immunity were independent of bacterial burden, dendritic cell co-stimulation, or innate immune cell infiltrate. Instead, we observed prostaglandin E2 (PGE2) production following L. monocytogenes is critical for the formation of an antigen-specific CD8+ T-cell response. Use of the alternative analgesic acetaminophen did not impair immunity. Taken together, our results suggest that COX-2 is necessary for optimal CD8+ T-cell responses to L. monocytogenes while COX-1 is detrimental. Use of pharmacotherapies that spare COX-2 activity and the production of PGE2 like acetaminophen will be critical for the generation of optimal anti-tumor responses using L. monocytogenes.
Introduction
Recent advances in understanding the immune system have led to new, powerful immunotherapy approaches against cancer including check-point blockade therapies, adoptive cellular immunotherapies, and pathogen-based immunotherapies (1). One promising approach uses pathogens, such as the Gram-positive intracellular bacteria Listeria monocytogenes, that naturally trigger robust cell-mediated immune responses to program tumor targeting antigen specific CD8+ T-cells (2). As is common with many childhood vaccines (3), immunotherapies, including L. monocytogenes immunotherapy, include NSAIDs as part of their clinical trial protocol to limit unwanted side effects of immune activation such fever and general malaise (4–8). Despite this inclusion as standard of care, how NSAIDs ultimately influence the efficacy of an adaptive immune response in the context L. monocytogenes stimulated immunity, or many other vaccinations and immunotherapies, remains largely unknown.
NSAIDs modulate cyclooxygenase (COX) production to limit the processing of arachidonic acid into prostaglandins and thromboxane (9), collectively known as eicosanoids. Eicosanoids have broad impacts on both homeostatic physiology and inflammatory responses and have been implicated in both anti-inflammatory and pro-inflammatory signaling effects depending on the cell type that produces and/or responds to the eicosanoid, the receptor that the eicosanoid signals through, and the other inflammatory mediators present (10, 11). The eicosanoid prostaglandin E2 (PGE2) can inhibit T-cell proliferation and cytotoxic function via signaling through EP2 and EP4 receptors (12). In the context of chronic infections such as LCMV this eicosanoid signaling has profound effects such that PGE2 inhibition can improve the clearance of chronic infection (13). Similarly, targeted inhibition of PGE2 or the EP2/EP4 receptors improves antigen presentation, CD8+ T-cell mediated immunity and ultimately survival following influenza A infection (14). In contrast, imbalance in eicosanoid production, specifically loss of PGE2, results in impairments in Mycobacterium tuberculosis mediated immunity, while administration of exogenous PGE2 improves acute survival (15). These results suggest that eicosanoids modulate immunity to pathogens and that they may modulate immunity in the context of L. monocytogenes immunotherapy.
Eicosanoids have previously been implicated in regulating acute infection by L. monocytogenes. Treatment of mice with the non-specific COX inhibitor indomethacin increased susceptibility to acute L. monocytogenes infection, potentially through inhibition of thromboxane A2 synthesis (16). PGE2 can additionally limit macrophage phagocytosis of L. monocytogenes (17) further suggesting that eicosanoids modulate L. monocytogenes pathogenesis. Despite these studies on the impact of eicosanoids on acute infection with pathogenic L. monocytogenes, little is known about how eicosanoids influence adaptive immune responses to L. monocytogenes. The use of L. monocytogenes as a platform for tumor immunotherapy and the explicit treatment of patients in these trials with NSAIDs demands an understanding how COX inhibition may influence L. monocytogenes-stimulated antigen specific cytotoxic T-cell responses (3, 7, 8). To address this question, we measured the activation of antigen specific T-cells and their function in protection from lethal challenge following L. monocytogenes immunization in the presence of various COX inhibitors and knock-out mouse models. Our data suggest that COX-2, and specifically PGE2, is critical for optimal L. monocytogenes-stimulated immune responses, while COX-1 inhibits generation of an antigen specific CD8+ T-cell mediated protective immune response.
Materials and Methods
Ethics statement
This work was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All protocols were reviewed and approved by the University of Wisconsin—Madison Institutional Animal Care and Use Committee.
Bacterial and mouse strains
All L. monocytogenes strains used in this study were in the 10403s background and were cultured in brain heart infusion (BHI) media. The immunizing strain Lm (referred to as attenuated Lm in the figure legends) was constructed in the ΔactA and ΔinlB background, as previously described (18) and expressed full length OVA and the B8R20-27 epitope. The B8R20-27 epitope and full length OVA were engineered as a single fusion protein behind the actA promoter and in-frame with the secretion signal of the amino terminal 300bp of the ActA gene (ActAN100) in the site-specific pPL2e vector as previously described (19).
6–8 week old C57Bl/6 male and female mice were obtained from NCI/Charles River NCI facility. Ptgs1−/− (COX-1−/−) mice (20) were obtained from Taconic and were back-crossed to a C57Bl/6 background for 6 generations and maintained as heterozygote breeding pairs for use in this study. Mice were age and sex matched. mPGES1−/− mice, encoded by Ptges, that lack microsomal prostaglandin E synthase, the enzyme critical for prostaglandin E2 (PGE2) production, have been previously described (21–23). Indomethacin (Sigma) was dosed at 2mg/kg intraperitoneally (16) at 2 hours before immunization and 24 hours after immunization. Celecoxib (Cayman Chemical) was milled into the standard mouse chow (Envigo) at 100mg/kg (13) and fed ad lib for 48 hours before and after immunization. Prostaglandin E2 (Cayman Chemical) was diluted in PBS, and administered to mice intraperitoneally 8 (0.125μg) and 12 (0.25μg) hours post immunization. Acetaminophen (Infant’s Tylenol) was diluted into water at 1.1mg/mL so that mice received approximately 200mg/kg per day as previously described for 48 hours before and after immunization (24). Water consumption was measured to ensure accurate dosing.
In vivo Infections
All mice were immunized intravenously with logarithmic phase ΔactA/ΔinlB L. monocytogenes (attenuated Lm) diluted in 200μL of phosphate buffered saline (PBS) at the doses indicated. For bacterial burden analysis, mice were sacrificed at 24 or 48 hours post infection. For challenge studies, mice immunized 30 days prior were challenged with 2LD50 (2×105 cfu) of virulent L. monocytogenes expressing full length OVA and the B8R20-27 and burdens were analyzed 68–72 hours post infection. Organs were homogenized in 0.1% Nonidet P-40 in PBS and plated on LB plates containing streptomycin to quantify bacterial loads.
T-Cell and Immune Cell Analysis
Spleens taken at the indicated time points post immunization were made into a single cell suspension, and red blood cells were lysed in ACK buffer. Total splenocytes were counted with a Z1 Coulter Counter and a total of 1.8×106 splenocytes were stained for analysis. For primary T-cell responses measured by intracellular cytokine staining, splenocytes were stimulated ex vivo for 5 hours with B8R20-27 (TSYKFESV) or OVA257-264 (SIINFEKL) in the presence of brefeldin A (eBioscience). Stimulated cells were surface stained with anti-CD3 (clone 145-2C11) and anti-CD8α (clone 53-6.7) antibodies, fixed and permeabilized using IC fixation and permeabilization buffer (Ebioscience), then stained intracellularly for IFNγ (clone XMG1.2), TNFα (clone MP6-XT22), and IL-2 (clone JES6-5H4). For primary and memory tetramer analysis, cells were stained with B8R-tetramer (NIH Tetramer Facility, Atlanta, Georgia, contract HHSN272201300006C), followed by surface staining with anti-CD3, anti-CD8α, and anti-CD44 (clone IM7) antibodies. Innate immune cells were stained with markers anti-CD3, anti-CD8α, anti-CD11c (clone N418), anti-CD86 (clone GL1), anti-CD11b (clone M1/70), anti-CD40 (clone 1C10), anti-Ly6C (clone HK1.4), anti-Ly6G (clone 1A8-Ly6G), and anti-NK1.1 (clone PK136). All fluorophore-conjugated antibodies were obtained from Ebioscience. Samples were acquired using a LSRII flow cytometer (BD Biosciences) with FACS DIVA software (BD Biosciences). Data were analyzed using FlowJo software (Treestar, Ashland, OR).
Eicosanoid measurement
Spleens from infected mice were flash frozen and stored at −80° until solid phase extraction. Spleens were homogenized in 1mL cold methanol and centrifuged to remove debris. Samples were processed as previously described (25). Briefly, samples were rapidly acidified with water pH 3.5 and loaded into conditioned solid phase C18 cartridges. Columns were washed with neutral pH water, followed by hexanes and then eluted with methyl acetate followed by methanol. Samples were concentrated using a nitrogen manifold and suspended in a final solution of 55:45 MeOH:H2O. Samples were analyzed on a HPLC coupled to a mass spectrometer (Thermo Scientific Q Exactive) using a C18 Aquity BEH column (100 mm × 2.1mm × 1.7 μm) operated in negative ionization mode. Lipid mediators were eluted with a mobile phase of 55:45:0.1 methanol:H2O:acetic acid that was shifted to a 98:2:0.1 over the course of 20 minutes. Scanning occurred from 3.5–20 minutes and included m/z’s between 100 and 800. Eicosanoid standards (Cayman Chemical) were run to ensure peak specificity. Data were analyzed via MAVEN (26, 27).
Cytokine analysis
Blood was obtained by cardiac puncture or retro-orbital bleed and serum was analyzed by cytokine bead array with the mouse inflammation kit (BD Biosciences, San Jose, CA) per the manufacturers’ instructions. Samples were acquired using a LSRII flow cytometer with FACS DIVA software.
Statistical analysis
Statistical analysis using was performed using GraphPad Prism Software (La Jolla, CA) and analyzed with a one-way ANOVA with Bonferroni’s correction or Mann-Whitney U-test unless otherwise indicated.
Results
Broad cyclooxygenase inhibition impairs L. monocytogenes stimulated immunity
Nonsteroidal anti-inflammatory drugs (NSAIDs) are routinely administered following vaccination to modulate eicosanoid production to prevent the unwanted effects of immune activation such as fever and muscle soreness (3, 6, 28). Attenuated L. monocytogenes is being developed as a cancer immunotherapeutic platform to stimulate anti-tumor responses; yet how eicosanoid inhibition impacts the effectiveness of L. monocytogenes immunotherapy is unknown. NSAIDs are routinely administered to patients receiving L. monocytogenes immunotherapy, therefore it is critical to understand how eicosanoids influence L. monocytogenes triggered immunity (7, 8). To address this question, we assessed activation of cell-mediated immunity following immunization with the attenuated ΔactA/ΔinlB strain of L. monocytogenes (attenuated Lm) in the presence or absence of the non-selective NSAID indomethacin. The ΔactA/ΔinlB strain of L. monocytogenes, currently in clinical trials as an immunotherapeutic platform, contains two genomic deletions that limit the spread of L. monocytogenes from cell to cell (ΔactA) and limit invasion of hepatocytes (ΔinlB), thus leaving L. monocytogenes to invade primarily antigen-presenting immune cells (18). CD8+ T-cells comprise the majority of the adaptive immune response generated by L. monocytogenes and are the cell type responsible for controlling L. monocytogenes infection, providing long-lasting immunity, and inducing an anti-tumor response (29). As eicosanoids have been shown to directly impact CD8+ T-cell functions (12), we hypothesized that cyclooxygenase inhibition through NSAID use would influence the generation of immunity to L. monocytogenes.
We dosed mice with a non-specific cyclooxygenase inhibitor, indomethacin, 2 hours before immunization and 24 hours post immunization with 1×107 attenuated Lm. This dosing strategy inhibits cyclooxygenase activity for at least 48 hours (16), the critical time of T-cell priming following L. monocytogenes infection and mimics the real-world application of cyclooxygenase inhibition following pathogen based immunotherapies (8). Seven days post immunization there were significant decreases in both antigen specific single positive IFNγ-producing CD8+ T-cells (Fig. 1A) as well as impaired multifunctional antigen-specific T-cell responses (Fig. 1B). These multifunctional cells that produce IFNγ, TNFα, and IL-2 simultaneously are the cell type capable of forming memory cells (30–32), suggesting that administration of NSAIDs may cause defects in long term protective immunity. To test this hypothesis, we immunized mice with a low-dose of attenuated Lm (1×103) in the presence or absence of indomethacin, and 30 days later challenged mice with a lethal dose of virulent L. monocytogenes. Following challenge, mice immunized in the presence of indomethacin had significantly impaired protective immune responses as evidenced by elevated bacterial burdens in their spleens (Fig. 1C) and livers (Fig. 1D). Taken together, these data suggest that non-specific cyclooxygenase inhibition following administration of NSAIDs results in impairments in cell-mediated immune responses to L. monocytogenes based immunotherapy.
Figure 1. Indomethacin impairs cell-mediated immunity to L. monocytogenes.

C57Bl/6 mice were immunized with 1×107 cfu attenuated Lm in the presence of 2mg/kg indomethacin i.p. or vehicle control. Splenocytes were examined for B8R-specific antigen responses 7 days post immunization. Cells were gated for CD3+ and CD8+ followed by IFNγ (A) or IFNγ, TNFα, and IL-2 (B). Mice were immunized with 1×103 cfu attenuated Lm in the presence of 2mg/kg indomethacin or vehicle control and challenged 30 days later with a lethal dose of virulent L. monocytogenes. Bacterial burdens in the spleens (C) and livers (D) were enumerated 68–72 hours post challenge. Data are representative of at least 3 independent experiments of 5 mice per group. Significance was determined by a one-way ANOVA with Bonferroni’s correction (A,B), or a Mann-Whitney U-test (C,D). *p<0.05, **p<0.01, ****p<0.0001
Cyclooxygenase-1 is detrimental while cyclooxygenase-2 is critical for immunity to L. monocytogenes
Cyclooxygenases exist in two forms throughout the body. COX-1 is a constitutive enzyme largely responsible for maintenance of physiological functions throughout the body including maintenance of the stomach lining and proper blood clotting functions (9). COX-2, in contrast, is an inducible enzyme largely expressed by immune cells that is largely responsible for the production of fever and malaise (33). Though COX-2 is thought to be the critical mediator for inflammatory signals, COX-1 has recently been implicated in inflammatory responses (34), suggesting that either COX enzyme may influence immunity.
Treatment with the non-selective COX inhibitor indomethacin (35) inhibited L. monocytogenes stimulated immunity. To understand whether COX-1 or COX-2 differentially affects the generation of immune responses to L. monocytogenes we used both genetic and pharmacologic approaches to isolate the role of each cyclooxygenase. COX-1 selective inhibitors exist against purified enzymes (36), however in vivo these inhibitors are non-selective (37), therefore we utilized COX-1−/− mice (20) to isolate the role of COX-1. In contrast to treatment with indomethacin, immunization of COX-1−/− mice resulted in improved antigen specific CD8+ T-cell responses 7 days post immunization, both in the effector subset (Fig. 2A) as well as the multifunctional CD8+ T-cell subset (Fig. 2B). Similar effects were seen with the use of a B8R-specific tetramer (Fig. S1A) suggesting an improved T-cell priming and overall CD8+ T-cell effector response as opposed to just an increased functional response. Additionally, similar increases in T-cell activation were observed in response to the SIINFEKL peptide of ovalbumin, suggesting that this increased response is not antigen-specific (Fig. S1B, C). Consistent with increased T-cell priming in the absence of COX-1, COX-1−/− immunized mice were more protected from a secondary lethal challenge of virulent L. monocytogenes, harboring lower bacterial burdens in their spleens (Fig. 2C) while burdens in the liver were similar to COX-1 sufficient mice (Fig. 2D). These results are in direct contrast to the inhibitory effect of indomethacin and suggest that COX-1 activity is inhibitory to the development of L. monocytogenes stimulated immunity.
Figure 2. COX-2 is critical while COX-1 is detrimental for the development of immunity to L. monocytogenes.

The indicated strains of mice were immunized with 1×107 cfu attenuated Lm in the presence or absence of a celecoxib-laden chow. Splenocytes were examined for B8R-specific antigen responses 7 days post immunization. Cells were gated for CD3+ and CD8+ followed by IFNγ (A) or IFNγ, TNFα, and IL-2 (B). Mice were immunized with 1×103 cfu attenuated Lm in the presence or absence of celecoxib-laden chow and challenged 30 days later with a lethal dose of virulent L. monocytogenes. Bacterial burdens in the spleens (C) and livers (D) were enumerated 68–72 hours post challenge. Data are representative of at least 3 independent experiments of 3–5 mice per group. Significance was determined by a one-way ANOVA with Bonferroni’s correction (A,B), or a Mann-Whitney U-test (C,D). *p<0.05, **p<0.01
Given our unexpected observations that COX-1 activity is detrimental to immunity generated by L. monocytogenes, we hypothesized that inhibition of COX-2 was the basis of the inhibitory effects of indomethacin. To assess the role of COX-2 we used a COX-2 specific inhibitor, celecoxib, that has previously been shown to specifically impair COX-2 at the dose used (13). Mice given celecoxib had impaired antigen-specific effector and multifunctional CD8+ T-cell responses (Fig. 2A, B, black diamonds), similar to the impairments seen in mice dosed with indomethacin. Mice treated with celecoxib also had decreased B8R-specific tetramer responses (Fig. S1A) and OVA-specific responses (Fig. S1B, C). Consistent with decreased CD8+ T-cell activation, inhibition of COX-2 also impaired long-term protective immunity, as celecoxib-treated animals had increased burdens following lethal challenge (Fig. 2C, D, black diamonds) suggesting that inhibition of COX-2 impairs immune responses to L. monocytogenes.
Our findings suggest that COX-1 is detrimental to the immune response generated to L. monocytogenes while COX-2 is critical to forming an effective immune response. Most NSAIDs, like indomethacin, are non-selective (35); therefore, we asked which effect would be dominant by treating COX-1−/− mice with the COX-2 specific inhibitor. Similar to what we observed with the non-selective COX inhibitor, indomethacin, COX-1−/− mice treated with celecoxib had impaired effector and multifunctional antigen-specific responses (Fig. 2A, B, gray diamonds), B8R-tetramer responses (Fig. S1A), and OVA-specific responses (Fig. S1B, C), though not quite to the level of COX-2 inhibition alone. Consistent with the poor effector and multifunctional T-cell responses following immunization, we also observed impairments in clearance of the secondary challenge, (Fig. 2C, D, gray diamonds). Taken together, our results suggest that COX-1 activity negatively impacts the development of immunity to L. monocytogenes while COX-2 activity is critical. Further, non-specific NSAIDs administered following immunization with L. monocytogenes based immunotherapeutics likely inhibit optimal T-cell responses, suggesting that the use of COX-1 specific inhibitors or non-NSAID analgesics may improve the immune response to L. monocytogenes in clinical trials.
Modulation of cyclooxygenases does not influence the innate immune response
Consistent with the way that NSAIDs are traditionally used following vaccination, our experimental approach examines the effect of cyclooxygenases during the first 48 hours post immunization with L. monocytogenes, the critical period when the majority of T-cell priming occurs (38). We hypothesized that altering the eicosanoid milieu at this critical time point could influence the three signals needed for T-cell priming, namely antigen presentation, co-stimulation, or inflammation (39), particularly as eicosanoids can increase dendritic cell (DC) maturation states (40). We did not observe any differences in viable bacterial burdens in the spleens or livers of mice following cyclooxygenase modulation throughout the first 48 hours post immunization (Fig. S1D–G). We also examined the total number of DCs, both conventional CD11c+ cells and the critical cross-presenting CD8α+ CD11c+ cells, and their maturation state with both CD86 and CD40 expression. We observed slight differences in the number of DCs or their co-stimulatory expression, but these differences did not correlate with either T-cell responses or protective immunity (Fig. S1H–K) suggesting that modulation of cyclooxygenases was not significantly altering either of the first two steps of T-cell priming.
Eicosanoids are known to influence chemoattractant signals as well as the production of cytokines from innate immune cells which can ultimately modulate T-cell responses (40–44). Thus, we analyzed the innate immune response consisting of both cellular infiltrate and cytokine production at 24 and 48 hours post infection. We did not observe any changes in immune cell recruitment, including monocytes, inflammatory monocytes, neutrophils or NK cells, into the spleen following COX modulation (Fig. S2A–D). We next assessed a variety of pro-inflammatory cytokines in the serum following various COX manipulations and saw no significant differences in levels of MCP-1, IL-12p70, IL-6, or TNFα (Fig. S2E–H). We observed a significant elevation in IFNγ at 24 post infection when either COX-1 or COX-2 were inhibited, which persisted only in the COX-2 inhibition group at 48 hours post infection (Fig. S2I). We also observed significantly lower levels of the anti-inflammatory cytokine IL-10 when COX-2 was inhibited (Fig. S2J). These results suggest that, similar to previous reports, COX-2 promotes an immunoregulatory environment.
Prostaglandin E2 is upregulated following L. monocytogenes immunization
To determine what eicosanoids are essential for optimal L. monocytogenes triggered immunity we first assessed what eicosanoids are present. Many eicosanoids act locally, so we assessed the eicosanoid milieu in the spleen using the highly specific and sensitive method of LC-MS over the first 48h post-immunization (25). We examined PGE2, an eicosanoid with pleotropic effects on the immune system, PGD2, an eicosanoid with largely immunosuppressive effects, thromboxane B2 (TXB2, the stable break-down product of thromboxane A2), and leukotriene B4 (LTB4), a major by-product lipoxygenase activity, the other arm of arachidonic acid breakdown. We observed a trend for increased PGE2 by 8 hours post immunization that became significant by 12 hour post immunization resulting in approximately a 6–8 fold increase in PGE2 levels over uninfected mice (Fig. 3A). PGE2 quickly declined to baseline levels by 24 hours post immunization and remained there throughout 48 hours post immunization. Additionally, we observed significant increases in PGD2 at 8 and 12 hours post immunization of 2–3 fold over uninfected mice, though this was not consistent in every animal (Fig. 3B). Throughout the first 48 hours post immunization we did not observe any changes in TXB2 (Fig. 3C), or LTB4 (Fig. 3D) suggesting that prostaglandins are specifically induced downstream of cyclooxygenase function following L. monocytogenes immunization. Taken together, these results suggest that L. monocytogenes immunization transiently, but specifically increases the levels of PGE2 and PGD2, with a more pronounced increase in PGE2.
Figure 3. PGE2 is elevated following L. monocytogenes infection.

C57Bl/6 mice were immunized with 1×107 cfu attenuated Lm. At the indicated time point spleens were removed for eicosanoid extraction. Following extraction, samples were analyzed by LC-MS. Data are the combination of 2 independent experiments of 3 mice per group. Significance was determined by a one-way ANOVA with Bonferroni’s correction. *p<0.05, *p<0.01
Prostaglandin E2 is necessary and sufficient for generating an optimal immune response to L. monocytogenes
Prostaglandin E2 is one of the major prostanoids produced downstream of COX-2 stimulation (45). Given that we found that COX-2 is critical for immunity to L. monocytogenes, and that PGE2 is induced following L. monocytogenes immunization, we hypothesized that PGE2 is critical for generation of an immune response to L. monocytogenes. To test this hypothesis we immunized mice deficient in microsomal prostaglandin synthase E1 (mPGES1−/−) that lack the major enzyme responsible for PGE2 synthesis in the mouse (21). Seven days post immunization we observed impaired antigen-specific IFNγ production (Fig. 4A) and a significant impairment for multifunctional CD8+ T-cell responses (Fig. 4B), similar to what we observed following treatment with the COX-2 specific inhibitor celecoxib. Similarly, mPGES1−/− mice had a trend for impaired B8R-specific tetramer responses (Fig. S3A), consistent with a decreased effector CD8+ T-cell response, and decreased OVA-specific responses indicating an antigen non-specific response (Fig. S3B, C). PGE2-dependency is conserved independent of immunizing dose as mPGES1−/− mice immunized with attenuated Lm at a low-dose (1×103 cfu) similarly had impaired IFNγ B8R-specific response (Fig. S3E) and multifunctional B8R-specific CD8+ T-cell response (Fig. S3F). mPGES1−/− mice immunized with a low dose of attenuated Lm also had impaired B8R-specific tetramer (Fig. S3D) and OVA-specific responses (Fig. S3G, H), suggesting that the observed effects occur independent of immunizing dose and antigen. Consistent with these defects in primary T-cell activation, mPGES1−/− mice were unable to clear a secondary challenge of virulent L. monocytogenes and harbored increased bacterial burdens in their spleens and livers when compared to wild-type mice (Fig. 4C, D). mPGES1−/− mice were not innately more sensitive to L. monocytogenes infection as naïve mPGES1−/− harbored similar burdens to mPGES1+/+ mice in both their spleens (Fig. 4C) and livers (Fig. 4D). Finally, deficits in effector T-cell function could result in impaired memory responses and ultimately contribute to loss of protective immunity. To assess memory responses, we examined B8R-specific CD44+ CD8+ T-cells present 30 days post immunization with a high dose (1×107 cfu) of attenuated Lm. mPGES1−/− mice strongly trended towards having impaired B8R-specific memory responses at thirty days post immunization when compared to wild-type mice (Fig. S3I), suggesting that deficits in effector T-cell function are maintained throughout the memory period. Consistent with the elevated inflammatory cytokines and diminished anti-inflammatory cytokines observed in COX-2 inhibited mice, IL6 and TNFα were elevated in mPGES1−/− mice while IL-10 was diminished (Fig. S4C, D, F). These data suggest a role for PGE2 in preventing hyperinflammation to promote T-cell priming following L. monocytogenes immunization, consistent with previous reports that PGE2 promotes an immunosuppressive environment (40). Taken together, these results suggest that PGE2 is critical for generation of cell-mediated immunity to L. monocytogenes.
Figure 4. Prostaglandin E2 is critical for immunity to L. monocytogenes.

The indicated strain of mice was immunized with 1×107 cfu attenuated Lm, and splenocytes were examined for B8R-specific antigen responses 7 days post immunization. Cells were gated for CD3+ and CD8+ followed by IFNγ (A) or IFNγ, TNFα, and IL-2 (B). The indicated strain of mice was immunized with 1×103 cfu attenuated Lm, challenged 30 days later with a lethal dose of virulent L. monocytogenes. Bacterial burdens in the spleens (C) and livers (D) were enumerated 68–72 hours post challenge. Wild-type or celecoxib-treated mice were immunized with 1×107 cfu attenuated Lm and subsequently dosed with 0.125μg PGE2 8 hours post immunization, followed by 0.25μg PGE2 at 12 hours post immunization. Splenocytes were assessed for B8R-specific T cell responses seven days post immunization. CD8+ T-cells were assessed for IFNγ (E) or IFNγ, TNFα, and IL-2 (F). Data are representative of at least 3 independent experiments of 4–5 mice per group. Significance was determined by a one-way ANOVA with Bonferroni’s correction (A,B), or a Mann-Whitney U-test (C,D). *p<0.05, **p<0.01
Given that PGE2 is critical for generating immunity to L. monocytogenes and PGE2 is one of major eicosanoid products produced downstream of COX-2 (45), we next asked whether PGE2 is sufficient for rescuing immunity during COX-2 inhibition. Celecoxib-treated mice were immunized with 1×107 cfu of attenuated Lm and subsequently treated with 0.125μg PGE2 at 8 hours post immunization followed by an additional 0.25μg PGE2 at 12 hours post immunization, dosing that was based on the amount of PGE2 present following immunization with attenuated Lm in a wild-type mouse (Fig. 3A). Treatment with PGE2 led to a significant rescue to wild-type levels of both B8R-specific IFNγ production as well as multifunctional T-cell responses at seven days post immunization (Fig. 4E, F). Consistent with a rescue of T-cell responses in celecoxib-treated mice, tetramer staining revealed a trend towards restoration of the number of B8R-specific CD8+ T-cells when PGE2 was supplemented following celecoxib treatment (Fig. S4G) and treatment with PGE2 also significantly improved OVA-specific responses, suggesting that the observed results were independent of antigen (Fig. S4H, I). These results suggest that PGE2 downstream of COX-2 is both necessary and sufficient for generating cell-mediated immunity to L. monocytogenes.
The non-NSAID drug acetaminophen prevents impairments in immunity
Commercially available NSAIDs are non-selective in vivo (35) and thus likely limit the production of the critical molecule, PGE2. We therefore asked whether a non-NSAID analgesic capable of limiting fever and malaise, such as acetaminophen, would be a better alternative to NSAIDs during L. monocytogenes immunotherapy. We administered acetaminophen orally in the drinking water at a concentration (200mg/kg) that is routinely used to provide pain relief for mice (24, 46) and found that it did not impair the generation of antigen-specific effector (Fig. 5A) or multifunctional (Fig. 5B) CD8+ T-cell responses following L. monocytogenes immunization. Consistent with the lack of effect on the primary CD8+ T-cell response, immunized acetaminophen treated mice had bacterial burdens similar to untreated immunized mice (Fig. 5C, D), further suggesting that acetaminophen does not negatively impact the cell mediated immune response to L. monocytogenes. Taken together, our results suggest that the use of non-NSAIDs such as acetaminophen may be a suitable alternative to mitigate the unwanted side effect of L. monocytogenes immunization while promoting the immunogenicity of this immunotherapy.
Figure 5. Acetaminophen administration maintains CD8+ T-cell generation following L. monocytogenes immunization.
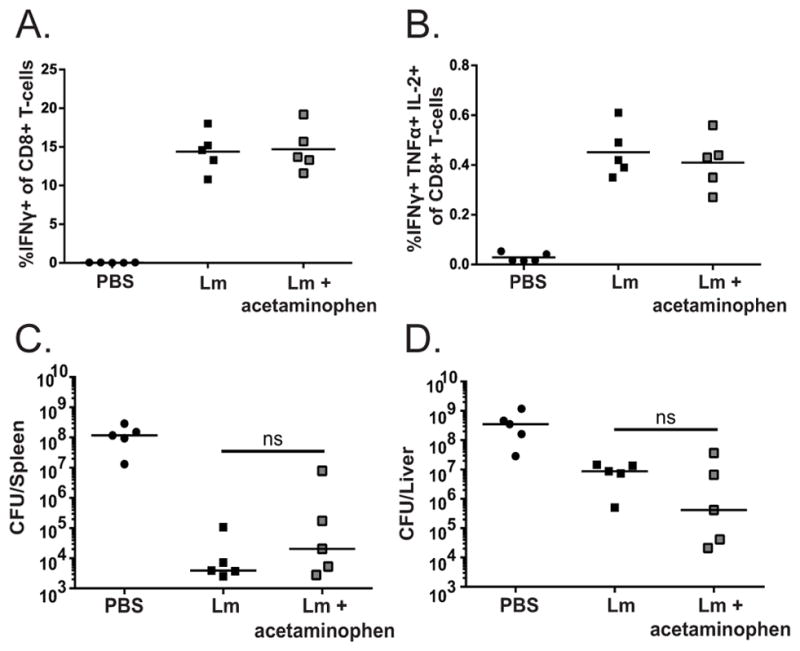
C57Bl/6 mice were treated with approximately 200mg/kg acetaminophen diluted in the drinking water. Mice were immunized with 1×107 cfu attenuated Lm and splenocytes were examined for B8R-specific antigen responses 7 days post immunization. Cells were gated for CD3+ and CD8+ followed by IFNγ (A) or IFNγ, TNFα, and IL-2 (B). Mice were immunized with 1×103 cfu attenuated Lm, challenged 30 days later with a lethal dose of virulent L. monocytogenes and bacterial burdens in the spleens (C) and livers (D) were enumerated 68–72 hours post challenge. Data are representative of at least 3 independent experiments of 4–5 mice per group (A,B). Data are representative of at least 2 independent experiments of 5 mice per group (C,D). Significance was determined by a one-way ANOVA with Bonferroni’s correction (A,B), or a Mann-Whitney U-test (C,D).
Discussion
NSAIDs are routinely given with childhood vaccinations, as well as cancer immunotherapies (3, 8), however, how they influence the generation of cell-mediated immunity remains unknown. Here we show that eicosanoids, particularly prostaglandin E2 downstream of COX-2, play a crucial role in the immune response generated to the intracellular pathogen and immunotherapeutic platform, L. monocytogenes. Surprisingly, we observed that the two forms of the cyclooxygenase enzyme that convert arachidonic acid into prostaglandins and thromboxane have inverse effects on immune response to L. monocytogenes: normal levels of COX-1 activity associated with L. monocytogenes immunization are detrimental to the development of cell-mediated immunity while L. monocytogenes induction of COX-2 activity is critical for generating robust antigen-specific CD8+ T-cell dependent immunity to L. monocytogenes. Our results suggest that it would be beneficial to use analgesics that spare COX-2 such as acetaminophen for purposes of L. monocytogenes immunotherapy, and that more studies should be done to understand the effects of NSAIDs in the context of other pathogen-based vaccines and immunotherapies.
COX-1 is a constitutive enzyme with homeostatic functions, but whose role in inflammation and immunity has often been overlooked in favor of its physiologic functions. We and others have begun to elucidate its critical role in modulating immune responses (34). In our model, we see that COX-1 is detrimental and impairs development of CD8+ T-cell mediated immunity. Other groups have shown detrimental effects of COX-1 following inflammasome activation (47) and some forms of arthritis (48). What downstream product mediating these detrimental effects in our system is unclear. COX-1 has been shown to preferentially, though not exclusively, couple with the enzymes responsible for thromboxane A2 synthesis and prostaglandin D2 (49, 50). Thromboxane A2 (TXA2) has potent vasoconstrictive properties that can negatively modulate DC-T-cell interactions (51). TXA2 produced from neutrophils can also limit T-cell migration into the lymph nodes and ultimately a recall response suggesting that the overall role of TXA2 is immunosuppressive (43). We were unable to observe any changes in TXB2 synthesis (the stable breakdown product of TXA2) over the course of L. monocytogenes infection; however, it is possible that examining TXA2 production from a certain cell type, as opposed to the entire spleen may elucidate differences. Additionally, use of TXA2 specific inhibitors (43) or thromboxane receptor deficient mice (52) may illuminate roles for TXA2 downstream of COX-1 following L. monocytogenes immunization. Though we were unable to observe changes in TXB2, we did observe increases in PGD2 expression following L. monocytogenes immunization to 2–3 fold levels over mock-infected controls. PGD2 has largely been implicated in allergic responses as its production largely comes from mast cells and can result in the hallmark symptoms of an asthma exacerbation including bronchoconstriction and airway eosinophil accumulation (53, 54). PGD2 can also be produced by dendritic cells and Th2 cells where it can limit antigen presentation and DC migration (55) to decrease T-cell activation. Whether this minimally transient increase in PGD2 is dependent on COX-1 and can explain our observed increases in immunity in COX-1−/− mice remains to be determined.
In contrast to COX-1, COX-2 preferentially couples with the enzymes responsible for prostacyclin (PGI) and PGE2 expression (45). Consistent with our observations of impaired immunity following COX-2 inhibition, we similarly observed impairments in immunity when PGE2 was absent suggesting that our phenotypes due to COX-2 inhibition are dependent on PGE2. PGE2 has been implicated in many immunosuppressive, as well as inflammatory effects, affecting both the innate and adaptive immune responses. PGE2 can promote regulatory T-cell formation (56), modulate T-cell cytokine production towards a Th2 profile (57) and promote immunosuppressive cytokine production from DCs (40). In contrast, PGE2 when found in an already inflammatory environment can enhance DC co-stimulatory expression, migration, and lymph node homing allowing for more efficient T-cell priming (58, 59). The pleiotropic effects of PGE2 are in part determined by which of the 4 receptors through which it signals. EP2 and EP4 signal through the G-protein coupled receptor Gs to result in activation of adenylate cyclase (60, 61), while EP3 signals through Gi to inhibit adenylate cyclase (62), and EP1 works through calcium release (63). Each receptor has different affinities for PGE2 leading to another level of regulation: EP3 and EP4 are high affinity receptors that are rapidly desensitized following ligand binding, while EP1 and EP2 are low-affinity receptors that respond to prolonged PGE2 signaling (64). We observe only a transient increase in PGE2 levels throughout the first 48 hours post immunization, suggesting that a high-affinity receptor may be responsible for our effects. The EP4 receptor is abundantly expressed in the spleen and can promote anti-inflammatory cytokine responses (65). Consistent with this, we observe increased pro-inflammatory cytokine production and decreased IL-10 production when we inhibit COX-2 or PGE2. Recent work with L. monocytogenes suggests that, although inflammation is critical for generation of effective CD8+ T-cell immunity (66), excessive amounts of inflammation or increased sensitivity to inflammatory mediators such as TNFα, IFNγ, or type I IFNs actually impairs CD8+ T-cell immunity (67, 68). We suggest that the role of COX-2 and PGE2 in our system may be immunoregulatory to limit excessive innate immune responses and inflammation that have previously been shown to be detrimental in the context of L. monocytogenes immunization (67–69).
In addition to promoting anti-inflammatory cytokines, PGE2 can limit type I IFN production during infections such as influenza and M. tuberculosis (14, 15). Whether PGE2 is critical or detrimental in these infections ultimately is determined by the role for type I IFN: in influenza infection, type I IFNs are critical for controlling viral replication and mounting anti-viral responses; addition of PGE2 is detrimental in this system as type I IFN levels are decreased. In contrast, in an M. tuberculosis infection, type I IFN promotes bacterial pathogenesis (70) and limits immunity (15), and PGE2 is critical for immunity by limiting the detrimental effects of type I IFN. Similar to M. tuberculosis, type I IFN production is detrimental to the host both acutely (71, 72) and for generation of an adaptive immune response (68) following L. monocytogenes immunization, suggesting that the critical role for PGE2 in generating cell-mediated immune response may be through control of type I IFN. Whether type I IFNs are altered following COX modulation in a PGE2 dependent manner and how they ultimately influence the immune response will be critical to address in the future.
Interestingly, we observe tissue specific effects for each COX-1 and COX-2: lack of COX-1 improves protective immunity in the spleen while lack of COX-2 impairs protective immunity in the liver. COX-2 has been implicated in multiple liver-specific pathologies, including hepatocellular carcinoma, fibrosis, non-alcoholic steatohepatitis (NASH), and non-alcoholic fatty liver disease (NAFLD) (73). Hepatocytes contain receptors for many of the COX-2 specific stimulators including TNFα, IL-1, and LPS, and liver specific Kuppfer and stellate cells have specifically been shown to upregulate COX-2 expression following inflammatory stimuli (73, 74). Similarly, following LPS administration, COX-2 pathway activity increases in the liver (75), while COX-1 pathway activity has been shown to increase in the spleen and not the liver (76) similar to the phenotypes we see. Though the mechanisms governing these distinct tissue-specific differences remain unclear, it will be critical in the future to elucidate the tissue-specific differences to ensure the most therapeutic outcome pending the tissue affected.
Almost all commercially available NSAIDS exhibit non-specific COX inhibition in vivo (35); thus using pharmacotherapies that are able to limit the unwanted side effects of L. monocytogenes immunotherapy while preserving the critical role of PGE2 will be critical. In contrast to NSAIDs such as indomethacin and celecoxib, acetaminophen is a common non-NSAID pharmacotherapy for pain relief and fever reduction that lacks a well-defined mechanism of action (77). Acetaminophen metabolites have been shown to modulate endogenous cannabinoid receptor activity centrally to provide pain relief (78) suggesting that it acts centrally to exhibit its effects. Additionally, similar to NSAIDs acetaminophen can inhibit COX enzymes, though its inhibition is specific to central nervous system COX activity while it lacks an effect against peripheral COX activity (79). These contrasting sites of action, centrally in the case of acetaminophen and peripherally in the case of NSAIDs, likely explain why in our system acetaminophen administration does not impair the development of CD8+ T-cell immunity while it still maintains effective anti-pyretic activity.
We and others have previously shown that the inflammatory environment in which T-cell priming occurs following L. monocytogenes immunization modulates the magnitude of the response (66, 67, 69). Here we present evidence that in addition to the traditional cytokine mediators of inflammation lipid mediators play significant roles in the generation of cell-mediated immunity. Understanding how cyclooxygenase therapy modulates inflammation and which inflammatory mediators are detrimental will be critical as cyclooxygenase inhibitors are routinely given with vaccinations and immunotherapies, and are specifically given during L. monocytogenes immunotherapy (7, 8). We suggest that development of novel, specific COX-1 inhibitors; therapies that spare COX-2 and PGE2 production; or use of non-COX acting pharmacotherapies such as acetaminophen that limit alterations in peripheral inflammation will be critical for optimal immune responses to L. monocytogenes, as well as other platforms that require the generation of cell-mediated immunity.
Supplementary Material
Acknowledgments
This work was funded by the National Institutes of Health R01CA188034 (JDS) and F30 CA210912 (ET).
The authors would like to thank Dr. Charles Serhan for technical assistance and protocols for eicosanoid extraction. We acknowledge the NIH Tetramer Core Facility for provision of MHC-I B8R tetramers.
References
- 1.Lizée G, Overwijk WW, Radvanyi L, Gao J, Sharma P, Hwu P. Harnessing the Power of the Immune System to Target Cancer. Annu Rev Med. 2013;64:71–90. doi: 10.1146/annurev-med-112311-083918. [DOI] [PubMed] [Google Scholar]
- 2.Brockstedt DG, Dubensky TW. Promises and challenges for the development of Listeria monocytogenes-based immunotherapies. Expert Rev Vaccines. 2008;7:1069–84. doi: 10.1586/14760584.7.7.1069. [DOI] [PubMed] [Google Scholar]
- 3.Manley J, Taddio A. Acetaminophen and ibuprofen for prevention of adverse reactions associated with childhood immunization. Ann Pharmacother. 2007;41:1227–32. doi: 10.1345/aph.1H647. [DOI] [PubMed] [Google Scholar]
- 4.Rubin K. Understanding Immune Checkpoint Inhibitors for Effective Patient Care. Clin J Oncol Nurs. 2015;19:709–717. doi: 10.1188/15.CJON.709-717. [DOI] [PubMed] [Google Scholar]
- 5.Bryant R. Managing side effects of childhood cancer treatment. J Pediatr Nurs. 2003;18:113–125. doi: 10.1053/jpdn.2003.11. [DOI] [PubMed] [Google Scholar]
- 6.Mavroukakis SA, Muehlbauer PM, White RL, Schwartzentruber DJ. Clinical pathways for managing patients receiving interleukin 2. Clin J Oncol Nurs. 5:207–17. [PubMed] [Google Scholar]
- 7.An Assessment of an Attenuated Live Listeria Vaccine in CIN 2+ - Full Text View - ClinicalTrials.gov.
- 8.Herzog T, Backes FJ, Del Pilar Estevez Diz M, Hare TW, Huh W, Kim B-G, Moore KN, Oaknin A, Small WJ, Tewari KS, Monk BJ. AIM2CERV: a randomized phase 3 study of adjuvant AXAL immunotherapy following chemoradiation in patients who have high-risk locally advanced cervical cancer (HRLACC) Society for Cancer Immunotherapy 2016 [Google Scholar]
- 9.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: Structural, Cellular, and Molecular Biology. Annu Rev Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 10.Harizi H, Corcuff JB, Gualde N. Arachidonic-acid-derived eicosanoids: roles in biology and immunopathology. Trends Mol Med. 2008;14:461–469. doi: 10.1016/j.molmed.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 11.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011;31:986–1000. doi: 10.1161/ATVBAHA.110.207449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nataraj C, Thomas DW, Tilley SL, Nguyen MT, Mannon R, Koller BH, Coffman TM. Receptors for prostaglandin E(2) that regulate cellular immune responses in the mouse. J Clin Invest. 2001;108:1229–35. doi: 10.1172/JCI13640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen JH, Perry CJ, Tsui YC, Staron MM, Parish IA, Dominguez CX, Rosenberg DW, Kaech SM. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat Med. 2015;21:327–334. doi: 10.1038/nm.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Coulombe F, Jaworska J, Verway M, Tzelepis F, Massoud A, Gillard J, Wong G, Kobinger G, Xing Z, Couture C, Joubert P, Fritz JH, Powell WS, Divangahi M. Targeted prostaglandin E2 inhibition enhances antiviral immunity through induction of type I interferon and apoptosis in macrophages. Immunity. 2014;40:554–568. doi: 10.1016/j.immuni.2014.02.013. [DOI] [PubMed] [Google Scholar]
- 15.Mayer-Barber KD, Andrade BBB, Oland SD, Amaral EP, Barber DL, Gonzales J, Derrick SC, Shi R, Kumar NP, Wei W, Yuan X, Zhang G, Cai Y, Babu S, Catalfamo M, Salazar AM, Via LE, Barry CE, 3rd, Sher A, Barry CE, III, Sher A. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. 2014;511:99–103. doi: 10.1038/nature13489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tripp CS, Needleman P, Unanue ER. Indomethacin in vivo increases the sensitivity to Listeria infection in mice. A possible role for macrophage thromboxane A2 synthesis. J Clin Invest. 1987;79:399–403. doi: 10.1172/JCI112825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hutchison DL, Myers RL. Prostaglandin-mediated suppression of macrophage phagocytosis of Listeria monocytogenes. Cell Immunol. 1987;110:68–76. doi: 10.1016/0008-8749(87)90102-x. [DOI] [PubMed] [Google Scholar]
- 18.Brockstedt DG, Giedlin MA, Leong ML, Bahjat KS, Gao Y, Luckett W, Liu W, Cook DN, Portnoy DA, Dubensky TW. Listeria-based cancer vaccines that segregate immunogenicity from toxicity. Proc Natl Acad Sci U S A. 2004;101:13832–13837. doi: 10.1073/pnas.0406035101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sauer JD, Pereyre S, Archer KA, Burke TP, Hanson B, Lauer P, Portnoy DA. Listeria monocytogenes engineered to activate the Nlrc4 inflammasome are severely attenuated and are poor inducers of protective immunity. Proc Natl Acad Sci U S A. 2011;108:12419–12424. doi: 10.1073/pnas.1019041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD, Kim HS, Smithies O. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:483–92. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 21.Trebino CE, Stock JL, Gibbons CP, Naiman BM, Wachtmann TS, Umland JP, Pandher K, Lapointe JM, Saha S, Roach ML, Carter D, Thomas NA, Durtschi BA, McNeish JD, Hambor JE, Jakobsson PJ, Carty TJ, Perez JR, Audoly LP. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc Natl Acad Sci U S A. 2003;100:9044–9. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakanishi M, Montrose DC, Clark P, Nambiar PR, Belinsky GS, Claffey KP, Xu D, Rosenberg DW. Genetic Deletion of mPGES-1 Suppresses Intestinal Tumorigenesis. Cancer Res. 2008;68:3251–3259. doi: 10.1158/0008-5472.CAN-07-6100. [DOI] [PubMed] [Google Scholar]
- 23.Nakanishi M, Menoret A, Tanaka T, Miyamoto S, Montrose DC, Vella AT, Rosenberg DW. Selective PGE(2) suppression inhibits colon carcinogenesis and modifies local mucosal immunity. Cancer Prev Res (Phila) 2011;4:1198–208. doi: 10.1158/1940-6207.CAPR-11-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christy AC, Byrnes KR, Settle TL. Evaluation of medicated gel as a supplement to providing acetaminophen in the drinking water of C57BL/6 mice after surgery. J Am Assoc Lab Anim Sci. 2014;53:180–4. [PMC free article] [PubMed] [Google Scholar]
- 25.Colas RA, Shinohara M, Dalli J, Chiang N, Serhan CN. Identification and signature profiles for pro-resolving and inflammatory lipid mediators in human tissue. AJP Cell Physiol. 2014;307:C39–C54. doi: 10.1152/ajpcell.00024.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melamud E, Vastag L, Rabinowitz JD. Metabolomic Analysis and Visualization Engine for LC–MS Data. Anal Chem. 2010;82:9818–9826. doi: 10.1021/ac1021166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clasquin MF, Melamud E, Rabinowitz JD. Current Protocols in Bioinformatics. Unit14.11. Chapter 14. John Wiley & Sons, Inc; Hoboken, NJ, USA: 2012. LC-MS Data Processing with MAVEN: A Metabolomic Analysis and Visualization Engine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, Lee L, Isakson P. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc Natl Acad Sci U S A. 1994;91:12013–7. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pamer EGEG. Immune responses to Listeria monocytogenes. Nat Rev Immunol. 2004;4:812–823. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 30.Freeman MM, Ziegler HK. Simultaneous Th1-type cytokine expression is a signature of peritoneal CD4+ lymphocytes responding to infection with Listeria monocytogenes. J Immunol. 2005;175:394–403. doi: 10.4049/jimmunol.175.1.394. [DOI] [PubMed] [Google Scholar]
- 31.Wherry EJ, Teichgräber V, Becker TC, Masopust D, Kaech SM, Antia R, von Andrian UH, Ahmed R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol. 2003;4:225–234. doi: 10.1038/ni889. [DOI] [PubMed] [Google Scholar]
- 32.Schiemann M, Busch V, Linkemann K, Huster KM, Busch DH. Differences in maintenance of CD8+ and CD4+ bacteria-specific effector-memory T cell populations. Eur J Immunol. 2003;33:2875–2885. doi: 10.1002/eji.200324224. [DOI] [PubMed] [Google Scholar]
- 33.O’Sullivan MG, Chilton FH, Huggins EM, McCall CE. Lipopolysaccharide priming of alveolar macrophages for enhanced synthesis of prostanoids involves induction of a novel prostaglandin H synthase. J Biol Chem. 1992;267:14547–50. [PubMed] [Google Scholar]
- 34.Teeling JL, Cunningham C, Newman TA, Perry VH. The effect of non-steroidal anti-inflammatory agents on behavioural changes and cytokine production following systemic inflammation: Implications for a role of COX-1. Brain Behav Immun. 2010;24:409–419. doi: 10.1016/j.bbi.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitchell JA, Akarasereenont P, Thiemermann C, Flower RJ, Vane JR. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc Natl Acad Sci U S A. 1993;90:11693–7. doi: 10.1073/pnas.90.24.11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith CJ, Zhang Y, Koboldt CM, Muhammad J, Zweifel BS, Shaffer A, Talley JJ, Masferrer JL, Seibert K, Isakson PC. Pharmacological analysis of cyclooxygenase-1 in inflammation. Proc Natl Acad Sci U S A. 1998;95:13313–8. doi: 10.1073/pnas.95.22.13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brenneis C, Maier TJ, Schmidt R, Hofacker A, Zulauf L, Jakobsson PJ, Scholich K, Geisslinger G. Inhibition of prostaglandin E2 synthesis by SC-560 is independent of cyclooxygenase 1 inhibition. FASEB J. 2006;20:1352–1360. doi: 10.1096/fj.05-5346com. [DOI] [PubMed] [Google Scholar]
- 38.Mercado R, Vijh S, Allen SE, Kerksiek K, Pilip IM, Pamer EG. Early programming of T cell populations responding to bacterial infection. J Immunol. 2000;165:6833–9. doi: 10.4049/jimmunol.165.12.6833. [DOI] [PubMed] [Google Scholar]
- 39.Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK, Mescher MF. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J Immunol. 1999;162:3256–62. [PubMed] [Google Scholar]
- 40.Kaliński P, Hilkens CM, Snijders A, Snijdewint FG, Kapsenberg ML. IL-12-deficient dendritic cells, generated in the presence of prostaglandin E2, promote type 2 cytokine production in maturing human naive T helper cells. J Immunol. 1997;159:28–35. [PubMed] [Google Scholar]
- 41.Yu Y, Chadee K. Prostaglandin E2 Stimulates IL-8 Gene Expression in Human Colonic Epithelial Cells by a Posttranscriptional Mechanism. J Immunol. 1998;161:3746–3752. [PubMed] [Google Scholar]
- 42.Yang CW, Strong BSI, Miller MJ, Unanue ER. Neutrophils influence the level of antigen presentation during the immune response to protein antigens in adjuvants. J Immunol. 2010;185:2927–34. doi: 10.4049/jimmunol.1001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang CW, Unanue ER. Neutrophils control the magnitude and spread of the immune response in a thromboxane A 2 -mediated process. J Exp Med. 2013;210:375–387. doi: 10.1084/jem.20122183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang M, Stolina M, Sharma S, Mao JT, Zhu L, Miller PW, Wollman J, Herschman H, Dubinett SM. Non-small cell lung cancer cyclooxygenase-2-dependent regulation of cytokine balance in lymphocytes and macrophages: up-regulation of interleukin 10 and down-regulation of interleukin 12 production. Cancer Res. 1998;58:1208–16. [PubMed] [Google Scholar]
- 45.Brock TG, McNish RW, Peters-Golden M. Arachidonic acid is preferentially metabolized by cyclooxygenase-2 to prostacyclin and prostaglandin E2. J Biol Chem. 1999;274:11660–6. doi: 10.1074/jbc.274.17.11660. [DOI] [PubMed] [Google Scholar]
- 46.Kolstad AM, Rodriguiz RM, Kim CJ, Hale LP. Effect of pain management on immunization efficacy in mice. J Am Assoc Lab Anim Sci. 2012;51:448–57. [PMC free article] [PubMed] [Google Scholar]
- 47.von Moltke J, Trinidad NJ, Moayeri M, Kintzer AF, Wang SB, van Rooijen N, Brown CR, Krantz BA, Leppla SH, Gronert K, Vance RE. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012;490:107–111. doi: 10.1038/nature11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen M, Boilard E, Nigrovic PA, Clark P, Xu D, FitzGerald GA, Audoly LP, Lee DM. Predominance of cyclooxygenase 1 over cyclooxygenase 2 in the generation of proinflammatory prostaglandins in autoantibody-driven K/BxN serum–transfer arthritis. Arthritis Rheum. 2008;58:1354–1365. doi: 10.1002/art.23453. [DOI] [PubMed] [Google Scholar]
- 49.Naraba H, Murakami M, Matsumoto H, Shimbara S, Ueno A, Kudo I, Oh-ishi S. Segregated Coupling of Phospholipases A2, Cyclooxygenases, and Terminal Prostanoid Synthases in Different Phases of Prostanoid Biosynthesis in Rat Peritoneal Macrophages. J Immunol. 1998:160. [PubMed] [Google Scholar]
- 50.Murakami M, Matsumoto R, Austen KF, Arm JP. Prostaglandin endoperoxide synthase-1 and -2 couple to different transmembrane stimuli to generate prostaglandin D2 in mouse bone marrow-derived mast cells. 1994:22269–75. [PubMed] [Google Scholar]
- 51.Kabashima K, Murata T, Tanaka H, Matsuoka T, Sakata D, Yoshida N, Katagiri K, Kinashi T, Tanaka T, Miyasaka M, Nagai H, Ushikubi F, Narumiya S. Thromboxane A2 modulates interaction of dendritic cells and T cells and regulates acquired immunity. Nat Immunol. 2003;4:694–701. doi: 10.1038/ni943. [DOI] [PubMed] [Google Scholar]
- 52.Kabashima K, Murata T, Tanaka H, Matsuoka T, Sakata D, Yoshida N, Katagiri K, Kinashi T, Tanaka T, Miyasaka M, Nagai H, Ushikubi F, Narumiya S. Thromboxane A2 modulates interaction of dendritic cells and T cells and regulates acquired immunity. Nat Immunol. 2003;4:694–701. doi: 10.1038/ni943. [DOI] [PubMed] [Google Scholar]
- 53.Lewis RA, Soter NA, Diamond PT, Austen KF, Oates JA, Roberts LJ. Prostaglandin D2 generation after activation of rat and human mast cells with anti-IgE. J Immunol. 1982:129. [PubMed] [Google Scholar]
- 54.Hardy CC, Robinson C, Tattersfield AE, Holgate ST. The Bronchoconstrictor Effect of Inhaled Prostaglandin D2 in Normal and Asthmatic Men. N Engl J Med. 1984;311:209–213. doi: 10.1056/NEJM198407263110401. [DOI] [PubMed] [Google Scholar]
- 55.Hammad H, de Heer HJ, Soullie T, Hoogsteden HC, Trottein F, Lambrecht BN. Prostaglandin D2 inhibits airway dendritic cell migration and function in steady state conditions by selective activation of the D prostanoid receptor 1. J Immunol. 2003;171:3936–40. doi: 10.4049/jimmunol.171.8.3936. [DOI] [PubMed] [Google Scholar]
- 56.Baratelli F, Lin Y, Zhu L, Yang SC, Heuzé-Vourc’h N, Zeng G, Reckamp K, Dohadwala M, Sharma S, Dubinett SM. Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J Immunol. 2005;175:1483–90. doi: 10.4049/jimmunol.175.3.1483. [DOI] [PubMed] [Google Scholar]
- 57.Betz M, Fox BS. Prostaglandin E2 inhibits production of Th1 lymphokines but not of Th2 lymphokines. J Immunol. 1991;146:108–13. [PubMed] [Google Scholar]
- 58.Rieser C, Böck G, Klocker H, Bartsch G, Thurnher M. Prostaglandin E2 and tumor necrosis factor alpha cooperate to activate human dendritic cells: synergistic activation of interleukin 12 production. J Exp Med. 1997;186:1603–8. doi: 10.1084/jem.186.9.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scandella E, Men Y, Gillessen S, Förster R, Groettrup M. Prostaglandin E2 is a key factor for CCR7 surface expression and migration of monocyte-derived dendritic cells. Blood. 2002;100:1354–61. doi: 10.1182/blood-2001-11-0017. [DOI] [PubMed] [Google Scholar]
- 60.Honda A, Sugimoto Y, Namba T, Watabe A, Irie A, Negishi M, Narumiya S, Ichikawa A. Cloning and expression of a cDNA for mouse prostaglandin E receptor EP2 subtype. J Biol Chem. 1993;268:7759–62. [PubMed] [Google Scholar]
- 61.Regan JW, Bailey TJ, Pepperl DJ, Pierce KL, Bogardus AM, Donello JE, Fairbairn CE, Kedzie KM, Woodward DF, Gil DW. Cloning of a novel human prostaglandin receptor with characteristics of the pharmacologically defined EP2 subtype. Mol Pharmacol. 1994;46:213–20. [PubMed] [Google Scholar]
- 62.Sugimoto Y, Namba T, Honda A, Hayashi Y, Negishi M, Ichikawa A, Narumiya S. Cloning and expression of a cDNA for mouse prostaglandin E receptor EP3 subtype. J Biol Chem. 1992;267:6463–6. [PubMed] [Google Scholar]
- 63.Funk CD, Furci L, FitzGerald GA, Grygorczyk R, Rochette C, Bayne MA, Abramovitz M, Adam M, Metters KM. Cloning and expression of a cDNA for the human prostaglandin E receptor EP1 subtype. J Biol Chem. 1993;268:26767–72. [PubMed] [Google Scholar]
- 64.Abramovitz M, Adam M, Boie Y, Carrière MC, Denis D, Godbout C, Lamontagne S, Rochette C, Sawyer N, Tremblay NM, Belley M, Gallant M, Dufresne C, Gareau Y, Ruel R, Juteau H, Labelle M, Ouimet N, Metters KM. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim Biophys Acta - Mol Cell Biol Lipids. 2000;1483:285–293. doi: 10.1016/s1388-1981(99)00164-x. [DOI] [PubMed] [Google Scholar]
- 65.Yokoyama U, Iwatsubo K, Umemura M, Fujita T, Ishikawa Y. The Prostanoid EP4 Receptor and Its Signaling Pathway. Pharmacol Rev. 2013;65:1010–1052. doi: 10.1124/pr.112.007195. [DOI] [PubMed] [Google Scholar]
- 66.Richer MJ, Nolz JC, Harty JT. Pathogen-specific inflammatory milieux tune the antigen sensitivity of CD8(+) T cells by enhancing T cell receptor signaling. Immunity. 2013;38:140–152. doi: 10.1016/j.immuni.2012.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Alice AF, Kramer G, Bambina S, Baird JR, Bahjat KS, Gough MJ, Crittenden MR. Amplifying IFN-γ Signaling in Dendritic Cells by CD11c-Specific Loss of SOCS1 Increases Innate Immunity to Infection while Decreasing Adaptive Immunity. J Immunol. 2018;200:177–185. doi: 10.4049/jimmunol.1700909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Archer KA, Durack J, Portnoy DA. STING-Dependent Type I IFN Production Inhibits Cell-Mediated Immunity to Listeria monocytogenes. PLoS Pathog. 2014;10:e1003861. doi: 10.1371/journal.ppat.1003861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Theisen E, Sauer JD. Listeria monocytogenes-Induced Cell Death Inhibits the Generation of Cell-Mediated Immunity. Infect Immun. 2017;85:e00733–16. doi: 10.1128/IAI.00733-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Manca C, Tsenova L, Bergtold A, Freeman S, Tovey M, Musser JM, Barry CE, Freedman VH, Kaplan G. Virulence of a Mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce Th1 type immunity and is associated with induction of IFN-alpha/beta. Proc Natl Acad Sci U S A. 2001;98:5752–7. doi: 10.1073/pnas.091096998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Auerbuch V, Brockstedt DG, Meyer-Morse N, O’Riordan M, Portnoy DA. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J Exp Med. 2004;200:527–33. doi: 10.1084/jem.20040976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.O’Connell RM, Saha SK, Vaidya SA, Bruhn KW, Miranda GA, Zarnegar B, Perry AK, Nguyen BO, Lane TF, Taniguchi T, Miller JF, Cheng G. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med. 2004;200:437–45. doi: 10.1084/jem.20040712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martín-Sanz P, Casado M, Boscá L. Cyclooxygenase 2 in liver dysfunction and carcinogenesis: Facts and perspectives. World J Gastroenterol. 2017;23:3572–3580. doi: 10.3748/wjg.v23.i20.3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ledwith BJ, Pauley CJ, Wagner LK, Rokos CL, Alberts DW, Manam S. Induction of cyclooxygenase-2 expression by peroxisome proliferators and non-tetradecanoylphorbol 12,13-myristate-type tumor promoters in immortalized mouse liver cells. J Biol Chem. 1997;272:3707–14. doi: 10.1074/jbc.272.6.3707. [DOI] [PubMed] [Google Scholar]
- 75.Leach M, Hamilton LC, Olbrich A, Wray GM, Thiemermann C. Effects of inhibitors of the activity of cyclo-oxygenase-2 on the hypotension and multiple organ dysfunction caused by endotoxin: A comparison with dexamethasone. Br J Pharmacol. 1998;124:586–592. doi: 10.1038/sj.bjp.0701869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Steiner AA, Hunter JC, Phipps SM, Nucci TB, Oliveira DL, Roberts JL, Scheck AC, Simmons DL, Romanovsky AA. Cyclooxygenase-1 or -2—which one mediates lipopolysaccharide-induced hypothermia? Am J Physiol Integr Comp Physiol. 2009;297:R485–R494. doi: 10.1152/ajpregu.91026.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ghanem CI, Pérez MJ, Manautou JE, Mottino AD. Acetaminophen from liver to brain: New insights into drug pharmacological action and toxicity. Pharmacol Res. 2016;109:119–131. doi: 10.1016/j.phrs.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Högestätt ED, Jönsson BAG, Ermund A, Andersson DA, Björk H, Alexander JP, Cravatt BF, Basbaum AI, Zygmunt PM. Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system. J Biol Chem. 2005;280:31405–12. doi: 10.1074/jbc.M501489200. [DOI] [PubMed] [Google Scholar]
- 79.FLOWER RJ, VANE JR. Inhibition of Prostaglandin Synthetase in Brain explains the Anti-pyretic Activity of Paracetamol (4-Acetamidophenol) Nature. 1972;240:410–411. doi: 10.1038/240410a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.