

Abstract
The association between obesity and breast cancer risk and prognosis is well established in ER-positive disease but less clear in HER2-positive disease. Here, we report preclinical evidence suggesting weight maintenance through calorie restriction may limit risk of HER2-positive breast cancer. In female MMTV-HER2/neu transgenic mice, we found that ERα and ERβ expression, mammary tumorigenesis and survival are energy balance-dependent in association with epigenetic reprogramming. Mice were randomized to receive a calorie restriction (CR), overweight (OW)-inducing, or diet-induced obesity (DIO) regimen (n = 27/group). Subsets of mice (n = 4/group/time point) were euthanized after 1, 3 and 5 months to characterize diet-dependent metabolic, transcriptional, and epigenetic perturbations. Remaining mice were followed up to 22 months. Relative to the OW and DIO regimens, CR decreased body weight, adiposity, and serum metabolic hormones as expected, and also elicited an increase in mammary ERα and ERβ expressioñ Increased DNA methylation accompanied this pattern, particularly at CpG dinucleotides located within binding or flanking regions for the transcriptional regulator CCCTC-binding factor (CTCF) of ESR1 and ESR2, consistent with sustained transcriptional activation of ERα and ERβ. Mammary expression of the DNA methylation enzyme DNMT1 was stable in CR mice but increased over time in OW and DIO mice, suggesting CR obviates epigenetic alterations concurrent with chronic excess energy intake. In the survival study, CR elicited a significant suppression in spontaneous mammary tumorigenesis. Overall, our findings suggest a mechanistic rationale to prevent or reverse excess body weight as a strategy to reduce HER2-positive breast cancer risk.
Keywords: Breast cancer, DNA methylation/epigenetics, hormone receptors and diagnosis/prognosis, preclinical studies of endocrine-related cancers, receptors and signal transduction, animal/transgenic models in promotion and prevention, biological and biochemical mechanisms in prevention, diet and cancer, molecular markers in prevention research
INTRODUCTION
Breast cancer is the most frequently diagnosed noncutaneous neoplasm among women in the United States and is the leading cause of cancer death among women worldwide (1,2). Approximately 30% of all breast cancers lack estrogen receptor alpha (ERα), which confers a worse prognosis in comparison to ERα-positive breast tumors (3). Furthermore, the expression of estrogen receptor beta (ERβ), a putative tumor suppressor, is also lost in most ERα-negative breast tumors (4). ERβ is the more prevalent ER in normal mammary tissue, but its expression is reduced during tumor formation. Numerous studies have linked greater breast tumor ERβ expression with an improved prognosis (5, 6). However, the degree to which ERα and ERβ expression is impacted by dietary energy balance and/or controlled epigenetically in breast tumorigenesis remains unclear.
Obesity, a result of chronic positive dietary energy balance, is an established risk factor for postmenopausal breast cancer and may also enhance risk in premenopausal women with additional breast cancer risk factors, including type 2 diabetes (7, 8). In addition, excess energy intake and increased adiposity have been associated with greater breast tumor size, increased progression markers, and therapy resistance in both pre- and postmenopausal women (9–11). In contrast, the maintenance of a negative energy balance via calorie restriction (CR) prevents weight gain and inhibits the development of several types of cancer, including ER-positive and ER-negative breast cancers, in numerous animal models (12, 13). CR also has been associated with changes in several serum and tissue biomarkers in women causally linked with reduced breast cancer risk (14, 15).
Although links between energy metabolism, epigenetic regulation of gene expression, and several chronic diseases have been previously established, the relationship between dietary energy balance, epigenetics and breast cancer is poorly understood (16, 17). DNA methylation levels are regulated in part by DNA (cytosine-5)-methyltransferase 1 (DNMT1), which predominantly serves to maintain genomic DNA methylation during DNA replication (17). Thus, during times of high cell proliferation such as during development, DNMT1 is highly expressed. However, DNMT1 can become deregulated throughout the life course in response to metabolic, inflammatory and environmental disturbances, and this dysregulation has been linked with aberrant DNA methylation and cancer (16, 18–20).
The effects of DNA methylation are dependent upon the location of methyl groups in the genomic landscape. In general, promoter methylation results in transcriptionally silent genes. However, methylation in the transcription region of genes is often positively correlated with gene expression (21, 22). In addition, methylation that prevents a repressor from binding DNA can correspond to increased gene expression (23). CCCTC-binding factor (CTCF) is an 11-zinc finger protein and highly conserved transcription factor with enhancer-blocking activity (24). DNA methylation at CpG (5′-C-phosphate-G-3′) dinucleotides is inversely correlated with CTCF occupancy (23, 25). We posit that the diet-methylation-CTCF axis may serve as a critical sensory system that regulates gene transcription via energy balance-associated changes in DNA methylation at or near CTCF binding sites.
The genetically engineered Mouse Mammary Tumor Virus (MMTV)-neu mouse model is characterized by mammary gland overexpression of the oncogene human epidermal growth factor receptor (HER2). MMTV-neu mice initially have histologically normal, ERα-positive mammary glands that subsequently develop regions of ductal carcinoma in situ with ERα expression lost in most cells. If untreated, these mice ultimately develop ERα-negative, HER2-positive mammary adenocarcinomas before 24 months of age (26). In the present study, we tested the hypothesis that dietary energy balance modulation alters ERα and/or ERβ expression, DNA methylation and tumor incidence in female MMTV-neu mice.
MATERIALS AND METHODS
In vivo studies in MMTV-neu transgenic mice
All animal studies and procedures were approved and monitored by the University of Texas Institutional Animal Care and Use Committee. Female 6- to 8-week-old MMTV-neu mice (JAX stock #002376, n = 86) were purchased from Jackson Laboratory (Bar Harbor, ME, USA) and fed a modified AIN-93G semipurified diet, defined as the overweight (OW)-inducing diet for this study (catalog #D12450B, Research Diets, Inc., New Brunswick, NJ, USA) ad libitum for 1 week of acclimation.
Baseline mice
Following acclimation, a subset of mice (n = 5) were fasted for 6 hours and then euthanized by CO2 asphyxiation followed by cervical dislocation. Blood was collected by cardiac puncture, allowed to coagulate for 30 minutes at room temperature, and centrifuged at 10,000 × g for 5 minutes; serum was removed and stored at −80°C for subsequent analyses. Mammary tissues were collected for further molecular and pathological analysis.
Time point study
A subset of 36 mice were singly housed and randomized (n = 12/diet group) to one of the following three diet treatment groups (each modified from the OW diet, which is AIN-93G semipurified diet formulation) for a 5-month time point study: 1) calorie restriction regimen (CR), a low-fat, low-carbohydrate diet (#D0302702); 2) OW diet, a high-carbohydrate, low-fat diet providing 3.8 kcal/g (#D12450B); or 3) diet-induced obesity regimen (DIO), a high-carbohydrate, high-fat diet providing 5.2 kcal/g (#D12492), all from Research Diets, Inc. CR mice were administered their diet formulation as daily aliquots of food that provided 70% of the kcal but 100% of the vitamins, minerals, essential fatty acids and amino acids relative to the OW group. Mice were weighed weekly. After 1, 3 and 5 months on diet, mice were analyzed for percent body fat using quantitative magnetic resonance spectroscopy (Echo Medical Systems, Houston, TX, USA). At each of these same time points 4 mice per diet group were killed, and serum and nontumor-bearing mammary tissue were collected as described above. No tumors developed in any mice in the time point study.
Survival study
The remaining subset of 45 mice were singly housed and randomized (n = 15/diet group) to the CR, OW or DIO diet regimens and were followed for survival for up to 22 months. The survival curve for each diet group illustrates time-to-event data, with the event consisting of either death or the presence of a mammary tumor > 1.0 cm in any direction, the IACUC approved maximal tumor size (27). Non-mammary tumor-related deaths were censored. Mice were palpated for mammary tumors weekly. Once detected, tumor diameters were measured in two dimensions twice weekly with electronic calipers. When tumor diameter reached 1.0 cm in either dimension (or after 22 months of study in the absence of tumor), mice were killed. Tumor and/or distal mammary tissue were collected and processed. One half of each collected tissue sample was fixed in 10% neutral buffered formalin for 24 hours, transferred to 70% ethanol for at least 24 hours, embedded in paraffin, and cut into 4 μm thick sections for hematoxylin and eosin (H&E) staining or immunohistochemical analysis. The other half was placed in a cryotube, flash frozen in liquid nitrogen and stored at −80°C for subsequent molecular analyses. Blood was collected by cardiac puncture and serum was isolated for analysis.
Analyses of circulating energy balance-related hormones and 17β-estradiol
Serum samples from all 5 mice at baseline, and all 4 mice per diet group at the 1-, 3- and 5-month time points were collected after mice were fasted to reduce variability of metabolic hormones. Estrous cycle was not assessed. Serum samples were analyzed for leptin, insulin, insulin-like growth factor (IGF)-1, and adiponectin by Luminex-based bead array assay (Millipore, Billerica, MA, USA) read on MagPix multianalyte detection system (BioRad, Hercules, CA, USA). The mean inter-assay coefficient of variation (C.V.) of multiplexed bead-based assays for metabolic hormone detection has been shown to be <15% in published studies (28). Serum 17β-estradiol was measured by ELISA (Alpha Diagnostics, San Antonio, TX, USA).
Real-time quantitative reverse transcription (qRT)-PCR analyses of ERα and ERβ
Total RNA was extracted using TRI-Reagent (Sigma-Aldrich, St. Louis, MO, USA) according to manufacturer’s instructions from the flash-frozen mammary tissue samples collected at baseline (n = 5 mice) and each of the 3 time points (n = 4 mice/diet group/time point). RNA was also extracted from nontumor-bearing, flash-frozen mammary tissue collected from mice in the survival study upon their termination (between 14 and 22 months). RNA concentration was spectrophotometrically determined using a nanodrop (Thermo Scientific, Logan, UT, USA), and quality was confirmed using an Agilent 2100 Bioanalyzer (Santa Clara, CA, USA). RNA was reverse transcribed with Multiscribe RT (Applied Biosystems, Carlsbad, CA, USA) and resulting cDNA were assayed in triplicate using Taqman® Gene Expression Assays for ERα, ERβ and DNMT1 (Applied Biosystems). PCR reactions were monitored by a ViiA™7 Real time PCR system (Applied Biosciences). Gene expression data were normalized to the housekeeping gene β-actin and analyzed using the delta delta cycle threshold method.
Histopathologic and immunohistochemical analyses
Tumors were examined for histopathological markers of tumor progression, including vascularity (presence of blood vessels) and proliferation (number of mitotic figures per field) in H&E sections by a board-certified veterinary pathologist. Vascularity was graded in a blinded fashion on a categorical score for the entire slide (0 = no intratumoral blood vessels present, 1 = low number of vessels present, 2 = medium number of vessels present, 3 = high number of vessels present). Mitotic figures were counted in 5 non-overlapping fields of view, and a mean number of mitotic figures was determined for each mouse. Values from each mouse were used to calculate mean vascularity score and mitotic figures for each diet group.
Immunohistochemical staining of mammary tissue was performed (n = 4 mice/diet group) using a primary antibody for ERα (Catalog #sc542, Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 1:500 and ERβ (Abcam #3576, Cambridge, MA, USA) at 1:100. The secondary antibody was horseradish peroxidase-labeled anti-rabbit antibody (DAKO Cytomation, Carpinteria, CA, USA).
DNA methylation analysis
DNA was extracted from a random sample (n = 3/group) of mammary tissues from baseline mice and CR and OW mice in the 5-month time point and survival study using UltraPure™ phenol:chloroform:isoamyl alcohol per manufacturer’s instructions (Life Technologies). Library preparation and sequencing for baseline, CR and OW in the 5-month time point and survival study were performed at UT MD Anderson Cancer Center’s DNA Methylation Analysis Core and Science Park Next-Generation Sequencing Facility, according to published protocols as previously described (29) (Supplementary Table S1). Samples from DIO mice were not analyzed given the cost of RRBS and the similarities between OW and DIO mice in ERα and ERβ mRNA and protein expression. Gene promoter regions were calculated based on RefSeq gene annotations with regions starting 1 kb upstream of the annotated transcription start site (TSS) and extending 500 base pairs downstream of TSS. Differential methylation was calculated by filtering samples based on read coverage ≥ 20, then performed at the single base level. Methylkit R package was used to apply logistic regression and the likelihood ratio test. Observed p-values were adjusted with the success likelihood index method (SLIM). CpG dinucleotides that exhibited differential methylation patterns between CR and OW groups were cross-referenced with annotated gene regulatory regions within and surrounding ESR1 and ESR2 in Mus musculus outlined by Ensembl (30). To generate a heat map, we identified CpG dinucleotides with significantly higher methylation in CR survival vs. OW survival mice that also had percent methylated DNA values available for baseline and CR and OW mice at the 5-month time point. Differentially methylated CpG dinucleotides were clustered using hierarchical clustering with complete linkage and a Euclidian distance measure. Corresponding dendrogram and heat maps for promoter, intron, exon and other were produced using the heatmap.2 function from the gplots package in R (version 3.3.1).
Differential expression analysis using RNA-seq and Ingenuity Pathway Analysis (IPA)
RNA was extracted as described above. RNA libraries were prepared using the Illumina TruSeq Stranded Total RNA Sample Preparation kit according to manufactures instructions. The libraries were sequenced using a 2×76 bases paired end protocol on the Illumina HiSeq 2000 instrument. The reads were mapped to mouse genome (mm10) by TopHat (version 2.0.7) (31). The number of fragments in each known gene from RefSeq database (32) (UCSC Genome Browser 2013) was enumerated using HTSeq-count from HTSeq package (version 0.5.3p9) (HTSeq). The differential expression between conditions was statistically assessed by R/Bioconductor package EdgeR (version 1.10.1). Genes with false discovery rate ≤ 0.05 were called significant. For pathway analysis, genes with differential expression in tissue from CR vs. OW mice after 5 months on diet and/or in survival study mice were integrated into IPA pathway designer (Qiagen, Venlo, Netherlands) to draw connections of regulatory relationships using validated scientific findings.
Statistical analyses
Values are presented as mean ± standard deviation (s.d.). For all tests, GraphPad Prism software was used (GraphPad Software Inc., La Jolla, CA), and P < 0.05 was considered statistically significant. Differences between diet groups in body weight were analyzed by repeated measures analysis of variance (ANOVA) followed by Tukey’s post hoc test. Differences between diet groups in insulin, leptin, adiponectin, IGF-1, 17β-estradiol and mammary ERα and ERβ mRNA and protein expression at each time point were analyzed by one-way ANOVA followed by Tukey’s post hoc test. Kaplan-Meier survival curves were plotted, and the difference in overall survival between the groups was analyzed by the log-rank test.
RESULTS
Dietary energy balance modulation impacts body weight, body composition and serum metabolic hormones
Female MMTV-neu mice were randomized to receive dietary energy balance modulation via CR, overweight-inducing (OW), or diet-induced obesity (DIO) diet regimens, and were monitored as part of a 1, 3 and 5 month time point study (n = 4 mice/diet for each time point) or for up to 22 months in a survival study (n = 15 mice/diet; hereafter referred to as “survival study mice”). CR, OW, and DIO diet regimens resulted in lean, overweight, and obese phenotypes, respectively. After 1 month of diet treatment, differences in average caloric intake (Supplementary Figure S1) produced differences in mean body weight (Figure 1A), with CR < OW < DIO. This continued for up to 15 months in the survival study mice, after which tumor development in OW and DIO mice made body weight measurements unstable. CR mice had significantly lower percent body fat (Figure 1B) compared with DIO mice after 1, 3 and 5 months on diet. OW mice had intermediate percent body fat between CR and DIO.
Figure 1.
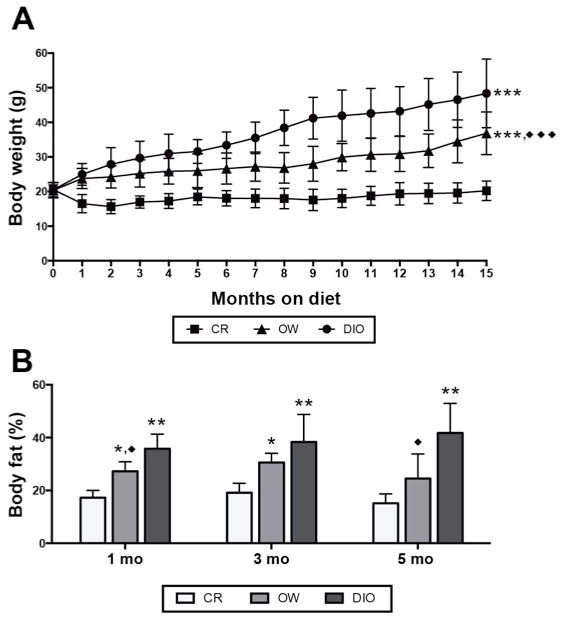
Dietary energy balance modulation affects body weight and percent body fat in MMTV-neu mice. (A) Body weight (mean ± s.d.) in mice receiving calorie restriction (CR), overweight-inducing (OW), or diet-induced obesity (DIO) diet regimens over 15 months (n = 27 mice/diet). Heterogeneity in body weights after 15 months increased as mice became moribund therefore weight data beyond this point are not shown. Statistical differences in body weights between groups were determined by repeated measures analysis of variance (ANOVA). (B) Percent body fat assessed by quantitative magnetic resonance spectroscopy (mean ± s.d.) at 1, 3 and 5 months on diet (n = 4 mice/diet/time point). Statistical differences in body fat between groups were determined by one-way ANOVA. OW or DIO vs. CR: *P < 0.05, **P < 0.01, ***P < 0.001. OW vs. DIO: ◆P < 0.05, ◆◆P < 0.01, ◆◆◆P < 0.001
Compared with CR mice, OW and DIO mice were metabolically dysregulated as assessed by energy balance-related hormones (Table 1). DIO mice, compared with CR mice, had higher serum levels of insulin, leptin, insulin-like growth factor (IGF-1) and 17β-estradiol, and lower serum levels of adiponectin, consistent with obesity-associated metabolic perturbations. OW mice generally displayed intermediate levels of these metabolic factors after 1, 3 and 5 months on diet.
Table 1.
Energy balance impacts serum metabolic hormones
| Months on Diet | CR | OW | DIO | ||
|---|---|---|---|---|---|
| Insulin (ng/mL) | Baseline | 0.83 ± 0.40 | |||
| 1 | 0.5 ± 0.3 | 0.7 ± 0.1 | 1.4 ± 0.7 | ||
| 3 | 0.3 ± 0.1 | 0.8 ± 0.6 | 1.0 ± 0.6 | ||
| 5 | 0.2 ± 0.1 | 0.7 ± 0.2 | 1.8 ± 0.5***,◆◆ | ||
| Leptin (ng/mL) | Baseline | 0.94 ± 0.38 | |||
| 1 | 0.7 ± 0.5 | 2.8 ± 2.7 | 4.4 ± 3.4 | ||
| 3 | 0.6 ± 0.5 | 4.1 ± 2.4* | 6.0 ± 2.7* | ||
| 5 | 0.2 ± 0.2 | 3.2 ± 2.0 | 9.2 ± 6.1* | ||
| Adiponectin (ng/mL) | Baseline | 2.02 ± 0.32 | |||
| 1 | 3.8 ± 0.5 | 3.1 ± 0.7 | 2.8 ± 0.3* | ||
| 3 | 4.5 ± 0.9 | 3.1 ± 0.3* | 2.4 ± 0.6** | ||
| 5 | 4.3 ± 0.9 | 2.6 ± 0.2** | 2.3 ± 0.5** | ||
| IGF-1 (ng/mL) | Baseline | 463.2 ± 88.7 | |||
| 1 | 232.2 ± 96.7 | 396.5 ± 70.3* | 425.4 ± 36.8* | ||
| 3 | 187.8 ± 26.5 | 344.4 ± 56.4* | 503.9 ± 112.2***,◆ | ||
| 5 | 194.9 ± 26.4 | 376.2 ± 28.5*** | 452.0 ± 57.7***, ◆ | ||
| 17 β -estradiol(pg/mL) | Baseline | 209.8 | |||
| 1 | 155.6 ± 50.9 | 180.4 ± 68.0 | 227.9 ± 36.7 | ||
| 3 | 148.7 ± 25.2 | 262.4 ± 42.1* | 260.1 ± 58.8* | ||
| 5 | 117.4 ± 17.4 | 198.1 ± 22.3* | 210.4 ± 54.5* |
Differences between diet groups in insulin, leptin, adiponectin, IGF-1, and 17β-estradiol at each time point were analyzed by one-way ANOVA followed by Tukey’s post hoc test. OW or DIO vs. CR:
P < 0.05,
P < 0.01,
P < 0.001.
OW vs. DIO:
P < 0.05,
P < 0.01,
P < 0.001.
Dietary energy balance modulation impacts mammary ERα and ERβ expression
Mammary tissue was collected from time point study mice after 1, 3 and 5 months, and from survival study mice between 14 and 22 months upon their termination. Tissues were analyzed for ERα and ERβ expression and values are reported as relative to expression in mammary tissues collected from a baseline sample of 5 mice killed before initiation of dietary modulation. CR mice, compared with OW and DIO mice, had significantly higher mammary mRNA levels of ERα after 3 and 5 months on diet and in survival study mice (Figure 2A). In addition, mammary ERα protein levels were decreased in OW and DIO mice, compared to CR mice, by 1 month and continued throughout study (Figures 2B and F).
Figure 2.

Dietary energy balance modulation affects mammary ERα and ERβ gene and protein expression. Relative mammary ERα (A) mRNA and (B) protein levels (mean ± s.d.) in CR, OW, or DIO mice at the 1-, 3- and 5-month time point and at survival (n = 4 mice/diet/time point). Data are presented as relative to baseline levels (n = 5 mice). Relative mammary ERβ (C) mRNA and (D) protein levels (mean ± s.d.) in CR, OW, and DIO mice at 1, 3 and 5 months on diet and at survival (n = 4 mice/diet/time point). Protein levels were analyzed by IHC, with percent ERα and ERβ positive nuclei determined and results presented as relative to baseline levels (n = 5 mice). (E) The ERα to ERβ ratio was calculated by dividing the percent positive nuclei for ERα by percent positive nuclei for ERβ. (F) Representative images of ERα and ERβ IHC at baseline and for each diet group at 5 months on diet. Statistical differences in ERα or ERβ mRNA and protein between diet groups were determined by one-way ANOVA. OW or DIO vs. CR: *P < 0.05, **P < 0.01, ***P < 0.001.
Relative to baseline, mammary ERβ mRNA levels were increased (at least 4-fold, on average) in tissues from CR mice, but decreased in tissues from OW and DIO mice (Figure 2C). At the 1-month time point and every time point thereafter, CR mice had significantly greater levels of both ERβ mRNA and ERβ protein compared with OW and DIO mice (Figures 2C, D, and F). No differences between OW and DIO mice in ERα expression, ERβ expression, or the ratio of ERα to ERβ were found. Significant diet-dependent differences in the ERα to ERβ ratio were detected at each time point in tissues collected from CR vs. OW mice, as well CR vs. DIO mice (Figure 2E).
Dietary energy balance modulation impacts DNA methylation within ESR1 and near ESR2
Using reduced representation bisulfite sequencing (RRBS), we analyzed diet effects on the methylation status of DNA isolated from the mammary tissue of baseline, CR and OW mice at the 5-month time point and in survival study mice. Due to the similarities between OW and DIO mice in ERα and ERβ mRNA and protein expression, samples from DIO mice were not analyzed.
Mammary DNA methylation was generally higher in CR mice than OW mice, particularly in CpG dinucleotides at CTCF binding sites or flanking regions within ESR1 and near ESR2 (Supplementary Table S2). In mammary tissue collected in the survival study from CR mice, compared with OW mice, the percentage of methylated DNA was significantly different (CR > OW) at 3 distinct intronic CpG dinucleotides (Chr10:4710028, Chr10:4710036, Chr10:4710084) at an annotated CTCF binding site within ESR1, which encodes ERα. Diet-dependent effects on ERα DNA methylation were not observed in samples from the 5-month time point (Figure 3A and Supplementary Table S3).
Figure 3.

Dietary energy balance modulation affects DNA methylation of the intron regions of ESR1 and upstream and downstream of ESR2. (A) ESR1 intron methylation (mean percent DNA methylation ± s.d.) in the mammary tissue of CR and OW mice at the 5-month time point and survival study mice. DNA methylation at three distinct CpG dinucleotides (Chr 10: 4710028, Chr 10: 4710036, and Chr 10: 4710084), which all fall within a CTCF binding site, are shown. (B) Mean percent DNA methylation ± s.d. at CpG dinucleotides upstream (Chr 12: 76184418, Chr 12: 76184436) and downstream (Chr 12: 76080926, Chr 12: 76086754, Chr 12: 76086771) of ESR2 in the mammary tissue of mice maintained on CR or OW diet regimens for 5 months or through survival are shown. These all fall within a CTCF binding site or CTCF flanking region as indicated. Statistical differences determined by logistic regression and the likelihood ratio test, p-values were adjusted with the success likelihood index method. *P < 0.05, **P < 0.01, ***P < 0.001.
In mammary tissue from CR mice compared with OW mice, collected at either the 5-month time point or the survival study, 5 CpG dinucleotides near ESR2 (the gene encoding ERβ) had significantly higher methylation within or near a CTCF binding site (Figure 3B). Specifically, significant diet-dependent effects on DNA methylation were detected in samples from the survival study at 3 CpG dinucleotides near ESR2 (Chr 12:76080926, downstream of ESR2; and Chr 12:76184418 and Chr 12:76184436, both upstream) that all fell ± 1 kb of a CTCF binding site, which we define as a CTCF flanking region. We also observed significantly higher methylation in CR mice at the 5-month time point at 2 other sites (Chr 12:76086754 and Chr 12:76086771, both downstream) that fell within an annotated CTCF binding site (Figure 3B and Supplementary Table S4).
Dietary energy balance modulation impacts ERα and ERβ expression in part through epigenetic mechanisms
We investigated global diet-dependent differences in mammary DNA methylation in CR versus OW mice. After 5 months on diet, OW mice had 511 CpG dinucleotides with significantly higher methylation and 248 CpG dinucleotides with significantly lower methylation compared with CR mice. Furthermore, survival study mice possessed more pronounced diet-dependent differences in mammary DNA methylation. OW survival study mice had 651 CpG dinucleotides with significantly higher methylation and 6901 with significantly lower methylation compared with CR. Often, these genes with higher methylation in CR survival compared to OW survival had higher methylation levels in baseline and in CR and OW mice at the 5-month time point, supporting a deviance in OW survival methylation (Figures 4A and B).
Figure 4.

Differences in dietary energy balance impact genome wide methylation patterns in the mammary tissue. (A) Heat map and clustering dendrogram of mammary tissue DNA methylation. CpG dinucleotides with significant differences in methylation (CR survival vs. OW survival) are shown and clustered within genomic location, i.e. promoter, exon, intron, other. Baseline, CR and OW after 5 months on diet and CR and OW survival study mice are each represented (n = 3/group, with one column per n shown). The methylation frequency percentage ranges from 0 to 100. A value of ‘0’ is completely unmethylated and ‘100’ is fully methylated. Statistical differences (CR vs. OW survival) determined by logistic regression and the likelihood ratio test, p-values were adjusted with the success likelihood index method. (B) Representation of genomic locations of differentially methylated CpG dinucleotides, 6.5% mapped to a promoter region, 34.1% mapped to a gene exon, 25.6% mapped to an intron region, and 33.8% did not map to a promoter, exon or intron and are classified as ‘other.’ (C) Mammary DNMT1 mRNA levels, measured by qualitative real time PCR, in mice maintained on CR, OW, and DIO diet regimens for 1, 3 and 5 months or through survival. Data are presented as relative to baseline levels (n = 4 mice). Statistical differences in DNMT1 expression determined by one-way ANOVA *P < 0.05, **P < 0.01, ***P < 0.001.
Mammary gland RNA-seq analysis demonstrated that DNA (cytosine-5)-methyltransferase 1 (DNMT1) expression was significantly higher in OW compared with CR survival study mice (Supplementary Table S5). To validate RNA-seq results, we analyzed DNMT1 mRNA levels by qRT-PCR in the mammary tissue of CR, OW and DIO mice at all time points (Figure 4C). DNMT1 mRNA levels were not significantly different among CR, OW and DIO mice after 1, 3 and 5 months on diet. However, in survival study mice, mammary tissues from OW and DIO mice had significantly higher DNMT1 mRNA levels compared with mammary tissues from CR mice.
RNA-seq analysis in non-tumor mammary tissue from the survival study also identified several components of the signal transducer and activator of transcription 3 (STAT3), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), E2F transcription factor (E2F), and insulin signaling pathways, that can impact expression of DNMT1 and were significantly different in OW than CR mice (Supplementary Table S5). Each of these pathways can impact expression of DNMT1 (33–35). Thus, the metabolic and inflammation-related perturbations measured in OW and DIO mice (relative to CR mice) may underlie the observed diet-dependent changes in expression of DNMT1.
CR inhibits MMTV-neu mammary tumor development
Compared with OW and DIO diet regimens, CR was associated with significantly increased survival (Figure 5A). Survival was comparable between OW and DIO mice. After 22 months of study, only 1 OW mouse and 2 DIO mice, compared with 11 CR mice, remained alive and tumor-free. Among the tumor-bearing mice, the mammary tumors from OW (n = 12) and DIO (n = 11) mice, compared with CR mice (n = 3), were generally more vascular and consisted of more proliferating cells (Figure 5B).
Figure 5.

Dietary energy balance modulation affects survival, tumor vascularity and proliferation. In MMTV-neu mice (A) Kaplan-Meier survival curves for CR, OW and DIO mice (n = 15 mice/diet) over a 22-month period. Mice with nontumor-related deaths (1 CR, 2 OW, and 2 DIO mice) were censored. Statistical differences in survival rate determined by log-rank test (B) Photomicrographs of mammary tumor sections stained with hematoxylin and eosin and displayed at 10x or 40x magnification. Among the tumor-bearing mice, the mammary tumors from OW (n = 12) and DIO (n = 11) mice, compared with CR mice (n = 3) were more vascular (group mean ± s.d. vascularity scores: 2.6 ± 0.18, 2.2 ± 0.25, and 1.5 ± 1.5, respectively) and consisted of more proliferating cells (group mean ± s.d. number of mitotic figures, indicated by black arrowhead: 2.6 ± 0.5, 2.4 ± 0.3 and 0.5 ± 0.4, respectively). Statistical differences in vascularity and mitotic figures determined by one-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001.
DISCUSSION
This study assessed whether dietary energy balance modulation, ranging from lean (CR) to overweight (OW) to obesity (DIO), alters mammary ER expression, epigenetic reprogramming and/or mammary tumor development in female MMTV-neu transgenic mice. We first characterized diet-dependent metabolic perturbations in subsets of diet-treated mice euthanized at baseline, 1, 3 and 5 months on study. CR mice, relative to OW and DIO mice, had decreased body weight, adiposity, and obesity-associated serum metabolic hormones, including insulin, leptin, IGF-1, and 17β-estradiol, as well as increased adiponectin, after 1 month of diet treatment, consistent with previous studies (36, 37).
We show for the first time that CR (but not OW and DIO regimens) preserves mammary ERα and ERβ expression in MMTV-neu mice. Loss of ERα and ERβ, and increased ERα to ERβ ratio, as observed in OW and DIO mice, have each been linked with poor breast cancer prognosis in clinical studies (38, 39). Our finding that the ERα to ERβ ratio, a prognostic indicator in breast cancer, can be manipulated by energy balance modulation has not, to our knowledge, been previously reported.
We and others have shown that obesity can induce aberrant DNA methylation of genes involved in growth factor and inflammatory signaling (29, 40). To assess whether the energy balance-dependent effects on ERα and ERβ expression are controlled epigenetically, we characterized epigenetic alterations in mammary tissue DNA from baseline mice, from CR and OW mice after 5 months on diet, and from survival study mice after 14 to 22 months of diet. In survival study mice differentially methylated CpG dinucleotides were observed in a CTCF binding site within the ESR1 gene and in CTCF binding sites or flanking regions (± 1kb of a CTCF binding site) upstream and downstream of ESR2, consistent with sustained transcriptional activation of ERα and ERβ. Mammary expression of DNMT1 was stable in CR mice but increased over time in OW and DIO mice, suggesting CR prevented epigenetic reprogramming of DNMT1 that occurs with excess energy intake and weight gain. The effect of altered DNMT1 expression likely impacts the expression of many genes, including ESR1 and ESR2. However, a plausible mechanism for the sustained expression of the ESR1 and ESR2 genes in response to CR is the maintenance of DNMT1 expression and DNA methylation (Figure 6). This can prohibit CTCF binding, thereby preventing allosteric repression and decreased interactions between enhancers and promoters (24). Previous studies have shown that DNA methylation can impede CTCF binding, positively influencing transcription via a loss of repression of specific genes, such as GAD1 (41) and XAF1 (42), and increased enhancer-promoter interactions in IGF2 (43) and c-MYC (44).
Figure 6.

Proposed model of an energy balance-responsive network associated with DNMT1 regulation, DNA methylation and transcriptional regulation of ERα and ERβ. Obesity and associated energy excess results in several metabolic perturbations that are ameliorated by calorie restriction. To identify regulatory relationships between DNMT1 and other genes that were differentially expressed in our RNA-seq analysis, we used the Path Designer function of Ingenuity Pathway Analysis (IPA). We found transcription factors STAT3, NF-kB and E2F to have direct relationships with DNMT1 activation. Upstream of these relationships, IPA also linked growth factors such as insulin and EGF, as well as cytokines including TNF, IL-1β and IL-8, to the E2F, STAT3 and NF-kB signaling pathways. We propose that one consequence of obesity-associated growth factor and cytokine signaling is increased DNMT1 activation, which in turn can modulate DNA methylation and impact transcription of important genes in breast cancer such, as ERα and ERβ. Methylated CpG dinucleotides are indicated by red circles, unmethylated CpG dinucleotides are indicated by white circles.
We also assessed the effects of dietary energy balance modulation on mammary tumor development in a cohort of mice randomized to the three diets (n = 15/diet). CR, relative to the OW and DIO regimens, resulted in significantly increased survival in MMTV-neu mice in association with increased mammary ERα and ERβ expression and DNA methylation at or near CTCF binding sites. Specifically, of the 15 MMTV-neu mice fed the CR diet for up to 22 months, only 3 developed spontaneous mammary tumors, while the median survival of the OW and DIO groups was less than 15 months (Figure 5A). To our knowledge, this study is the first to demonstrate the anticancer effects of a chronic CR regimen (compared with OW or DIO) in MMTV-neu mice. However, Mizuno et al. found that intermittent calorie restriction decreased mammary tumor incidence in MMTV-neu mice (45). Our findings of the anticancer effects of CR, compared with OW or DIO regimens, are consistent with reports of a link between dietary energy balance modulation and mammary tumorigenesis in other preclinical models of mammary cancer (46, 47).
Two previous publications compared DIO versus chow diets (similar to our OW regimen, Supplementary Table S6) on spontaneous mammary tumorigenesis in MMTV-neu mice. Cleary et al. reported that DIO and chow-fed mice had similar mammary tumor development and survival, consistent with our observation of no significant difference in tumor development or survival in OW versus DIO mice (48). In contrast, Chen et al. reported a significant diet-dependent difference (chow > DIO) in survival rates (49). Our findings with CR may help reconcile these apparently conflicting results, since we found highly significant differences in survival in CR mice relative to OW and DIO mice. Chen et al. linked the procancer effects of DIO, relative to chow, in their study to increased signaling through the IKKβ, mTOR, and VEGF pathways which stimulate proliferation and survival. We have previously established that DIO increases, and CR decreases, circulating IGF-1, insulin and their downstream signals through the IGF-1 and insulin receptor tyrosine kinases (36, 47). IGF-1 is a potent mitogen, which promotes signaling through the IKKβ, mTOR, and VEGF pathways, ultimately promoting growth and also inhibiting apoptosis. In women circulating IGF-1 is positively associated with terminal duct lobular unit involution, mammographic density, and breast cancer risk (50, 51).
In the present study, we found that after 1 month of diet treatment CR mice, relative to OW and DIO mice, had decreased serum IGF-1 and insulin (Table 1). However, the DIO and chow-fed mice in the Cleary study did not differ in IGF-1. Thus, one possible explanation for the observed differential tumor responses may involve the diet-dependent effect (or lack thereof) on systemic metabolism, particularly growth factors and their downstream signals. Differential tumor latencies across the studies may also contribute to the apparent study-specific differences in tumor responses to DIO, as the median survival for the Cleary study chow-fed mice and our OW mice were comparable (~18 months and ~15 months, respectively), in contrast to the Chen study chow-fed mice (~7.5 months).
Interactions between dietary energy balance modulation and DNA methylation may influence the expression of key breast cancer-related genes, such as ERα, ERβ, via diet-induced changes in DNMT1 expression and DNA methylation at or near CTCF binding sites. Figure 6 integrates findings from RNA-seq analysis (Supplementary Table S5) using IPA to illustrate a proposed model of a diet responsive network contributing to altered gene expression related to several transcription factor pathways (e.g. E2F, STAT3, and NF-κB) that serve as regulators of DNMT1. Inflammation promotes DNMT1 expression, which has been shown to positively correlate with IL-6 expression in tumors and blood (52, 53). Overexpression of DNMT1 may be a mechanism utilized by cancer cells to evade regulation by rendering tumor suppressors transcriptionally inactive (17, 54). Thus, mediators of obesity-associated inflammation may promote increased expression of DNMT1, thereby contributing to aberrant DNA methylation and transcription of breast cancer-related genes such as ERα and ERβ.
To our knowledge, there are no previous reports regarding the maintenance of mammary ERα and ERβ positivity with a CR regimen. As mentioned previously our CR mice had lower levels of energy balance-related metabolic factors, including IGF-1, insulin, and leptin, consistent with numerous studies in the literature (12, 15, 47). As illustrated in Figure 6, these factors and their downstream signaling pathways can also impact DNMT1, but our study is limited in the ability to establish their precise role. Future studies are planned to assess the possible links between systemic metabolic factors, such as insulin, IGF-1, leptin, and 17β-estradiol and the increased mammary ERα- and ERβ-associated DNA methylation at or near CTCF binding sites in CR mice. Alternatively, the decreased levels of 17β-estradiol observed in CR mice could contribute to prolonged survival, independent of epigenetic events, as high levels of 17β-estradiol can promote cell proliferation and tumor progression (39). Additional study limitations include: a) restricting RRBS and RNA-seq analysis to CR and OW mice (although OW and DIO mice were similar regarding ERα and ERβ mRNA and protein expression and survival); and b) not measuring physical activity (although we have previously shown that CR, relative to control or DIO diets, does not increase locomotor activity in mice) (55).
In conclusion, we found in MMTV-neu transgenic mice that mammary tumor development and ERα and ERβ expression are dietary energy balance-dependent in association with epigenetic reprogramming. Specifically, a diet-DNMT1-methylation axis may serve as a primary regulator of gene transcription via diet-induced changes in DNA methylation at or near CTCF binding sites. These preclinical findings suggest that interventions reducing the impact of excess weight on epigenetic dysregulation of ER may represent a new strategy for the prevention or control of HER2-positive breast cancer in overweight and obese women.
Supplementary Material
Acknowledgments
Financial Support: This study was supported by the American Institute for Cancer Research (8A049), Breast Cancer Research Foundation and R35 (CA197627) from the National Cancer Institute. The mouse RNA-seq and methylation analysis was supported by the UT-MD Anderson Cancer Center Science Park NGS Core, supported by CPRIT Core Facility Support Grant RP120348. E.L. Rossi and L.W. Bowers were supported by a training grant from the National Cancer Institute (R25CA057726), S.M. Dunlap was supported by a USAMRMC Breast Cancer Research Program Postdoctoral Fellowship (W81XWH-09-1-0720) and N.A. Ford was supported by an American Institute for Cancer Research Postdoctoral Fellowship.
We want to wish a special thanks to Lucia J. Chiao, Jennifer Lim and Stephanie Chen for the animal care, feeding and monitoring. Audrey J. Rasmussen and Lauren Harrison provided organizational help and animal care supervision. We thank Dr. Jianjun Shen, the Director of the Molecular Biology Facility at MD Anderson Cancer Center-Science Park for assistance with the mouse RNA-seq and methylation analysis.
Footnotes
Conflict of interest: The authors have no conflicts of interest to report.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11. International Agency for Research on Cancer; 2013. [Google Scholar]
- 3.Jemal A, Bray F, Center MM, Giacinti L, Claudio PP, Lopez M, Giordano A. Epigenetic information and estrogen receptor alpha expression in breast cancer. Oncologist. 2006;11:1–8. doi: 10.1634/theoncologist.11-1-1. [DOI] [PubMed] [Google Scholar]
- 4.Skliris GP, Munot K, Bell SM, Carder PJ, Lane S, Horgan K, et al. Reduced expression of oestrogen receptor beta in invasive breast cancer and its re-expression using DNA methyl transferase inhibitors in a cell line model. J Pathol. 2003;201:213–20. doi: 10.1002/path.1436. [DOI] [PubMed] [Google Scholar]
- 5.Honma N, Horii R, Iwase T, Saji S, Younes M, Takubo K, et al. Clinical importance of estrogen receptor-beta evaluation in breast cancer patients treated with adjuvant tamoxifen therapy. J Clin Oncol. 2008;26:3727–34. doi: 10.1200/JCO.2007.14.2968. [DOI] [PubMed] [Google Scholar]
- 6.Iwase H, Zhang Z, Omoto Y, Sugiura H, Yamashita H, Toyama T, et al. Clinical significance of the expression of estrogen receptors alpha and beta for endocrine therapy of breast cancer. Cancer Chemother Pharmacol. 2003;52(Suppl 1):S34–38. doi: 10.1007/s00280-003-0592-1. [DOI] [PubMed] [Google Scholar]
- 7.Morimoto LM, White E, Chen Z, Chlebowski RT, Hays J, Kuller L, et al. Obesity, body size, and risk of postmenopausal breast cancer: the Women’s Health Initiative (United States) Cancer Causes Control. 2002;13:741–51. doi: 10.1023/a:1020239211145. [DOI] [PubMed] [Google Scholar]
- 8.Vona-Davis L, Rose DP. Type 2 diabetes and obesity metabolic interactions: common factors for breast cancer risk and novel approaches to prevention and therapy. Curr Diabetes Rev. 2012;8:116–30. doi: 10.2174/157339912799424519. [DOI] [PubMed] [Google Scholar]
- 9.Alokail MS, Al-Daghri NM, Al-Attas OS, Hussain T. Combined effects of obesity and type 2 diabetes contribute to increased breast cancer risk in premenopausal women. Cardiovasc Diabetol. 2009;8:33. doi: 10.1186/1475-2840-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Healy LA, Ryan AM, Carroll P, Ennis D, Crowley V, Boyle T, et al. Metabolic syndrome, central obesity and insulin resistance are associated with adverse pathological features in postmenopausal breast cancer. Clin Oncol. 2010;22:281–8. doi: 10.1016/j.clon.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Litton JK, Gonzalez-Angulo AM, Warneke CL, Buzdar AU, Kau SW, Bondy M, et al. Relationship between obesity and pathologic response to neoadjuvant chemotherapy among women with operable breast cancer. J Clin Oncol. 2008;26:4072–7. doi: 10.1200/JCO.2007.14.4527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hursting SD, Smith SM, Lashinger LM, Harvey AE, Perkins SN. Calories and carcinogenesis: Lessons learned from 30 years of calorie restriction research. Carcinogenesis. 2009;31:83–9. doi: 10.1093/carcin/bgp280. [DOI] [PubMed] [Google Scholar]
- 13.Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325:201–4. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fabian CJ, Kimler BF, Donnelly JE, Sullivan DK, Klemp JR, Petroff BK, et al. Favorable modulation of benign breast tissue and serum risk biomarkers is associated with > 10 % weight loss in postmenopausal women. Breast Cancer Res Treat. 2013;142:119–32. doi: 10.1007/s10549-013-2730-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harvie M, Wright C, Pegington M, McMullan D, Mitchell E, Martin B, et al. The effect of intermittent energy and carbohydrate restriction v. daily energy restriction on weight loss and metabolic disease risk markers in overweight women. Br J Nutr. 2013;110:1534–47. doi: 10.1017/S0007114513000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donohoe DR, Bultman SJ. Metaboloepigenetics: Interrelationships between energy metabolism and epigenetic control of gene expression. J Cell Physiol. 2012;227:3169–77. doi: 10.1002/jcp.24054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–68. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 18.Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr. 2007;27:363–88. doi: 10.1146/annurev.nutr.27.061406.093705. [DOI] [PubMed] [Google Scholar]
- 19.Sharma NK, Varma V, Ma L, Hasstedt SJ, Das SK. Obesity associated modulation of miRNA and co-regulated target transcripts in human adipose tissue of non-diabetic subjects. MicroRNA. 2015;4:194–204. doi: 10.2174/2211536604666151103121817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mutze K, Langer R, Schumacher F, Becker K, Ott K, Novotny A, et al. DNA methyltransferase 1 as a predictive biomarker and potential therapeutic target for chemotherapy in gastric cancer. Eur J Cancer. 2011;47:1817–25. doi: 10.1016/j.ejca.2011.02.024. [DOI] [PubMed] [Google Scholar]
- 21.Yu D-H, Ware C, Waterland RA, Zhang J, Chen M-H, Gadkari M, et al. Developmentally programmed 3′ CpG island methylation confers tissue- and cell-type-specific transcriptional activation. Mol Cell Biol. 2013;33:1845–58. doi: 10.1128/MCB.01124-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones PA. The DNA methylation paradox. Trends Genet. 1999;15:34–7. doi: 10.1016/s0168-9525(98)01636-9. [DOI] [PubMed] [Google Scholar]
- 23.Lai AY, Fatemi M, Dhasarathy A, Malone C, Sobol SE, Geigerman C, et al. DNA methylation prevents CTCF-mediated silencing of the oncogene BCL6 in B cell lymphomas. J Exp Med. 2010;207:1939–50. doi: 10.1084/jem.20100204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ong C-T, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet. 2014;15:234–46. doi: 10.1038/nrg3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H, Maurano MT, Qu H, Varley KE, Gertz J, Pauli F, et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012;22:1680–8. doi: 10.1101/gr.136101.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rowse GJ, Tempero RM, VanLith ML, Hollingsworth MA, Gendler SJ. Tolerance and immunity to MUC1 in a human MUC1 transgenic murine model. Cancer Res. 1998;58:315–21. [PubMed] [Google Scholar]
- 27.Thomas DG, Breslow N, Gart JJ. Trend and homogeneity analysis of proportions and life table data. Comput and Biomed Res. 1977;10:373–81. doi: 10.1016/0010-4809(77)90006-4. [DOI] [PubMed] [Google Scholar]
- 28.Browne RW, Kantarci A, LaMonte MJ, Andrews CA, Hovey KM, Falkner KL, et al. Performance of multiplex cytokine assays in serum and saliva among community-dwelling postmenopausal women. PLoS ONE. 2013;8:e59498. doi: 10.1371/journal.pone.0059498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rossi EL, de Angel RE, Bowers LW, Khatib SA, Smith LA, Van Burren E, et al. Obesity-associated alterations in inflammation, epigenetics, and mammary tumor growth persist in formerly obese mice. Cancer Prev Res. 2016;9:339–48. doi: 10.1158/1940-6207.CAPR-15-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, et al. Ensembl 2016. Nucleic Acids Res. 2016;44:D710–6. doi: 10.1093/nar/gkv1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:1–13. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pruitt KD, Tatusova T, Brown GR, Maglott DR. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40:130–5. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Q, Wang HY, Woetmann A, Raghunath PN, Odum N, Wasik MA. STAT3 induces transcription of the DNA methyltransferase 1 gene (DNMT1) in malignant T lymphocytes. Blood. 2006;108:1058–64. doi: 10.1182/blood-2005-08-007377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang BG, Hu L, Zang MD, Wang HX, Zhao W, Li JF, et al. Helicobacter pylori CagA induces tumor suppressor gene hypermethylation by upregulated DNMT1 via AKT-NFκB pathway in gastric cancer development. Oncotarget. 2016;7:9788–9800. doi: 10.18632/oncotarget.7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kimura H, Nakamura T, Ogawa T, Tanaka S, Shiota K. Transcription of mouse DNA methyltransferase 1 (Dnmt1) is regulated by both E2F-Rb-HDAC-dependent and -independent pathways. Nucleic Acids Res. 2003;31:3101–13. doi: 10.1093/nar/gkg406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nogueira LM, Dunlap SM, Ford NA, Hursting SD. Calorie restriction and rapamycin inhibit MMTV-Wnt-1 mammary tumor growth in a mouse model of postmenopausal obesity. Endocr Relat Cancer. 2012;19:57–68. doi: 10.1530/ERC-11-0213. [DOI] [PubMed] [Google Scholar]
- 37.Olivo-Marston SE, Hursting SD, Perkins SN, Schetter A, Khan M, Croce C, et al. Effects of calorie restriction and diet-induced obesity on murine colon carcinogenesis, growth and inflammatory factors, and MicroRNA expression. PLoS One. 2014;9 doi: 10.1371/journal.pone.0094765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madeira M, Mattar A, Logullo AF, Soares FA, Gebrim LH. Estrogen receptor alpha/beta ratio and estrogen receptor beta as predictors of endocrine therapy responsiveness—a randomized neoadjuvant trial comparison between anastrozole and tamoxifen for the treatment of postmenopausal breast cancer. BMC Cancer. 2013;13 doi: 10.1186/1471-2407-13-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nadal-Serrano M, Sastre-Serra J, Pons DG, Miró AM, Oliver J, Roca P. The ERalpha/ERbeta ratio determines oxidative stress in breast cancer cell lines in response to 17Beta-estradiol. J Cell Biochem. 2012;113:3178–85. doi: 10.1002/jcb.24192. [DOI] [PubMed] [Google Scholar]
- 40.Barres R, Kirchner H, Rasmussen M, Yan J, Kantor F, Krook A, et al. Weight Loss after Gastric Bypass Surgery in Human Obesity Remodels Promoter Methylation. Cell Rep. 2013;3:1020–7. doi: 10.1016/j.celrep.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 41.Yan H, Tang G, Wang H, Hao L, He T, Sun X, et al. DNA methylation reactivates GAD1 expression in cancer by preventing CTCF-mediated polycomb repressive complex 2 recruitment. Oncogene. 2015;35:1–14. doi: 10.1038/onc.2015.423. [DOI] [PubMed] [Google Scholar]
- 42.Victoria-Acosta G, Vazquez-Santillan K, Jimenez-Hernandez L, Muñoz-Galindo L, Maldonado V, Martinez-Ruiz GU, et al. Epigenetic silencing of the XAF1 gene is mediated by the loss of CTCF binding. Sci Rep. 2015;5:14838. doi: 10.1038/srep14838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hark AT, Schoenherr CJ, Katz DJ, Ingram RS, Levorse JM, Tilghman SM. CTCF mediates methylation-sensitive enhancer blocking activity at the H19/Igf2 locus. Nature. 2000;405:486–90. doi: 10.1038/35013106. [DOI] [PubMed] [Google Scholar]
- 44.Filippova GN, Fagerlie S, Klenova EM, Myers C, Dehner Y, Goodwin G, et al. An exceptionally conserved transcriptional repressor, CTCF, employs different combinations of zinc fingers to bind diverged promoter sequences of avian and mammalian c-myc oncogenes. Mol Cell Biol. 1996;16:2802–13. doi: 10.1128/mcb.16.6.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mizuno NK, Rogozina OP, Seppanen CM, Liao DJ, Cleary MP, Grossmann ME. Combination of Intermittent calorie restriction and eicosapentaenoic acid for inhibition of mammary tumors. Cancer Prev Res. 2013;6:540–7. doi: 10.1158/1940-6207.CAPR-13-0033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Engelman RW, Day NK, Good RA. Calorie intake during mammary development influences cancer risk: Lasting inhibition of C3H/HeOu mammary tumorigenesis by peripubertal calorie restriction. Cancer Res. 1994;54:5724–30. [PubMed] [Google Scholar]
- 47.Nogueira LM, Lavigne JA, Chandramouli GV, Lui H, Barrett JC, Hursting SD. Dose-dependent effects of calorie restriction on gene expression, metabolism, and tumor progression are partially mediated by insulin-like growth factor-1. Cancer Med. 2012;1:275–88. doi: 10.1002/cam4.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cleary MP, Grande JP, Juneja SC, Maihle NJ. Diet-induced obesity and mammary tumor development in MMTV-neu female mice. Nutr Cancer. 2004;50:174–80. doi: 10.1207/s15327914nc5002_7. [DOI] [PubMed] [Google Scholar]
- 49.Chen CT, Du Y, Yamaguchi H, Hsu JM, Kuo HP, Hortobagyi GN, et al. Targeting the IKKbeta/mTOR/VEGF signaling pathway as a potential therapeutic strategy for obesity-related breast cancer. Mol Cancer Ther. 2012;11:2212–21. doi: 10.1158/1535-7163.MCT-12-0180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.The Endogenous Hormones and Breast Cancer Collaborative Group. Insulin-like growth factor 1 (IGF1), IGF binding protein 3 (IGFBP3), and breast cancer risk: pooled individual data analysis of 17 prospective studies. Lancet Oncol. 2010;11:530–42. doi: 10.1016/S1470-2045(10)70095-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Horne HN, Sherman ME, Pfeiffer RM, Figueroa JD, Khodr ZG, Falk RT, et al. Circulating insulin-like growth factor-1, insulin-like growth factor binding protein-3 and terminal duct lobular unit involution of the breast: a cross-sectional study of women with benign breast disease. Breast Cancer Res. 2016;18 doi: 10.1186/s13058-016-0678-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen MF, Lin PY, Wu CF, Chen WC, Wu CT. IL-6 expression regulates tumorigenicity and correlates with prognosis in bladder cancer. PLoS ONE. 2013;8:e61901. doi: 10.1371/journal.pone.0061901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murphy TM, O’Donovan A, Mullins N, O’Farrelly C, McCann A, Malone K. Anxiety is associated with higher levels of global DNA methylation and altered expression of epigenetic interleukin-6 genes. Psychiatry Genet. 2015;25:71–8. doi: 10.1097/YPG.0000000000000055. [DOI] [PubMed] [Google Scholar]
- 54.Roy DM, Walsh LA, Chan TA. Driver mutations of cancer epigenomes. Protein Cell. 2014;5:265–96. doi: 10.1007/s13238-014-0031-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Padovani M, Lavigne JA, Chandramouli GV, Perkins SN, Barrett JC, Hursting SD, et al. Distinct effects of calorie restriction and exercise on mammary gland gene expression in C57BL/6 mice. Cancer Prev Res. 2009;2:1076–87. doi: 10.1158/1940-6207.CAPR-09-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.