

Abstract
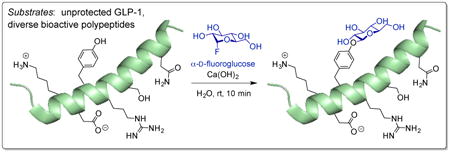
Glycosylated natural products and synthetic glycopeptides represent a significant and growing source of biochemical probes and therapeutic agents. However, methods that enable the aqueous glycosylation of endogenous amino acid functionality in peptides without the use of protecting groups are scarce. Here, we report a transformation that facilitates the efficient aqueous O-glycosylation of phenolic functionality in a wide range of small molecules, unprotected tyrosine, and tyrosine residues embedded within a range of complex, fully unprotected peptides. The transformation, which uses glycosyl fluoride donors and is promoted by Ca(OH)2, proceeds rapidly at room temperature in water with good yields and selective formation of unique anomeric products depending on the stereochemistry of the glycosyl donor. High functional group tolerance is observed, and the phenol glycosylation occurs selectively in the presence of virtually all side chains of the proteinogenic amino acids with the singular exception of Cys. This method offers a highly selective, efficient, and operationally simple approach for the protecting group-free synthesis of O-aryl glycosides and Tyr-O-glycosylated peptides in water.
Glycosylation can modulate the physiological properties of small molecules and peptides, with specific impacts such as improved metabolic stability,1 membrane permeability,2 biodistribution,3 and ligand-target interactions.4,5 Thus, numerous glycosylated natural products and synthetic glycopeptides are extant as important biochemical probes and therapeutic agents (some examples are shown in Figure 1a).6,7,8,9,10 While methods for the glycosylation of organic compounds are numerous, glycosylation methods typically require protection of glycosyl donors and acceptors.11,12,13 The most common exceptions generally involve the application of glycosyl transferases in the enzymatic context.14,15,16,17 The optimization of glycosylation methodology for reactions conducted in organic solvents has made applications on highly polar compounds such as native oligopeptides rare.18 Perhaps for related reasons, despite the introduction of many new methods for selective peptide/protein synthesis, strategies for the selective glycosylation of endogenous amino acid functionality in unprotected peptides, and in aqueous solvents, remain scarce.19,20,21,22,23,24 To date, such transformations have been limited to S-glycosylation of cysteine (Cys)25,26,27,28 and amide coupling of aspartic acid (Asp) to form N-glycosides.29 Reported herein is an efficient O-glycosylation of phenolic functionality in a wide range of small molecules, the amino acid tyrosine (Tyr), and of the Tyr residue embedded within a range of complex, water-soluble, fully unprotected peptides. Features of this method include rapid reaction rates and good yields with high anomeric selectivity, even as the glycosylation occurs in bulk aqueous solvent. At the same time, high functional group tolerance is observed such that the desired reaction occurs in the presence of virtually all side chains of the proteinogenic amino acids (with the singular, but potentially exploitable exception of Cys), with attendant high chemoselectivity for phenolic O-glycosylation, including Tyr-O-glycosylation, even in the presence of other nucleophilic, potential acceptor functionality.
Figure 1.

Natural products containing phenolic O-glycosyl moieties and strategies to access phenolic O-glycosyl linkages in small and complex molecules (a) Selected examples of glycosylated natural products exhibiting varied biological activity. (b) Schematic representation of direct vs. convergent synthetic methodologies to access glycopeptides. Coloured circles represent amino acids. (c) Conditions for previously reported aromatic O-glycosylation methods, discussed and referenced in the text; those require a protecting group for the oxygen sites (OPG). (d) Previously reported protecting-group-free aqueous glycosylation of sucrose promoted by Ca2+ and NMe3 (ref. 56). The OH groups required for reaction are highlighted in green, the OH reaction site is shown in blue. (e) This work: O-glycosylation in aqueous solvent.
Naturally occurring Tyr-O-glycosides have been observed in the active site of glycogenin,30 certain prokaryotic surface-layer glycoproteins,31 and amyloid β-peptides detected in human cerebrospinal fluid.32,33 Currently, strategies to access Tyr-O-glycopeptides entail solid phase peptide synthesis incorporating protected-glycosyl Tyr residues that are prepared via aromatic O-glycosylation prior to peptide chain assembly (Figure 1b).34,35,36 Biosynthetic approaches with synthetically preglycosylated amino acids have also been reported.37 A wide range of methods for aromatic O-glycosylation have been developed since the coupling of peracetyl glucopyranosyl chloride and potassium phenolate was reported by Michael in 1879,38 and many of these methods reflect mild reaction conditions, tolerance of various functional groups, and high anomeric selectivity (Figure 1c) so long as the chemistry occurs in organic solvents with protected substrates.39,40,41,42,43,44 Major advances in catalytic control of glycosylations, the complexity of oligosaccharides that are available, and our mechanistic understanding of key reactions are all emerging.45,46,47 At the same time, current aromatic methods share two features that might be viewed as limitations for the goal of convergent Tyr-O-glycosylation of complex peptides: each requires multiple protecting groups to ensure reaction specificity, and most are based on chemistry that functions well only in a non-aqueous solvent or co-solvent. The development of new, phenolic O-glycosylation strategies for substrates soluble only in water demand reactions that proceed with high efficiency in water, which is a potentially competitive nucleophile for most activated glycosyl donors. An additional, highly desirable feature would be efficient glycosylation that occurs in the presence of the diverse functionality characterizing unprotected polypeptide chains of natural or synthetic origin.
To enable aqueous glycosylation, glycosyl donors that do not undergo hydrolysis or hydrolyze slowly are required. Glycosyl fluorides, developed and studied by Mukaiyama and Nicolaou, exhibit relative stability toward hydrolysis and have emerged as useful glycosyl donors in conventional chemical glycosylation.48,49,50 Glycosyl fluorides have also seen wide usage as transition state-analogue substrates (TSAS) for hydrolases51,52 and as agents for proteomic profiling.53 From the physical organic perspective, Jencks and co-workers reported the aqueous substitution of α-fluoroglucose by sodium azide, notably avoiding hydrolysis and observing complete inversion of stereochemistry to produce the β-anomer.54,55 Capitalizing on this observation, our laboratories developed a method to effect the regioselective glycosylation of unprotected sucrose and sucrose analogues in an aqueous medium. This reaction employed α-fluoroglycosides as the glycosyl donors and proceeded efficiently for a range of sucrose analogues as glycosyl acceptors in the presence of Ca2+ additives (Figure 1d).56 Mechanistic studies of this intriguing process, including nuclear magnetic resonance (NMR) studies and examination of the exhaustive set of mono-deoxysucrose analogues, revealed that the glycosylation of sucrose requires a discrete array of substrate hydroxyl groups that presumably engage both Ca2+ and the glycosylation substrates with multiple contacts. The reaction is remarkably specific. However, inspired once again by the stability of glycosyl fluorides, their classical reactivity toward anionic nucleophiles, and our prior results, we investigated whether these glycosyl donors could be utilized for the fully aqueous O-glycosylation of phenols, including peptide-embedded Tyr.
Results and Discussion
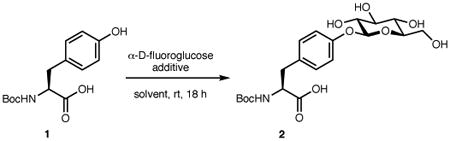
Our experiments began by examining the glycosylation of N-Boc-tyrosine (1) under conditions previously developed for the selective glycosylation of sucrose (Table 1, entry 1). The reaction proceeded with complete conversion to generate N-Boc-O-β-d-glucopyranosyl-l-tyrosine (2). NMR analysis of the reaction mixture indicated glycosylation exclusively at the hydroxyl group of 1, with no products of aromatic C-glycosylation or carboxylic acid O-glycosylation. We then investigated whether conditions without trimethylamine could be developed in order to improve compatibility with more complex substrates. However, exclusion of trimethylamine, in aqueous solvent and Ca(OTf)2 as a promoter, led to no conversion, highlighting the need for alkaline conditions (Table 1, entry 2). Exchanging Ca(OTf)2 with Ca(OH)2 proved effective, generating the O-glucosyl (O-Glc) product with near complete conversion (95%; Table 1, entry 3). Interestingly, investigation of other alkali and alkaline earth metal hydroxides underscored the special reactivity enabled by Ca2+ (Table 1, entries 4–9). Omitting a metal salt altogether preempted product formation (Table 1, entry 10), and only slow hydrolysis of the glycosyl fluoride was observed. Optimization of the reagent loading and concentration (entries 11-17) culminated in efficient conditions to produce products in nearly quantitative yield (entry 16). Finally, time-course studies revealed the fast rate of the reaction, showing that the reaction is nearly complete after only 10 minutes (Table 1, entries 16 and 17).
Table 1.
Optimization of reaction conditions for the O-glycosylation of Boc-L-Tyr-OH (1).
| ||||
|---|---|---|---|---|
| Entry | Solvent (conc.) | α-D-fluoroglucose (equiv.) | Additive | NMR yield (%) |
| 1 | 45% NMe3/H2O (0.2 M) | 6 | Ca(OTf)2 (6 equiv.) | 100 |
| 2 | H2O (0.2 M) | 6 | Ca(OTf)2 (6 equiv.) | 0 |
| 3 | H2O (0.2 M) | 6 | Ca(OH)2 (6 equiv.) | 95 |
| 4 | H2O (0.2 M) | 6 | LiOH (6 equiv.) | 8 |
| 5 | H2O (0.2 M) | 6 | NaOH (6 equiv.) | 6 |
| 6 | H2O (0.2 M) | 6 | KOH (6 equiv.) | 6 |
| 7 | H2O (0.2 M) | 6 | CsOH (6 equiv.) | 5 |
| 8 | H2O (0.2 M) | 6 | Mg(OH)2 (6 equiv.) | 0 |
| 9 | H2O (0.2 M) | 6 | Ba(OH)2 (6 equiv.) | 4 |
| 10 | H2O (0.2 M) | 6 | none | 0 |
| 11 | H2O (0.2 M) | 3 | Ca(OH)2 (3 equiv.) | 73 |
| 12 | H2O (0.5 M) | 3 | Ca(OH)2 (3 equiv.) | 92 |
| 13 | H2O (1.0 M) | 3 | Ca(OH)2 (3 equiv.) | 97 |
| 14 | H2O (1.0 M) | 2 | Ca(OH)2 (2 equiv.) | 93 |
| 15 | H2O (1.0 M) | 1 | Ca(OH)2 (1 equiv.) | 16 |
| 16a | H2O (1.0 M) | 3 | Ca(OH)2 (3 equiv.) | 95 |
| 17b | H2O (1.0 M) | 3 | Ca(OH)2 (3 equiv.) | 94 |
Reaction time = 1 h.
Reaction time = 10 min.
Notably, the optimized glycosylation conditions generate exclusively the O-β-glucosyl product, providing evidence for a SN2-type mechanism. This observation is analogous to those recorded in the Ca2+/NMe3-promoted sucrose glycosylation using α-D-fluoroglycosides under aqueous conditions;56 so too, this observation recalls that of Jencks in the study of displacement of α-D-fluoroglucose by azide ion, later studied computationally.55,57,58,59 The observed increase in conversion as a function of increased salt concentration is also consistent with the ionic-strength dependence of a generic SN2 reaction mechanism, also observed by Jencks.55 The stereochemical purity of the product is independent of reaction duration; under 1.0 M reaction conditions, only the β-anomer product was observed after 10 min and after 24 h reaction times. No evidence of epimerization of the Tyr α-stereocenter after 24 h was detected under these conditions (see Supplementary Pages 18–22).
With suitable reaction conditions for Tyr O-glycosylation established, we evaluated a range of phenols as glycosyl acceptors (Figure 2). Protected O-glucosyl tyrosine products 2 and 3, generated from Boc-Tyr-OH and Ac-Tyr-NH2, respectively, were each isolated in 91% yield. Fully unprotected O-glucosyl tyrosine 4 was isolated in 92% yield, with exclusive glycosylation of the phenolic hydroxyl group of tyrosine despite the presence of a primary amine. Conversion of phenol to 5 proceeded in 92% yield, and substituted aryl glucosides including 4-methoxy (6; 92% yield), 4-chloro (7; 93% yield), 4-cyano (8; 80% yield), and 4-trifluoromethyl (9, 60% yield) were all isolated in good to excellent yields. Higher yields with more electron-rich (less acidic) phenols may result from greater nucleophilicity of conjugate bases as well as more rapid in situ hydrolysis of electron-deficient aryl glucoside products. Substituted aryl glucosides bearing aniline (10; 72% yield), primary amine (11; 83% yield), primary alcohol (12; 82% yield), and ketone (13; 94% yield) functionality were isolated in good yields, as well, and in each case glycosylation was only observed at the phenolic hydroxyl group. The impact of sterics on the glycosylation of substituted phenols was evaluated through formation of 2,6-dimethyl (14) and 3,5-dimethyl (15) phenyl glucosides. Significantly diminished yield for 14 (42%) compared to 15 (77%), despite similar substrate solubility, revealed the lower reactivity of substrates with ortho substituents; this effect parallels diminished isolated yields obtained for O-β-aryl glucosides resulting from thymol (16; 23% yield) and 2,3-dimethoxyphenol (17; 80% yield). In addition to ortho substitution, substrate insolubility under the reaction conditions significantly decreased yield; benzyloxy (18, 10% yield) and alkenyl (19, 10% yield) substituted O-β-aryl glucosides were each isolated in low yield due to low substrate solubility. In addition to α-D-fluoroglucose, the glycosyl donors α-D-fluoromaltose and α-D-fluorogalactose underwent glycosylation with Boc-Tyr-OH under the reaction conditions, providing O-glycosyl tyrosine products 20 and 21, respectively, in good yields (74% yield and 46% yield, respectively). The case of α-D-fluoromannose is also notable in that, while 22 can be isolated (∼25% yield), the reaction is a bit less efficient and the major product is the α-anomer (see Supplementary Pages 32–33 and 114–115), consistent with a double inversion process involving the axial C2-OH group. We also examined the reaction of β-D-fluoroglucose under analogous reaction conditions, employing 4-methoxyphenol as the glycosyl acceptor. Once again, this reaction takes place with net retention of anomeric configuration, and 6 is formed in 26% yield, also consistent with a double inversion. In this case, the greater instability of β-fluoroglucose may account for the lower yield of 6, and its reactivity at the anomeric position. Double inversion with β-fluoroglucose may involve intramolecular displacement as a prelude to glycosylation,60 proceeding through glycosyl epoxide intermediates when the vicinal 2-hydroxyl group and 1-fluoride are disposed trans in the starting material61 (see Supplementary Page 90 for details.) In any event, taken together, these results above reflect a significant scope of phenolic compounds that may be subjected to the rapid, stereoselective O-glycosylation reaction in bulk aqueous solvent.
Figure 2.

Evaluating substrate scope. The reactions were carried out at a substrate concentration of 1.0 M, the yields shown refer to isolated products. Reaction conditions: 0.5 mmol substrate, α-D-fluoroglycoside (1.5 mmol, 3 equiv.), Ca(OH)2 (1.5 mmol, 3 equiv.), H2O (1.0 M in substrate), rt, 1 h. The products were purified via reversed-phase flash column chromatography.
We then turned our attention to the question of biocompatible functional group tolerance. The interrogation of chemoselective Tyr-O-glycosylation within fully unprotected peptides seemed a compelling context to explore. To examine reactions on a scale of relevance to the study of complex bioactive peptides, practical reaction conditions for smaller quantities of substrate were developed. Optimization of dilute reaction conditions for the glycosylation of Boc-Tyr-OH revealed that a 1.0 M loading of both α-D-fluoroglucose and Ca(OH)2 allowed complete conversion to 2 at 100 mM, 10 mM, and 1.0 mM substrate loading (see Supplementary Pages 11–17).
We then applied these conditions to a range of bioactive peptides. Glucagon-like peptide-1 (GLP-17-37; 23; Figure 3)62 is a 30-residue peptide hormone bearing a single Tyr residue, and functions to stimulate and inhibit the secretion of insulin and glucagon, respectively.63 GLP-1 also possesses a host of other side chain functionalities, setting up in situ evaluation of functional group tolerance during the projected Tyr-O-glycosylation. Upon reaction of 23 for 10 min, high-performance liquid chromatography (HPLC) and liquid chromatography–mass spectrometry (LCMS) analysis indicated 46% conversion to a mono-glucosyl product (24) with 31% remaining 23 (Figure 3). Of the remaining 23%, HPLC analysis of the unpurified reaction mixture shows a mixture of various mono- and bis-glucosyl products, indicating that a small amount of non-phenolic glycosylation as well as peptide decomposition may be occurring under the reaction conditions. Purification of the major component is straightforward, and MS/MS analysis of the isolated mono-glucosyl product supports assignment of Tyr as the site of glycosylation (see Supplementary Pages 37–48). A GLP-1 mutant lacking the phenolic hydroxyl, Y19F GLP-17-37 (25), was also prepared by solid-phase peptide synthesis. Upon reaction of 25 for 10 min, HPLC and LCMS analysis revealed 86% remaining starting material along with minor mono-glucosyl product peaks (Figure 3), further supporting Tyr as the site of glycosylation.
Figure 3.

Tyrosine-selective glycosylation of glucagon-like peptide 1. HPLC yield reported (calculated by integrating peak corresponding to monosubstituted peptide at 214 nm). Reaction conditions: 0.5 or 1.0 mg substrate, α-D-fluoroglucose (1.0 M, 1000 equiv.), Ca(OH)2 (1.0 M, 1000 equiv.), H2O (1.0 mM in substrate), rt, 10 min. Reactions quenched with 0.5 M EDTA solution (pH = 8.0) and purified using desalting and buffer exchange column (SpinOUT™ GT-100).
Next, we assessed the generality of the aqueous Tyr-O-glycosylation reaction with additional biologically active peptides that each carried a single Tyr residue (Figure 4). Endomorphin-2, a highly selective agonist for the μ-opiod receptor,64 was glycosylated to generate 26 in 100% HPLC yield (40% isolated yield). While the crude reaction mixture once again revealed the presence of some bis-glycosylated material (as indicated by LCMS), this side product could be removed such that a sample of mono-glycosylated endomorphin-2 could be characterized by 1H and 13C NMR spectroscopy. Interestingly, endomorphin-1, when subjected to the same reaction conditions delivered 27 with near quantitative conversion. In this case, however, the isolated material revealed the presence of a second species, which could not be removed. While racemization was not observed in the control experiments described above, it cannot not be excluded in this case. Since the only difference between endomorphin-2 and endomorphin-1 is the swap of a tryptophan (Trp) for a phenylalanine (Phe) residue, we performed a control/competition experiment in which N-Boc-Tyr-OH and N-Boc-Trp-OH were exposed to glycosylation conditions, and only phenolic glycosylation was observed (see Supplementary Page 85 for details; conformational isomerism may be responsible for the observation of a second species in the case of glycosylated endomorphin-1.)65 Nevertheless, the opioid receptor ligand leu-enkephalin was an excellent substrate for the glycosylation, with 28 generated in 81% HPLC yield (36% isolated yield). Compound 28 could be characterized as a mono-glycosylated species by 1H and 13C NMR spectroscopy, as in the case of endomorphin-2.66 We then returned to examination of some more complicated peptides. Luteinizing-hormone releasing-hormone (LH-RH) was glycosylated to generated analogue 29 in 83% HPLC yield.67 The neurohypophysial hormones vasopressin and oxytocin were glycosylated in 67% HPLC yield and 68% HPLC yield, respectively, to generate analogues 30 and 31.68 Finally, the 32-amino acid polypeptide hormone, calcitonin, was glycosylated in 49% HPLC yield to generate 32.69 In all cases, varying amounts of off-target glycosylation and/or decomposition were observed by HPLC analysis. We note that the identification of off-target glycosylation products by tandem mass spectrometry (MS/MS) was nontrivial based on the small quantities observed and due to inhomogeneity of the peaks observed in the LC traces for these minor products. For example, we analyzed the byproducts obtained during glycosylation of GLP-1 and its Phe mutant. Manually searching for glycosylated fragments lacking a tyrosine residue revealed multiple such species (see Supplementary Page 48). While we were not able to determine the definitive sites of glycosylation of these side products, this additional level of MS/MS analysis revealed that the minor levels of glycosylation were likely to the N-terminal side of the peptide. Notably, there are three serine (Ser) and two threonine (Thr) residues in GLP-1 to the N-terminal side of Tyr, in addition to the N-terminus itself. We speculate that these could be the off-target glycosylation sites. Like GLP-1, calcitonin incorporates multiple Ser, Thr, and lysine (Lys) residues, and, like vasopressin and oxytocin, calcitonin features a disulfide bond, demonstrating the high chemoselectivity of aqueous Tyr-O-glycosylation mediated by Ca2+ salts and glucosyl fluorides.
Figure 4.

Tyrosine-selective glycosylation of biologically active peptides. *Isolated yield reported (Reaction conditions specified in Supplementary Page 49). **HPLC yield reported (calculated by integrating peak corresponding to monosubstitutated peptide at 214 nm), reaction conditions: 0.5 or 1.0 mg substrate, α-D-fluoroglucose (1.0 M, 1000 equiv.), Ca(OH)2 (1.0 M, 1000 equiv.), H2O (1.0 mM in substrate), rt, 10 min. Reactions quenched with 0.5 M EDTA solution (pH = 8.0) and purified using desalting and buffer exchange column (SpinOUT™ GT-100).
These experiments imply that the side chains of Ser, Thr, Lys, Trp, Asp, glutamic acid (Glu), asparagine (Asn), glutamine (Gln), histidine (His), arginine (Arg), as well as disulfide bonds, are relatively inert under these glycosylation conditions. None of the bioactive peptides evaluated contained a free Cys side chain, and one might expect reactivity from Cys due to its high nucleophilicity and pKa not wildly different than that of Tyr.70 While Cys S-glycosides are rare in nature, chemists have prepared and utilized synthetic S-glycosides for various purposes.71,72,73,74 Indeed, under the glycosylation conditions optimized for Boc-Tyr-OH, we observed full conversion of Boc-Cys-OH (33) to N-Boc-S-β-d-glucopyranosyl-l-cysteine (34), and the product was isolated in 98% yield (Figure 5a). In light of future applications toward complex molecule glycosylation, a model dipeptide 35 bearing both Tyr and Cys was prepared. Under the standard reaction conditions, HPLC analysis revealed 36% conversion to the bis-glycosylated product 36, 33% conversion to Boc-Tyr-Cys(S-Glc)-OH 37, 10% conversion to Boc-Tyr(O-Glc)-Cys-OH 38, and 21% starting material (35), indicating low chemoselectivity between Tyr and Cys (Figure 5b). Ongoing efforts seek to better differentiate the reactivity of Tyr and Cys and explore strategies for site-selective glycosylation.
Figure 5.

Reactivity of cysteine under the aqueous glycosylation conditions. (a) Glycosylation of Boc-Cys-OH (33). (b) Glycosylation of Boc-Tyr-Cys-OH (35).
Conclusion
The experiments described above culminate in a highly efficient phenolic O-glycosylation method of significant potential. Fundamentally, these results extend the versatility of Ca2+-promoted glycosylation reactions that employ glycosyl fluorides beyond the idiosyncratic case of sucrose and its carefully adjusted derivatives. Indeed, through extension of the classical literature, we found that the lower pKa phenols provide a strong analogy to the results of Jencks concerning nucleophilic azide, and that wide generality of phenolic structural types may undergo rapid glycosylation in water under these conditions. Moreover, the reactions often occur with a predictable stereochemical outcome. The protocol is straightforward, and the reactions proceed generally with high efficiency, within ten minutes, and at ambient temperature. The reactions also proceed in entirely aqueous solvent. Substrates, including highly complex oligopeptides, may be employed without the use of protecting groups. The functional group tolerance of the reaction appears to be quite high, as exemplified by a number of complex, bioactive oligopeptide substrates. Numerous amino acid side chains are spectators when a Tyr residue is present in the sequence, and macrocyclic rings including side-chain disulfide bridges are tolerated, with no significant degradation through either hydrolysis or β-elimination pathways. Cys residues, on the other hand, appear to react competitively at the –SH in addition to the –OH position of Tyr, revealing a mode of reactivity to be considered when applying this method. On the one hand, this side reaction represents a current shortcoming of the approach. On the other hand, numerous applications of the present chemistry seem possible, and the site-selective Tyr versus Cys glycosylation points to one challenge we now hope to address, among others such as the site-selective glycosyl transfer to unique Tyr (or other residues) in polyfunctional cases where more than one Tyr or Cys is present. Of course, we remain interested in whether the chemistry described herein may be adapted to the selective glycosylation of polypeptides at other, for the moment-unreactive positions, such as Ser, Thr, and the other functionalized amino acid side chains.
Data availability
The characterization data for new chemical compounds are given in Supplementary Information.
Supplementary Material
Acknowledgments
This work was initially supported by the W. M. Keck Foundation and later by National Institutes of Health (NIH GM068649). The authors would like to thank Aaron Zwicker for insightful discussions. A. Steinauer was a Howard Hughes Medical Institute International Student Research fellow. G.P. is grateful to NSERC (PDF) and FQRNT (B3) for postgraduate fellowships. L.H. acknowledges support from a National Institutes of Health postdoctoral fellowship (F32-GM-122204).
Footnotes
Author contributions: T.J.W., A. Steinauer, G.P., A. Schepartz, and S.J.M. conceived and designed the study. G.P. performed initial experiments. T.J.W., A. Steinauer, and L.H. performed the synthetic experiments and analysed data for all compounds. T.J.W., A. Steinauer, L.H., G.P., A. Schepartz, and S.J.M. co-wrote the paper.
Additional information: Supplementary Information, including a description of Methods, and chemical compound information accompany this paper at www.nature.com/naturechemistry.
Competing financial interests: The authors declare no competing financial interests.
References
- 1.Bednarska NG, Wren BW, Willcocks SJ. The importance of the glycosylation of antimicrobial peptides: natural and synthetic approaches. Drug Discov Today. 2017;22:919–926. doi: 10.1016/j.drudis.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 2.Varamini P, et al. Synthesis and biological evaluation of an orally active glycosylated endomorphin-1. J Med Chem. 2012;55:5859–5867. doi: 10.1021/jm300418d. [DOI] [PubMed] [Google Scholar]
- 3.Polt R, Dhanasekaran M, Keyari CM. Glycosylated neuropeptides: A new vista for neuropsychopharmacology? Med Res Rev. 2005;25:557–585. doi: 10.1002/med.20039. [DOI] [PubMed] [Google Scholar]
- 4.Ho HH, Gilbert MT, Nussenzveig DR, Gershengorn M. C. Glycosylation is important for binding to human calcitonin receptors Biochemistry. 1999;38:1866–1872. doi: 10.1021/bi981195e. [DOI] [PubMed] [Google Scholar]
- 5.Herzner H, Reipen T, Schultz M, Kunz H. Synthesis of glycopeptides containing carbohydrate and peptide recognition motifs. Chem Rev. 2000;100:4495–4538. doi: 10.1021/cr990308c. [DOI] [PubMed] [Google Scholar]
- 6.Moradi SV, Hussein WM, Varamini P, Simerska P, Toth I. Glycosylation, an effective synthetic strategy to improve the bioavailability of therapeutic peptides. Chem Sci. 2016;7:2492–2500. doi: 10.1039/c5sc04392a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paradís-Bas M, Tulla-Puche J, Albericio F. The road to the synthesis of “difficult peptides”. Chem Soc Rev. 2016;45:631–654. doi: 10.1039/c5cs00680e. [DOI] [PubMed] [Google Scholar]
- 8.Westerlind U. Synthetic glycopeptides and glycoproteins with applications in biological research. Beilstein J Org Chem. 2012;8:804–818. doi: 10.3762/bjoc.8.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pratt MR, Bertozzi CR. Synthetic glycopeptides and glycoproteins as tools for biology. Chem Soc Rev. 2005;34:58–68. doi: 10.1039/b400593g. [DOI] [PubMed] [Google Scholar]
- 10.Seitz O. Glycopeptide synthesis and the effects of glycosylation on protein structure and activity. ChemBioChem. 2000;1:214–246. doi: 10.1002/1439-7633(20001117)1:4<214::AID-CBIC214>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 11.Downey AM, Hocek M. Strategies toward protecting group-free glycosylation through selective activation of the anomeric center. Beilstein J Org Chem. 2017;13:1239–1279. doi: 10.3762/bjoc.13.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor MS. Catalyst-controlled, regioselective reactions of carbohydrate derivatives. Top Curr Chem. 2016;372:125–155. doi: 10.1007/128_2015_656. [DOI] [PubMed] [Google Scholar]
- 13.Lee D, Taylor MS. Catalyst-controlled regioselective reactions of carbohydrate derivatives. Synthesis. 2012;44:3421–3431. doi: 10.1007/128_2015_656. [DOI] [PubMed] [Google Scholar]
- 14.Wang LX, Amin MN. Chemical and chemoenzymatic synthesis of glycoproteins for deciphering functions. Chem Biol. 2014;21:51–66. doi: 10.1016/j.chembiol.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nakayama A, et al. Enzymatic glycosylation of vancomycin aglycon: completion of a total synthesis of vancomycin and N- and C-terminus substituent effects of the aglycon substrate. Org Lett. 2014;16:3572–3575. doi: 10.1021/ol501568t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li TL, Liu YC, Lyu SY. Combining biocatalysis and chemoselective chemistries for glycopeptide antibiotics modification. Curr Opin Chem Biol. 2012;16:170–178. doi: 10.1016/j.cbpa.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 17.Zhang C, et al. Exploiting the reversibility of natural product glycosyltransferase-catalyzed reactions. Science. 2006;313:1291–1294. doi: 10.1126/science.1130028. [DOI] [PubMed] [Google Scholar]
- 18.Demchenko AV. Handbook of chemical glycosylation: advances in stereoselectivity and therapeutic relevance. Wiley-VCH; 2008. [Google Scholar]
- 19.deGruyter JN, Malins LR, Baran PS. Residue-specific peptide modification: a chemist's guide. Biochemistry. 2017;56:3863–3873. doi: 10.1021/acs.biochem.7b00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gunnoo SB, Madder A. Bioconjugation - using selective chemistry to enhance the properties of proteins and peptides as therapeutics and carriers. Org Biomol Chem. 2016;14:8002–8013. doi: 10.1039/c6ob00808a. [DOI] [PubMed] [Google Scholar]
- 21.Koniev O, Wagner A. Developments and recent advancements in the field of endogenous amino acid selective bond forming reactions for bioconjugation. Chem Soc Rev. 2015;44:5495–5551. doi: 10.1039/c5cs00048c. [DOI] [PubMed] [Google Scholar]
- 22.Boutureira O, Bernardes GJL. Advances in chemical protein modification. Chem Rev. 2015;115:2174–2195. doi: 10.1021/cr500399p. [DOI] [PubMed] [Google Scholar]
- 23.Gamblin DP, Scanlan EM, Davis BG. Glycoprotein synthesis: an update. Chem Rev. 2009;109:131–163. doi: 10.1021/cr078291i. [DOI] [PubMed] [Google Scholar]
- 24.Davis BG. Synthesis of glycoproteins. Chem Rev. 2002;102:579–602. doi: 10.1021/cr0004310. [DOI] [PubMed] [Google Scholar]
- 25.Lamandé-Langle S, et al. ‘Click’ glycosylation of peptides through cysteine propargylation and CuAAC. Bioorg Med Chem. 2014;22:6672–6683. doi: 10.1016/j.bmc.2014.09.056. [DOI] [PubMed] [Google Scholar]
- 26.Dondoni A, Marra A. Recent applications of thiolene coupling as a click process for glycoconjugation. Chem Soc Rev. 2012;41:573–586. doi: 10.1039/c1cs15157f. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez-Gonzalez M, et al. Site-selective chemoenzymatic construction of synthetic glycoproteins using endoglycosidases. Chem Sci. 2010;1:709–715. [Google Scholar]
- 28.Bernardes GJL, et al. From disulfide- to thioether-linked glycoproteins. Angew Chem Int Ed. 2008;47:2244–2247. doi: 10.1002/anie.200704381. [DOI] [PubMed] [Google Scholar]
- 29.Krauss IJ, et al. Fully synthetic carbohydrate HIV antigens designed on the logic of the 2G12 antibody. J Am Chem Soc. 2007;129:11042–11044. doi: 10.1021/ja074804r. [DOI] [PubMed] [Google Scholar]
- 30.Mu J, Roach PJ. Characterization of human glycogenin-2, a self-glucosylating initiator of liver glycogen metabolism. J Biol Chem. 1998;273:34850–34856. doi: 10.1074/jbc.273.52.34850. [DOI] [PubMed] [Google Scholar]
- 31.Zarschler K, et al. Protein tyrosine O-glycosylation—A rather unexplored prokaryotic glycosylation system. Glycobiology. 2010;20:787–798. doi: 10.1093/glycob/cwq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halim A, et al. Site-specific characterization of threonine, serine, and tyrosine glycosylations of amyloid precursor protein/amyloid β-peptides in human cerebrospinal fluid. Proc Natl Acad Sci U S A. 2011;108:11848–11853. doi: 10.1073/pnas.1102664108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lafite P, Daniellou R. Rare and unusual glycosylation of peptides and proteins. Nat Prod Rep. 2012;29:729–738. doi: 10.1039/c2np20030a. [DOI] [PubMed] [Google Scholar]
- 34.Wang P, Nilsson J, Brinkmalm G, Larson G, Huang X. Synthesis aided structural determination of amyloid-β(1-15) glycopeptides, new biomarkers for Alzheimer's disease. Chem Comm. 2014;50:15067–15070. doi: 10.1039/c4cc05085a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fichna J, et al. Novel glycosylated endomorphin-2 analog produces potent centrally-mediated antinociception in mice after peripheral administration. Bioorg Med Chem Lett. 2013;23:6673–6676. doi: 10.1016/j.bmcl.2013.10.041. [DOI] [PubMed] [Google Scholar]
- 36.Jansson AM, et al. Solid-phase glycopeptide synthesis of tyrosine-glycosylated glycogenin fragments as substrates for glucosylation by glycogenin. J Chem Soc, Perkin Trans. 1996;1:1001–1006. [Google Scholar]
- 37.Fahmi NE, Dedkova L, Wang B, Golovine S, Hecht SM. Site-specific incorporation of glycosylated serine and tyrosine derivatives into proteins. J Am Chem Soc. 2007;129:3586–3597. doi: 10.1021/ja067466n. [DOI] [PubMed] [Google Scholar]
- 38.Michael A. On the synthesis of helicon and phenolglucoside. Am Chem J. 1879;1:305–312. [Google Scholar]
- 39.Brito-Arias M. Synthesis and characterization of glycosides. Springer International Publishing; Cham: 2016. pp. 81–168. [Google Scholar]
- 40.Capicciotti CJ, et al. O-aryl-glycoside ice recrystallization inhibitors as novel cryoprotectants: a structure–function study. ACS Omega. 2016;1:656–662. doi: 10.1021/acsomega.6b00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Henderson AS, Medina S, Bower JF, Galan MC. Nucleophilic aromatic substitution (SNAr) as an approach to challenging carbohydrate–aryl ethers. Org Lett. 2015;17:4846–4849. doi: 10.1021/acs.orglett.5b02413. [DOI] [PubMed] [Google Scholar]
- 42.Li Y, Mo H, Lian G, Yu B. Revisit of the phenol O-glycosylation with glycosyl imidates, BF3·OEt2 is a better catalyst than TMSOTf. Carbohydr Res. 2012;363:14–22. doi: 10.1016/j.carres.2012.09.025. [DOI] [PubMed] [Google Scholar]
- 43.Jacobsson M, Malmberg J, Ellervik U. Aromatic O-glycosylation. Carbohydr Res. 2006;341:1266–1281. doi: 10.1016/j.carres.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 44.Jensen KJ. O-Glycosylations under neutral or basic conditions. J Chem Soc, Perkin Trans. 2002;1:2219–2233. [Google Scholar]
- 45.Park Y, et al. Macrocyclic bis-thioureas catalyze stereospecific glycosylation reactions. Science. 2017;355:162–166. doi: 10.1126/science.aal1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hahm HS, et al. Automated glycan assembly of complex oligosaccharides related to blood group determinants. J Org Chem. 2016;81:5866–5877. doi: 10.1021/acs.joc.6b00554. [DOI] [PubMed] [Google Scholar]
- 47.Mydock LK, Demchenko AV. Mechanism of chemical O-glycosylation: from early studies to recent discoveries. Org Biomol Chem. 2010;8:497–510. doi: 10.1039/b916088d. [DOI] [PubMed] [Google Scholar]
- 48.Nicolaou KC, Mitchell HJ. Adventures in carbohydrate chemistry: new synthetic technologies, chemical synthesis, molecular design, and chemical biology. Angew Chem Int Ed. 2001;40:1576–1624. [PubMed] [Google Scholar]
- 49.Toshima K. Glycosyl fluorides in glycosidations. Carbohydrate Res. 2000;327:15–26. doi: 10.1016/s0008-6215(99)00325-0. [DOI] [PubMed] [Google Scholar]
- 50.Shimizu M, Togo H, Yokoyama M. Chemistry of glycosyl fluorides. Synthesis. 1998;1998:799–822. [Google Scholar]
- 51.Willems LI, et al. From covalent glycosidase inhibitors to activity-based glycosidase probes. Chem Eur J. 2014;20:10864–10872. doi: 10.1002/chem.201404014. [DOI] [PubMed] [Google Scholar]
- 52.Rempel BP, Withers SG. Covalent inhibitors of glycosidases and their applications in biochemistry and biology. Glycobiology. 2008;18:570–586. doi: 10.1093/glycob/cwn041. [DOI] [PubMed] [Google Scholar]
- 53.Evans MJ, Cravatt BF. Mechanism-based profiling of enzyme families. Chem Rev. 2006;106:3279–3301. doi: 10.1021/cr050288g. [DOI] [PubMed] [Google Scholar]
- 54.Banait NS, Jencks WP. Reactions of anionic nucleophiles with α-D-glucopyranosyl fluoride in aqueous solution through a concerted, ANDN (SN2) mechanism. J Am Chem Soc. 1991;113:7951–7958. [Google Scholar]
- 55.Banait NS, Jencks WP. General-acid and general-base catalysis of the cleavage of α-D-glucopyranosyl fluoride. J Am Chem Soc. 1991;113:7958–7963. [Google Scholar]
- 56.Pelletier G, Zwicker A, Allen CL, Schepartz A, Miller SJ. Aqueous glycosylation of unprotected sucrose employing glycosyl fluorides in the presence of calcium ion and trimethylamine. J Am Chem Soc. 2016;138:3175–3182. doi: 10.1021/jacs.5b13384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bohé L, Crich D. A propos of glycosyl cations and the mechanism of chemical glycosylation; the current state of the art. Carbohydrate Res. 2015;403:48–59. doi: 10.1016/j.carres.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chan J, Sannikova N, Tang A, Bennet AJ. Transition-state structure for the quintessential SN2 reaction of a carbohydrate: reaction of α-glucopyranosyl fluoride with azide ion in water. J Am Chem Soc. 2014;136:12225–12228. doi: 10.1021/ja506092h. [DOI] [PubMed] [Google Scholar]
- 59.Stubbs JM, Marx D. Glycosidic Bond formation in aqueous solution: on the oxocarbenium intermediate. J Am Chem Soc. 2003;125:10960–10962. doi: 10.1021/ja035600n. [DOI] [PubMed] [Google Scholar]
- 60.Micheel F, Klemer A. Über den Reaktionsmechanismus der Glycosidbildung aus alpha- und beta-1-Fluor-Derivaten der D-Glucose und D-Mannose. Chem Ber. 1958;91:663–667. [Google Scholar]
- 61.Halcomb RL, Danishefsky SJ. On the direct epoxidation of glycals: application of a reiterative strategy for the synthesis of b-linked oligosaccharides. J Am Chem Soc. 1989;111:6661–6666. [Google Scholar]
- 62.Underwood CR, et al. Crystal structure of glucagon-like peptide-1 in complex with the extracellular domain of the glucagon-like peptide-1 receptor. J Biol Chem. 2010;285:723–730. doi: 10.1074/jbc.M109.033829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kieffer TJ, Habener JF. The glucagon-like peptides. Endocr Rev. 1999;20:876–913. doi: 10.1210/edrv.20.6.0385. [DOI] [PubMed] [Google Scholar]
- 64.Zadina JE, et al. Endomorphins: Novel endogenous mu-opiate receptor agonists in regions of high mu-opiate receptor density. Ann N Y Acad Sci. 1999;897:136–144. doi: 10.1111/j.1749-6632.1999.tb07885.x. [DOI] [PubMed] [Google Scholar]
- 65.Podlogar BL, et al. Conformational analysis of the endogenous μ-opioid agonist endomorphin-1 using NMR spectroscopy and molecular modeling. FEBS Lett. 1998;439:13–20. doi: 10.1016/s0014-5793(98)01202-2. [DOI] [PubMed] [Google Scholar]
- 66.Noda M, et al. Isolation and structural organization of the human preproenkephalin gene. Nature. 1982;297:431–434. doi: 10.1038/297431a0. [DOI] [PubMed] [Google Scholar]
- 67.Burgus R, et al. Primary structure of the ovine hypothalamic luteinizing hormone-releasing factor (LRF) Proc Nat Acad Sci U S A. 1972;69:278–282. doi: 10.1073/pnas.69.1.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Donaldson ZR, Young LJ. Oxytocin, vasopressin, and the neurogenetics of sociality. Science. 2008;322:900–904. doi: 10.1126/science.1158668. [DOI] [PubMed] [Google Scholar]
- 69.Austin LA, Heath H., 3rd Calcitonin physiology and pathophysiology. N Engl J Med. 1981;304:269–278. doi: 10.1056/NEJM198101293040505. [DOI] [PubMed] [Google Scholar]
- 70.Haynes WM, editor. CRC Handbook of chemistry and physics. 97th. CRC Press/Taylor & Francis; Boca Raton, FL: Internet Version 2017. [Google Scholar]
- 71.Lian G, Zhang X, Yu B. Thioglycosides in carbohydrate research. Carbohydr Res. 2015;403:13–22. doi: 10.1016/j.carres.2014.06.009. [DOI] [PubMed] [Google Scholar]
- 72.Brimble MA, et al. Synthesis of the antimicrobial S-linked glycopeptide, glycocin F. Chem Eur J. 2015;21:3556–3561. doi: 10.1002/chem.201405692. [DOI] [PubMed] [Google Scholar]
- 73.Rojas-Ocáriz V, et al. Design of α-S-neoglycopeptides derived from MUC1 with a flexible and solvent-exposed sugar moiety. J Org Chem. 2016;81:5929–5941. doi: 10.1021/acs.joc.6b00833. [DOI] [PubMed] [Google Scholar]
- 74.Kahne D, Walker S, Cheng Y, Van Engen D. Glycosylation of unreactive substrates. J Am Chem Soc. 1989;111:6881–6882. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The characterization data for new chemical compounds are given in Supplementary Information.