

ABSTRACT
Streptococcus pneumoniae is a major cause of invasive pneumococcal disease, septicemia, and meningitis that can result in high morbidity rates in children under 5 years old. The current polysaccharide-based vaccines can provide type-specific immunity, but a broad-spectrum vaccine would provide greater coverage. Therefore, developing pneumococcal-protein-based vaccines that can extend to more serum types is highly important. In this study, we vaccinated mice via the subcutaneous (s.c.) route with a systemic vaccine that is a mixture of fusion protein PsaA-PspA23 and a single protein, PspA4, with aluminum hydroxide as an adjuvant. As a comparison, mice were immunized intranasally with a mucosal vaccine that is a mixture of PspA2-PA-BLP (where PA is protein anchor and BLP is bacterium-like particle) and PspA4-PA-BLP, via the intranasal (i.n.) route. The two immunization processes were followed by challenge with Streptococcus pneumoniae bacteria from two different PspA families. Specific IgG titers in the serum and specific IgA titers in the mucosa were determined following immunizations. Bacterial loads and survival rates after challenge were compared. Both the systemic vaccine and the mucosal vaccine induced a significant increase of IgG against PspAs. Only the mucosal vaccine also induced specific IgA in the mucosa. The two vaccines provided protection, but each vaccine showed an advantage. The systemic vaccine induced higher levels of serum antibodies, whereas the mucosal vaccine limited the bacterial load in the lung and blood. Therefore, coimmunizations with the two types of vaccines may be implemented in the future.
KEYWORDS: pneumococcal protein vaccine, bacterium-like particles (BLPs), PspA, PsaA, Streptococcus pneumoniae, PsaA, PspA
INTRODUCTION
Streptococcus pneumoniae is a human nasopharyngeal bacterium that can normally invade sterile sites to cause invasive pneumococcal disease (IPD), including bacteremia and meningitis (1). In the year 2000, IPD accounted for more than 800,000 deaths in children under 5 years old (2). All currently commercially available pneumococcal vaccines, including mainly the polysaccharide vaccine and the conjugate vaccine, are designed on the basis of the serotype-specific polysaccharide capsule of the bacterium. However, the 23-valent polysaccharide vaccine (PPV23) is not effective in children younger than 5 years old (3), and the pneumococcal conjugate vaccines are effective in children but have limited serotype coverage (4). Therefore, development of a new kind of S. pneumoniae vaccine is critical and significant. Much research effort is currently invested in searching for pneumococcal proteins with protective potential to be included in future protein-based vaccines. The objective is to develop a protein-based pneumococcal vaccine that confers serotype-independent protection in all age groups (5–7). Several pneumococcal toxic proteins have been investigated as potential antigen candidates, such as pneumococcal surface protein A (PspA), pneumococcal surface adhesion A (PsaA), pneumolysin (Ply), pneumococcal choline-binding protein A (PcpA), and pneumococcal surface protein C (PspC) (8), among others. The wide-spectrum protein-based vaccines are low cost, substantially immunogenic, and highly conserved.
PspA is a pneumococcal virulence factor and a choline-binding protein. It has three major domains: an alpha-helical amino-terminal (N-terminal) domain, which exhibits a pattern of sequence variation that was used to classify PspA molecules into clades, a proline-rich region (PRR), and a choline-binding domain to anchor the protein across the cell wall (9). PspA shows variability in different isolates. Sequence-based classification divides PspA variants into three families, which are further subdivided into six clades: family 1 (clades 1 and 2), family 2 (clades 3, 4, and 5), and family 3 (clade 6) (10). To achieve complete coverage, it was suggested that a PspA-based vaccine should contain at least one PspA from each of the two major families (1 and 2) (11). Our group has previously revealed that parenteral immunization of mice with a recombinant PspA from family 2 (clade 2, clade 3, or clade 4) induced protection against challenge of lethal pneumococcal strains expressing PspA from families 1 and 2 (12). Antibodies generated against PspA are highly cross-reactive and cross-protective (13). The major cross-protective epitopes are located in the N-terminal alpha-helical sequence of PspA, especially the first and last 100 amino acids (14). Pneumococcal surface adhesion A (PsaA) is another antigen candidate that has been evaluated against S. pneumoniae infection in both animal models and human clinical trials with encouraging results. Therefore, we used it as part of the fusion protein of PsaA-PspA23 (15) to evaluate its immunogenicity and protection potential.
Compared to stimulating local immunity, mucosal vaccination has extra benefits such as needle-free administration, reduced side effects, and easy boosting. Bacterium-like particles (BLPs), once called Gram-positive enhancer matrix (GEM), are based on nongenetically modified Gram-positive bacteria and can be used to potentially enhance mucosal vaccines. They consist of nonliving bacterium-shaped delivery particles with adjuvant properties, which can easily be loaded with antigens containing a cell wall binding domain, called protein anchor (PA) (16). The BLPs are made from acid-pretreated Lactococcus lactis bacteria, with their original size and structures of about 1 μm retained, and are thus ideally sized for uptake by the M cells on the mucosal surface. The PA domain is composed of three LysM motifs of about 45 amino acids separated by spacer regions and can be added to antigens as a recombinant fusion protein (17–19).
In this study, two types of vaccines were generated (our unpublished data): a systemic vaccine, including the fusion protein PsaA-PspA23 and the single protein PspA4 as antigens, and a mucosal vaccine, using fusion proteins of PspA2-PA and PspA4-PA antigens attached to BLPs. We compared the antibody types and levels, bacterial colonization levels, and survival rates conferred by these vaccines after challenge with two strains from different families.
RESULTS
Preparation of antigens.
The fusion protein PsaA-PspA23 includes portions of PsaA and PspA domains, in which the PspA23 portion contains both the N-terminal clade 2- and N-terminal clade 3-defining regions of PspA (15). The PspA4 antigen contains the N-terminal domain and the proline-rich region domain of PspA clade 4. The proteins were all expressed in Escherichia coli with a histidine tag in order to be purified by a nickel affinity chromatographic column. Analysis by SDS-PAGE gel and Western blotting (using mouse anti-His tag monoclonal antibody as the primary antibody) showed the expected molecular masses for PspA-PspA23 and PspA4 of approximately 90 kDa and 66 kDa, respectively (Fig. 1).
FIG 1.

SDS-PAGE and Western blot analysis of antigens. Lanes 1 and 5, systemic antigen 449 protein PspA4, 66 kDa (KD); lanes 2 and 6, systemic antigen protein PsaA-PspA23, 90 kDa; lanes 3 and 7, 450 BLP-bound mucosal antigen PspA2-PA, 67 kDa; lanes 4 and 8, BLP-bound mucosal antigen 451 PspA4-PA, 90 kDa. For the integrity of results and performance in this figure, we cut the space lane in gels and spliced them together.
The fusion proteins PspA2-PA and PspA4-PA were individually expressed in E. coli and mixed with BLPs after ultrasonication. After binding with BLPs, PspA2-PA-BLP and PspA4-PA-BLP were washed, centrifuged, resuspended, and then analyzed by SDS-PAGE and Western blotting (with PA polyvalent antibody as the primary antibody), which showed molecular masses for PspA2-PA and PspA4-PA of approximately 67 kDa and 90 kDa, respectively (Fig. 1). The BLP vaccines show a background of degraded lactococcal components in SDS-PAGE. Binding efficiency of PspA2 or PspA4 with BLPs was calculated from SDS-PAGE. Results showed that PspA2-PA and PspA4-PA bound with BLPs efficiently, with a capacity of about 30 μg PspA2-PA per mg BLPs and 60 μg PspA4-PA per mg BLPs.
Induction of antigen-specific IgG and IgA antibody titers.
Two weeks after the third immunization, the antigen-specific IgG and IgA levels in mouse sera and bronchoalveolar lavage fluids were determined. Mice immunized with systemic vaccine [PsaA-PspA23and PspA4 with Al(OH)3 as adjuvant] showed a statistically significant increase in anti-PspA23 and anti-PspA4 IgG titers (Fig. 2A and B). Meanwhile, animals immunized with mucosal vaccine (BLP-PspA2 and BLP-PspA4) showed statistically significant increases in anti-PspA2 and anti-PspA4 IgG antibodies (Fig. 2C and D), although the IgG titers were lower than that with the systemic vaccine. The mice immunized with mucosal vaccine showed statistically significant increases in anti-PspA2 and anti-PspA4 IgA antibodies (Fig. 2E and F) in bronchoalveolar washes. These results indicate that both mucosal and systemic immunizations induced IgG antibodies but the mucosal vaccine was also able to elicit nasal IgA antibodies.
FIG 2.
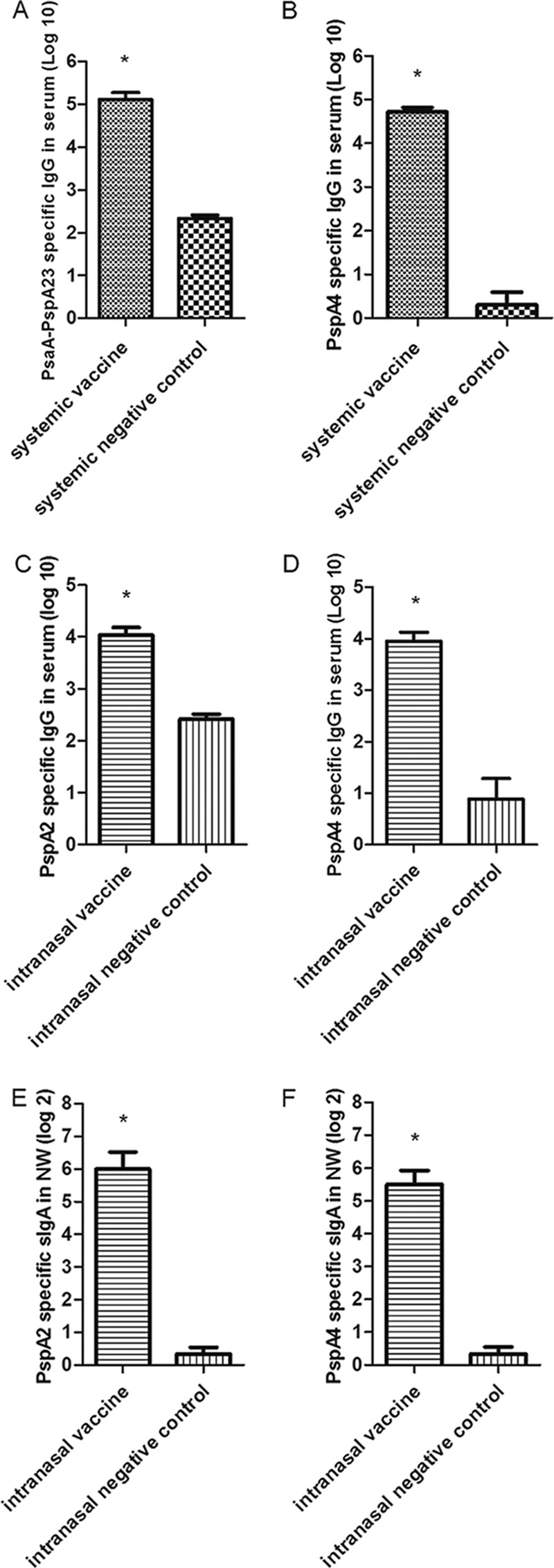
Analysis of immunogenicity of systemic and mucosal vaccines. (A to D) IgG titers specific for PsaA-PspA23 (A) and PspA4 (B) in the serum of the systemic vaccine group or specific for PspA2 (C) or PspA4 (D) in the serum of the mucosal vaccine group. (E and F) IgA titers for PspA2 (E) or PspA4 (F) in the nasal washes (NW) of the mucosal vaccine group. Statistical difference of test vaccine groups from the negative controls: *, P < 0.05.
Analysis of bacterial colonization in mouse blood and lung after challenge.
Mice immunized with different vaccines were challenged intranasally with two clades of S. pneumoniae strains from family 1 and family 2, which were ATCC 10813 (family 1, clade 2) and ATCC 6303 (family 2, clade 5). Bacteria were recovered from the blood and lung 24 h after challenge. Animals immunized with the systemic vaccine showed statistically significant reductions (P < 0.001) of ATCC 10813 and ATCC 6303 bacterial colonization in the blood and lung samples compared to the systemic vaccine control group of phosphate-buffered saline (PBS) with 10% Al(OH)3 (Fig. 3A to D). Mice immunized with systemic vaccine also showed a statistically significant reduction of ATCC 10813 (P < 0.01 in the blood and lung) and ATCC 6303 (P < 0.05 in the blood and P < 0.01 in the lung) bacterial colonization compared to the positive-control group PPV23.
FIG 3.

Recovery of pneumococcus bacterial loads in blood and lung of mice after challenge. Comparison of bacterial loads in mouse lung (A, C) and blood (B, D) samples after challenge with two bacterial strains for the systemic vaccine, systemic negative control, intranasal vaccine, intranasal negative control, and positive control (PPV23) groups. The density of pneumococcal colonization, expressed in log10 of total CFU in the lung and blood, was determined for individual mice at 24 h after challenge. Recovery of strain ATCC 10813 (family 1, clade 2) (A and B) and strain ATCC 6303 (family 2, clade 5) (C and D) in blood and lung, respectively. Symbols indicate statistically significant differences compared with corresponding controls by one-way ANOVA, with Tukey's multicomparison test. For comparison of the PPV23 group to the systemic control group: *, P < 0.05; **, P < 0.01; ***, P < 0.001. For comparison with the positive-control group, PPV23: #, P < 0.05; ##, P < 0.01; ###, P < 0.001. Results obtained from two independent experiments.
Animals immunized with the intranasal vaccine showed statistically significant reductions in ATCC 10813 and ATCC 6303 (P < 0.05) bacterial colonization compared to the intranasal negative-control group of only BLPs. There is no statistical difference in bacterial colonization reduction between the intranasal vaccine and the positive-control group PPV23 except with ATCC 6303 in the lung (P < 0.01). When we compared the effects of the intranasal and systemic vaccine immunizations, the systemic vaccine group showed higher bacterial colonization reduction in the blood and lung, meaning that the systemic immunization provided a higher bacterial clearance rate than the intranasal vaccine group. Without effective antigen, inoculation of BLPs alone (the intranasal negative-control group) showed a lower colonization level in the blood and lung than that of the systemic negative-control group, suggesting that mucosal application of only BLPs might block the colonization of pneumococci in the lung, which subsequently reduced bacterial amplification in the blood compared to the systemic control group.
Mouse survival rate after challenge with two clades of S. pneumoniae strains.
As shown in Fig. 4A, when challenged with a lethal dose of ATCC 10813 (PspA family 1, clade 2, serotype 3), mice vaccinated with the mucosal vaccine (with antigens of PspA clades 2 and 4) showed a 90% survival rate, whereas the systemic-vaccine group of mice (with antigens PsaA and PspA clades 2, 3, and 4) showed 100% survival. All the immunized groups showed significant higher protection than their own controls (P < 0.001 and P < 0.05 for systemic and mucosal vaccine groups, respectively). Consistent with the colonization study, mice immunized by BLPs alone provided some protection toward the challenge, which suggested that mucosal application of BLPs might block the infection of S. pneumoniae. The results indicated that both vaccines can provide a high level of protection against challenge from PspA family1, clade 2 bacteria if the vaccine contains the same PspA-specific clade strain. The positive control of PPV23 also provided 100% protection to the mice after challenge.
FIG 4.

Vaccines induced protection against lethal i.n. challenge. Protection levels of systemic vaccine group against challenge with ATCC 10813 (family 1, clade 2) and ATCC 6303 (family 2, clade 5) (P < 0.001) and that of the intranasal vaccine group against challenge with ATCC 10813 (P < 0.05) and ATCC 6303 (P < 0.01) were statistically significant. Results were obtained from two independent experiments.
As shown in Fig. 4B, when the challenge strain was ATCC 6303 (PspA family 2, clade 5, serotype 3), the commercial positive-control vaccine (PPV23) surprisingly did not provide any protection to the challenge, although this serotype 3 challenge strain, ATCC 6303, is covered in PPV23's protection spectrum. The reasons could be the high 90% lethal dose (LD90) challenge doses that we applied or the unstable behavior of this vaccine. A similar discrepancy was observed for negative control of pure BLPs. No protection against challenge of strain ATCC 6303 was obtained, compared to more than 50% protection against strain ATCC 10813. The application of ATCC 6303 is probably more lethal than that of strain ATCC 10813. However, for our tested vaccines, although they did not contain any clade 5 component, 80% of mucosally vaccinated mice and 100% of systemically vaccinated mice were protected. All the immunized groups showed significantly higher protection than did their own controls (P < 0.001).
We suggest that a high contribution in the cross-clade protection of the two tested vaccines can be assigned to PspA4, which is the component in both vaccines. PspA4 is in the same family (family 2) as ATCC 6303 but in a different clade. Similar results were observed in Darrieux et al.'s research (20), which showed that the antiserum of rPspA4 provided a broad cross-reactivity, recognizing pneumococcal strains containing PspAs of all clades from families 1 and 2. Other studies (10) suggest that antibodies generated against PspA protein showed enough cross-protection only to PspA antigens from the same family. Therefore, researchers tried to overcome this limitation by combining PspAs from different clades and families (11, 21) into the vaccine. This is also the design principle of vaccines in this study.
DISCUSSION
PspA is a preferred candidate for a protein-based vaccine against pneumococcal infections. However, its sequence variability might restrict the protection coverage of a PspA-based vaccine. Therefore, some studies aiming to investigate the level of cross-reactivity among PspAs in mice indicated that antibodies generated against PspA showed higher cross-reactivity with the strains expressing PspAs of the same family than with those that bear PspAs of different families (22, 23). In addition, since the pneumococci infect humans through mucosal tissues, which they colonize at the nasopharynx, induction of protective immunity at mucosal tissues is a primary strategy to prevent infectious diseases. Therefore, a mucosal vaccine has been considered an ideal form for prevention of and protection from infectious diseases. Oliveira et al. (24), for example, used a whole-cell pertussis vaccine to combine with the single PspA protein as a new mucosal pneumococcal vaccine, enhancing the protection against the strains in different clades.
By forming an antigen mixture of PspA proteins, we produced two pneumococcal vaccines in different vaccination principles, which are based on subcutaneous and mucosal immunization. PspA2 and PspA4 proteins are present in the two vaccines as major protective components, while PspA3 and PsaA components in the vaccines, in our understanding, were in charge of enhancing the immunization effects (15). This choice of antigen combination was based on their protection data obtained in our previous studies (data not shown). We compared the two vaccines in different immunization routes, with alum and BLPs used as adjuvants. Subcutaneous immunization with the systemic vaccine induced a significant increase in the PspA-specific IgG titer compared to its negative control. Nasal immunization with the mucosal vaccine induced both serum anti-PspA IgG antibodies and mucosal IgA antibodies. Furthermore, in the colonization studies after challenge with two S. pneumoniae strains from families 1 and 2, mice immunized by both vaccines showed a statistically significant reduction of bacterial colonization in blood and lung compared to their negative controls. In accordance to our previous data showing cross-protection of alum adjuvant-PspA4 protein-based-vaccine (15), cross-protection of PspA-based systemic and mucosal vaccines against S. pneumoniae from different PspA families was observed in this study. The systemic vaccine showed comparable or even improved behavior compared to the commercial PPV23 vaccine in the aspect of protection against the two challenge strains. These results suggest that the attribution to cross-clade protection of the vaccines is possibly due to the PspA4 component, which is present in both tested vaccines.
It was shown that the bacterial colonization reduction and vaccine protection of systemic vaccines are higher than those of intranasal vaccines. However, immunization with BLP-based intranasal vaccine led to the induction of not only serum but also mucosal antibodies against PspAs. Therefore, the BLP-conjugated vaccine provided both humoral and cellular responses for protection (25, 26). Negative control of mucosal vaccine (only BLPs) surprisingly showed reduction in bacterial colonization, suggesting that mucosal application of BLPs might help to reduce the colonization of lung by pneumococci, which subsequently reduced the bacterial amplification in blood. Accordingly, similar data were observed in other studies reporting that BLP-based pneumococcal mucosal vaccines showed mucosal and systemic responses in adult mice (27) and significant protection against lethal respiratory pneumococcal challenge (28).
Since soluble antigens are generally less immunogenic than antigens in particular formulations when mucosal routes are used (29), BLPs were widely used as adjuvants to develop mucosal vaccines to cause both humoral and cellular responses. The nasal application route of BLP-based vaccines offers easy administration compared to subcutaneous immunization. Binding of PA fusion proteins to BLP is fast and easy without the need for chemical treatments or extensive purification steps. The BLP-based vaccines are ideal for mass immunization: they enable needle-free administration, they allow easy immunization of large populations, and most likely they can be transported and stored for a long period without the need for a cold chain.
In summary, both the systemic and mucosal vaccinations tested in this study induced antigen-specific IgG and provided high protection levels. The systemic vaccine produced higher antigen-specific antibody levels, whereas the nasal vaccine limited the bacterial loads by inducing IgA. Our future study will focus on the immunization strategy of combining the systemic and mucosal vaccines to achieve better and broader protection.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
All S. pneumoniae strains used in the study were purchased from ATCC (American Type Culture Collection, Manassas, VA, USA). ATCC 10813 (serotype 3, PspA clade 2, family 1) and ATCC 6303 (serotype 3, PspA clade 5, family 2) are from different PspA families but have the same serotype. They were cultured in Todd-Hewitt broth with 0.5% yeast extract (THY) and used for bacterial challenge.
Mice.
Four- to 6-week-old female BALB/c mice, weighing 16 to 20 g, were obtained from the Chang Chun Institute of Biological Products. All animal experiments were approved by the Institutional Animal Care and Use Committee of Jilin University.
Protein antigens, adjuvant, and reagents.
The systemic vaccine antigens consisted of a mix of the fusion protein PsaA-PspA23 and PspA4, which were recombinantly expressed in Escherichia coli and purified by nickel affinity chromatography column, yielding a purity of about 85% as evaluated by SDS-PAGE. The adjuvant was a sterile PBS solution with 10% Al(OH)3 (alum). Details of the preparation of antigens have been described elsewhere (15).
The mucosal vaccine antigens were recombinant proteins PspA2-PA and PspA4-PA, which were expressed in Escherichia coli BL21. BLPs were mixed with the antigens to bind with the PA anchor on the antigen for 30 min at room temperature. The particles were then washed with sterile PBS three times to remove unbound proteins, resulting in PspA2-BLP and PspA4-BLP vaccines. BLPs were prepared as described previously (27). The concentration of the vaccines was calculated with the weight of BLPs by optical density at 600 nm (OD600). Prior to the animal experiments, the prepared PspA-BLP pneumococcal vaccine was stored at 4°C as single-use aliquots.
Immunizations.
Mice were immunized with three doses of vaccines at 14-day intervals. Systemic antigens were prepared from 5 μg of PsaA-PspA23 and 20 μg of PspA4 in 100 μl buffer for subcutaneous (s.c.) administration per dose. Mucosal antigens were prepared from 0.15 mg of PspA2-BLP and 0.3 mg of PspA4-BLP in 20 μl for intranasal (i.n.) administration per dose. Nasal immunizations were conducted in mice, which were previously anesthetized with ether. The commercial 23-valent polysaccharide vaccine (PPV23) used as a positive control was given only once, with blood samples that were obtained after 1 month and prior to challenge. PBS with 10% Al(OH)3 and 0.3 mg BLPs per dose were used as negative controls for the s.c. group and the i.n. group, respectively. Every group consisted of 16 immunized mice, 6 for colonization and 10 for protection.
Determination of antibody titers.
Serum and bronchoalveolar lavage fluid (BALF) samples were collected 2 weeks after the third immunization for evaluation of antibody levels. Antigen-specific IgG antibodies were measured by indirect enzyme-linked immunosorbent assay (ELISA) as follows: 96-well immunoassay plates (JFT Biofil) were coated overnight at room temperature (RT) with 0.5 μg/well of antigens in coating buffer (0.05 M carbonate bicarbonate buffer, pH 9.6). The next day, plates were blocked with PBS containing 3% bovine serum albumin (BSA; Genview) for 1 h prior to applying mouse sera. Individual mouse sera were diluted at a 1/1,000 dilution (in PBS–1% BSA) followed by 8 serial 3-fold dilutions. The bound antibodies were detected with horseradish peroxidase (HRP)-conjugated goat-anti-mouse IgG conjugate (Beijing Dingguo Changsheng Biotech), followed by the enzyme substrate 3,3′,5,5′-tetramethylbenzidine (TMB)/H2O2 (Tiangen), and the reaction was terminated after 15 min at RT with 2 M H2SO4. Optical density was measured at 450 nm using an automated plate reader (Bio-Rad) and analyzed with ELISA reader software (Micro plate manager 6). Results were expressed as the titers. The IgA titer was similar to that of IgG. The bound antibodies were detected with goat-anti-rabbit IgA conjugate (Beijing Dingguo Changsheng Biotech) in 8 serial 2-fold dilutions.
Determination of bacterial colonization.
Fourteen days after the last immunization, mice were anesthetized with ether, and PBS containing strains of ATCC 10813 (family 1, clade 2) or ATCC 6303 (family 2, clade 5) (2 × 105 CFU each) was inoculated i.n. in a volume of 20 μl into both nostrils. Mice were sacrificed 24 h later. None of the animals showed any sign of illness after challenge. Pneumococcal loads were determined in blood and lung homogenates by plating serial dilutions of the samples on blood agar as previously described (30).
Streptococcus pneumoniae challenge.
Two weeks after the third immunization with antigens or PBS, mice were anesthetized in ether and challenged intranasally with an LD90 dose of S. pneumoniae strains of ATCC 10813 (2 × 105 CFU/mouse) or ATCC 6303 (4 × 105 CFU/mouse) in a volume of 20 μl each into both nostrils. LD90 is the bacterial challenge dose concentration that results in the survival of 10% of the challenged mice compared to the blank group. Since LD90 is specific to each batch of tested mice, it was tested for each new batch of mice. For survival experiments, mice were monitored for 14 days, and differences between survival rates in each group were analyzed.
Statistical analysis.
Statistical analysis was performed using Graph Pad Prism Software (Graph Pad Software, La Jolla, CA, USA). Differences were determined using one-way analysis of variance (ANOVA) with t tests.
ACKNOWLEDGMENTS
We gratefully acknowledge Phuong Thi Sarkis and Fei Xu for editorial support in the preparation of the manuscript.
REFERENCES
- 1.Mitchell TJ. 2006. Streptococcus pneumoniae: infection, inflammation and disease. Adv Exp Med Biol 582:111–124. doi: 10.1007/0-387-33026-7_10. [DOI] [PubMed] [Google Scholar]
- 2.Black S, Shinefield H, Fireman B, Lewis E, Ray P, Hansen JR, Elvin L, Ensor KM, Hackell J, Siber G, Malinoski F, Madore D, Chang I, Kohberger R, Watson W, Austrian R, Edwards K. 2000. Efficacy, safety and immunogenicity of heptavalent pneumococcal conjugate vaccine in children. Northern California Kaiser Permanente Vaccine Study Center Group. Pediatr Infect Dis J 19:187–195. [DOI] [PubMed] [Google Scholar]
- 3.O'Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, Lee E, Mulholland K, Levine OS, Cherian T, Hib and Pneumococcal Global Burden of Disease Study Team. 2009. Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374:893–902. doi: 10.1016/S0140-6736(09)61204-6. [DOI] [PubMed] [Google Scholar]
- 4.Vila-Corcoles A, Ochoa-Gondar O, Guzmán JA, Rodriguez-Blanco T, Salsench E, Fuentes CM, EPIVAC Study Group. 2010. Effectiveness of the 23-valent polysaccharide pneumococcal vaccine against invasive pneumococcal disease in people 60 years or older. BMC Infect Dis 10:73. doi: 10.1186/1471-2334-10-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyaji EN, Oliveira ML, Carvalho E, Ho PL. 2013. Serotype-independent pneumococcal vaccines. Cell Mol Life Sci 70:3303–3326. doi: 10.1007/s00018-012-1234-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moffitt KL, Malley R. 2011. Next generation pneumococcal vaccines. Curr Opin Immunol 23:407–413. doi: 10.1016/j.coi.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feldman C, Anderson R. 2014. Review: current and new generation pneumococcal vaccines. J Infect 69:309–325. doi: 10.1016/j.jinf.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 8.Hanage WP. 2008. Serotype-specific problems associated with pneumococcal conjugate vaccination. Future Microbiol 3:23–30. doi: 10.2217/17460913.3.1.23. [DOI] [PubMed] [Google Scholar]
- 9.Roche H, Hakansson A, Hollingshead SK, Briles DE. 2003. Regions of PspA/EF3296 best able to elicit protection against Streptococcus pneumoniae in a murine infection model. Infect Immun 71:1033–1041. doi: 10.1128/IAI.71.3.1033-1041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hollingshead SK, Becker R, Briles DE. 2000. Diversity of PspA: mosaic genes and evidence for past recombination in Streptococcus pneumoniae. Infect Immun 68:5889–5900. doi: 10.1128/IAI.68.10.5889-5900.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Briles DE, Hollingshead SK, King J, Swift A, Braun PA, Park MK, Ferguson LM, Nahm MH, Nabors GS. 2000. Immunization of humans with recombinant pneumococcal surface protein A (rPspA) elicits antibodies that passively protect mice from fatal infection with Streptococcus pneumoniae bearing heterologous PspA. J Infect Dis 182:1694–1701. doi: 10.1086/317602. [DOI] [PubMed] [Google Scholar]
- 12.Moreno AT, Oliveira ML, Ferreira DM, Ho PL, Darrieux M, Leite LC, Ferreira JM Jr, Pimenta FC, Andrade AL, Miyaji EN. 2010. Immunization of mice with single PspA fragments induces antibodies capable of mediating complement deposition on different pneumococcal strains and cross-protection. Clin Vaccine Immunol 17:439–446. doi: 10.1128/CVI.00430-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darrieux M, Goulart C, Briles D, Leite LC. 2015. Current status and perspectives on protein-based pneumococcal vaccines. Crit Rev Microbiol 41:190–200. doi: 10.3109/1040841X.2013.813902. [DOI] [PubMed] [Google Scholar]
- 14.Arulanandam BP, Lynch JM, Briles DE, Hollingshead S, Metzger DW. 2001. Intranasal vaccination with pneumococcal surface protein A and interleukin-12 augments antibody-mediated opsonization and protective immunity against Streptococcus pneumoniae infection. Infect Immun 69:6718–6724. doi: 10.1128/IAI.69.11.6718-6724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu J, Sun T, Wang D, Dong Y, Xu M, Hou H, Kong FT, Liang C, Gu T, Chen P, Sun S, Lv X, Jiang C, Kong W, Wu Y. 2015. Protective immune responses elicited by fusion protein containing PsaA and PspA fragments. Immunol Invest 44:482–496. doi: 10.3109/08820139.2015.1037956. [DOI] [PubMed] [Google Scholar]
- 16.Riepe HR, Pillidge CJ, Gopal PK, McKay LL. 1997. Characterization of the highly autolytic Lactococcus lactis subsp. cremoris strains CO and 2250. Appl Environ Microbiol 63:3757–3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joris B1, Englebert S, Chu CP, Kariyama R, Daneo-Moore L, Shockman GD, Ghuysen JM. 1992. Modular design of the Enterococcus hirae muramidase-2 and Streptococcus faecalis autolysin. FEMS Microbiol Lett 70:257–264. doi: 10.1111/j.1574-6968.1992.tb05218.x. [DOI] [PubMed] [Google Scholar]
- 18.Bateman A, Bycroft M. 2000. The structure of a LysM domain from E. coli membrane-bound lytic murein transglycosylase D (MltD). J Mol Biol 299:1113–1119. doi: 10.1006/jmbi.2000.3778. [DOI] [PubMed] [Google Scholar]
- 19.Steen A, Palumbo E, Deghorain M, Cocconcelli PS, Delcour J, Kuipers OP, Kok J, Buist G, Hols P. 2005. Autolysis of Lactococcus lactis is increased upon D-alanine depletion of peptidoglycan and lipoteichoic acids. J Bacteriol 187:114–124. doi: 10.1128/JB.187.1.114-124.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Darrieux M, Moreno AT, Ferreira DM, Pimenta FC, de Andrade AL, Lopes AP, Leite LC, Miyaji EN. 2008. Recognition of pneumococcal isolates by antisera raised against PspA fragments from different clades. J Med Microbiol 57:273–278. doi: 10.1099/jmm.0.47661-0. [DOI] [PubMed] [Google Scholar]
- 21.Piao Z, Akeda Y, Takeuchi D, Ishii KJ, Ubukata K, Briles DE, Tomono K, Oishi K. 2014. Protective properties of a fusion pneumococcal surface protein A (PspA) vaccine against pneumococcal challenge by five different PspA clades in mice. Vaccine 32:5607–5613. doi: 10.1016/j.vaccine.2014.07.108. [DOI] [PubMed] [Google Scholar]
- 22.Virolainen A, Russell W, Crain MJ, Rapola S, Käyhty H, Briles DE. 2000. Human antibodies to pneumococcal surface protein A in health and disease. Pediatr Infect Dis J 19:134–138. doi: 10.1097/00006454-200002000-00011. [DOI] [PubMed] [Google Scholar]
- 23.Nabors GS, Braun PA, Herrmann DJ, Heise ML, Pyle DJ, Gravenstein S, Schilling M, Ferguson LM, Hollignshead SK, Briles DE, Becker RS. 2000. Immunization of healthy adults with a single recombinant pneumococcal surface protein A (PspA) variant stimulates broadly cross-reactive antibodies. Vaccine 18:1743–1754. doi: 10.1016/S0264-410X(99)00530-7. [DOI] [PubMed] [Google Scholar]
- 24.Oliveira ML, Miyaji EN, Ferreira DM, Moreno AT, Ferreira PC, Lima FA, Santos FL, Sakauchi MA, Takata CS, Higashi HG, Raw I, Kubrusly FS, Ho PL. 2010. Combination of pneumococcal surface protein A (PspA) with whole cell pertussis vaccine increases protection against pneumococcal challenge in mice. PLoS One 5:1–14. doi: 10.1371/journal.pone.0010863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nganou-Makamdop K, van Roosmalen ML, Audouy SA, van Gemert GJ, Leenhouts K, Hermsen CC, Sauerwein RW. 2012. Bacterium-like particles as multi-epitope delivery platform for Plasmodium berghei circumsporozoite protein induce complete protection against malaria in mice. Malar J 11:50. doi: 10.1186/1475-2875-11-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruna-Romero O, Rocha CD, Tsuji M, Gazzinelli RT. 2004. Enhanced protective immunity against malaria by vaccination with a recombinant adenovirus encoding the circumsporozoite protein of Plasmodium lacking the GPI-anchoring motif. Vaccine 22:3575–3584. doi: 10.1016/j.vaccine.2004.03.050. [DOI] [PubMed] [Google Scholar]
- 27.Audouy SA, van Roosmalen ML, Neef J, Kanninga R, Post E, van Deemter M, Metselaar H, van Selm S, Robillard GT, Leenhouts KJ, Hermans PW. 2006. Lactococcus lactis GEM particles displaying pneumococcal antigens induce local and systemic immune responses following intranasal immunization. Vaccine 24:5434–5441. doi: 10.1016/j.vaccine.2006.03.054. [DOI] [PubMed] [Google Scholar]
- 28.Audouy SA, van Selm S, van Roosmalen ML, Post E, Kanninga R, Neef J, Estevao S, Nieuwenhuis EE, Adrian PV, Leenhouts K, Hermans PW. 2007. Development of lactococcal GEM-based pneumococcal vaccines. Vaccine 25:2497–2506. doi: 10.1016/j.vaccine.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 29.Grangette C, Muller-Alouf H, Hols P, Goudercourt D, Delcour J, Turneer M, Mercenier A. 2004. Enhanced mucosal delivery of antigen with cell wall mutants of lactic acid bacteria. Infect Immun 72:2731–2737. doi: 10.1128/IAI.72.5.2731-2737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu J, Sun T, Hou H, Xu M, Gu T, Dong Y, Wang D, Chen P, Wu C, Liang C, Sun S, Jiang C, Kong W, Wu Y. 2014. Detoxified pneumolysin derivative Plym2 directly protects against pneumococcal infection via induction of inflammatory cytokines. Immunol Invest 43:717–726. doi: 10.3109/08820139.2014.930478. [DOI] [PubMed] [Google Scholar]