

ABSTRACT
Novel detrimental functions of senescent cells have been recently uncovered in the context of cancer development and progression, which they mainly exert through the secretion of several pro-tumorigenic factors. Here we discuss how cellular senescence and its secretory phenotype can be involved in the widely unexplored phenomenon of paracrine tumorigenesis.
KEYWORDS: β-catenin, adamantinomatous craniopharyngioma, pituitary, senescence, SASP, stem cells
Cellular senescence is widely considered an archetypal mechanism of tumor suppression. In particular, Oncogene-Induced Senescence (OIS) is of great interest for cancer research as it is caused by the activation of oncogenic signaling such as that mediated by the β-catenin/WNT pathway.1 OIS prevents the proliferation of transformed cells by establishing a permanent cell cycle arrest but also elicits non-cell autonomous activities through the Senescence-Associated Secretory Phenotype (SASP), such as inducing senescence in neighboring cells and activating an immune response.2 However, the SASP is composed of such a broad variety of potent signaling factors that it can be detrimental in chronic and pathologic contexts. This can also be the case in cancer, as the SASP can have paradoxical pro-tumorigenic effects such as: directly promoting cancer cell growth, facilitating the emergence of Cancer Stem Cells (CSCs) and generating tumor-permissive microenvironments (Fig. 1).3
Figure 1.
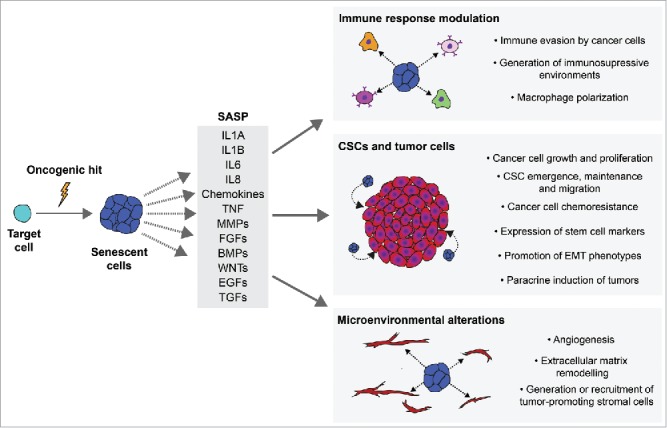
The pro-tumorigenic effects of oncogene-induced senescence and the senescence-associated secretory phenotype. The activation of oncogenes can lead to the onset of senescence and the activation of a Senescence-Associated Secretory Phenotype (SASP). The SASP is composed by a wide variety of factors with pleiotropic functions. Examples of common SASP factors are shown. The pro-tumorigenic activities of the SASP can be categorized as involved in 1) modulation of the immune response, 2) pro-oncogenic signaling to Cancer Stem Cells (CSCs) and/or tumor cells, 3) generating microenvironmental alterations that promote tumorigenesis. IL1A: Interleukin 1 alpha; IL1B: Interleukin 1 beta; IL6: Interleukin 6; IL8: Interleukin 8; TNF: Tumor necrosis factor; MMPs: metalloproteinases; FGFs: fibroblast growth factors; BMPs: bone morphogenetic proteins; WNTs: Wingless-related integration site proteins; EGFs: epidermal growth factor family of proteins; TGFs: transforming growth factor superfamily of proteins; EMT: epithelial-mesenchymal transition.
Adamantinomatous craniopharyngiomas (ACPs) are benign, but aggressive, tumors of the pituitary gland which contain activating mutations of the β-catenin gene (CTNNB1).4 We previously described two mouse models for ACP which contain a functionally identical mutation to that found in humans (hereafter known as oncogenic β-catenin). In the embryonic ACP model, the mutation was targeted to pituitary progenitor cells at embryonic stages, whilst in the inducible ACP model, the targeted cells were adult pituitary stem cells expressing the SRY-box 2 (SOX2) transcription factor. Importantly, these models showed that activating this oncogene in pituitary progenitor/stem cells leads to the formation of “clusters” of nucleocytoplasmic β-catenin accumulating cells, a histological and diagnostic hallmark of human ACP.5,6
We initially hypothesized that the β-catenin clusters were the cell-of-origin of pituitary tumors in both mice and humans, as they displayed several features of Cancer Stem Cells (CSCs) such as the expression of stem cell markers and a quiescent-like state.5 Unexpectedly, lineage tracing experiments in both ACP models showed the tumors were not formed by descendants of the targeted cells.6,7 Further sequencing studies not only confirmed this, but showed a distinct set of mutations present in these tumors. These findings suggested an underlying mechanism that greatly differs from the well-established CSC model of tumorigenesis. Such mechanism should prevent cluster cells from overproliferating and forming tumors themselves, while at the same time inducing tumorigenesis in non-cell autonomous fashion through some paracrine signal. In this sense, cellular senescence and the SASP cover both requirements.
We recently demonstrated that the oncogenic β-catenin clusters undergo senescence in both murine ACP models, as well in human tumors.7 Due to the known complexity of the senescent phenotype, we evaluated numerous stress-related biological processes shared by all senescent cells, such as: loss of proliferative capacity, absence of apoptosis, increased lysosomal activity, acquisition of DNA damage and a DNA-damage-response (DDR), as well as activation of the Nuclear Factor of Kappa-light-chain-enhancer of B cells pathway (NF-κB) and the SASP.8 These results were corroborated through unbiased transcriptomic analyses, which showed that mouse and human clusters display conserved OIS and SASP signatures. We also observed evidence of the clusters´ paracrine activities in the form of drastic alterations of their microenvironment. In particular, the clusters appear to closely interact with non-targeted stromal cells expressing SRY-box 9 (SOX9) and the endothelial marker endomucin (EMCN), which we hypothesize contains the cell-of-origin of mouse ACP tumors.
In our study, we showed that inducing the mutation in pituitary stem cells of aging mice still leads to the formation of senescent β-catenin clusters, although these are smaller and the expression of SASP factors is reduced. Notably, these aging mice developed tumors at much lower rates in comparison to young mice. This is an interesting finding since ACPs are primarily pediatric tumors with initial diagnosis at a median age of 8.8 years.4 We further explored the consequence of a dampened SASP signature by removing the adenomatosis polyposis coli (Apc) gene in pituitary progenitor/stem cells. Crucially, Apc-null pituitaries also contain small senescent β-catenin clusters with a diminished SASP signature, but these do not modify their microenvironment and tumors do not develop.
Our results in the murine models have important implications for future research on human ACP pathogenesis. Our data supports the notion that the clusters´ paracrine activities may underlie their ability to influence tumor invasion, growth and recurrence. This is supported by computer-based 3D reconstructions of human ACP cellular architecture, which showed the presence of clusters along finger-like epithelial tumor protrusions that invade nearby brain tissue.9 Additionally, the activation of a senescent phenotype in human clusters could make them impervious to conventional anti-cancer therapies (e.g. radiotherapy), thus explaining the tendency of human ACP to relapse even after radiotherapy.
Our study is the first to point at senescence and the SASP as causative agents of non-cell-autonomous tumorigenesis in vivo. In terms of the applicability of our model beyond ACP, it will be particularly interesting to study the role of senescence and the SASP in other models where tumors/cancers arise non-cell autonomously, especially in those driven by oncogenic β-catenin/WNT signaling.10 Our results also warn against the idea that OIS has exclusively beneficial roles in tumourigenesis, as this view has mostly derived from the observation that premalignant cancers display high numbers of senescent cells, while fully malignant cancers do not.1 Finally, we propose that future studies addressing the role of OIS and the SASP in the origin of cancer should be conducted in light of lineage-tracing experiments, as senescent cells carrying driver mutations might not be related to the cancer cell-of-origin, leading to important consequences in the future development of targeted therapies for human cancers.
Funding Statement
Medical Research Council (MRC) (MR/M000125/1) Great Ormond Street Hospital Charity (W1055).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
J.M.G.M. was supported by a CONACYT-UCL postgraduate fellowship (CVU: 316764) and a CONACYT-REDESD Travel fellowship (MR-225-17). This work was supported by the Medical Research Council (MRC) (Grants MR/M000125/1), Great Ormond Street Hospital for Children Charity/Children with Cancer UK (GOSHCC/CWCUK) (Grant W1055) and by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children National Health Service Foundation Trust and University College London. J.P.M.-B. is a Great Ormond Street Hospital for Children's Charity Principal Investigator.
References
- 1.Collado M, Serrano M. Senescence in tumours: evidence from mice and humans. Nat Rev Cancer. 2010;10:51–57. doi: 10.1038/nrc2772. PMID:20029423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pérez-Mancera PA, Young ARJ, Narita M. Inside and out: the activities of senescence in cancer. Nat Rev Cancer. 2014;14;547–558. doi: 10.1038/nrc3773. PMID:25030953. [DOI] [PubMed] [Google Scholar]
- 3.Lecot P, Alimirah F, Desprez PY, Campisi J, Wiley C. Context-dependent effects of cellular senescence in cancer development. Br J Cancer. 2016;114;1180–1184. doi: 10.1038/bjc.2016.115. PMID:27140310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Müller HL, Merchant TE, Puget S, Martinez-Barbera JP. New outlook on the diagnosis, treatment and follow-up of childhood-onset craniopharyngioma. Nat Rev Endocrinol. 2017;13;299–312. doi: 10.1038/nrendo.2016.217. PMID:28155902. [DOI] [PubMed] [Google Scholar]
- 5.Gaston-Massuet C, Andoniadou CL, Signore M, Jayakody SA, Charolidi N, Kyeyune R, Vernay B, Jacques TS, Taketo MM, Le Tissier P, et al.. Increased Wingless (Wnt) signaling in pituitary progenitor/stem cells gives rise to pituitary tumors in mice and humans. Proc Natl Acad Sci U S A. 2011;108:11482–11487. doi: 10.1073/pnas.1101553108. PMID:21636786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andoniadou CL, Matsushima D, Mousavy Gharavy SN, Signore M, Mackintosh AI, Schaeffer M, et al.. Sox2+ Stem/Progenitor Cells in the Adult Mouse Pituitary Support Organ Homeostasis and Have Tumor-Inducing Potential. Cell Stem Cell. 2018;13:433–45. doi: 10.1016/j.stem.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez-Meljem JM, Haston S, Carreno G, Apps JR, Pozzi S, Stache C, et al.. Stem cell senescence drives age-attenuated induction of pituitary tumours in mouse models of paediatric craniopharyngioma. Nat Commun. 2017;8:1819. doi: 10.1038/s41467-017-01992-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15;397–408. doi: 10.1038/nrc3960. PMID:26105537. [DOI] [PubMed] [Google Scholar]
- 9.Apps JR, et al.. Imaging Invasion: Micro-CT imaging of adamantinomatous craniopharyngioma highlights cell type specific spatial relationships of tissue invasion. Acta Neuropathol Commun. 2016;4:57. doi: 10.1186/s40478-016-0321-8. PMID:27260197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kode A, Manavalan JS, Mosialou I, Bhagat G, Rathinam CV, Luo N, Khiabanian H, Lee A, Murty VV, Friedman R, et al.. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature. 2014;506;240–244. doi: 10.1038/nature12883. PMID:24429522. [DOI] [PMC free article] [PubMed] [Google Scholar]