

Abstract
Halogenation is commonly used in medicinal chemistry to improve the potency of pharmaceutical leads. While synthetic methods for halogenation present selectivity and reactivity challenges, halogenases have evolved over time to perform selective reactions under benign conditions. The optimization of halogenation biocatalysts has utilized enzyme evolution and structure-based engineering alongside biotransformation in a variety of systems to generate stable site-selective variants. The recent improvements in halogenase-catalyzed reactions has demonstrated the utility of these biocatalysts for industrial purposes, and their ability to achieve a broad substrate scope implies a synthetic tractability with increasing relevance in medicinal chemistry.
Introduction
Enzymes have evolved to catalyze reactions with high efficiency and selectivity, and can overcome inherent chemical biases with complex substrates. Halogenation is particularly important in medicinal chemistry and drug design, and with the aid of biocatalysts, chemists can implement efficient methods for halogenation that are environmentally benign. Since the discovery of halogenating enzymes (halogenases), an effort has been made to engage their selective reactivity in syntheses of complex molecules such as natural products. Thus, the collective knowledge gained from the characterization of these enzymes has led to a major effort in halogenase discovery and engineering for biocatalyst development.1–4 This has included methodologies in structure-based engineering5,6 and directed evolution7 where biocatalysts with scalable capabilities8 have been generated. Additionally, halogenases have been implemented in vivo using various expression systems to optimize biotransformation of complex molecules.
Most halogenases are currently grouped into four classes including 1) haloperoxidases (heme-containing and vanadium-containing), 2) iron (II)/2-(oxo)-glutarate (FeII/2OG)-dependent halogenases, 3) flavin-dependent halogenases (FDHs), and 4) fluorinases. Haloperoxidases tend to be nonselective due to the freely diffusing hypohalous acid that acts as the halogenating agent, whereas FeII/2OG-dependent halogenases proceed through a radical mechanism to halogenate aliphatic carbons on a variety of substrates. FDHs utilize hypohalous acid similarly to the haloperoxidases, but they implement a selectivity mechanism involving the formation of chloramine on a catalytic lysine in the enzyme active site.9 Enzymatic fluorination requires unique conditions that enable desolvation of the fluoride ion, which subsequently performs a nucleophilic attack on the substrate. Numerous studies relating to the mechanism and function of FDHs have been reported, from which a foundation for halogenase engineering has been built. Recent studies have included the first application of computational chemistry to analyze the mechanism of FDH-catalyzed chlorination.10
Bioactive halogenated natural products and halogenating enzymes
Many of the over 4,000 known halogenated natural products have therapeutic potential. The antitumor agent rebeccamycin,11 antibiotic vancomycin,12,13 and vasorelaxant malbrancheamide14,15 are structurally complex compounds, and strategies toward their synthesis would benefit from new halogenation methods (Fig. 1). The challenges involved in preparing these materials through synthetic methods have motivated researchers to investigate the biosynthetic machinery involved in the assembly and late-stage tailoring of these secondary metabolites. Current chemical halogenation methods are rarely selective and tend to involve the use of toxic reagents. The rebeccamycin halogenase RebH has been extensively characterized9,16,17 and the wealth of biochemical information has been utilized toward enzyme engineering efforts. RebH binds tryptophan, a biosynthetic precursor, and catalyzes chlorination at the Trp C7 position prior to incorporation into the metabolite by additional biosynthetic enzymes (Fig. 1a). The halogenase involved in vancomycin biosynthesis (VhaA) performs a late-stage dihalogenation of the carrier protein bound hexapeptide precursor prior to cyclization and offloading of the final product (Fig. 1b).18 Late-stage halogenation of complex molecules is particularly challenging, but tethering a substrate to a carrier protein presents its own biosynthetic demands. Numerous fungal FDHs that react with freestanding complex molecules have been identified,19–24 including MalA which is a late-stage halogenase involved in malbrancheamide biosynthesis (Fig. 1c).10 Chloramphenicol is a well-known antibiotic that is chlorinated by the FDH CmlS, which bears a rare covalently bound flavin cofactor, and its biosynthetic halogenation remains under investigation.25–28 Molecular dissection has enabled engineering strategies directed toward improving stability, expanding substrate scope, and altering site-selectivity of FDHs.
Figure 1.
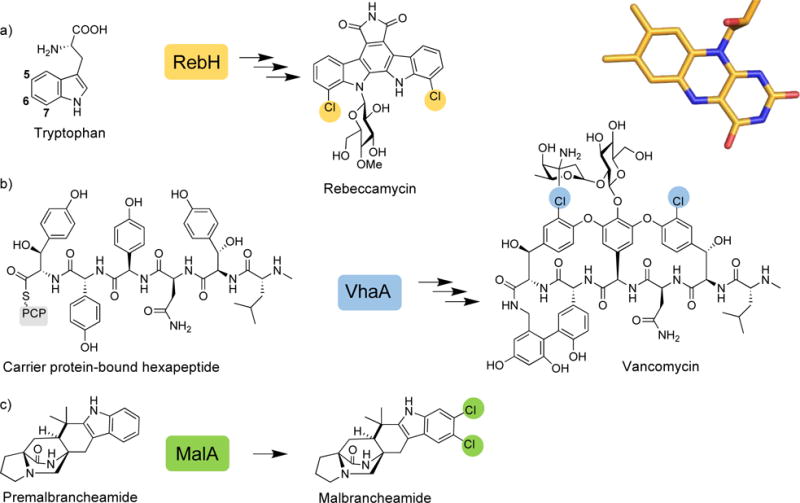
Flavin-dependent halogenases involved in the production of therapeutically relevant natural products. PCP denotes peptidyl carrier protein.
Halogenation and drug design
One third of the drugs currently in clinical trials are halogenated, which underscores the significance of methods for introducing halogen atoms into pharmaceuticals.29 Halogens can aid in controlling metabolism and improving pharmacological properties such as lipophilicity and permeability. Additionally, the halogen functionality can facilitate target binding which was originally thought to be a consequence of electron-withdrawing and steric effects. More recently, investigators have begun studying the “halogen bond” and its utility in generating more selective therapeutics.30 Due to the anisotropic nature of the halogen atom, a region of positive potential (sigma hole) can interact with a negatively charged region on the protein to form a halogen bond,31,32 the strength of which directly correlates with the size of the halogen itself. In fact, the strongest halogen bond (between iodine and a backbone carbonyl) is comparable in energy to the weakest hydrogen bond.33 Typically, halogen bonds have been observed either between the ligand halogen and aromatic amino acids or a backbone carbonyl.32 The interactions with aromatic side chains are considered either edge-on or face-on, where the halogen approaches the aromatic ring from the periphery (edge-on) or interacts directly with the center of the aromatic ring (face-on).29,30 Structural analysis of proteins bound to halogenated ligands has indicated that the edge-on interaction is more common and tends to involve phenylalanine and histidine side chains. In contrast, the face-on interaction frequently involves the side chains of tryptophan and tyrosine residues, potentially due to their higher π densities.30 When the halogen atom is attached to an aromatic region of the ligand, these bond strengths are potentiated.
The pharmacokinetic properties of drugs can be modulated by the insertion of a halogen substituent such as fluorine, which is commonly substituted for aromatic hydrogens that are prone to metabolism. In a study of N-benzylamide-based thrombin inhibitors, para substitution of fluorine showed the highest increase in potency presumably due to the decrease in metabolism.34 The halogen bond has also been utilized to facilitate the selective binding of certain cytochrome P450 enzymes (CYPs).29 Cl-π interactions between the cardiovascular therapeutics ticlopidine and amlodipine and CYP2B4 stabilize a conformation conducive to metabolism that is not observed when the molecules bind CYP2B6. The Cl-π interaction is not only significant for therapeutic targets, but it can also potentiate reactivity of some biosynthetic halogenases involved in producing halogenated natural products. Recent work that focused on the characterization of an iterative halogenase, MalA, found that a Cl-π interaction facilitated the binding of the monochlorinated ligands, leading to a higher catalytic efficiency for the second halogenation reaction (Fig. 2).10 Detailed mechanistic and structural analyses of halogenases have been instrumental in the development of these enzymes as biocatalysts. With insight into how these proteins work, engineering efforts have produced selective halogenases that perform chemical transformations on therapeutically relevant small molecules.
Figure 2.
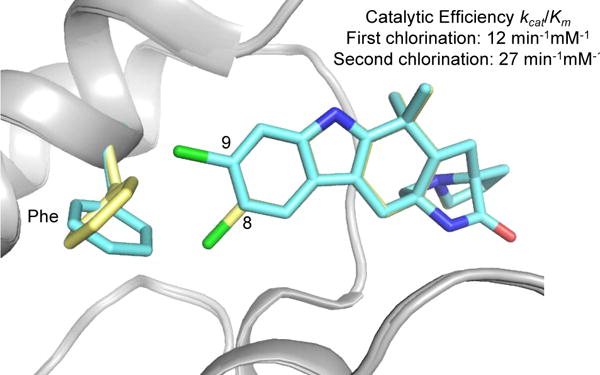
Cl-π interaction in MalA halogenase. The C9 chlorinated substrate is shown in blue (PDB ID: 5WGW) and the C8 chlorinated substrate is shown in yellow (PDB ID: 5WGZ).
Halogenase engineering
Enzyme engineering has long been employed in efforts to provide robust and selective biocatalysts for applications in synthetic chemistry. Engineering strategies have included directed evolution35 as well as structure-guided protein engineering,5 both of which have been utilized to generate improved biocatalysts for halogenation. Directed evolution involves selecting a parent enzyme that performs a reaction of interest and randomly mutating the polypeptide to produce a library of variants. In contrast, a structure-guided approach relies on knowledge of the protein structure and mechanism for rational selection of mutations that are predicted to alter the reactivity. Although unpredictable, directed evolution has produced biocatalysts with increased thermostability,36 varied substrate scope,37 and altered site-selectivity.7 Structure-guided engineering presents a more rational approach that can be used for well-characterized proteins. This strategy has produced biocatalysts with reactivity comparable to traditional synthetic methods for halogenation,8 as well as with improved reactivity38 and site-selectivity.5,6 A key difference between these two methods is that developing a hind-site rationale for enhancement in enzyme activity is often difficult for directed evolution.
Enzyme stability and optimal activity
In order to utilize biocatalysts for chemical transformations, the enzymes must be highly efficient. In principle, a more efficient biocatalyst would exhibit both increased stability and higher reactivity. Based on this concept, initial studies using directed evolution to develop a thermostable FDH provided a RebH variant with a melting temperature 18 °C higher and an optimal reaction temperature 5 °C higher than the wild-type enzyme.36 The thermostable RebH variant also had improved conversion and a longer half-life at higher reaction temperatures. At lower reaction temperatures, which were optimal for the wild-type enzyme, the catalytic efficiency of the variant was much lower than that of wild-type RebH. This work demonstrated that the stability of an enzyme is not necessarily a reflection of its activity. In this example, the thermostable RebH variant may have been less active at physiological temperature due to constrained conformational flexibility.36 Flavin-dependent proteins in general have been reported to exhibit broad conformational flexibility between the oxidized and reduced cofactor bound forms. The reduced flavin cofactor is required for halogenase activity, and it has been supplemented into the system using a variety of methods,8,39 as described below.
The use of FDHs as biocatalysts can be challenging due to the enzymatic systems required to generate the reduced flavin (FADH2) that is necessary for their activity. A nonspecific NAD(P)H-dependent flavin reductase (FR), such as RebF in rebeccamycin biosynthesis,16 often provides the FADH2 for these reactions. In the most efficient scenarios, the NAD(P)H is regenerated through a recycling system consisting of either glucose dehydrogenase39 or alcohol dehydrogenase (Fig. 3).8 One hypothesis is that bringing the enzymes in this system into close proximity would allow for a faster rate of cofactor exchange. Frese, et al. immobilized these macromolecular components into aggregates and achieved gram-scale bromination of tryptophan by RebH.8 The aggregates could not only be reused up to ten times, but they were also amenable to storage for up to four months at 4°C with no apparent decrease in activity. Immobilization of the enzyme components also streamlined the purification of the brominated product. Moreover, in contrast to in vivo bioconversion methods, chloride ion contamination could be avoided in the immobilized system, limiting the production of chlorinated molecules when bromination was desired.
Figure 3.
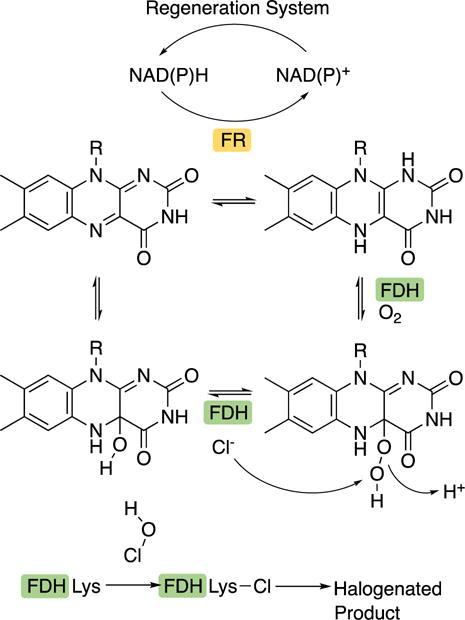
Flavin-dependent halogenase cofactor regeneration.
Rather than link all of the enzymes in the system together, Andorfer et al.40 applied a technique previously shown to be successful in generating productive P450/reductase fusion enzymes.41 The autologous FR was linked to RebH with the expectation that the increased local concentration of FADH2 would increase enzyme activity. While reductase activity was maintained, the kinetic parameters of the halogenation reaction for the fusion protein were diminished. When excess reductase was added to the reaction mixture, the halogenation catalyzed by the fusion protein increased, indicating that the FR must be present in a higher concentration than the halogenase to provide adequate levels of FADH2 for efficient catalysis. When the halogenase and reductase were present in a 1:1 ratio the activity was decreased in both the fusion and the standalone systems, indicating that the local concentration of reductase in proximity to the halogenase was not pertinent to increasing the activity. While in vitro reactions of the fusion protein did not produce higher conversion rates, the in vivo fusion protein assays demonstrated higher conversion, which may have been due to the presence of an endogenous FR enhancing generation of the reduced cofactor.
Cofactor regeneration can also be achieved by using an in vivo system for biotransformation. By expressing Trp 7-halogenase RebH and Trp 5-halogenase PyrH,42 alongside FR in Madagascar periwinkle (Catharanthus roseus), the incorporation of halogenated indole into various medicinally relevant alkaloids was observed (Fig. 4).43 The halogenated Trp was converted to the tryptamine precursor by the endogenous Trp-decarboxylase for the targeted indole alkaloid products. While wild-type C. roseus produced minor amounts of 19,20-dihydroakuammicine, the chlorinated analog 12-chloro-19,20-dihydroakuammicine was produced in high quantities in the RebH/RebF16 variant, demonstrating the inherent flexibility of the downstream enzymes to accept the 7-chlorotryptamine precursor. A similar study where RebH and Trp-6-halogenase SttH44 were expressed in Nicotiana benthamiana revealed that the localization of the FDH/FR system to the chloroplasts could aid cofactor regeneration.45 Localization of halogenases to the chloroplasts in the absence of exogenous FR led to comparable product formation indicating that the plastids provide a sufficient supply of reducing agents from native metabolic pathways.46 The targeted coexpression of the Trp decarboxylase was required for production of halogenated tryptamine derivatives as the chloroplasts are devoid of this enzyme. Although most of the FDHs discussed are derived from bacterial hosts, the concept of transferring these biocatalysts to plants is particularly intriguing with respect to their less well-studied fungal counterparts, as these may be more effectively expressed in a eukaryotic host.
Figure 4.
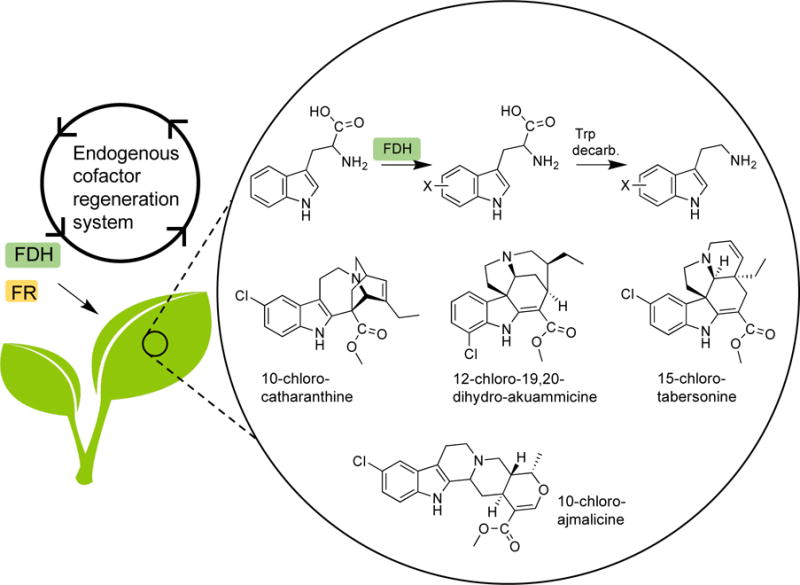
Incorporation of FDHs into plants to produce halogenated precursors to complex indole alkaloids.
Structure-based halogenase engineering
The halogenases involved in preparation of bioactive natural products are most synthetically useful when a clear understanding of their function has been determined. The first structure of the FDH PrnA47 led to a mechanistic proposal that continues to be refined, and initial attempts to modulate the selectivity of these enzymes were structure-based. In the case of PrnA, a structural analysis of substrate complexes led to the mutation of a phenylalanine residue that appeared to interact directly with the indole ring of the tryptophan substrate.6 This mutation induced a shift in the site-selectivity of the chlorination and bromination reactions from solely C7-halogenation to both C7 and C5 due to differential substrate positioning near the chloramine adduct that the enzyme is thought to employ for selective halogenation.9
Further structure-based engineering of RebH resulted in a mutant form that preferentially accepted tryptamine over its native tryptophan substrate.48 The introduction of this mutant into C. roseus led to the selective chlorination of tryptamine and its incorporation into 12-chloro-19,20-dihydroakuammicine. The in vivo biosynthetic applications of the chlorinated tryptamine precursor included preparation of halogenated analogs of molecules with therapeutic potential (Fig. 4). The further utility of the tryptamine-selective mutant in this system depended on the flexibility of the downstream biosynthetic machinery toward accepting the halogenated precursor. Rational engineering of halogenase function has also been applied to the Trp 7-FDH PrnA and Trp 5-FDH PyrH.38 The substrate scope of these two halogenases was assessed on small molecules that were aromatic, but lacked the indole ring present in tryptophan. A key substrate with more mobility in the active site, anthranilic acid, was selected to query engineered PrnA. Basic residues were substituted into the active site to coordinate the molecule and position it for selective halogenation. Single point mutations led to an increase in activity and selectivity between the para and ortho positions. By combining two of the most significant mutations, a large increase in activity was observed. X-ray crystal structures of the mutants displayed shifts in residues important for substrate binding, but the mechanism of altered selectivity remained ambiguous due to the lack of bound substrate in the structures. The structure of the first Trp 6-halogenase, SttH, guided rational engineering efforts toward more selective halogenation.5 The factors contributing to site-selective halogenation were determined by comparing the structures of the Trp 7-halogenases (RebH and PrnA) and the Trp 5-halogenase PyrH to that of SttH (Fig. 5). A triple mutant was prepared by targeting residues in a region close to the core of the Trp substrate (L460, P461, and P462). Introduction of these mutations tuned the selectivity of the Trp 6-halogenase, which was then able to chlorinate at the C5 position as well. Upon utilizing a substrate with more flexibility in the active site (3- indolepropionic acid), the SttH triple mutant produced primarily 5-chloro-3-indole-propionic acid. Alternatively, when the corresponding triple mutant of PyrH was produced in an attempt to shift its selectivity from the C5 to C6 position, the protein was inactive. This work provided a rational strategy for switching the site-selectivity of a FDH without decreasing the catalytic efficiency of the enzyme. The observed differences between the different FDHs were exploited to design halogenation biocatalysts with tailored site-selectivity. This approach was applied to additional substrates and, combined with the immobilization methods of Frese et al.,8 demonstrated the synthetic utility of these biocatalysts on a preparative scale.
Figure 5.
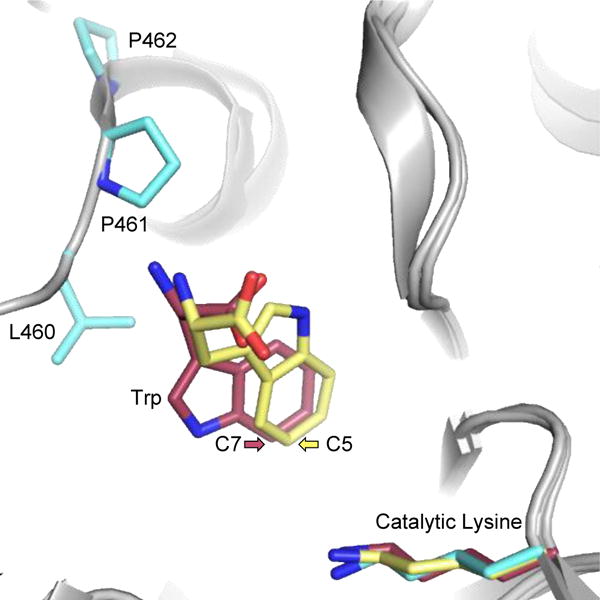
Overlay of Trp 7-halogenase PrnA (pink, PDB ID: 2JKC), Trp 6-halogenase SttH (cyan, PDB ID: 5HY5), and Trp 5-halogenase PyrH (yellow, PDB ID: 2WET). Residues 460-462 of SttH were mutated to modulate the regioselectivity toward the Trp substrate.
A similar comparison of the crystal structures of the tetrahalogenase Bmp2 and dihalogenase Mpy16 led to the identification of structural elements responsible for controlling the degree of pyrrole halogenation.49 A Bmp2 triple mutant with the corresponding residues from Mpy16 produced a monohalogenated product, indicating that the substrate may have been repositioned. The same substitution in Mpy16 led to insoluble protein, thus the ability to shift the selectivity of FDHs may require modulation of amino acids outside the immediate vicinity of the substrate.
Structure-based engineering efforts have also been implemented to change the type of C–H functionalization reaction performed by an enzyme. After acquiring a detailed mechanistic understanding of FeII/2OG-dependent oxygenases and halogenases, an effort to convert an oxygenase to the latter was undertaken.50 FeII/2OG halogenases are members of the oxygenase family and are highly similar to related enzymes that catalyze hydroxylation reactions. Unlike FDHs, FeII/2OG-dependent halogenases perform chlorination or bromination reactions on aliphatic carbons through a radical-based mechanism. The structure of the halogenase WelO5 provided information about substrate binding that was critical to this engineering effort.51,52 The mechanism in both FeII/2OG-dependent oxygenases and halogenases proceeds through a ferryl (FeIV-O) intermediate, which abstracts a hydrogen atom from the substrate, leaving behind a substrate carbon-centered radical and FeIII-OH. In hydroxylases, the substrate radical reacts with the FeIII-OH species to yield a hydroxylated product. The iron coordination differs between hydroxylases, which utilize a carboxylate residue, and halogenases where this residue is substituted with an alanine/glycine. In the latter, an additional coordination site is open for a halide ion to bind and react with the substrate radical. With the aid of the WelO5 structure, it was determined that in order for halogenation to occur, the substrate must be located away from the ferryl intermediate and in a conformation unique to FeII/2OG-dependent halogenases. A hydroxylase with high structural similarity to WelO5 was identified and the FeII carboxylate ligand was mutated to glycine. With a structurally similar protein, it was hypothesized that the substrate would bind optimally for halogenation, and only the single amino acid substitution would be required for conversion to a halogenase. Both chlorinated and brominated products were acquired using this mutant, while some hydroxylation was still observed. This work provided a proof of concept that a thorough understanding of reaction mechanism could be exploited to achieve a switch in reaction type.
Directed evolution for engineered halogenases
In contrast to structure-based protein engineering strategies, RebH has also been subjected to a thorough directed evolution campaign. The pioneering effort to stabilize the enzyme toward a more tractable biocatalyst led to an increased melting temperature, and a more active enzyme.36 The substrate scope of the thermostable RebH was expanded, and the mutants were shown to chlorinate a variety of complex unnatural substrates.37 Previous structure-based work had demonstrated the ability of a RebH mutant (Y455W) to selectively halogenate tryptamine over tryptophan.48 This idea was utilized in subsequent work where RebH was engineered for optimal activity on tryptamine (N470S) to facilitate the employment of a high-throughput screening strategy for assessing halogenation selectivity.7 The method involved assaying RebH mutants (generated through directed evolution) with 7-deuterotryptamine to assess halogenation at alternative sites by mass spectrometry. Error-prone PCR was employed to generate a library of RebH mutants with excellent selectivity for chlorinating the C6- and C5-positions of the tryptamine indole ring. Mutations identified from previous work were incorporated into the evolution process,36 and degenerate codons were utilized at sites that showed particular selectivity when varied. Although the process appears relatively straightforward, the system became more complex when the authors found that reverting some of the original mutations in the newer variants led to higher reactivity while maintaining selectivity. After preparation of the selective mutants, the authors attempted to rationalize the mechanism of selectivity through docking models. For each mutant, the docking simulations produced a variety of substrate poses, some of which were consistent with the observed selectivity. For the C7- and C5-selective halogenases, the docking results included poses that were highly similar to those found in the crystal structures of the Trp 7-halogenase RebH and Trp 5-halogenase PyrH substrate complexes. The engineered C6-halogenase provided mixed results, with no clear indication of the mechanism of selectivity. Shortly after this work was performed, the first structure of a Trp 6-halogenase (SttH) was solved, further aiding this analysis.5
To expand the inquiry into FDH utility in synthetic approaches, a collaborative academic-industry partnership enabled a large survey of substrate scope for a number of natural and engineered FDHs.53 The biocatalysts included wild-type RebH (Trp 7-halogenase), ThaI (Trp 6-halogenase), several engineered RebH mutants, and a set of marginally characterized fungal FDHs. The RebH mutants were selected based on previous work that identified enzymes exhibiting increased activity on nonnative substrates37,38 as well as altered site selectivity on tryptamine.7 Through this screen, the investigators determined that fungal FDHs preferentially halogenated substrates that were less nucleophilic than bacterial Trp FDHs. Using computational methods, the authors predicted that there was a minimal electronic activation (HalA) required for halogenation by FDHs through the proposed electrophilic aromatic substitution. Accordingly, potential substrates could be more accurately predicted. In previous directed evolution studies with these proteins, it was observed that the electronic activation is not the only factor contributing to their site-selectivity. Docking studies were useful for developing hypotheses regarding substrate binding, but developments using these methods were not always insightful. While a broad range of substrates were analyzed in this work,53 FDH activity could only be correlated with the calculations of HalA and the key component of structural validation remained to be analyzed.
An interesting analysis was conducted between the chlorination reactions catalyzed by the FDHs compared to the common small-molecule reagent N-chlorosuccinimide (NCS).53 The halogenases mediated a site-selective reaction, whereas NCS typically produced mixtures of regioisomers. Additionally, the use of NCS sometimes led to dihalogenated molecules, which was not observed for the corresponding FDHs.16 The authors concluded that this was due to the decreased activity on halogenated substrates. Monohalogenation has been uniquely overcome by the fungal FDH MalA which displayed a higher catalytic efficiency for the second of two chlorination reactions on the complex indole substrate premalbrancheamide.10 The surprising variation within this class of enzymes indicates that discovery of new FDHs will continue to broaden the repertoire of halogenating biocatalysts.
Fluorination biocatalysts
The elusive fluorinase is of particular interest in medicinal chemistry due to the frequent use of fluorine to improve potency and selectivity of pharmaceutical lead compounds. Fluorine is frequently substituted for hydrogen in therapeutic molecules during the development phase because it tends to maintain or enhance target binding. Fluorine can also effectively modulate the metabolic profile and pharmacokinetic properties of the molecule.34,54,55 Synthetic methods for fluorination represent a discrete focus of medicinal chemistry because of the challenges associated with fluoride ion desolvation. Without the use of harsh synthetic strategies, a fluorinase enzyme from Streptomyces cattleya was found to produce the free F− ion, which could perform a substitution reaction on S-adenosyl-L-methionine (SAM) to produce 5′-fluorodeoxyadenosine (5′-FDA) and 4-fluoro-threonine.56–58 S. cattleya also produced fluoroacetate which could then be incorporated into therapeutically relevant natural products such as polyketides and steroids, via fluoromalonyl-CoA (Fig. 6).55 Novel fluorinated natural products have been produced through feeding fluorinated precursors into biosynthetic systems, as well as by genetically engineering fluorinases into the producing organism of interest.59 The fluorinase gene from S. cattleya was genetically incorporated into Salinospora tropica to produce a fluorinated salinosporamide A, which is a naturally chlorinated anticancer agent.59,60 These strategies are only favorable when the downstream biosynthetic machinery can accommodate the fluorinated intermediates. In the S. tropica system, endogenous enzymes were found to prefer the natural chlorinated substrates over the unnatural fluorinated molecules, and a disruption of the chlorinase gene was generated for optimal fluorine incorporation. Following discovery of the first fluorinase, an in silico genome mining effort was conducted to identify four additional enzymes within this family.61,62
Figure 6.
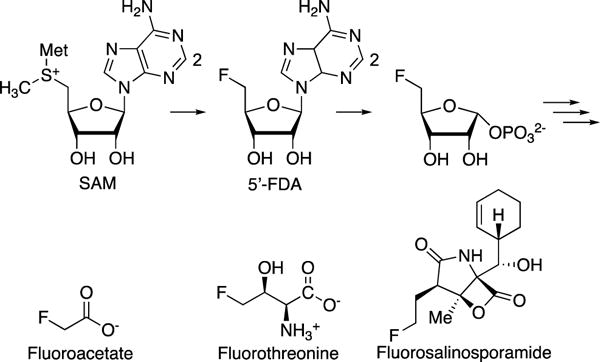
Biosynthesis of fluorinated molecules.
The fluorinase from S. cattleya has also been used in the preparation of 18F-labeled imaging reagents for medical applications.63,64 Biocatalytic fluorination presents a significant advancement in 18F chemistry because this radioisotope is usually prepared using a cyclotron with [18O]-H2O, leaving the final 18F− ion in aqueous conditions that are not suitable for the subsequent chemistry.65 Fluorinase-catalyzed functionalization can utilize the 18F− ion under aqueous conditions efficiently without having to deviate from ambient temperature and neutral pH. In an application of biocatalytic 18F fluorination, a fluorinase was paired with a nucleoside hydrolase to produce 5-[18F]-fluoro-5-deoxyribose which was then used in tumor imaging.63
Initial efforts to engineer fluorinase enzymes started with a comparison of the chlorinase in S. tropica to the fluorinase in S. cattleya which have 35% amino acid identity. Upon comparison of the halide ion binding site, an attempt was made to convert the chlorinase to a fluorinase by substituting the respective halide-binding residues.57,66 Structural67,68 and computational studies69 of the fluorinase and chlorinase provided insights into the mechanism of halide selection. Crystal structures of the fluorinase in complex with chloride ion (since F− is indistinguishable from water) showed that the halide coordination is reliant on S158 and T80 in the fluorinase which are substituted with G131 and Y70 in the chlorinase, respectively (Fig. 7).56,58,67
Figure 7.
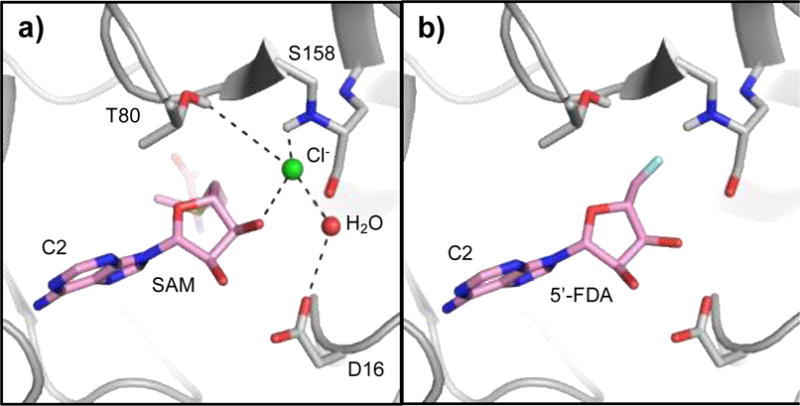
a) The mechanism of halide binding was demonstrated in crystal structures of the S. cattleya fluorinase with SAM and Cl−. The halide is proposed to be further coordinated by S158 to desolvate the F− ion for fluorination activity (PDB ID: 2V7U). b) Product complex with 5′-FDA. (PDB ID: 2V7V).
Although, unsuccessful, this endeavor provided evidence that the halide binding site was not the only mechanism of halide selectivity. In recent advances in biocatalytic fluorination, more complex analogs of the traditional 5′-FDA were prepared through late-stage fluorination of C2-substituted molecules, including those bearing click-chemistry handles.66,70 One shortcoming of biocatalysis is that enzymes tend to exhibit reduced catalytic efficiency with unnatural substrates, which has been observed in the previously discussed halogenases, and fluorinases are no exception. Saturation mutagenesis was used to generate more active mutants, while reaction conditions such as temperature and leaving group options on the substrate were optimized.70 Further understanding of the molecular determinants of substrate specificity based on the work of Sun, et al. led to an expanded substrate scope and higher reactivity on unnatural substrates.66 The field of fluorine chemistry has benefited greatly from advances in fluorination biocatalysis, adding to the significance of biohalogenation as a whole.
Conclusions
Halogenases have been engineered to perform a variety of reactions on molecules relevant to medicinal chemistry and drug development. Enzyme engineering efforts have produced halogenases with greater stability and reactivity bringing the field closer to industrial applicability. The work toward plant-based systems for in vivo halogenation of small molecules has succeeded in bringing the complexity of the system (including cofactors and multiple accessory proteins) down to a practical level. This has also been realized through the use of cross-linked enzyme aggregates and fusion proteins that produced a more viable system for large-scale catalysis. As biocatalytic halogenation systems become more robust and scalable, they will become increasingly attractive to academic, medicinal, and process chemists working on drug development as well as total syntheses.
Acknowledgments
The authors thank the National Science Foundation under the CCI Center for Selective C–H Functionalization (CHE-1700982), NIH grant R35 GM118101, and the Hans W. Valteich Professorship (to D.H.S.) We are also indebted to LSI Multimedia Designer Stephanie King for contributions in preparation of the graphical abstract.
References
- 1.a Latham J, Brandenburger E, Shepherd SA, Menon BRK, Micklefield J. Development of halogenase enzymes for use in synthesis. Chem Rev. 2018;118:232–269. doi: 10.1021/acs.chemrev.7b00032. [DOI] [PubMed] [Google Scholar]; b Andorfer MC, Lewis JC. Understanding and improving the activity of flavin-dependent halogenases by random and targeted mutagenesis. Ann Rev Biochem. doi: 10.1146/annurev-biochem-062917-012042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agarwal V, Miles ZD, Winter JM, Eustáquio AS, El Gamal AA, Moore BS. Enzymatic halogenation and dehalogenation reactions: pervasive and mechanistically diverse. Chem Rev. 2017;117:5619–5674. doi: 10.1021/acs.chemrev.6b00571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schnepel C, Sewald N. Enzymatic halogenation: a timely strategy for regioselective C–H activation. Chemistry. 2017;23:12064–12086. doi: 10.1002/chem.201701209. [DOI] [PubMed] [Google Scholar]
- 4.Weichold V, Milbredt D, van Pée K-H. Specific enzymatic halogenation-from the discovery of halogenated enzymes to their applications in vitro and in vivo. Angew Chem Int Ed. 2016;55:6374–6389. doi: 10.1002/anie.201509573. [DOI] [PubMed] [Google Scholar]
- 5.Shepherd SA, Menon BRK, Fisk H, Struck A-W, Levy C, Leys D, Micklefield J. A structure-guided switch in the regioselectivity of a tryptophan halogenase. ChemBioChem. 2016;17:821–824. doi: 10.1002/cbic.201600051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lang A, Polnick S, Nicke T, Williams P, Patallo EP, Naismith JH, van Pée K-H. Changing the regioselectivity of the tryptophan 7-halogenase PrnA by site-directed mutagenesis. Angew Chem Int Ed. 2011;50:2951–2953. doi: 10.1002/anie.201007896. [DOI] [PubMed] [Google Scholar]
- 7.Andorfer MC, Park HJ, Vergara-Coll J, Lewis JC. Directed evolution of RebH for catalyst-controlled halogenation of indole C–H bonds. Chem Sci. 2016;7:3720–3729. doi: 10.1039/c5sc04680g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frese M, Sewald N. Enzymatic halogenation of tryptophan on a gram scale. Angew Chem Int Ed. 2015;54:298–301. doi: 10.1002/anie.201408561. [DOI] [PubMed] [Google Scholar]
- 9.Yeh E, Blasiak LC, Koglin A, Drennan CL, Walsh CT. Chlorination by a long-lived intermediate in the mechanism of flavin-dependent halogenases. Biochemistry. 2007;46:1284–1292. doi: 10.1021/bi0621213. [DOI] [PubMed] [Google Scholar]
- 10.Fraley AE, Garcia-Borràs M, Tripathi A, Khare D, Mercado-Marin EV, Tran H, Dan Q, Webb GP, Watts KR, Crews P, Sarpong R, Williams RM, Smith JL, Houk KN, Sherman DH. Function and Structure of MalA/MalA’, iterative halogenases for late-stage C–H functionalization of indole alkaloids. J Am Chem Soc. 2017;139:12060–12068. doi: 10.1021/jacs.7b06773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pruddhomme M. Recent developments of rebeccamycin analogues as topoisomerase I inhibitors and antitumor agents. Curr Med Chem. 2000;7:1189–1212. doi: 10.2174/0929867003374138. [DOI] [PubMed] [Google Scholar]
- 12.Nitanai Y, Kikuchi T, Kakoi K, Hanamaki S, Fujisawa I, Aoki K. Crystal structures of the complexes between vancomycin and cell-wall precursor analogs. J Mol Biol. 2009;385:1422–1432. doi: 10.1016/j.jmb.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 13.Yim G, Thaker MN, Koteva K, Wright G. Glycopeptide antibiotic biosynthesis. J Antibiot. 2014;67:31–41. doi: 10.1038/ja.2013.117. [DOI] [PubMed] [Google Scholar]
- 14.Madariaga-Mazón A, Hernández-Abreu O, Estrada-Soto S, Mata R. Insights on the vasorelaxant mode of action of malbrancheamide. J Pharm Pharmacol. 2015;67:551–558. doi: 10.1111/jphp.12346. [DOI] [PubMed] [Google Scholar]
- 15.Martínez-Luis S, Rodríguez R, Acevedo L, González MC, Lira-Rocha A, Mata R. Malbrancheamide, a new calmodulin inhibitor from the fungus Malbranchea aurantiaca. Tetrahedron. 2006;62:1817–1822. [Google Scholar]
- 16.Yeh E, Ganeau S, Walsh CT. Robust in vitro activity of RebF and RebH, a two-component reductase/halogenase, generating 7-chloro-tryptophan during rebeccamycin biosynthesis. Proc Natl Acad Sci. 2005;102:3960–3965. doi: 10.1073/pnas.0500755102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bitto E, Huang Y, Bingman CA, Singh S, Thorson JS, Phillips GN., Jr The structure of flavin-dependent tryptophan 7-halogenase RebH. Proteins. 2007:289–293. doi: 10.1002/prot.21627. [DOI] [PubMed] [Google Scholar]
- 18.Schmartz PC, Zerbe K, Abou-Hadeed K, Robinson JA. Bis-chlorination of a hexapeptide-PCP conjugate by the halogenase involved in vancomycin biosynthesis. Org Biomol Chem. 2014;12:5574–5577. doi: 10.1039/c4ob00474d. [DOI] [PubMed] [Google Scholar]
- 19.Cacho RA, Chooi Y-H, Zhou H, Tang Y. Complexity generation in fungal polyketide biosynthesis: a spirocycle-forming P450 in the concise pathway to the antifungal drug griseofulvin. ACS Chem Biol. 2013;8:2322–2330. doi: 10.1021/cb400541z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chankhamjon P, Tsunematsu Y, Ishida-Ito M, Sasa Y, Meyer F, Boettger-Schmidt D, Urbansky B, Menzel K-D, Scherlach K, Watanabe K, Hertweck C. Regioselective dichlorination of a non-activated aliphatic carbon atom and phenolic bismethylation by a multifunctional fungal flavoenzyme. Angew Chem Int Ed. 2016;55:11955–11959. doi: 10.1002/anie.201604516. [DOI] [PubMed] [Google Scholar]
- 21.Ferrara M, Perrone G, Gambacorta L, Epifani F, Solfrizzo M, Gallo A. Identification of a halogenase involved in the biosynthesis of ochratoxin A in Aspergillus carbonarius. 2016;82:5631–5641. doi: 10.1128/AEM.01209-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Menon BRK, Brandenburger E, Sharif HH, Klemstein U, Shepherd SA, Greaney MF, Micklefield J. RadH: a versatile halogenase for integration into synthetic pathways. Angew Chem Int Ed. 2017;56:11841–11845. doi: 10.1002/anie.201706342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wick J, Heine D, Lackner G, Misiek M, Tauber J, Jagusch H, Hertweck C, Hoffmeister D. A fivefold parallelized biosynthetic process secures chlorination of Armillaria mellea (honey mushroom) toxins. Appl Environ Microbiol. 2016;82:1196–1204. doi: 10.1128/AEM.03168-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zeng J, Zhan J. A novel fungal flavin-dependent halogenase for natural product biosynthesis. ChemBioChem. 2010;11:2119–2123. doi: 10.1002/cbic.201000439. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki T, Honda H, Katsumata R. Production of antibacterial compounds analogous to chloramphenicol by a n-paraffin-grown bacterium. Agr Biol Chem. 1972;36:2223–2228. [Google Scholar]
- 26.Schlünzen F, Zarivach R, Harms J, Bashan A, Tocilj A, Albrecht R, Yonath A, Franceschi F. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature. 2001;413:814–821. doi: 10.1038/35101544. [DOI] [PubMed] [Google Scholar]
- 27.Piraee M, White RL, Vining LC. Biosynthesis of the dichloroacetyl component of chloramphenicol in Streptomyces venezuelae ISP5230: genes required for halogenation. Microbiol. 2004;150:85–94. doi: 10.1099/mic.0.26319-0. [DOI] [PubMed] [Google Scholar]
- 28.Podzelinska K, Latimer R, Bhattacharya A, Vining LC, Zechel DL, Jia Z. Chloramphenicol biosynthesis: the structure of CmlS, a flavin-dependent halogenase showing a covalent flavin-aspartate bond. J Mol Biol. 2010;397:316–331. doi: 10.1016/j.jmb.2010.01.020. [DOI] [PubMed] [Google Scholar]
- 29.Shah MB, Liu J, Zhang Q, Stout CD, Halpert JR. Halogen-π interactions in cytochrome P450 active site: structural insights into human CYP2B6 substrate selectivity. ACS Chem Biol. 2017;12:1204–1210. doi: 10.1021/acschembio.7b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Imai YN, Inoue Y, Nakanishi I, Kitaura K. Cl-π interactions in protein-ligand complexes. Protein Sci. 2008;17:1129–1137. doi: 10.1110/ps.033910.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Politzer P, Murray JS, Clark T. Halogen bonding: an electrostatically-driven highly directional noncovalent interaction. Phys Chem Chem Phys. 2010;12:7748–7757. doi: 10.1039/c004189k. [DOI] [PubMed] [Google Scholar]
- 32.Xu Z, Yang Z, Liu Y, Lu Y, Chen K, Zhu W. Halogen bond: its role beyond drug-target binding affinity for drug discovery and development. J Chem Inf Model. 2014;54:69–78. doi: 10.1021/ci400539q. [DOI] [PubMed] [Google Scholar]
- 33.Bissantz C, Kuhn B, Stahl M. A medicinal chemist’s guide to molecular interactions. J Med Chem. 2010;53:5061–5084. doi: 10.1021/jm100112j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gillis EP, Eastman KJ, Hill MD, Donnely DJ, Meanwell NA. Applications of fluorine in medicinal chemistry. J Med Chem. 2015;58:8315–8359. doi: 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- 35.Renata H, Wang J, Arnold FH. Expanding the enzyme universe: accessing non-natural reactions by mechanism-guided directed evolution. Angew Chem Int Ed. 2015;54:3351–3367. doi: 10.1002/anie.201409470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poor CB, Andorfer MC, Lewis JC. Improving the stability and catalyst lifetime of the halogenase RebH by directed evolution. ChemBioChem. 2014;15:1286–1289. doi: 10.1002/cbic.201300780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Payne JT, Poor CB, Lewis JC. Directed evolution of RebH for site-selective halogenation of large biologically active molecules. Angew Chem Int Ed. 2015;54:4226–4230. doi: 10.1002/anie.201411901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shepherd SA, Karthikeyan C, Latham J, Struck A-W, Thompson ML, Menon BRK, Styles MQ, Levy C, Leys D, Micklefield J. Extending the biocatalytic scope of regiocomplementary flavin-dependent halogenase enzymes. Chem Sci. 2015;6:3454–3460. doi: 10.1039/c5sc00913h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Payne JT, Andorfer MA, Lewis JC. Regioselective arene halogenation using the FAD-dependent halogenase RebH. Angew Chem Int Ed. 2013;52:5271–5274. doi: 10.1002/anie.201300762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Andorfer MC, Belsare KD, Girlich AM, Lewis JC. Aromatic halogenation by using bifunctional flavin reductase-halogenase fusion enzymes. ChemBioChem. 2017;18:1–6. doi: 10.1002/cbic.201700391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li S, Podust LM, Sherman DH. Engineering and analysis of a self-sufficient biosynthetic cytochrome P450 PikC fused to the RhFRED reductase domain. J Am Chem Soc. 2007;129:12940–12941. doi: 10.1021/ja075842d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zehner S, Kotzsch A, Bister B, Süssmuth RD, Méndez C, Salas JS, van Pée K-H. A regioselective tryptophan 5-halogenase is involved in pyrroindomycin biosynthesis in Streptomyces rugospora LL-42D005. Chem Biol. 2005;12:445–452. doi: 10.1016/j.chembiol.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 43.Runguphan W, Qu X, O’Connor SE. Integrating carbon-halogen bond formation into medicinal plant metabolism. Nature. 2010;468:461–464. doi: 10.1038/nature09524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeng J, Zhan J. Characterization of a tryptophan 6-halogenase from Streptomyces toxytricini. Biotechnol Lett. 2011;33:1607–1613. doi: 10.1007/s10529-011-0595-7. [DOI] [PubMed] [Google Scholar]
- 45.Fräbel S, Krischke M, Staniek A, Warzecha H. Recombinant flavin-dependent halogenases are functional in tobacco chloroplasts without co-expression of flavin reductase genes. Biotechnol. 2016;11:1586–1594. doi: 10.1002/biot.201600337. [DOI] [PubMed] [Google Scholar]
- 46.Sandoval FJ, Zhang Y, Roje S. Flavin nucleotide metabolism in plants. J Biol Chem. 2008;283:30890–30900. doi: 10.1074/jbc.M803416200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dong C, Flecks S, Unversucht S, Haupt C, van Pée K-H, Naismith JH. Tryptophan halogenase (PrnA) structure suggests a mechanism for regioselective chlorination. Science. 2005;309:2216–2219. doi: 10.1126/science.1116510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Glenn WS, Nims E, O’Connor SE. Reengineering a tryptophan halogenase to preferentially chlorinate a direct alkaloid precursor. J Am Chem Soc. 2011;133:19346–19349. doi: 10.1021/ja2089348. [DOI] [PubMed] [Google Scholar]
- 49.El Gamal A, Agarwal V, Diethelm S, Rahman I, Schorn MA, Sneed JM, Louie GV, Whalen KE, Mincer TJ, Noel JP, Paul VJ, Moore BS. Biosynthesis of coral settlement cue tetrabromopyrrole in marine bacteria by a uniquely adapted brominase-thioesterase enzyme pair. Proc Natl Acad Sci. 2016;113:3797–3802. doi: 10.1073/pnas.1519695113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mitchell AJ, Dunham NP, Bergman JA, Wang B, Zhu Q, Chang W-c, Liu X, Boal AK. Structure-guided reprogramming of a hydroxylase to halogenate its small molecule substrate. Biochemistry. 2017;56:441–444. doi: 10.1021/acs.biochem.6b01173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hillwig LM, Liu X. A new family of iron-dependent halogenases acts on freestanding substrates. Nat Chem Biol. 2014;10:921–923. doi: 10.1038/nchembio.1625. [DOI] [PubMed] [Google Scholar]
- 52.Mitchell AJ, Zhu Q, Maggiolo AO, Ananth N, Hillwig ML, Liu X, Boal AK. Structural basis for halogenation by iron- and 2-oxo-glutarate-dependent enzyme WelO5. Nat Chem Biol. 2016;12:636–640. doi: 10.1038/nchembio.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andorfer MC, Grob JE, Hajdin CE, Chael JR, Siuti P, Lilly J, Tan KL, Lewis JC. Understanding flavin-dependent halogenase reactivity via substrate activity profiling. ACS Catal. 2017;7:1897–1904. doi: 10.1021/acscatal.6b02707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weeks AM, Coyle SM, Jinek M, Doudna JA, Chang MCY. Structural and biochemical studies of a fluoroacetyl-CoA-specific thioesterase reveal a molecular basis for fluorine selectivity. Biochemistry. 2010;49:9269–9279. doi: 10.1021/bi101102u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Walker MC, Thuronyi BW, Charkoudian LK, Lowry B, Khosla C, Chang MCY. Expanding the fluorine chemistry of living systems using engineered polyketide synthase pathways. Science. 2013;341:1089–1094. doi: 10.1126/science.1242345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Deng H, Cobb SL, McEwan AR, McGlinchey RP, Naismith JH, O’Hagan D, Robinson DA, Spencer SB. The fluorinase from Streptomyces cattleya is also a chlorinase. Angew Chem Int Ed Engl. 2006;45:759–762. doi: 10.1002/anie.200503582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deng H, O’Hagan D. The fluorinase, the chlorinase and the duf-62 enzymes. Cur Opin Chem Biol. 2008;12:582–59. doi: 10.1016/j.cbpa.2008.06.036. [DOI] [PubMed] [Google Scholar]
- 58.Zhu X, Robinson DA, McEwan AR, O’Hagan D, Naismith JH. Mechanism of enzymatic fluorination in Streptomyces cattleya. J Am Chem Soc. 2007;129:14597–14604. doi: 10.1021/ja0731569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eustáquio AS, O’Hagan D, Moore BS. Engineering fluorometabolite production: fluorinase expression in Salinospora tropica yields fluorosalinosporamide. J Nat Prod. 2010;73:378–382. doi: 10.1021/np900719u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Williams PG, Buchanon GO, Feling RH, Kauffman CA, Jensen PR, Fenical W. New cytotoxic salinosporamides from the marine actinomycete Salinispora tropica. J Org Chem. 2005;70:6196–6203. doi: 10.1021/jo050511+. [DOI] [PubMed] [Google Scholar]
- 61.Deng H, Ma L, Bandaranayaka N, Qin Z, Mann G, Kyeremeh K, Yu Y, Shepherd T, Naismith JH, O’Hagan D. Identification of fluorinases from Streptomyces sp MA37, Norcardia brasiliensis, and Actinoplanes sp N902-109 by genome mining. ChemBioChem. 2014;15:364–368. doi: 10.1002/cbic.201300732. [DOI] [PubMed] [Google Scholar]
- 62.O’Hagan D, Deng H. Enzymatic fluorination and biotechnological developments of the fluorinase. Chem Rev. 2015;115:634–649. doi: 10.1021/cr500209t. [DOI] [PubMed] [Google Scholar]
- 63.Dall’Angelo S, Bandaranayaka N, Windhorst AD, Vugts DJ, van der Born D, Onega M, Schweiger LF, Zanda M, O’Hagan D. Tumour imaging by positron emission tomography using fluorinase generated 5-[18F]fluoro-5-deoxyribose as a novel tracer. Nucl Med Biol. 2013;40:464–470. doi: 10.1016/j.nucmedbio.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 64.Thompson S, Onega M, Ashworth S, Fleming IN, Passchier J, O’Hagan D. A two-step fluorinase enzyme mediated 18F labelling of an RGD peptide for positron emission tomography. Chem Commun. 2015;51:13542–13545. doi: 10.1039/c5cc05013h. [DOI] [PubMed] [Google Scholar]
- 65.Thompson S, Fleming IN, O’Hagan D. Enzymatic transhalogenation of dendritic RGD peptide constructs with the fluorinase. Org Biomol Chem. 2016;14:3120–3129. doi: 10.1039/c6ob00239k. [DOI] [PubMed] [Google Scholar]
- 66.Yeo WL, Chew X, Smith DJ, Chan KP, Sun H, Zhao H, Lim YH, Ang EL. Probing the molecular determinants of fluorinase specificity. Chem Commun. 2017;53:2559–2562. doi: 10.1039/c6cc09213f. [DOI] [PubMed] [Google Scholar]
- 67.Dong C, Huang F, Deng H, Schaffrath C, Spencer JB, O’Hagan D, Naismith JH. Crystal structure and mechanism of a bacterial fluorinating enzyme. Nature. 2004;427:561–565. doi: 10.1038/nature02280. [DOI] [PubMed] [Google Scholar]
- 68.Zhu X, Robinson DA, McEwan AR, O’Hagan D, Naismith JH. Mechanism and enzymatic fluorination in Streptomyces cattleya. J Am Chem Soc. 2007;129:14597–14604. doi: 10.1021/ja0731569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Senn HM. Insight into enzymatic halogenation from computational studies. Front Chem. 2014;2:1–15. doi: 10.3389/fchem.2014.00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun H, Yeo WL, Lim YH, Chew X, Smith DJ, Xue B, Chan KP, Robinson RC, Robins EG, Zhao H, Ang EL. Directed evolution of a fluorinase for improved fluorination efficiency with a non-native substrate. Angew Chem Int Ed. 2016;55:14277–14280. doi: 10.1002/anie.201606722. [DOI] [PubMed] [Google Scholar]