

ABSTRACT
The binding of bacteria to platelets is thought to be a central event in the pathogenesis of infective endocarditis. The serine-rich repeat (SRR) glycoproteins of viridans group streptococci have been shown to mediate platelet binding in vitro and to contribute to virulence in animal models. However, it is not known whether SRR adhesins can mediate streptococcal binding under the high fluidic shear stress conditions present on the endocardial surface. We found that three streptococcal SRR adhesins (GspB, Hsa, and SrpA) with differing structures and sialoglycan binding specificities nevertheless exhibited similar biomechanical properties. All three adhesins mediated shear-enhanced streptococcal binding to immobilized platelets through the platelet receptor GPIbα. Shear-enhanced adhesion was manifested in three ways. First, the number of circulating streptococci binding via SRR adhesins to immobilized platelet receptors peaked at 1 dyn/cm2. Second, bound streptococci switched from weak rolling to strong stationary adhesion as shear stress increased to 10 dyn/cm2. Third, while a few streptococci detached each time the flow was increased, the majority of streptococci bound to platelets remained firmly attached through 20 to 80 dyn/cm2 (shear levels typical of arteries and the endocardium). Thus, all three adhesins mediated shear-enhanced streptococcal binding to platelets under the flow conditions found in heart valves. The ability of the SRR adhesins to mediate shear-enhanced binding strongly suggests that they form catch bonds that are activated by tensile force and provides a mechanism for the selective targeting of bacteria to platelet receptors immobilized on the endocardial surface.
KEYWORDS: Streptococcus, adhesins, biomechanics, bloodstream infections, infective endocarditis, platelets, shear stress
INTRODUCTION
Infective endocarditis is a life-threatening infection of the heart valves. Patients with underlying rheumatic heart disease, congenital cardiac defects, prosthetic heart valves, or intravenous drug use are at high risk for this illness. The viridans group streptococci (including Streptococcus gordonii and Streptococcus sanguinis) are a leading cause of infective endocarditis, accounting for 17 to 45% of all cases (1–3). Despite prompt therapy, patients with streptococcal endocarditis frequently develop complications such as progressive valve destruction, heart failure, or stroke. In view of the increasing resistance of these organisms to conventional antimicrobial agents (4, 5), new therapeutic modalities are needed.
A central event in the pathogenesis of endocarditis is bacterial binding to platelets (6–14). S. gordonii surface protein B (GspB) is a lectin-like, serine-rich repeat (SRR) adhesin that mediates bacterial binding to human platelets through its interaction with the trisaccharide Neu5Acα2-3Galβ1-3GalNAc (sialyl-T antigen [sTa]) on the platelet membrane receptor GPIbα (15–18). This interaction has been linked to increased virulence, as measured by animal models of endocarditis (11, 18). GspB is part of an expanding family of SRR glycoprotein adhesins (19) that have been associated with pathogenicity in a variety of infections (11, 12, 18, 20–23). These adhesins are highly prevalent in strains of S. gordonii and S. sanguinis and include Hsa of S. gordonii strain Challis and SrpA of S. sanguinis strain SK36 (both oral isolates). Like GspB, Hsa and SrpA bind platelets through sialoglycans on GPIbα (16, 17, 24–27). The crystal structures of the GspB (18, 28) and SrpA (29, 30) ligand-binding regions have been determined in complex with sialoglycans and show a common Siglec-like fold of the binding domain, which is also predicted for Hsa (19). This family of adhesins is therefore referred to as Siglec-like SRR adhesins because of this structural homology to the sialic acid-binding immunoglobulin-type lectins (Siglecs) of eukaryotic cells. The selectivity of all three adhesins has been characterized thoroughly using glycan microarrays and other binding assays (31). GspB is highly selective for sTa, Hsa recognizes a broad range of sialyl glycans, including sTa, and SrpA has low affinity for sTa and other defined sialyl glycans (31) but shows high levels of sialic acid-dependent binding to GPIbα and platelets (30).
These results collectively demonstrate that the lectin-like SRR adhesins of viridans group streptococci can bind platelets through sialoglycans under static conditions. However, they do not address whether or how they mediate adhesion to platelets under the flow conditions present on endocardial surfaces. Bacterial vegetations (the characteristic anatomic lesions of endocarditis) typically form on the upstream surface of heart valves (32), which are exposed to rapidly flowing blood. The effect of fluid flow on bound cells is dependent on the fluidic wall shear stress, which is the product of the viscosity of the fluid and the velocity gradient of the fluid parallel to the wall. This fluidic shear stress applies a drag force on adherent bacteria, which in turn applies tensile force to the adhesin-receptor bonds that mediate adhesion. Because adhesion molecules often form “slip bonds” that are overpowered by tensile force (33), fluid flow is traditionally seen as a natural defense mechanism that inhibits bacterial adhesion (34). However, some bacteria bind better at higher flow, at least in part because some adhesins form “catch bonds” that are activated by tensile force (34). Heart valves are exposed to the highest level of fluidic wall shear stress of all niches in the circulatory system (20 to 80 dyn/cm2) (35), raising the question of how streptococci can bind in the high shear stress found in heart valves.
Two prior studies draw opposing conclusions as to whether flow enhances or inhibits streptococcal binding via SRR adhesins and whether SRR adhesins can mediate adhesion at the wall shear stress reported in heart valves. Ding et al. demonstrated that Hsa-mediated adhesion of streptococci to salivary glycoproteins was enhanced by shear stress and withstood up to 50 dyn/cm2 (36). In contrast, Plummer et al. concluded that the SrpA-mediated adhesion of streptococci to platelets could occur only under low shear stress, because no such binding was detectable above 15 dyn/cm2 (24). These findings raise two questions. First, do only some Siglec-like SRR adhesins mediate shear-enhanced adhesion, or are certain receptors or conditions required for this property? Second, can Siglec-like SRR adhesins mediate streptococcal adhesion to platelets in the high flow characteristic of heart valves? Specifically, we ask whether the ability of SRR adhesins to mediate shear-enhanced adhesion that tolerates high flow depends on which Siglec adhesin is involved, which receptor is involved, or the hydrodynamic conditions.
To further explore these issues, we compared the abilities of GspB, Hsa, and SrpA to mediate bacterial adhesion to platelets, the platelet GPIbα receptor, and immobilized sTa, using in vitro flow conditions that replicate key hydrodynamic conditions present within the heart. We found that both shear-enhanced adhesion and the ability to support adhesion at high flow were fundamental properties of the interaction between Siglec-like SRR adhesins and their receptors.
RESULTS
Specificity of binding in flow.
We first assessed whether SRR adhesins can mediate binding of streptococci to immobilized platelets or platelet receptors under conditions of shear. Streptococci suspended in phosphate-buffered saline with bovine serum albumin (PBS-BSA) were flowed over receptor-coated surfaces at 1.4 dyn/cm2, and the number of bacteria that had attached to the surface after 3 min was determined by microscopy. This shear stress was chosen because it supported optimal attachment (see below). S. gordonii strain M99 expresses GspB (37), S. gordonii strain PS798 expresses Hsa (17), and S. sanguinis strain SK36 expresses SrpA (24), and for clarity these strains are here referred to as GspB+, Hsa+, and SrpA+, respectively (Table 1). We also tested an isogenic variant of each strain that lacks the SRR adhesin (ΔGspB, ΔHsa, and ΔSrpA) (Table 1). We measured the binding of all six strains to three receptor-coated surfaces: immobilized platelets, the immobilized extracellular region of GPIbα (ecGPIbα), and biotinylated sTa that is immobilized on streptavidin. As a negative control, we also measured binding to immobilized BSA.
TABLE 1.
Strains used in the present study
| Strain | Description or relevant genotype | Source or reference |
|---|---|---|
| M99 | S. gordonii endocarditis isolate | Sullam et al. (37) |
| PS846 | M99 ΔgspB::pEVP3 | Bensing et al. (49) |
| PS798 | S. gordonii Challis CH1 secA2::pM993′A2 | Bensing et al. (17) |
| PS779 | S. gordonii Challis CH1 Δhsa::pEVP3 | Bensing et al. (17) |
| SK36 | S. sanguinis oral isolate | Plummer et al. (24) |
| PS1071a | SK36 srpA::pB1060flag; pVA891 insertion in srr2 region | This study |
Secretes C-terminally truncated, 3XFLAG-tagged SrpA; lacks cell wall-anchored SrpA.
GspB+ streptococci bound well to immobilized platelets, ecGPIbα, and sTa, but not to BSA, while ΔGspB streptococci did not bind measurably to any of the four surfaces (Fig. 1A). Hsa+ streptococci also bound to platelets, ecGPIbα, and sTa at significantly higher levels than to BSA, while ΔHsa streptococci failed to bind measurably to any of the four surfaces. Finally, SrpA+ streptococci bound well to platelets and ecGPIbα, but not to sTa or BSA, while ΔSrpA streptococci failed to bind measurably to any of the four surfaces (Fig. 1A). This demonstrates that all three SRR adhesins can mediate streptococcal binding to platelets or GPIbα under flow and that sTa alone is a sufficient ligand for binding by GspB or Hsa. The inability of SrpA to recognize sTa in flow is consistent with its lower affinity for sTa than that of the other two SRR adhesins (31) and supports the conclusion that SrpA must recognize an alternate or more complex sialic acid-containing carbohydrate moiety on GPIbα for binding to occur (30).
FIG 1.

Attachment of streptococci in flow. (A) Binding of the indicated streptococcal strains to the indicated immobilized receptors at 1.4 dyn/cm2. Each bar shows the mean and standard deviation of the number bound for duplicate samples. (B to D) Number of bacteria from each indicated streptococcal strain binding to surfaces coated with platelets (B), ecGPIbα (C), and sTa (D) at the indicated levels of fluidic shear stress. All data were normalized to the number of bacteria of the indicated strain binding to the indicated receptor at 1.4 dyn/cm2. Each data point shows the average and standard deviation of the normalized data taken on two (B) or three (C and D) days.
Effect of flow on attachment.
We then studied the effect of fluidic shear stress on binding for each combination of bacterial strain and target receptor for which specific binding was observed in the above-described experiments. Bacteria in PBS-BSA were pumped over receptor-coated surfaces at seven shear stresses ranging from 0.1 to 10 dyn/cm2. We then divided the number of bound bacteria at each shear stress by the number bound at 1.4 dyn/cm2 for the same strain-receptor pair, to allow better comparisons of their responses to flow. In contrast to the large differences in the total number of bacteria bound by the various strain-receptor pairs (as described above), these strain-receptor combinations demonstrated remarkably similar biphasic responses to fluidic shear stress (Fig. 1). An optimal shear stress of 1.4 dyn/cm2 supported the highest number of bacteria being bound for all strain-receptor pairs, with fewer bacteria bound at both higher and lower shear stresses. That is, more streptococci bound through these SRR adhesins as shear stress increased from 0.1 to 1.4 dyn/cm2 (P value < 0.05 for all three wild-type strains), but fewer bound as shear stress increased further from 1.4 to 10 dyn/cm2 (P value < 0.05 for all three strains) as determined by Student's 1-tailed t test for paired samples. The finding that similar patterns are observed for bacterial binding to platelets, ecGPIbα, and sTa and that binding is abolished by deletion of the SRR adhesins indicates that the response of the bacteria to shear stress is due entirely to properties of the adhesin-receptor interaction, rather than other aspects of the bacteria, platelets, or the artificial-receptor-coated surfaces.
The peak of adhesion at 1.4 dyn/cm2 demonstrates that Siglec-like SRR adhesins of oral streptococci can mediate adhesion to sialylated glycoproteins under the flow conditions found on dental surfaces (1 dyn/cm2 [38]). Indeed, the shear-enhanced adhesion observed here for all three SRR adhesins appears to be ideal for the oral niche. However, binding was dramatically reduced at higher levels of shear stress, raising questions about whether or how Siglec-like SRR adhesins can mediate adhesion to platelets at the level of fluidic shear stress experienced by heart valve surfaces (20 to 80 dyn/cm2 [35]).
Strength of adhesion.
To address why bacteria bound in such low numbers at 10 dyn/cm2, we asked if SRR-mediated adhesion is too weak to withstand the drag force applied by this shear stress to bound streptococci. As an initial test of binding strength, we tracked the movement of individual bacteria over time to determine whether they bound in a weaker, less stationary manner at 10 dyn/cm2 than with lower shear stresses. At 1.4 dyn/cm2, all strain-receptor combinations tested demonstrated a “stick-and-roll” manner of adhesion, in which they sometimes bound in a strong, stationary mode of adhesion (Fig. 2A, track 1) and other times in a weak rolling mode (track 2), which allowed detachment (track 3). This binding phenotype was clearly due to two modes of adhesion, rather than two populations of bacteria, because bacteria sometimes switched from rolling to stationary (track 4), from stationary to rolling (track 5), or even repeatedly back and forth (track 6). (The stick-and-roll manner of adhesion is illustrated in Video S1 in the supplemental material.) To quantify the fraction of bound bacteria that were in the strong stationary mode, we counted the number of bacteria that did not move for at least 1 s and divided this by the total number of bound bacteria. All strain-receptor combinations tested demonstrated similar effects of shear stress by this measure. The fraction of stationary bacteria increased even between 1.4 and 10 dyn/cm2 (the P value was <0.05 for all strain-receptor pairs except for GspB+ over platelets, for which it was 0.07, using Student's 1-tailed t test for paired samples) (Fig. 2B to D). Thus, while the total number of bacteria bound peaked at 1.4 dyn/cm2 (Fig. 1), the fraction of attached bacteria exhibiting strong, stationary binding was maximal at or above 10 dyn/cm2.
FIG 2.

Effect of flow on mode of adhesion. (A) The results of the experiments described for Fig. 1 were further analyzed to classify movements of bound bacteria. The positions of individual GspB+ bacteria binding to ecGPIbα at 1.4 dyn/cm2 are shown over a 25-s period to illustrate how bacteria switch between stationary and rolling modes of adhesion. The “X” indicates where a bacterium detached. Each track is assigned a number to facilitate the description of these data in the text. (B to D) The fraction of stationary bacteria (defined as bacteria that do not move within 1 s) is shown for the indicated streptococcal strains binding to surfaces coated with platelets (B), ecGPIbα (C), and sTa (D) at the indicated levels of fluidic shear stress. Each data point shows the average and standard deviation of data taken on two (B) or three (C and D) days.
These observations suggest that binding via SRR adhesins may be sufficiently strong to maintain attachment to platelets and platelet receptors at a shear stress higher than 10 dyn/cm2, once the bacteria are already bound to surfaces. To test this directly, we allowed streptococci to bind immobilized platelets or receptors at 2 dyn/cm2, washed them at 5 dyn/cm2, and then subjected them to stepwise-increasing shear stress up to 80 dyn/cm2 (Fig. 3A; see also Video S2 in the supplemental material). Above a critical shear stress, a subpopulation of bacteria detached rapidly each time shear stress was stepped up, after which no more detached. This suggests that the individual bacteria vary in their ability to withstand high flow, perhaps due to variable adhesin expression or differences in bacterial chain length and thus drag force. We also subjected streptococci to stepwise-decreasing shear stress down to 0.1 dyn/cm2 (Fig. 3B; see also Video S3 in the supplemental material). Below a critical shear stress, streptococci detached slowly but continuously over the time observed. This suggests that detachment at low flow is slow and stochastic, rather than due to subpopulations of bacteria that differ in strength of adhesion at low shear stress. Because of the difference in detachment patterns at high versus low shear stress described above, it must be remembered that the fraction detached at high flow likely reflects shear stress, while the fraction detached at low flow may also reflect the duration of time spent below a critical threshold required for shear-enhanced binding.
FIG 3.

Effect of flow on detachment of streptococci. Bacteria were bound to the surface at 0.5 dyn/cm2 and washed at 5 dyn/cm2; then, shear stress was stepped up or down every 15 s, while the fractions of bacteria remaining bound were determined. (A and B) The fraction remaining bound over time is shown for one strain-receptor pair (GspB+ binding to ecGPIbα) as shear stress is increased (A) or decreased (B). (C to E) Similar time courses for all strains binding to platelets (C), ecGPIbα (D), and sTa (E) were analyzed to calculate the fraction of bacteria remaining bound at the end of each 15-s step. Each data point shows the average and standard deviation of data taken on 2 (C and D) or 3 (E) days.
To characterize the ability of the three SRR adhesins to maintain streptococcal adhesion to various receptors at high and low shear stresses, we repeated the above-described studies for all strain-receptor combinations that demonstrated specific binding and determined the fraction of streptococci remaining bound at the end of each stepwise decrease or increase in shear stress (Fig. 3C to E). For all strain-receptor combinations tested, most streptococci remained bound between about 1 dyn/cm2, near the optimal rates of attachment as shown in Fig. 1, and about 10 dyn/cm2, which mediated the most-stationary adhesion illustrated in Fig. 2. This confirms that streptococci can maintain adhesion via SRR adhesins to platelets and platelet receptors at much higher shear stress than is optimal for initiating attachment from flowing buffer. When shear stress was decreased stepwise below this level, GspB+ and SrpA+ detached significantly (P value < 0.05 for the 5 strain-receptor combinations). While detachment by Hsa+ was not statistically significant when any one receptor was considered (0.05 < P value < 0.2), Hsa+ detached at low shear stress in every run on every receptor, and the effect when all these conditions were considered together was significant (P value < 0.05). When shear stress was increased stepwise to 80 dyn/cm2, the maximum predicted for heart valves, a significant number of GspB+, Hsa+, and SrpA+ isolates detached from all surfaces (P value < 0.05 for the eight strain-receptor combinations), but a subpopulation of 50% to 80% of streptococci remained bound to platelets (Fig. 3C). Together, the tests for stationary adhesion and for detachment demonstrate that for a majority subpopulation of streptococci, SRR adhesins provide sufficient mechanical strength to withstand the flow conditions of heart valves, once binding has been established.
One possibility as to why only a subpopulation of streptococci detach at each shear stress is that streptococci can form chains of different lengths, which would be subjected to different drag forces at the same shear stress. Interestingly, the three strains in this study differed in their tendency to form chains. SrpA+ primarily took the form of chains, GspB+ appeared as both single bacteria and chains, and Hsa+ primarily took the form of single streptococci (Fig. 4A). Indeed, chains of streptococci detached more than single streptococci when shear stress was stepped up to 80 dyn/cm2 (Fig. 4B) (P value < 0.05, using Student's two-tailed t test for paired samples on the combined set of two runs each for the three strains). This supports the theory that the larger size is one of the critical properties of the subpopulations that detach when shear stress is stepped up. Remarkably, the chains also detached more than single streptococci when shear stress was stepped down to 0.1 dyn/cm2 (Fig. 4C) (P < 0.01), although it is hard to explain why this would be the case. In spite of the small but significant effects of chain size on detachment at high and low shear stresses, our key observations transcend chain size. That is, whether streptococci remain single or form chains, they exhibit shear-enhanced adhesion in which they detach at low flow, and a subpopulation can withstand the high flow found in heart valves.
FIG 4.

Effect of streptococcal chain length on binding. The results of the detachment experiments described for Fig. 3B were further analyzed to determine whether the length of the streptococcal strain affected shear-enhanced binding. The streptococci were identified as single if they were approximately round or as a chain if they were longer in one dimension, and each particle (single or chain) was counted as one streptococcus. (A) Numbers of single versus chain streptococci initially bound to ecGPIbα at 2 dyn/cm2 for the three strains are shown. (B, C) Fraction of the streptococci of the indicated strain and size that remained bound after shear stress was stepped down to 0.1 (B) or up to 80 (C) dyn/cm2.
In summary, all three Siglec-like SRR adhesins display similar biomechanical properties, in spite of different binding specificities and chain sizes. This suggests that the biomechanical properties must arise from common structural elements, while the chemical specificity is more likely to derive from structural differences.
Effect of pulsatile flow on adhesion.
The above-described studies demonstrate that binding via SRR adhesins is sufficiently strong to maintain adhesion at the highest shear stress found in heart valves but raise two questions critical to infective endocarditis. First, if suspended streptococci initiate adhesion to platelets more poorly at 10 dyn/cm2 than at 1 dyn/cm2, can they initiate any adhesion to platelets at higher shear stresses typical of heart valves? Second, how is bacterial adhesion affected by the cyclical changes in flow associated with valve opening and closing while blood is pumped through the heart? On the one hand, the gradual detachment of streptococci from platelet receptors that we observed at low shear stress could mean that streptococci would detach during the brief periods of diastolic low shear stress. That is, streptococcal adhesion could be strong enough to withstand the highest shear stress encountered in heart valves but not the lowest. On the other hand, it is possible that streptococci might attach to the endocardium during the brief periods of low shear stress present during cardiac diastole and, once bound, could withstand the subsequent periods of high systolic shear stress. That is, periodic changes in flow could increase the rate at which suspended streptococci initiate binding.
To address these questions, we created pulsatile flow in the flow chambers with a programmable pump, turning the flow on and then back off in a repeating cycle. To confirm that pulsatile flow was indeed obtained, we used particle imaging velocimetry to calculate the fluid speed and thus the shear stress near surfaces under these conditions. The flow in the chamber showed minor latency in response to each change, as would be expected from the resistance and compliance of our fluidic system, but we were nevertheless able to create a pulsatile flow that switched every 2 s (Fig. 5A). In spite of limitations with the pump used to create pulsatile flow, we were also able to create shear stresses as high as 30 dyn/cm2, which is well within the range experienced by heart valves. This allowed us to probe the ability of suspended bacteria to adhere to platelet receptors immobilized on a surface in the high pulsatile flow characteristic of the endocardium.
FIG 5.

Effect of pulsatile flow on binding of GspB+ to ecGPIbα. (A) Characterization of pulsatile flow. The pump velocity reflects, for all three SRR adhesins studied (GspB, Hsa, and SrpA), the pump turning on or off every 2 s. The measured fluid velocity is determined by the velocity of microspheres that were included in the bacterial suspension that was pumped through the chamber. The modeled velocity is fit to the data assuming that the pump, tubing, and chamber constitute a linear fluidic circuit with a single characteristic response time and no inertia. All velocities are normalized to the bead velocity that would be predicted if the pump were on the entire time. (B) Binding of GspB+ bacteria to ecGPIbα or BSA in constant or pulsatile flow. The abscissa indicates the level of shear stress that would be created if the pump were on the entire time. The numbers of bacteria were normalized to the optical density (concentration) of the sample. Similar results were observed in two additional experiments.
We then pumped GspB+ bacteria over immobilized ecGPIbα or BSA over a range of shear stresses (0.1 to 30 dyn/cm2) and compared the numbers of streptococci attaching under conditions of constant versus pulsatile shear stress. After normalizing for the concentration of bacteria in solution as measured by optical density, we plotted the number of bacteria attaching over time as a function of the shear stress at the peak of the constant or pulsatile flow. Remarkably, the number of attached bacteria was the same whether flow was continuous or pulsatile, as long as the peak shear stress was the same (Fig. 5B). Consistent with the data presented in Fig. 1, the number of suspended bacteria that bound to immobilized platelet receptors dramatically decreased with flow between 1 dyn/cm2 and 30 dyn/cm2 for both constant (P value < 0.05) and pulsatile (P value < 0.05) flows. Thus, short periods of low shear stress during pulsatile flow do not facilitate the initial attachment of bacteria to immobilized receptors. In both constant and pulsatile flow, significant numbers of suspended streptococci still bound to immobilized platelet receptors at 30 dyn/cm2, so short periods of low shear stress also did not reduce streptococcal binding. This demonstrates that a small population of suspended bacteria can initiate and maintain binding under the high pulsatile flow conditions characteristic of heart valves.
Adhesion in whole blood.
Blood contains both protein-associated and cell-associated sialic acid, which could act as competitive inhibitors to SRR adhesins in vivo. To address whether SRR adhesins mediate shear-enhanced binding under more biologically relevant conditions, we mixed the three strains of streptococci with whole blood and measured binding at 1 and 25 dyn/cm2. GspB+ and Hsa+ each bound to immobilized platelets in large numbers (Fig. 6). Moreover, binding was significantly higher at the higher shear stress (P value < 0.04 for Student's 1-tailed unpaired t test). While significant binding to a negative-control surface with no platelets was also observed, the difference between total and nonspecific binding, which provides an estimate of specific binding, was about 3.6-fold higher at 25 than at 1 dyn/cm2 for both strains. Interestingly, SrpA+ bound in low numbers in whole blood, and the effect of higher shear stress was not statistically significant (P value = 0.17). Therefore, GspB and Hsa mediate shear-enhanced binding to immobilized platelets in the presence of blood, demonstrating that these SRR adhesins are not competitively inhibited by sialylated glycoproteins or circulating blood cells. Interestingly, SrpA+ does not bind well in whole blood, suggesting that it is inhibited by one or more components of blood (Fig. 6). Although this difference among the three adhesins warrants further study, we can draw a key conclusion from our observations about Gsp+ and Hsa+. Whole blood contains both ecGPIbα and platelets, which are clearly receptors for Gsp+ and Hsa+, so shear-enhanced adhesion mediated by SRR adhesins can resist inhibition by circulating receptors.
FIG 6.
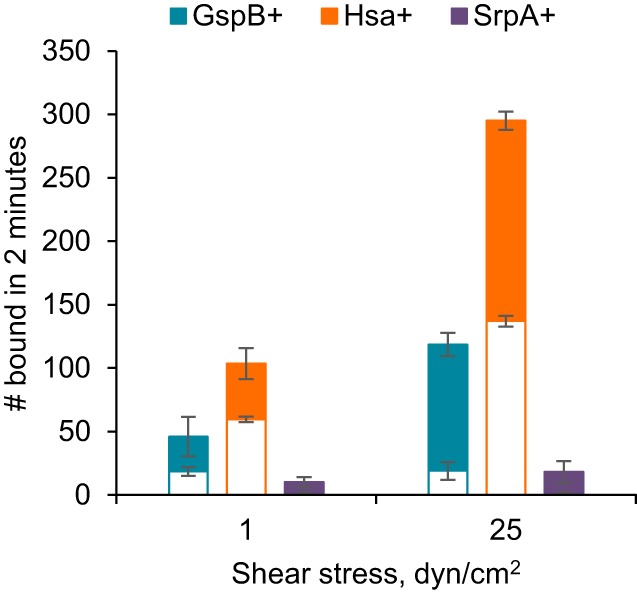
Shear-enhanced streptococcal binding in whole blood. Streptococci of the indicated strains were mixed in whole blood and tested for binding to immobilized fixed platelets at the indicated shear stresses (total bar heights). The open bars indicate the numbers binding to negative-control plates with no platelets, so the difference—the solid portion of the bars—provides an estimate for the amount of binding that is specific to immobilized platelets. The error bars indicate the standard deviations for two runs under each condition.
DISCUSSION
The SRR glycoproteins are highly prevalent among Gram-positive species and can interact with a variety of ligands. For many of the viridans group streptococci, they are Siglec-like adhesins that can bind sialoglycans on salivary proteins as well as on the platelet receptor GPIbα. The latter interaction appears to contribute to the pathogenesis of infective endocarditis, as measured by animal models of infection (11, 14, 18). The exact mechanisms by which the SRR glycoproteins enhance virulence are not entirely known, but these adhesins may be important both for the attachment of organisms in the bloodstream to platelets immobilized on valve surfaces and for the subsequent recruitment of circulating platelets to the infected endocardium, thereby leading to vegetation formation. To perform any of these functions during vegetation formation, SRR adhesins would need to bind in the high shear stress on the endocardial surface.
We found that the three SRR adhesins studied can mediate streptococcal binding to platelets and platelet receptors in a shear-enhanced manner, as indicated by three independent metrics. First, more streptococci expressing GspB, Hsa, or SrpA bound these immobilized targets at moderate shear stress (1 dyn/cm2) than at low shear stress (0.1 dyn/cm2) (Fig. 1). Second, streptococci bound in a stronger, more stationary manner as shear stress increased from 0.1 dyn/cm2 to 10 dyn/cm2 (Fig. 2). Third, once bound, streptococci detached at low shear stress (0.1 dyn/cm2) but not at moderate to high shear stress (1 dyn/cm2 to 10 dyn/cm2) (Fig. 3). This shear-enhanced adhesion is mediated by the SRR adhesins rather than other bacterial components, because isogenic variants that lack SRR adhesins did not bind significantly to platelets. Moreover, shear-enhanced adhesion was mediated by the sialoglycan receptors on platelets rather than other platelet components, since platelets themselves were not required for shear-enhanced adhesion. These observations are consistent with previous data showing that Hsa mediated shear-enhanced binding to salivary glycoproteins (36) and indicate that shear-enhanced adhesion is a conserved property of the Siglec-like SRR adhesins.
The shear-enhanced adhesion of the Siglec-like adhesins is most likely due to catch bonding, a process whereby tension-induced conformational changes increase the effective affinity. The common structural characteristics of GspB, Hsa, and SrpA suggest some possibilities for catch bonding. Shear stress could induce conformational changes within the sialyl glycan-binding Siglec domain. While the Siglec domains of the three adhesins share a semiconserved YTRY motif that is essential for binding, they vary significantly both in the overall shape of the glycan binding pockets surrounding the YTRY motifs (18, 29) and in their specific glycan targets (31), raising the question as to how they could respond so similarly to shear stress. GspB, Hsa, and SrpA each have a Unique domain that connects the Siglec domain to the cell wall-anchored domain of the adhesin (Fig. 7A). Although the role of the Unique domain has not been tested for other adhesins, it is essential for binding to sTa in GspB (19). Tension might align the Siglec and Unique domains into a conformation that has high affinity, due to allosteric interdomain regulation or through formation of an extended binding pocket (Fig. 7B). However, the isolated binding regions form high-affinity interactions even in static assays that provide no tension (19), suggesting that something in the full-length adhesin normally stabilizes the low-affinity conformation. All three adhesins have SRR1 and SRR2 regions that are heavily glycosylated. These might bend (Fig. 7C) or partially mask the binding site (Fig. 7D) in the two-domain binding region. Tension would then pull the SRR domains away to align or unmask the high-affinity binding site (Fig. 7E). This hypothesis is consistent with all available structural and functional data for these three adhesins and would provide a conserved mechanism for shear-enhanced adhesion and catch bond formation that is unique to the SRR adhesins.
FIG 7.
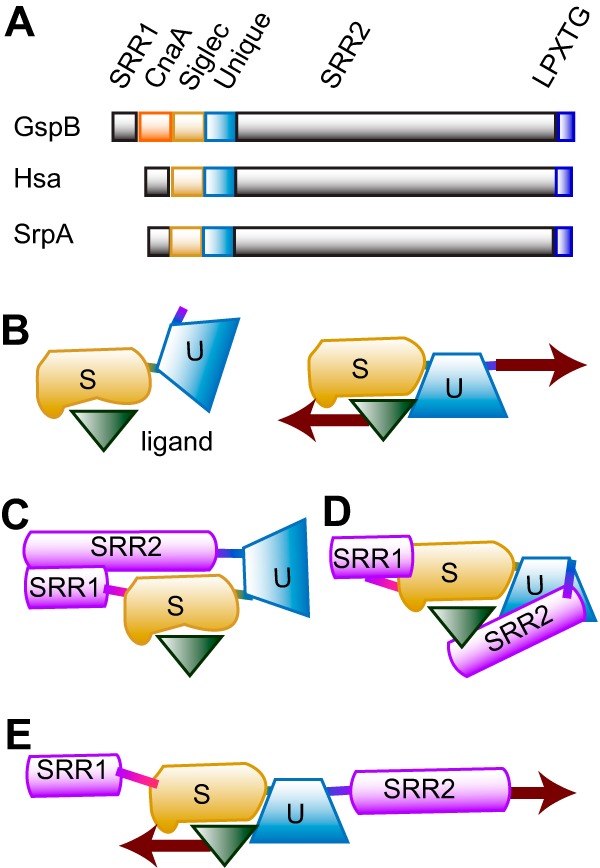
Model for shear-enhanced SRR-mediated adhesion through catch bonds. (A) Domain structure of GspB, Hsa, and SrpA from primary sequence analysis. (B to E) Speculative model for how mechanical force activates binding. The Unique domain (U) may require mechanical force to bring it into optimal alignment with the Siglec domain (S). Optimal alignment may create a combined binding pocket in which both domains contact the ligand or might enable correct contacts between the two domains for allosteric activation of the Siglec by the Unique domain. Without mechanical force, the alignment would be lost again, allowing the ligand to dissociate from a less-than-optimal binding pocket.
Regardless of the structural mechanism for the phenomenon, the shear-enhanced property of SRR adhesins suggests a key mechanism for selective attachment of streptococci to immobilized receptors such as platelet GPIbα, compared with sialoglycan targets within the bloodstream, such as fibrinogen, glycocalicin, or cell surface receptors. Shear-enhanced adhesion is generally resistant to inhibition by soluble molecules that cannot generate significant drag force (39, 40). Moreover, the shear-enhanced switch from rolling to stationary adhesion suggests that tensile force is required for long-lived interactions formed by SRR adhesins (41, 42). This would prevent long-lived interactions between SRR adhesins and soluble blood proteins and would reduce stable attachment of streptococci to blood cells in the low-flow regions of the circulation. The hypothesis that shear-enhanced adhesion enables selective attachment to immobilized receptors is supported by our observations that GspB+ and Hsa+ were able to bind to immobilized platelets at high flow even when mixed with whole blood.
Our results also address whether SRR adhesins can mediate adhesion to platelets and platelet receptors in the very high shear stress characteristic of heart valves. We found that streptococci suspended in buffer initiated adhesion poorly at high shear stress but were able to maintain adhesion once bound. This reduced initial binding at high flow is nearly universal in studies of bacterial adhesion and has been attributed to insufficient adhesive strength of the various adhesive mechanisms (43–46). Our results contradict that interpretation because the adhesive strength of SRR adhesins was sufficient to maintain adhesion at high shear (Fig. 3). A more likely explanation is that an effect called hydrodynamic lift, which is known to push microparticles away from the surface at high shear stress (47), also applies to bacteria. This concept would explain the inability of pulsatile flow to increase adhesion of suspended streptococci in our study, because streptococci that are driven away from the wall during high shear stress will not instantly return when flow is pulsed off. This small number of suspended streptococci that can bind to platelets in high pulsatile flow may be sufficient to initiate infective endocarditis. However, it is also possible that streptococci may bind to platelets before the platelets bind to heart valves. Regardless of how streptococci initially bind to the endocardium, our studies clearly demonstrate that SRR adhesins have the properties appropriate for maintaining adhesion in the hydrodynamic conditions on the surface of heart valves.
Finally, these findings have important implications for the development of novel therapies that target bacterial binding. Screening of inhibitors of bacterial binding is typically done under static conditions, using soluble compounds. However, candidates identified by this approach are unlikely to be effective against catch-bonding adhesins, since soluble inhibitors cannot generate the drag force needed to activate high-affinity catch bonds. For catch-bonding adhesins, a more effective approach may be to develop noncompetitive inhibitors that control the conformational changes associated with catch bonds (48). In combination with the comparative analysis provided above, our results suggest that the correctly glycosylated forms of the full extracellular region of the SRR adhesins will be an essential tool to study the conformational activation and inhibition of SRR adhesins. Our findings also suggest that inhibitors should be tested in early stages of development under more physiological conditions of shear stress.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
S. gordonii strain M99 expresses GspB (37) and is referred to as GspB+ in this study. Strain PS846 is derived from M99 by deletion of gspB as described previously (49) and is referred to as ΔGspB in this study. Strain PS798 is derived from S. gordonii strain Challis by repair of a stop codon in secA2, which is essential for Hsa export and surface expression (17) and so is referred to as Hsa+ in this study. Strain PS779 is derived from S. gordonii strain Challis by deletion of hsa as described previously (17) and so is referred to as ΔHsa in this study. S. sanguinis strain SK36 expresses SrpA (24) and is referred to as SrpA+ in this study. Strain PS1071 was derived from SK36 by transformation with pB1060flag. This plasmid carries a segment of the SRR2 region of gspB (49). The integration of pB1060flag into the SRR2 region of srpA results in a truncated form of SrpA that is secreted into the culture medium, rather than anchored to the bacterial cell wall. PS1071 is therefore referred to as SrpA− in this study. The six strains used in this study are summarized in Table 1. Bacteria were grown for 18 h in Todd-Hewitt broth in a 5% carbon dioxide environment, washed twice, and sonicated briefly to disrupt aggregated cells. In order to visualize the bacteria in the platelet studies, bacteria were treated for 30 min with 50 μM Syto-13 green fluorescent nucleic acid stain (Molecular Probes) in PBS and washed twice.
Synthesis of biotinylated sialyl-T-antigen.
Neu5Acα2-3Galβ1-3GalNAcαProN3 (50) (15 mg, 0.019 mmol) was dissolved in 4 ml of water, and palladium on carbon (Pd/C; 20 mg) was added. The mixture was stirred at room temperature under H2 atmosphere for 1 h. After filtration, the flowthrough was evaporated to dryness to provide the amine product Neu5Acα2-3Galβ1-3GalNAcαProNH2. To a solution of the amine, biotin-hexaethylene glycol linker (biotin-HEG) (51) (13.0 mg, 0.023 mmol) and N-hydroxybenzotriazole (HOBt; 1.1 equivalent) in 3 ml of dry dimethylformamide (DMF), 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDCI) (1.3 equivalent), and N,N-diisopropylethylamine (DIEA, 1.3 equivalent) were added at 0°C. The mixture was stirred at room temperature overnight. After removal of the solvent, the residue was purified by flash column chromatography (ethyl acetate-methanol-water [EtOAc-MeOH-H2O], 5:3:2 by volume) to produce the corresponding biotinylated sialyl T-antigen Neu5Acα2-3Galβ1-3GalNAcα-HEG-biotin (19 mg, 81%). 1H nuclear magnetic resonance (NMR) (800 MHz, D2O) δ 4.83 (d, 1H, J = 4.0 Hz, H-1′), 4.49 (d, 1H, J = 8.0 Hz, H-1″), 4.26 (dd, 1H, J = 4.0 and 10.4 Hz), 4.18 (d, 1H, J = 3.2 Hz), 4.03 to 3.52 (m, 7H), 3.79 (t, 1H, J = 10.4 Hz), 3.70 to 3.30 (m, 44H), 2.71 (t, 1H, J = 7.2 Hz), 2.70 (dd, 1H, J = 4.8 and 12.0 Hz), 2.39 (t, 2H, J = 6.4 Hz), 1.98 (s, 6H), 1.86 to 1.41 (m, 9H); 13C NMR (200 MHz, D2O) δ 174.80, 174.40, 174.12, 173.79, 172.25, 104.33, 99.56, 97.01, 77.26, 75.44, 74.59, 72.63, 71.67, 70.46, 70.11, 69.57, 69.50, 69.49, 69.47, 69.45, 69.43, 69.41, 69.40, 69.37, 69.25, 69.21, 69.19, 68.92, 68.65, 68.50, 68.25, 67.86, 67.22, 64.79, 62.30, 61.80, 61.11, 60.82, 52.26, 51.49, 48.47, 39.51, 38.81, 37.00, 35.83, 35.69, 34.09, 30.45, 28.12, 23.10, 21.90, 21.88. HRMS (ESI) m/z for C52H89N6O28S (M-H). Calculated: 1,277.5446. Found: 1,277.5460.
Preparation of platelet-coated surfaces.
Corning 35-mm tissue culture dishes were incubated with 50 μl 0.01% polylysine for 30 min at 37°C and then dried. Washed human platelets (37) were fixed with 1.6% formaldehyde at 37°C for 15 min and added to the polylysine-coated plates for 2 h at 37°C. Plates were washed with 0.2% PBS-BSA (phosphate-buffered saline with 0.2% bovine serum albumin) and incubated overnight to prevent nonspecific adhesion.
Preparation of ecGPIbα-coated surfaces.
The extracellular domain of GPIbα (referred to as ecGPIbα in this paper but also known as glycocalicin) was purified from human blood as described previously (52) and stored at −80°C. ecGPIbα was incubated at 10 μg/ml in PBS buffer on Corning 35-mm tissue culture dishes for 75 min at 37°C. The dishes were washed with 0.2% PBS–BSA and incubated in the same medium overnight at 4°C to block nonspecific adhesion.
Preparation of sTa-coated surfaces.
Corning 35-mm tissue culture dishes were incubated with 200 μg/ml biotinylated BSA for 75 min at 37°C and washed with 0.2% PBS-BSA. The plates were incubated for 30 min with 100 μg/ml streptavidin, washed, and incubated overnight with PBS-BSA. The plates were incubated for 30 min with 40 μg/ml biotinylated sTa and washed with PBS-BSA. Wells treated with 0.2% PBS-BSA overnight at 4°C served as negative controls.
Bacterial binding in flow.
A GlycoTech flow chamber with Corning dishes coated as described above for the lower surface was used to assess binding. Bacteria at an optical density of 0.3 at 600 nm in 0.2% PBS–BSA were pumped through the flow chamber using a syringe pump at the flow rate necessary to achieve the wall shear stress indicated. Bound bacteria were visualized using a Coolsnap charge-coupled-device (CCD) camera on a microscope with a 10× objective. The videos of bound bacteria were then analyzed using ImageJ. In attachment assays, the bacteria were pumped through the chamber at the indicated shear stress, and the bacteria were counted after 3 min. When bacterial adhesion was measured in whole blood, bacteria were added to whole citrated blood at the concentration that would provide an optical density (OD) of 0.3 in buffer, and the attachment assays were run for only 2 min (to minimize the amount of blood used flowing through the chamber). In the detachment assays, bacteria were pumped through the chamber at 2 dyn/cm2 to allow bacteria to adhere and then washed with PBS-BSA at 5 dyn/cm2 for 30 s. Thereafter, shear stress was increased or decreased every 15 s to obtain the indicated shear stresses. To characterize the mode of bacterial adhesion (rolling or stationary), the number of moving cells was determined by subtracting an image from one taken 1 s later. This causes stationary cells to blend into the background, while moving cells appear as pairs of black and white spots. The fraction of stationary cells was then calculated as 1 minus the ratio of moving to total cells.
Characterizing pulsatile flow.
The resistance created by the flow chamber and the compliance of the tubing create a characteristic slight delay in response time, due to the transient expansion of the tubing when the pump is turned on and its subsequent contraction after the pump is turned off. To know the actual flow in the chamber, we characterized the fluid velocity with particle-tracking velocimetry. That is, we tracked microspheres that are next to the surface while turning the pump on and off. The microspheres can be tracked only at low shear stresses, but the fluidic circuit is expected to be linear and thus would give the same normalized response at all pump velocities, as long as the on-off switching pattern is the same.
Statistical methods.
To determine which conclusions were statistically significant, we applied Student's t test. We used paired samples when the duplicate experiments were performed on different days (each day testing the two conditions being compared) or, when the duplicate experiments involved different runs, with each run testing the two shear conditions being compared. We used unpaired samples when the duplicates and different conditions being run all involved independent runs on the same day. We used a 2-tailed t test if we asked whether two things were different, and a one-tailed t test if we specifically asked if one was greater than the other. Each time a P value is provided, the choice of test is briefly described. Unless otherwise indicated, all experiments and statistical tests were performed with two samples for each condition.
Ethics statement.
Human blood was obtained from donors for this study with approval of the University of Washington's Institutional Review Board (IRB). Donors provided written informed consent, and all recruitment and consent processes and materials were IRB approved.
Supplementary Material
ACKNOWLEDGMENTS
We thank Adam Munday and José Lopéz for providing glycocalicin (ecGPIbα) and Tina Iverson for her insightful comments on the manuscript.
This work was funded by NIH AI106987, NIH AI41513, the Washington NASA Space Grant Consortium, NASA grant number NNX15AJ98H, and the University of Washington GenOM Project (NIH 5R25HG007153-03).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00160-18.
REFERENCES
- 1.Murdoch DR, Corey GR, Hoen B, Miro JM, Fowler VG Jr, Bayer AS, Karchmer AW, Olaison L, Pappas PA, Moreillon P, Chambers ST, Chu VH, Falco V, Holland DJ, Jones P, Klein JL, Raymond NJ, Read KM, Tripodi MF, Utili R, Wang A, Woods CW, Cabell CH. 2009. Clinical presentation, etiology, and outcome of infective endocarditis in the 21st century: the International Collaboration on Endocarditis-Prospective Cohort Study. Arch Intern Med 169:463–473. doi: 10.1001/archinternmed.2008.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tleyjeh IM, Steckelberg JM, Murad HS, Anavekar NS, Ghomrawi HM, Mirzoyev Z, Moustafa SE, Hoskin TL, Mandrekar JN, Wilson WR, Baddour LM. 2005. Temporal trends in infective endocarditis: a population-based study in Olmsted County, Minnesota. JAMA 293:3022–3028. doi: 10.1001/jama.293.24.3022. [DOI] [PubMed] [Google Scholar]
- 3.Thornhill MH, Dayer MJ, Forde JM, Corey GR, Chu VH, Couper DJ, Lockhart PB. 2011. Impact of the NICE guideline recommending cessation of antibiotic prophylaxis for prevention of infective endocarditis: before and after study. BMJ 342:d2392. doi: 10.1136/bmj.d2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poutanen SM, de Azavedo J, Willey BM, Low DE, MacDonald KS. 1999. Molecular characterization of multidrug resistance in Streptococcus mitis. Antimicrob Agents Chemother 43:1505–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsu RB, Lin FY. 2006. Effect of penicillin resistance on presentation and outcome of nonenterococcal streptococcal infective endocarditis. Cardiology 105:234–239. doi: 10.1159/000091821. [DOI] [PubMed] [Google Scholar]
- 6.Sullam PM, Payan DG, Dazin PF, Valone FH. 1990. Binding of viridans group streptococci to human platelets: a quantitative analysis. Infect Immun 58:3802–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herzberg MC, MacFarlane GD, Gong K, Armstrong NN, Witt AR, Erickson PR, Meyer MW. 1992. The platelet interactivity phenotype of Streptococcus sanguis influences the course of experimental endocarditis. Infect Immun 60:4809–4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herrmann M, Lai QJ, Albrecht RM, Mosher DF, Proctor RA. 1993. Adhesion of Staphylococcus aureus to surface-bound platelets: role of fibrinogen/fibrin and platelet integrins. J Infect Dis 167:312–322. doi: 10.1093/infdis/167.2.312. [DOI] [PubMed] [Google Scholar]
- 9.Sullam PM, Bayer AS, Foss WM, Cheung AL. 1996. Diminished platelet binding in vitro by Staphylococcus aureus is associated with reduced virulence in a rabbit model of infective endocarditis. Infect Immun 64:4915–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moreillon P, Que YA, Bayer AS. 2002. Pathogenesis of streptococcal and staphylococcal endocarditis. Infect Dis Clin North Am 16:297–318. doi: 10.1016/S0891-5520(01)00009-5. [DOI] [PubMed] [Google Scholar]
- 11.Xiong YQ, Bensing BA, Bayer AS, Chambers HF, Sullam PM. 2008. Role of the serine-rich surface glycoprotein GspB of Streptococcus gordonii in the pathogenesis of infective endocarditis. Microb Pathog 45:297–301. doi: 10.1016/j.micpath.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siboo IR, Chambers HF, Sullam PM. 2005. Role of SraP, a serine-rich surface protein of Staphylococcus aureus, in binding to human platelets. Infect Immun 73:2273–2280. doi: 10.1128/IAI.73.4.2273-2280.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oyen WJ, Boerman OC, Brouwers FM, Barrett JA, Verheugt FW, Ruiter DJ, Corstens FH, van der Meer JW. 2000. Scintigraphic detection of acute experimental endocarditis with the technetium-99m labelled glycoprotein IIb/IIIa receptor antagonist DMP444. Eur J Nucl Med 27:392–399. doi: 10.1007/s002590050521. [DOI] [PubMed] [Google Scholar]
- 14.Takahashi Y, Takashima E, Shimazu K, Yagishita H, Aoba T, Konishi K. 2006. Contribution of sialic acid-binding adhesin to pathogenesis of experimental endocarditis caused by Streptococcus gordonii DL1. Infect Immun 74:740–743. doi: 10.1128/IAI.74.1.740-743.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bensing BA, Sullam PM. 2002. An accessory sec locus of Streptococcus gordonii is required for export of the surface protein GspB and for normal levels of binding to human platelets. Mol Microbiol 44:1081–1094. doi: 10.1046/j.1365-2958.2002.02949.x. [DOI] [PubMed] [Google Scholar]
- 16.Takamatsu D, Bensing BA, Cheng H, Jarvis GA, Siboo IR, Lopez JA, Griffiss JM, Sullam PM. 2005. Binding of the Streptococcus gordonii surface glycoproteins GspB and Hsa to specific carbohydrate structures on platelet membrane glycoprotein Ibalpha. Mol Microbiol 58:380–392. doi: 10.1111/j.1365-2958.2005.04830.x. [DOI] [PubMed] [Google Scholar]
- 17.Bensing BA, Lopez JA, Sullam PM. 2004. The Streptococcus gordonii surface proteins GspB and Hsa mediate binding to sialylated carbohydrate epitopes on the platelet membrane glycoprotein Ibalpha. Infect Immun 72:6528–6537. doi: 10.1128/IAI.72.11.6528-6537.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pyburn TM, Bensing BA, Xiong YQ, Melancon BJ, Tomasiak TM, Ward NJ, Yankovskaya V, Oliver KM, Cecchini G, Sulikowski GA, Tyska MJ, Sullam PM, Iverson TM. 2011. A structural model for binding of the serine-rich repeat adhesin GspB to host carbohydrate receptors. PLoS Pathog 7(7):e1002112. doi: 10.1371/journal.ppat.1002112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bensing BA, Khedri Z, Deng L, Yu H, Prakobphol A, Fisher SJ, Chen X, Iverson TM, Varki A, Sullam PM. 2016. Novel aspects of sialoglycan recognition by the Siglec-like domains of streptococcal SRR glycoproteins. Glycobiology 26:1222–1234. doi: 10.1093/glycob/cww042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Sorge NM, Quach D, Gurney MA, Sullam PM, Nizet V, Doran KS. 2009. The group B streptococcal serine-rich repeat 1 glycoprotein mediates penetration of the blood-brain barrier. J Infect Dis 199:1479–1487. doi: 10.1086/598217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seo HS, Mu R, Kim BJ, Doran KS, Sullam PM. 2012. Binding of glycoprotein Srr1 of Streptococcus agalactiae to fibrinogen promotes attachment to brain endothelium and the development of meningitis. PLoS Pathog 8:e1002947. doi: 10.1371/journal.ppat.1002947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rose L, Shivshankar P, Hinojosa E, Rodriguez A, Sanchez CJ, Orihuela CJ. 2008. Antibodies against PsrP, a novel Streptococcus pneumoniae adhesin, block adhesion and protect mice against pneumococcal challenge. J Infect Dis 198:375–383. doi: 10.1086/589775. [DOI] [PubMed] [Google Scholar]
- 23.Orihuela CJ. 2009. Role played by psrP-secY2A2 (accessory region 34) in the invasive disease potential of Streptococcus pneumoniae. J Infect Dis 200:1180–1181, author reply 1181–1182. doi: 10.1086/605649. [DOI] [PubMed] [Google Scholar]
- 24.Plummer C, Wu H, Kerrigan SW, Meade G, Cox D, Ian Douglas CW. 2005. A serine-rich glycoprotein of Streptococcus sanguis mediates adhesion to platelets via GPIb. Br J Haematol 129:101–109. doi: 10.1111/j.1365-2141.2005.05421.x. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi Y, Yajima A, Cisar JO, Konishi K. 2004. Functional analysis of the Streptococcus gordonii DL1 sialic acid-binding adhesin and its essential role in bacterial binding to platelets. Infect Immun 72:3876–3882. doi: 10.1128/IAI.72.7.3876-3882.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jakubovics NS, Kerrigan SW, Nobbs AH, Stromberg N, van Dolleweerd CJ, Cox DM, Kelly CG, Jenkinson HF. 2005. Functions of cell surface-anchored antigen I/II family and Hsa polypeptides in interactions of Streptococcus gordonii with host receptors. Infect Immun 73:6629–6638. doi: 10.1128/IAI.73.10.6629-6638.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yajima A, Takahashi Y, Konishi K. 2005. Identification of platelet receptors for the Streptococcus gordonii DL1 sialic acid-binding adhesin. Microbiol Immunol 49:795–800. doi: 10.1111/j.1348-0421.2005.tb03659.x. [DOI] [PubMed] [Google Scholar]
- 28.Pyburn TM, Yankovskaya V, Bensing BA, Cecchini G, Sullam PM, Iverson TM. 2010. Purification, crystallization and preliminary X-ray diffraction analysis of the carbohydrate-binding region of the Streptococcus gordonii adhesin GspB. Acta Crystallogr Sect F Struct Biol Cryst Commun 66:1503–1507. doi: 10.1107/S1744309110036535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bensing BA, Loukachevitch LV, McCulloch KM, Yu H, Vann KR, Wawrzak Z, Anderson S, Chen X, Sullam PM, Iverson TM. 2016. Structural basis for sialoglycan binding by the Streptococcus sanguinis SrpA adhesin. J Biol Chem 291:7230–7240. doi: 10.1074/jbc.M115.701425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loukachevitch LV, Bensing BA, Yu H, Zeng J, Chen X, Sullam PM, Iverson TM. 2016. Structures of the Streptococcus sanguinis SrpA binding region with human sialoglycans suggest features of the physiological ligand. Biochemistry 55:5927–5937. doi: 10.1021/acs.biochem.6b00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng L, Bensing BA, Thamadilok S, Yu H, Lau K, Chen X, Ruhl S, Sullam PM, Varki A. 2014. Oral streptococci utilize a Siglec-like domain of serine-rich repeat adhesins to preferentially target platelet sialoglycans in human blood. PLoS Pathog 10:e1004540. doi: 10.1371/journal.ppat.1004540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fernandez Guerrero ML, Alvarez B, Manzarbeitia F, Renedo G. 2012. Infective endocarditis at autopsy: a review of pathologic manifestations and clinical correlates. Medicine (Baltimore) 91:152–164. doi: 10.1097/MD.0b013e31825631ea. [DOI] [PubMed] [Google Scholar]
- 33.Bell GI. 1978. Models for the specific adhesion of cells to cells. Science 200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- 34.Persat A, Nadell CD, Kim MK, Ingremeau F, Siryaporn A, Drescher K, Wingreen NS, Bassler BL, Gitai Z, Stone HA. 2015. The mechanical world of bacteria. Cell 161:988–997. doi: 10.1016/j.cell.2015.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weston MW, LaBorde DV, Yoganathan AP. 1999. Estimation of the shear stress on the surface of an aortic valve leaflet. Ann Biomed Eng 27:572–579. doi: 10.1114/1.199. [DOI] [PubMed] [Google Scholar]
- 36.Ding AM, Palmer RJ Jr, Cisar JO, Kolenbrander PE. 2010. Shear-enhanced oral microbial adhesion. Appl Environ Microbiol 76:1294–1297. doi: 10.1128/AEM.02083-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sullam PM, Valone FH, Mills J. 1987. Mechanisms of platelet aggregation by viridans group streptococci. Infect Immun 55:1743–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prakobphol A, Burdsal CA, Fisher SJ. 1995. Quantifying the strength of bacterial adhesive interactions with salivary glycoproteins. J Dent Res 74:1212–1218. doi: 10.1177/00220345950740051101. [DOI] [PubMed] [Google Scholar]
- 39.Thomas WE, Trintchina E, Forero M, Vogel V, Sokurenko EV. 2002. Bacterial adhesion to target cells enhanced by shear force. Cell 109:913–923. doi: 10.1016/S0092-8674(02)00796-1. [DOI] [PubMed] [Google Scholar]
- 40.Nilsson LM, Thomas WE, Sokurenko EV, Vogel V. 2006. Elevated shear stress protects Escherichia coli cells adhering to surfaces via catch bonds from detachment by soluble inhibitors. Appl Environ Microbiol 72:3005–3010. doi: 10.1128/AEM.72.4.3005-3010.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas W. 2008. Catch bonds in adhesion. Annu Rev Biomed Eng 10:39–57. doi: 10.1146/annurev.bioeng.10.061807.160427. [DOI] [PubMed] [Google Scholar]
- 42.Thomas WE. 2009. Mechanochemistry of receptor-ligand bonds. Curr Opin Struct Biol 19:50–55. doi: 10.1016/j.sbi.2008.12.006. [DOI] [PubMed] [Google Scholar]
- 43.Wang IW, Anderson JM, Jacobs MR, Marchant RE. 1995. Adhesion of Staphylococcus epidermidis to biomedical polymers: contributions of surface thermodynamics and hemodynamic shear conditions. J Biomed Mater Res 29:485–493. doi: 10.1002/jbm.820290408. [DOI] [PubMed] [Google Scholar]
- 44.Dickinson RB, Cooper SL. 1995. Analysis of shear-dependent bacterial adhesion kinetics to biomaterial surfaces. AIChE J 41:2160–2174. doi: 10.1002/aic.690410915. [DOI] [Google Scholar]
- 45.Nejadnik MR, van der Mei HC, Busscher HJ, Norde W. 2008. Determination of the shear force at the balance between bacterial attachment and detachment in weak-adherence systems, using a flow displacement chamber. Appl Environ Microbiol 74:916–919. doi: 10.1128/AEM.01557-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raya A, Sodagari M, Pinzon NM, He X, Newby BMZ, Ju LK. 2010. Effects of rhamnolipids and shear on initial attachment of Pseudomonas aeruginosa PAO1 in glass flow chambers. Environ Sci Pollut Res 17:1529–1538. doi: 10.1007/s11356-010-0339-6. [DOI] [PubMed] [Google Scholar]
- 47.Williams PS, Lee SH, Giddings JC. 1994. Characterization of hydrodynamic lift forces by field-flow fractionation—inertial and near-wall lift forces. Chem Eng Commun 130:143–166. doi: 10.1080/00986449408936272. [DOI] [Google Scholar]
- 48.Kisiela DI, Avagyan H, Friend D, Jalan A, Gupta S, Interlandi G, Liu Y, Tchesnokova V, Rodriguez VB, Sumida JP, Strong RK, Wu XR, Thomas WE, Sokurenko EV. 2015. Inhibition and reversal of microbial attachment by an antibody with parasteric activity against the FimH adhesin of uropathogenic E. coli. PLoS Pathog 11:e1004857. doi: 10.1371/journal.ppat.1004857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bensing BA, Takamatsu D, Sullam PM. 2005. Determinants of the streptococcal surface glycoprotein GspB that facilitate export by the accessory Sec system. Mol Microbiol 58:1468–1481. doi: 10.1111/j.1365-2958.2005.04919.x. [DOI] [PubMed] [Google Scholar]
- 50.Lau K, Yu H, Thon V, Khedri Z, Leon ME, Tran BK, Chen X. 2011. Sequential two-step multienzyme synthesis of tumor-associated sialyl T-antigens and derivatives. Org Biomol Chem 9:2784–2789. doi: 10.1039/c0ob01269f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chokhawala HA, Huang S, Lau K, Yu H, Cheng J, Thon V, Hurtado-Ziola N, Guerrero JA, Varki A, Chen X. 2008. Combinatorial chemoenzymatic synthesis and high-throughput screening of sialosides. ACS Chem Biol 3:567–576. doi: 10.1021/cb800127n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Romo GM, Dong JF, Schade AJ, Gardiner EE, Kansas GS, Li CQ, McIntire LV, Berndt MC, Lopez JA. 1999. The glycoprotein Ib-IX-V complex is a platelet counterreceptor for P-selectin. J Exp Med 190:803–814. doi: 10.1084/jem.190.6.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.