

Abstract
Background
Clostridium perfringens causes a range of diseases in animals and humans including necrotic enteritis in chickens and food poisoning and gas gangrene in humans. Necrotic enteritis is of concern in commercial chicken production due to the cost of the implementation of infection control measures and to productivity losses. This study has focused on the genomic analysis of a range of chicken-derived C. perfringens isolates, from around the world and from different years. The genomes were sequenced and compared with 20 genomes available from public databases, which were from a diverse collection of isolates from chickens, other animals, and humans. We used a distance based phylogeny that was constructed based on gene content rather than sequence identity. Similarity between strains was defined as the number of genes that they have in common divided by their total number of genes. In this type of phylogenetic analysis, evolutionary distance can be interpreted in terms of evolutionary events such as acquisition and loss of genes, whereas the underlying properties (the gene content) can be interpreted in terms of function. We also compared these methods to the sequence-based phylogeny of the core genome.
Results
Distinct pathogenic clades of necrotic enteritis-causing C. perfringens were identified. They were characterised by variable regions encoded on the chromosome, with predicted roles in capsule production, adhesion, inhibition of related strains, phage integration, and metabolism. Some strains have almost identical genomes, even though they were isolated from different geographic regions at various times, while other highly distant genomes appear to result in similar outcomes with regard to virulence and pathogenesis.
Conclusions
The high level of diversity in chicken isolates suggests there is no reliable factor that defines a chicken strain of C. perfringens, however, disease-causing strains can be defined by the presence of netB-encoding plasmids. This study reveals that horizontal gene transfer appears to play a significant role in genetic variation of the C. perfringens chromosome as well as the plasmid content within strains.
Keywords: Clostridium perfringens, Necrotic enteritis, Capsule, Adhesion, Prophage, Genome, Pangenome
Background
Clostridium perfringens is a bacterial pathogen that causes a wide variety of diseases ranging from gas gangrene and food poisoning in humans to necrotic enteritis in chickens [1]. Aside from host factors, the type and severity of disease is dependent on the production of a set of toxins and extracellular enzymes [1]. Most C. perfringens toxins are not produced by all strains; their structural genes are present as a part of the accessory genome encoded on large conjugative plasmids [2, 3]. These plasmids are found in various combinations within different strains [4–7]. Some plasmid-encoded toxins have been uniquely associated with a specific disease; for example, NetB is associated with necrotic enteritis. Other toxins, such as the beta2 and beta toxins, are encoded on highly variable plasmids and are produced by strains associated with several diseases [2, 8, 9]. By contrast, alpha toxin is chromosomally encoded and is carried by all C. perfringens strains.
Classification of C. perfringens strains has traditionally been based on the profile of four typing toxins. Currently, strains are classified as type A to type E based on presence or absence of the toxins. Necrotic enteritis is caused by type A strains that produce NetB, a pore-forming toxin that is essential for disease in chickens [10]. Type A strains, which carry the alpha toxin gene, but none of the other toxin typing genes, also cause several other diseases, including gas gangrene and bovine abomasitis. Type A strains are also normal members of the gut microbiota in animals and humans, as well as being found ubiquitously in the environment [11–13].
A genomic comparison of three type A strains showed that there is a significant degree of genomic variation between strains SM101, ATCC13124 and 13, with over 300 genomic islands variably present, almost all of which are chromosomally encoded [14]. This chromosomal variation is also evident from studies that have used pulse-field gel electrophoresis (PFGE) to examine genomes via restriction digest fingerprinting. Strains from similar origins and disease-causing backgrounds are highly variable and have unique fingerprints, suggesting a highly variable genomic content in strains even when they appear to cause the same disease [15–17]. A whole-genome comparison of 12 strains from different origins revealed that type A strains could not be differentiated from the types B to E by examining the chromosomal gene content. Only the presence of plasmid-encoded genes allowed a clear differentiation between the different toxinotypes [18].
Necrotic enteritis is a disease of concern to the poultry industry due to the negative impact that it has on production costs. Necrotic enteritis reduces the efficiency of feed conversion and in severe cases causes death [19–21]. The disease is prevalent worldwide and, in addition to its significant economic impact on the poultry industry, it represents an important animal welfare issue. The NetB toxin is an essential virulence factor in necrotic enteritis [10], and the gene encoding this toxin is located on a pathogenicity locus, NEloc-1, which is in turn encoded on a large conjugative plasmid [7, 10, 22–25]. Some necrotic enteritis strains also produce another toxin, TpeL, which is also encoded on large plasmids; however, the role of this toxin, if any, in necrotic enteritis is unknown [26, 27]. Necrotic enteritis strains also sometimes encode resistance to the antibiotics tetracycline and bacitracin on plasmids, once again emphasising the importance of plasmids in the phenotypic differentiation of C. perfringens strains [7, 28].
Sub-genomic analyses involving multi-locus sequence typing (MLST) has identified several subtypes or clonal complexes of poultry-associated C. perfringens isolates [15, 23]. Two of these subtypes are predominately composed of pathogenic strains, which are defined by the presence of netB [23]. A comparative genome hybridization (CGH) microarray study [23] showed that among the 54 chicken isolates examined, approximately 400 genes were variably present. The variable genes were organised into 142 genomic regions, 49 of which were associated with netB-positive isolates [23]. Some of the variation was associated with chromosomal regions with predicted roles in metabolism, fitness associated factors, and plasmid maintenance. The CGH study revealed clustering of necrotic enteritis-associated strains into two distinct subtypes, a CP4-like and JGS4143-like subtype [23].
In this study, whole genome sequencing (WGS) was performed on a diverse collection of poultry-derived C. perfringens isolates from both necrotic enteritis-affected and healthy birds. The genomes were compared using whole genome content analysis to investigate the level of chromosomal variation. Since the role of plasmids in C. perfringens has been well established, and they are known to be transferable between strains via lateral gene transfer, this investigation focused on chromosomal variation.
Results
Strain selection and sequence statistics
The strains sequenced were selected to cover a wide range of spatial and temporal variability. They were isolated from both diseased and healthy chickens at various time periods and from several geographical locations (Table 1). Sequencing statistics can be observed in Table 2. Of the strains examined in this study 38 were poultry strains and 21 were non-poultry strains. The estimated size of the genomes, as predicted from total contig size, ranged from 2,897,393 bp to 3,751,199 bp (Table 2), with an average length of 3,353,566 bp. The pangenome size (i.e. the total number of genes within a group of genomes) was determined to give a measure of the relative complexity within the C. perfringens genomes. With the newly sequenced genomes added to those already in the public databases, the pangenome for the strain collection totals 8102 predicted genes, of which 3619 have a predicted function, 2866 are hypothetical conserved protein-coding sequences, which have been found elsewhere, and 1617 are genes of unknown function (novel coding sequences). Between 2611 and 4897 (mean = 3316; s.d. = 509) open reading frames (ORFs) were identified within individual strains. No correlations above 0.03 (Pearson’s R) were observed between the number of ORFs and sequencing method or any of the assembly metrics (number of contigs, total assembly length, or N50) indicating that neither the assembly quality nor the sequencing method affected the analysis. The core genome conserved among all strains consisted of 1192 ORFs, which ranged from 31% (JGS1721) to 43% (SM101) of the total ORFs in a genome. 2454 ORFs were singletons, present in only one strain. An overview of the data generated from 56 strains of C. perfringens is presented in Fig. 1.
Table 1.
Strains analysed in this study
| Strain | Toxinotype | netB+ | Source | Associated Disease | Country | Genome sequence | Clade | Reference | Accession |
|---|---|---|---|---|---|---|---|---|---|
| 0522A_28_397 | A | – | Human | unknown | USA | + | NP | NCBI | MKXR0000000.1 |
| 1207_CPER | A | – | Hospital | unknown | USA | + | NP | [81] | JVYV0000000 |
| 2789STDY | A | – | Human | No Disease | – | + | NP | Sanger | CYYX0000000 |
| 94–5224 | A | + | Chicken | Necrotic enteritis | USA | – | – | This study | a |
| 96–7415 | A | + | Chicken | Necrotic enteritis | USA | + | 3 | This study | PJTF00000000 |
| 98.78718–2 | A | + | Chicken | Necrotic enteritis | DEN | + | 1B | [25] | PJTE00000000 |
| ATCC3626 | B | – | Lamb | – | – | + | NP | [18] | ABDV0000000 |
| ATCC13124 | A | – | Human | Gas Gangrene | – | + | NP | [14] | CP000246 |
| BER-NE332 | A | – | Chicken | Necrotic enteritis | AUS | + | 3 | [25] | PJTD00000000 |
| CP41 | A | + | Chicken | Necrotic enteritis | CAN | + | 1A | [23] | LIYI0000000 |
| EHE-NE3 | A | + | Chicken | Necrotic enteritis | AUS | – | – | [82] | a |
| EHE-NE4 | A | + | Chicken | Necrotic enteritis | AUS | – | – | [82] | a |
| EHE-NE51 | A | + | Chicken | Necrotic enteritis | AUS | – | – | [82] | a |
| EHE-NE7 | A | + | Chicken | Necrotic enteritis | AUS | + | 2 | [82] | PJTC00000000 |
| EHE-NE181 | A | + | Chicken | Necrotic enteritis | AUS | + | 1B | [10] | CP025501 |
| EHE-NE20 | A | + | Chicken | Necrotic enteritis | AUS | – | – | [10] | a |
| EUR-NE151 | A | + | Chicken | Necrotic enteritis | AUS | + | 1B | [10] | PJTB00000000 |
| F262 | A | – | Cow | Bovine abomastis | CAN | + | NP | [11] | AFES0000000 |
| F4969 | A | – | Human | non-food-borne diarrhoea | Europe | + | NP | [18] | ABDX0000000 |
| FC1 | A | + | Chicken | Necrotic enteritis | USA | – | – | This study | a |
| FC2 | A | + | Chicken | Necrotic enteritis | USA | + | 1B | This study | PJTA00000000 |
| FORC_003 | A | – | Human | Food poisoning | SKR | + | NP | FORC | CP009557 |
| FORC_025 | A | – | Human | Food poisoning | SKR | + | NP | FORC | CP013101 |
| GNP-1 | A | + | Chicken | Necrotic enteritis | USA | + | 1A | This study | PJSZ00000000 |
| ITX1105-12MP | A | + | Chicken | Necrotic enteritis | USA | + | 1A | This study | PJSY00000000 |
| JGS1495 | C | – | Pig | Diarrhoea | – | + | NP | [18] | ABDU0000000 |
| JGS1721 | D | – | Sheep | Enteritis | – | + | NP | [18] | ABOO0000000 |
| JGS1987 | E | – | Cow | Enteritis | – | + | NP | [18] | ABDW0000000 |
| JGS41431 | A | + | Chicken | Necrotic enteritis | USA | + | 2 | [23] | |
| JJC | A | – | Environment | No Disease | MY | + | NP | [18] | AWRZ0000000 |
| JP55 | A | – | Horse | Necrotizing enteritis | CAN | + | NP | [83] | CP010993 |
| JP838 | A | – | Dog | Necrotizing enteritis | CAN | + | NP | [83] | CP010994 |
| K473 | A | – | Chicken | Necrotic enteritis | AUS | + | 3 | This study | PJSX00000000 |
| Kendall1 | A | + | Chicken | Necrotic enteritis | USA | – | – | This study | a |
| Mugdale 5 | A | – | Chicken | Healthy | AUS | – | – | [30] | a |
| NAG-NE12 | A | – | Chicken | Necrotic enteritis | AUS | + | 3 | [82] | PJSW00000000 |
| NAG-NE252 | A | – | Chicken | Necrotic enteritis | AUS | + | 3 | [82] | |
| NAG-NE311 | A | + | Chicken | Necrotic enteritis | AUS | + | 1A | [25] | PJSV00000000 |
| NCTC 8239 | A | – | Human | Food poisoning | UK | + | NP | [18] | ABDY0000000 |
| NobL12 | C | – | Chicken | Necrotic enteritis | USA | + | 3 | This study | PJSU00000000 |
| PBD12 | A | – | Chicken | Healthy | AUS | + | 4 | [30] | PJST00000000 |
| PBS4 | A | – | Chicken | Healthy | AUS | – | – | [30] | a |
| PBS52 | A | – | Chicken | Healthy | AUS | + | 4 | [30] | PJSS00000000 |
| PC52 | A | – | Chicken | Healthy | AUS | + | 4 | [30] | PJSR00000000 |
| Pennington | A | + | Chicken | Necrotic enteritis | USA | + | 1A | This study | PJSQ00000000 |
| SAF-1 | A | – | Chicken | Healthy | AUS | + | 3 | This study | PJSP00000000 |
| SM101 | A | – | Human | Food poisoning | UK | + | NP | [14] | CP000312 |
| 112 | A | – | Chicken | Healthy | BEL | + | 3 | [35] | PJSO00000000 |
| 132 | A | – | Soil | Gas gangrene | – | + | NP | [66] | BA000016 |
| 371 | A | + | Chicken | Necrotic enteritis | BEL | + | 2 | [35] | PJSN00000000 |
| 482 | A | – | Chicken | Necrotic enteritis | BEL | + | 3 | [35] | PJTN00000000 |
| 671 | A | + | Chicken | Necrotic enteritis | BEL | + | 2 | [35] | PJSM00000000 |
| SOM-NE341 | A | + | Chicken | Necrotic enteritis | AUS | + | 2 | [25] | PJSL00000000 |
| SOM-NE351 | A | + | Chicken | Necrotic enteritis | AUS | + | 2 | [25] | PJTM00000000 |
| TAM-NE38 | A | + | Chicken | Necrotic enteritis | AUS | + | 1A | This study | PJTL00000000 |
| TAM-NE40 | A | + | Chicken | Necrotic enteritis | AUS | + | 1A | This study | PJSK00000000 |
| SYD-NE41 | A | + | Chicken | Necrotic enteritis | AUS | + | 1B | This study | PJUV00000000 |
| TAM-NE42 | A | + | Chicken | Necrotic enteritis | AUS | + | 3 | This study | PJSJ00000000 |
| TAM-NE43 | A | + | Chicken | Necrotic enteritis | AUS | + | 3 | This study | PJTK00000000 |
| TAM-NE46 | A | + | Chicken | Necrotic enteritis | AUS | + | 1A | This study | PJSI00000000 |
| TAM-NE47 | A | + | Chicken | Necrotic enteritis | AUS | – | 1A | This study | a |
| T3381 | A | – | Chicken | Necrotic enteritis | AUS | + | 3 | This study | PJTJ00000000 |
| UDE 95–6812 | A | + | Chicken | Necrotic enteritis | USA | + | 1A | This study | PJTI00000000 |
| W1319 | A | – | Chicken | Necrotic enteritis | AUS | + | 3 | This study | PJSH00000000 |
| WAL-14572 | A | – | Human | No Disease | – | + | NP | [18] | ADLP0000000 |
| Warren1 | A | + | Chicken | Necrotic enteritis | USA | + | 1A | This study | PJTH00000000 |
| WER-NE361 | A | + | Chicken | Necrotic enteritis | AUS | + | 2 | [84] | PJTG00000000 |
1Isolate has been tested in a disease induction model and demonstrated to be virulent
2Isolate has been tested in a disease induction model and demonstrated to be avirulent
aThese isolates were not sequenced. They were only used for PFGE analysis
Table 2.
Sequencing statistics
| Strain | Chromosomal Contigs | Total bp | Max contig length (bp) | Min contig length (bp) | N50 | Potential genes (Chromosomal genes) | Accessory genes | Unique genes | Exclusively absent genes |
|---|---|---|---|---|---|---|---|---|---|
| 0522A | 31 | 3,417,203 | 799,085 | 1716 | 250,061 | 3165 | 1680 | 81 | 0 |
| 1207_CPER | 95 | 3,206,961 | 279,830 | 204 | 85,634 | 2818 | 1511 | 28 | 0 |
| 2789STDY | 48 | 3,321,076 | 2,127,284 | 377 | 903,119 | 2979 | 1566 | 32 | 0 |
| 96–7415 | 49 | 3,388,438 | 797,628 | 1527 | 244,647 | 3932 | 1651 | 86 | 0 |
| 98.78718–2 | 36 | 3,417,550 | 410,887 | 2042 | 196,455 | 3963 | 1772 | 35 | 3 |
| ATCC3626 | 64 | 3,585,098 | 299,576 | 1616 | 91,764 | 4159 | 1760 | 83 | 5 |
| ATCC13124 | 1 | 3,256,683 | 3,256,683 | 3,256,683 | 3,256,683 | 3719 | 1536 | 38 | 11 |
| BER-NE33 | 30 | 3,349,933 | 3,345,877 | 4056 | 3,345,877 | 4107 | 1558 | 38 | 19 |
| CP4 | 96 | 3,590,658 | 289,484 | 1282 | 83,876 | 3371 | 1500 | 211 | 42 |
| EHE-NE7 | 11 | 3,257,283 | 2,079,954 | 4153 | 2,079,954 | 4368 | 1597 | 0 | 0 |
| EHE-NE18 | 1 | 3, 474,200 | 3, 474,200 | 3, 474,200 | 3, 474,200 | 3965 | 1783 | 0 | 0 |
| EUR-NE15 | 11 | 3,378,967 | 2,213,684 | 2319 | 2,213,684 | 4672 | 1755 | 21 | 4 |
| F262 | 1 | 3,333,039 | 3,333,039 | 3,333,039 | 3,333,039 | 3793 | 1667 | 46 | 0 |
| F4969 | 61 | 3,420,663 | 365,849 | 1632 | 113,358 | 3997 | 1605 | 85 | 8 |
| FC2 | 24 | 3,297,593 | 986,908 | 4358 | 966,190 | 3867 | 1615 | 27 | 0 |
| FORC_003 | 1 | 3,338,532 | 3,338,532 | 3,338,532 | 3,338,532 | 3060 | 1603 | 17 | 0 |
| FORC_025 | 1 | 3,343,822 | 3,343,822 | 3,343,822 | 3,343,822 | 3038 | 1632 | 43 | 0 |
| GNP-1 | 26 | 3,522,496 | 1,064003 | 2009 | 363,806 | 4185 | 1905 | 11 | 0 |
| ITX1105-12MP | 259 | 3,277,088 | 115,260 | 1547 | 22,062 | 4424 | 1866 | 0 | 0 |
| JGS1495 | 59 | 3,412,610 | 383,044 | 1664 | 117,588 | 4003 | 1572 | 41 | 5 |
| JGS1721 | 104 | 3,608,664 | 192,053 | 1519 | 80,514 | 4331 | 1660 | 50 | 8 |
| JGS1987 | 72 | 3,751,199 | 270,422 | 1671 | 96,857 | 4397 | 1772 | 255 | 5 |
| JGS4143 | 318 | 3,658,663 | 3,658,663 | 3,658,663 | 3,658,663 | 4124 | 1657 | 183 | 0 |
| JJC | 54 | 3,203,273 | 445,718 | 1595 | 98,246 | 3724 | 1533 | 24 | 1 |
| JP55 | 1 | 3,347,300 | 3,347,300 | 3,347,300 | 3,347,300 | 3266 | 1618 | 21 | 0 |
| JP838 | 1 | 3,530,414 | 3,530,414 | 3,530,414 | 3,530,414 | 3807 | 1734 | 114 | 4 |
| K473 | 46 | 3,328,345 | 724,340 | 2307 | 158,573 | 6564 | 1316 | 130 | 91 |
| NAG-NE1 | 106 | 3,315,837 | 2,659,148 | 2002 | 2,659,148 | 4994 | 1434 | 92 | 43 |
| NAG-NE25 | 247 | 3,408,683 | 3,408,683 | 3,408,683 | 3,408,683 | 3953 | 1707 | 50 | 2 |
| NAG-NE31 | 72 | 3371,019 | 1,031,391 | 2265 | 683,309 | 5198 | 1684 | 26 | 2 |
| NCTC 8239 | 45 | 3,313,110 | 524,606 | 1602 | 146,935 | 3784 | 1520 | 33 | 6 |
| NobL1 | 10 | 3,574,575 | 3,403,922 | 2168 | 3,403,922 | 4605 | 1679 | 53 | 5 |
| PBD1 | 13 | 3,176,182 | 1,454,083 | 2985 | 572,872 | 5498 | 1509 | 44 | 40 |
| PBS5 | 10 | 3,110,163 | 2,317,119 | 2322 | 2,317,119 | 4377 | 1578 | 18 | 7 |
| PC5 | 11 | 3,103,348 | 2,196,352 | 2335 | 2,196,352 | 4284 | 1602 | 24 | 4 |
| Pennington | 35 | 3,501,064 | 1,037,768 | 2204 | 228,190 | 4158 | 1903 | 4 | 0 |
| SAF-1 | 80 | 3,356,947 | 3,354,395 | 2552 | 3,354,395 | 3934 | 1605 | 37 | 0 |
| SM101 | 1 | 2,897,393 | 2,897,393 | 2,897,393 | 2,897,393 | 3299 | 1136 | 62 | 93 |
| Strain 11 | 35 | 3,197,244 | 515,326 | 4530 | 206,922 | 3662 | 1523 | 15 | 0 |
| Strain 13 | 1 | 3,031,430 | 3,031,430 | 3,031,430 | 3,031,430 | 3438 | 1642 | 15 | 7 |
| Strain 37 | 36 | 3,255,773 | 461,756 | 1501 | 148,687 | 3764 | 1593 | 11 | 0 |
| Strain 48 | 33 | 3,250,386 | 670,855 | 1575 | 245,313 | 3738 | 1552 | 52 | 0 |
| Strain 67 | 34 | 3,268,421 | 451,598 | 1501 | 243,534 | 3794 | 1626 | 21 | 0 |
| SOM-NE34 | 25 | 3,418,437 | 3,355,569 | 62,868 | 3,355,569 | 3952 | 1720 | 1 | 0 |
| SOM-NE35 | 35 | 3,364,015 | 1,180,705 | 2351 | 878,365 | 4722 | 1748 | 36 | 1 |
| TAM-NE38 | 13 | 3,447,589 | 3,395,713 | 2411 | 3,395,713 | 4063 | 1801 | 0 | 0 |
| TAM-NE40 | 15 | 3,452,011 | 3,395,271 | 2201 | 3,395,271 | 4086 | 1797 | 2 | 1 |
| SYD-NE41 | 15 | 3,542,115 | 3,502,074 | 2002 | 3,502,074 | 4557 | 1790 | 36 | 5 |
| TAM-NE42 | 96 | 3,355,605 | 3,353,147 | 2458 | 3,353,147 | 4015 | 1613 | 17 | 0 |
| TAM-NE43 | 18 | 3,272,035 | 1,846,259 | 4695 | 1,846,259 | 3780 | 1596 | 27 | 0 |
| TAM-NE46 | 33 | 3371,805 | 492,250 | 1589 | 278,745 | 4003 | 1791 | 0 | 0 |
| T3381 | 67 | 3,335,304 | 352,891 | 2762 | 119,157 | 7258 | 1220 | 125 | 161 |
| UDE 95–6812 | 25 | 3,366,442 | 1,428,911 | 2009 | 381,265 | 4004 | 1766 | 0 | 0 |
| W1319 | 11 | 3,236,902 | 2,023,168 | 2290 | 2,023,168 | 4667 | 1568 | 35 | 9 |
| WAL-14572 | 26 | 3,403,365 | 618,435 | 2272 | 307,728 | 3928 | 1721 | 50 | 1 |
| Warren | 14 | 3,561,701 | 3,425,489 | 2387 | 3,425,489 | 4408 | 1866 | 9 | 1 |
| WER-NE36 | 113 | 3,283,756 | 3,281,264 | 2492 | 3,281,264 | 4536 | 1602 | 12 | 3 |
Fig. 1.
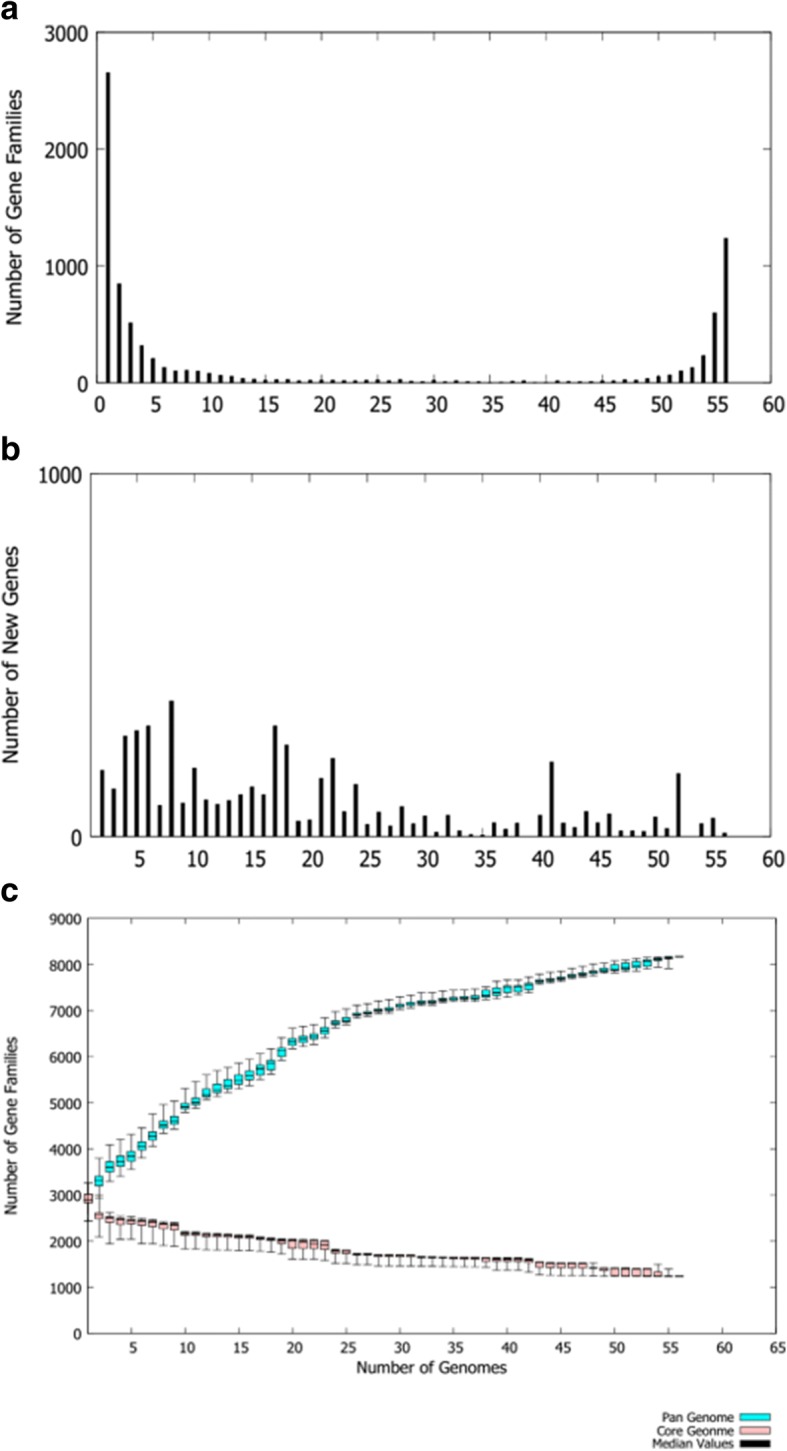
Presence/absence distribution of clusters. a. Histogram showing gene family distribution. b. Bar chart showing the number of new sequence clusters found in sequentially added C. perfringens genomes. c. Dot plot showing pan genome and core genome clusters in the C. perfringens genomes
To investigate the functionality of the pan-genome, a blast search was performed from representative sequences of each protein cluster against the COG database. As shown in Fig. 2, the functional categories of the proteins were assigned to core, accessory and singleton classes. The accessory and singleton genes encode proteins with a very large amount of functional diversity in comparison to the core proteins, suggesting that a wide range of functional variation is possible between strains based on the accessory genes that they carry.
Fig. 2.

COG functional analysis of pan-genome. The histogram shows the predicted functionality of proteins assigned to core, accessory and singleton clusters
Evolutionary relationships of isolates revealed by pan-genome analysis
A pan genome binary matrix was produced from the gene clusters, which were calculated using the UCLUST algorithm [29], and the presence or absence of a cluster was assigned to each strain. We found 8002 unique clusters, corresponding to the total gene content of the pan-genome. An average of 4039 clusters was assigned to each strain.
The pan genome binary matrix was used as input in a principal component analysis (PCA) in XLSTAT to reveal the dominant differences between strains. Major differences between isolates, with several distinct clusters of isolates, were defined (Fig. 3). Two clusters of non-pathogenic chicken isolates (dark green and light green) were seen, one being clustered with non-poultry strains in the bottom right of the plot and a second very different group at the top of the PCA plot (Fig. 3). Most non-poultry strains clustered closely together, while the pathogenic chicken strains fell into three distinct clusters (dark red, light red and orange) (Fig. 3). One of the pathogenic chicken clusters is closely related to the non-poultry strains and to one of the non-pathogenic chicken clusters (Fig. 3).
Fig. 3.

PCA on the pan-matrix. Each dot marks a genome in the first two principal components of the pan-matrix. Strains are coloured by clade designation and disease-causing capability. Shapes represent; triangle, netB positive from diseased birds; squares, netB negative poultry isolates; circle, non-poultry netB negative isolate
Since the PCA indicated some clustering that correlated with disease capability (Fig. 3), the genomes were investigated by producing a pan-genome tree of maximum likelihood to compare the total gene content of the strains. The pangenome tree showed two distinct clades of pathogenic chicken strains (Fig. 4). One clade of pathogenic strains (orange) has close relationships to the majority of non-pathogenic poultry and non-poultry strains, supporting the PCA observations. The other clade of pathogenic chicken strains shares a monophyletic branch on the tree (pink and red), however, there appears to be some divergence between strains within this group (Fig. 4). Following examination of the tree structure, pathogenic isolates were assigned to two major clades, pathogenic clades 1 and 2. Pathogenic clade 1 was split into sub-clades to account for both PCA diversity and divergence in the tree structure; two sub-clades were defined; pathogenic subclade 1A (red) and subclade 1B (pink). The other pathogenic poultry clade was designated pathogenic clade 2 – orange (Fig. 4). The non-pathogenic strains also fell into two clusters. The large clade, closely related to non-poultry strains, consisted of strains from both healthy and diseased poultry, but use of these strains in our necrotic enteritis disease induction model provided evidence that these strains were not capable of causing disease; this clade was designated non-pathogenic clade 3. A highly divergent group of strains formed non-pathogenic clade 4 (Fig. 4), and consisted of strains isolated from healthy chickens that were not capable of causing disease in a chicken necrotic enteritis infection model. Clades showed no correlation to country of origin or time of isolation, with the notable exception of non-pathogenic clade 4, which consists of only Australian isolates isolated in 2005.
Fig. 4.

Total gene content phylogeny. Total gene content phylogeny. Tree of maximum likelihood based on total gene content of the chromosome of various C. perfringens strains. Red, Pink represent pathogenic strains of C. perfringens clades 1A, and 1B respectively, while orange represent pathogenic clade 2. Dark Green and light green represent non-pathogenic strains in clades 3 and 4 respectively. Black, positive; white, negative
Variable chromosomal regions identified in C. perfringens poultry isolates
To further examine the basis of strain separation and divergence, the chromosomal content of each strain was investigated and compared. Several hundred genes were variably present in each strain. A binary matrix was used to determine which clusters were exclusively present or absent within and between the designated clades. Many of the genes responsible for the variation between strains were grouped within specific genomic loci or regions. These regions were mapped against the chromosome of each strain to establish their relative location within the genomes. These variable regions encoded proteins with putative and known roles in colonisation, phage integration, capsule polysaccharide biogenesis, motility, antimicrobial activity, and alternative metabolism pathways. Many of these regions encoded genes that were associated with mobile genetic elements (Fig. 5) such as transposase genes, recombinase genes and IS elements, or were flanked by highly conserved regions shared by all strains.
Fig. 5.

Presence of variable regions. Heatmap showing the presence and absence of variable chromosomal loci among 38 poultry strains and 21 non-poultry strains. Each column represents a different strain and each row represents a different chromosomal locus. Coloured sections represent a positive hit for that strain, grey represents a partial match (not entire locus but a single gene) and white is no match. Colours represent clade designation of a strains with pathogenic clade 1A (red), pathogenic clade 1B (pink) and pathogenic clade 2 (orange) while non-pathogenic clade 1 (dark green) and non-pathogenic clade 2 (light green). Non-poultry strains are coloured blue
Adhesion-associated locus
One region that varies between the pathogenic clades was an adhesion-associated locus (Fig. 6). This locus has three major variants and is located between two highly conserved genes present in all strains, which encode a sortase B enzyme and a CPR048-like protein. The first two variants have been previously described as VR-10A, encoding a vonWillebrand domain A protein, and VR-10B, encoding a collagen adhesion fimbriae-like protein [23, 30, 31]. Our work has provided further insights into this region, specifically; that a third variant, VR-10C, contains neither of the two previously described loci but instead only encodes the flanking genes (only in strains in non-pathogenic clade 4). VR-10B is exclusively found in pathogenic clade 1 and is highly conserved. VR-10A is the most common variant present in all other strains. VR-10A is, however, highly variable, with many mutations detected in the poultry strains, which may suggest gene decay due to its inessential function (Fig. 6). Major variations of VR-10A were observed in the non-poultry strains SM101, ATCC13124 and 13, each having a unique variant of the locus (Fig. 7).
Fig. 6.

Adhesion locus variations. Genetic map showing sequence alignment of the different adhesion associated loci. Alignment is shown for four strains TAM-NE43, WER-NE36, EHE-NE18 and PBD1. TAM-NE43 has the intact VR-10A (blue) variant, WER-NE36 shows a highly deleted variation of VR-10A, VR-10B is represented by EHE-NE18 (green) [30] and PBD1 represents the VR-10C variant. Flanking genes of the locus sortase (srtB), and CPR0488 are coloured grey, and the two variants of the response regulator and sensor histidine kinase (SKH) are coloured red. Abbreviations: Alt exp.; alternative export protein, sipW; signal peptidase, hyp; hypothetical proteins, RR; response regulator, SHK; sensor histidine kinase, vWBFa; vonWillebrand factor A-like protein
Fig. 7.

VR-10A variations. A genetic map showing sequence alignments of the different VR-10A adhesion loci. Alignment is shown for four strains: ATCC13124, SM101, TAM-NE43 and 13
Capsular polysaccharide synthesis locus (CpCap)
Directly upstream of the adhesion-associated locus is a large capsular polysaccharide synthesis locus (CpCap). One of the differentiating chromosomal regions between pathogenic clades is the variation within capsular polysaccharide synthesis genes. There are two major chromosomal regions predicted to play a role in capsular polysaccharide biosynthesis [14]. These regions vary in composition between strains, most likely resulting in different capsular polysaccharide structure (Fig. 8a and b).
Fig. 8.

Capsule polysaccharide synthesis locus variations. Genetic map comparing the three capsule polysaccharide synthesis (CpCap) loci in pathogenic strains of C. perfringens. Representative regions from EHE-NE18 (mannose), TAM-NE46 (rhamnose) and EHE-NE7 (glucosamine) and the CpCPSL-B variants. a. Cds regions are colored as follows: Dark Blue – regulation, light blue – glycosyltransferase, red - UDP-mannose, orange – dTDP-rhamnose, Brown – UDP-L-fucose, Green – UDP-glucosamine/UDP-galactose, Dark pink – polysaccharide polymerase, yellow- polysaccharide transporter/flippase, Black – acetyltransferase, grey – hypothetical/unknown, Light Pink – mobile genetic elements and light green UDP-glycerol. b. As above but also Light Pink – phosphocholine
In all strains the first large capsule region, designated “Cap A” (Fig. 8a), encodes genes required for the modulation and biosynthesis of the capsule, including homologs of the cpsCDE (wze, wzd, wchA) genes at the 5′ region and genes encoding UDP-galactose-4-epimerase (galE), two copies of UTP-glucose-phosphate uridyltransferase (galU), and an RNA polymerase sigma factor 70 at the 3′ end of the locus. However, individual strains carry a diverse set of glycosyltransferases and UDP-sugar synthesis genes that encode the production of one or a combination of UDP-fucose, UDP-mannose, UDP-glycerol and/or dTDP-rhamnose, to be assembled as a polysaccharide capsule. Strains also encode a varied set of capsule polymerases (Wzy-like) and flippases (Wzx-like), suggesting specificity for the encoded UDP-sugar pathway. In some strains, a duplication of the modulation region cpsCD is observed upstream of the first locus, resulting in a much larger capsule polysaccharide synthesis locus, however, this duplication has not been observed in pathogenic poultry strains.
The second capsule region, designated “Cap B”, is more conserved than capsule region A, in most strains the presence of this locus is correlated with capsule locus A. In all strains the second locus encodes another undecaprenyl-phosphate galactose phosphotransferase (CpsE/WchA) at the 5′ end of the locus and a homolog of the capsule regulator Wzg/CpsA (LytR response regulator) at the 3′ end (Fig. 8b). In all strains, this region contains a set of genes encoding phosphocholine biosynthesis and export and, like region A, encodes a capsule polymerase (Wzy-like), a flippase (Wzx-like) and a set of glycosyltransferases. Most strains that do not contain proteins required for dTDP-rhamnose synthesis in the first capsule region encode these proteins in the second region, except for some strains in pathogenic clade 2, which do not encode any dTDP-rhamnose biosynthesis genes.
Pathogenic clade 1 strains encode proteins required for the production of UDP-mannose, UDP-rhamnose and phosphocholine within these capsule regions. However, some strains within pathogenic clade 1 lack the UDP-mannose biosynthesis genes. Pathogenic clade 2 and some strains from non-pathogenic clade 3 appear to encode a simple capsule type, as they have neither UDP-mannose or UDP-rhamnose coding regions. In these strains, the presence of a glucose-dehydrogenase-encoding gene predicts that the pathogenic clade 3 strains encode a galactose/glucosamine capsule.
Many of the non-pathogenic clade 3 strains encode the most diverse capsules, with several different types observed. Strain 11, for example, encodes proteins in the capsule region A that are predicted to make fucose additions to the capsule while strains SAF-1 and 96–7415 have a duplication of the modulation genes in the capsule locus A as well as encoding glycerol modification proteins. Strains within pathogenic clade 2 and the non-pathogenic clades that do not encode a duplication of the modulation region only encode cpsE/WchA in the capsule region; instead, in region A they encode a UDP-N-acetyl-glucosamine 4,6-dehydratase (via the pseB/fnlA genes).
Overall, pathogenic poultry strains only appear to encode three different capsule polysaccharide synthesis loci while non-pathogenic strains and non-chicken strains have highly diverse capsule types.
NEloc-2 – Chromosomal necrotic enteritis-associated locus
NEloc-2, previously described by Lepp [22], contains 11 ORFs encoding proteins with various functions, including sigma factors, chitin synthases and CotH-like proteins. This locus was previously found to be highly prevalent in NE-causing C. perfringens strains, but in this study, we show that it also is present in non-chicken strains, with the locus observed in JGS1721, F262, WAL-14527, MUR7757A, and JP838. We found that all necrotic enteritis-causing strains carried the NEloc-2 region. Several non-pathogenic strains from healthy and diseased birds, including strain 11, BER-NE33, NAG-NE1 and TAM-NE42, did not encode this locus. In some strains, the NEloc-2 region of the chromosome is replaced by a gene predicted to encode a flavin adenine dinucleotide-binding protein. This study further confirms the association of NEloc-2 with necrotic enteritis causing C. perfringens isolates.
Prophage-like regions
A large amount of the chromosomal variation between strains results from prophage-like integration. The prophage-like regions are diverse in genomic content and vary considerably between strains. In this strain collection, there are approximately 16 unique prophage-like integrations (predicted to be intact by PHAST) in the various chromosomes (Fig. 5). Further prophage diversity was examined by the presence of partial fragments of what are presumed to be remnants of old integration events.
In some clades, the presence of prophage regions on the chromosome is less prominent (pathogenic clade 2 and non-pathogenic strains typically have one or two prophage elements), while in pathogenic clade 1, prophage elements are highly prevalent with multiple prophage regions characterised in each strain. Some of these prophages are co-linear, localised in the same region of the chromosome, with similar insertion sites. Some of the prophage regions are shared by otherwise chromosomally distinct strains. Most of the prophage integrations are distinct from previously characterised C. perfringens phage. One strain within pathogenic clade 1, FC2, encodes one phage associated region and is closer to pathogenic clade 2, which also typically contain a single prophage element; it therefore appears that FC2 may be more divergent from the other pathogenic clade 1 strains. One prophage element (Prophage-II) is uniquely associated with non-pathogenic clade 4, which includes strain PBD1. This prophage region is highly distinct from all the other prophage regions showing very little similarity to those in other strains. This region is a major driver for the clear separation observed between strains in non-pathogenic clade 4 and all other isolates. There are also many phage fragments (incomplete phage regions) usually present as singletons, which also contribute to strain diversity.
Inhibitory molecules
A chromosomal region encoding a putative antibiotic system/bacteriocin locus was found in some strains within pathogenic clade 2 (WER-NE36, EHE-NE7, SOM-NE34 and SOM-NE35). This locus contains genes predicted to encode two cytolysin-like bacteriocins (ClyM), an immunity protein, several membrane associated proteins and a cytolysin transport protein (Fig. 5). If functional, this locus could provide a selective advantage for these strains over other pathogenic strains within an infected host or within a common niche.
A secondary bacteriocin gene is highly prevalent in necrotic enteritis strains. Previously described as cpp (perfrin) [32], this bacteriocin-encoding gene is located within a variable phage-related locus only in some pathogenic necrotic enteritis causing strains. The gene is in several strains from pathogenic clade 1 but no strains in pathogenic clade 2 or any of the non-chicken or non-pathogenic chicken isolates (Fig. 5). Examples of strains that encode cpp are those with a phage-related locus with high identity to the EHE-NE18-prophage-III, including Pennington, Warren, GNP-1, SYD-NE41, TAM-NE38 and several others. It is important to note that some strains within pathogenic clade 1, including 98.78718–2 and FC2, do not encode the entire phage region and therefore do not contain the cpp gene. It has been shown that cpp plays an important role in the inhibition of C. perfringens strains [32] and therefore prophage regions are regarded as an important factor for virulence.
This analysis also discovered significant genome variability between strains with predicted functions in metabolism (fucose, citrate and glucoronate), motility (type IV pili), and bacterial immunity (CRISPR and type I restriction modification system). These regions were not observed to be strictly associated with pathogenesis in necrotic enteritis, but rather sporadically present across strains from varied hosts and disease-causing capability. Finally, there are also many singleton ORFs that contribute to the variation seen in the chromosomes of C. perfringens strains. The functional role of proteins encoded by these single genes is difficult to determine, but is likely to contribute or influence different metabolic pathways and virulence factors within the strains.
Plasmid carriage and clade designation
Sequence analysis in this study also provided information about the carriage of plasmids by each isolate. In the earlier analysis plasmid sequences were removed to focus on the chromosomal variation. However, a separate examination of the sequence data for known and unknown plasmids showed that all strains in pathogenic clades 1 and 2 carried netB and cpb2-encoding plasmids. All strains in pathogenic clade 1 carried a tetracycline resistance plasmid, like the well-characterised prototype plasmid pCW3. Only pathogenic sub-clade 1A strains carried a tpeL-encoding plasmid. By contrast, all non-pathogenic poultry strains isolated from healthy birds (SAF-1, strain 11, PBD1, PC5 and PBS5), and a single non-pathogenic strain isolated from a diseased bird (TAM-NE43), did not contain any plasmids. Most of the strains in non-pathogenic clade 3 contained a single plasmid which encoded both tetracycline resistance and the beta2 toxin, with similar composition to the published sequence pFORC03 (Accession Number: NZ_CP009558).
SNP analysis
The core genome (genes shared by all strains) consists of 1192 genes. Analysis of these genes using HARVEST tools v1.2 and Snippy v3.2 with ATCC13124 as the reference strain identified 119,657 core SNPs from 295,709 variant sites. The SNP occurrence in each strain was used to produce a relatedness tree of maximum likelihood (Fig. 9) using RAxML v8.0 general time reversible model with 1000 bootstraps [33]. A comparison of the SNP tree to the Pan-genome tree revealed distinct differences and similarities in the evolutionary relationships calculated using these different methods. The SNP tree indicates that the pathogenic poultry isolates are more widely dispersed than indicated by the pan-genome analysis. Isolates designated as pathogenic clade 1 and pathogenic clade 2 no longer cluster together, but rather are separated into multiple groups, suggesting that the core genome does not closely reflect the gene diversity seen in the pan-genome analysis. Some strains do, however, cluster closely together suggesting that they are highly related and possibly clonal, as clustering is conserved in both core gene SNPs and the pan-genome content. Overall, core genome phylogeny does not match disease causing capability.
Fig. 9.

Core genome phylogeny. Tree of maximum likelihood based on single nucleotide polymorphisms in the core chromosome of C. perfringens. Dark red and light red squares represent Pathogenic strains of C. perfringens clades 1 2 respectively while green triangles and diamonds represent non-pathogenic strains in clades 3 and 4 respectively. Black; positive, white; negative
PFGE fingerprinting
Pulsed-field gel electrophoresis (PFGE) was also used to examine the phylogenetic relationship between strains and to determine if this method could be used to categorize strains into the clades identified by whole genome sequencing. A chromosome digest followed by PFGE analysis revealed that among pathogenic necrotic enteritis-causing strains, three distinctly different PFGE patterns were observed. A dendrogram generated from the PFGE profiles placed all the pathogenic strains into similar groupings as that defined by the chromosome sequence comparison, demonstrating that PFGE gives a good representation of the pan-genome variation that has been observed within the C. perfringens strain collection examined here (Fig. 10). Several strains, including EHE-NE3, EHE-NE4, EHE-NE5, EHE-NE14, EHE-NE20, PBS4, Mugdale 5, TAM-NE47, FC1, Kendall, 94–5224, which had not been sequenced, were included in the PFGE analysis and these strains could be placed within the clade structure established for the sequenced strains. Most of the pathogenic strains examined clustered into three PFGE profiles (Fig. 10), supporting the designation of strains into these pathogenic clades.
Fig. 10.

PFGE. Tree of PFGE fingerprints of various C. perfringens strains. Colours are as follows; white; negative, black; positive, for features including netB, VR-10A and isolation from chicken. Colours referring to clade designation are as follows; blue; Non-poultry, Green (dark and light); Non-pathogenic clade 3 and 4 respectively, Pink; pathogenic clade 1B, Red; pathogenic clade 1A, Orange; pathogenic clade 2
Discussion
In this study, comparative genomic analysis showed that pathogenic strains of C. perfringens, which were isolated from birds with necrotic enteritis and contain the netB gene, can be classified into two distinct groups. The pathogenic clades are pathogenic clade 1, containing most netB-encoding strains and which is separated into subclades 1A (CP4-like) and 1B (NE18-like), and pathogenic clade 2, containing JGS4143/WER-NE36-like strains. Clades 3 and 4 comprise non-pathogenic isolates.
The presence of highly conserved clades of pathogenic necrotic enteritis-causing strains across distinct geographical locations and isolated across several decades suggests that there is persistence or transfer of strains between continents or that the isolates have originated from a common source, possibly from within the breeding stock used for global commercial production of chickens. Alternatively, perhaps these strains are under a common selection pressure and therefore retain or lose common genetic loci. These results expand on the existing knowledge of two pathogenic clades [23], and provide evidence that one of these clades appears to be diverging into subclades (1A and 1B). Pathogenic clade 2 is more closely related to non-pathogenic strains. The use of hybridisation methods such as comparative genome hybridization, as in earlier studies [23], would result in some of these distinct subclades being clustered together. The pathogenic clades 1A and 1B fit within the CHG cluster III and our pathogenic clade 2 and non-pathogenic clade 3 fit within CGH clusters I and II, as defined previously [23]. The clustering of clades observed in this study is supported by other published studies, in particular those involving the use of MLST and PFGE to examine strain diversity [34]. The high level of similarity between strains isolated many years apart suggests a strong selective pressure for their genome to be conserved. This observation is particularly true for pathogenic clade 1A, while other strains such as those in pathogenic clade 2 and the non-pathogenic clades are more diverse. In contrast, the high genomic diversity of the non-pathogenic strains is supported by PFGE studies, which is not surprising since healthy chickens are known to harbour a highly diverse C. perfringens population [16, 17, 35]. Our results have demonstrated that pathogenicity of necrotic enteritis does not correlate well to core genome phylogeny, or to the majority of variable genetic loci. Plasmids are known to encode the major factors required for virulence, e.g. toxins, but the variability in both plasmid carriage and chromosomal genetic islands show key regions associated to virulence and pathogenesis mechanisms are varied.
Most of the pathogenic poultry strains examined here are classified into pathogenic subclades 1A and B. These isolates share many of the variable chromosomal regions. The most significant regions separating these subclades from other C. perfringens isolates are the adhesion-associated loci and the capsule polysaccharide synthesis loci. They share the VR-10B locus [30], supporting the hypothesis that this adhesion locus is chicken specific and is highly prevalent and conserved in necrotic enteritis pathogenic strains. We previously [30, 31] demonstrated through gene deletion and virulence studies that this adhesion locus is a crucial factor for necrotic enteritis disease. Pathogenic clade 1 has a higher level of chromosomal variation compared to the other clades, hence the separation into subclades 1A and 1B. This variation is mostly due to the integration of various prophage genomes and other smaller genomic variations such as the presence of the variant motility locus and the type I RMS system. All strains within subclade 1B carry a capsule locus encoding proteins that catalyse UDP-mannose additions to the polysaccharide capsule. This region is also shared by approximately half the strains in subclade 1A, while the other 1A strains only encode proteins that facilitate dTDP-rhamnose and choline additions. Plasmid content also differentiates the subclades; clade 1A strains are the only pathogenic strains to carry four plasmids including the tpeL-encoding plasmid while clade 1B strains carry three plasmids, which encode Beta2 toxin production, tetracycline resistance and NetB toxin production. Pathogenic clade 1 appears to be diverging and evolving from a common ancestor into the two most prevalent necrotic enteritis-causing populations. From our analysis, we have noted that pathogenic clade 2 strains contain a more variable chromosome compared to the other pathogenic strains. Strains within this clade encode the VR-10A adhesion-associated locus and the simple galactose/glucosamine-based capsule locus. Other regions associated with pathogenic clade 2, and not found in other pathogenic strains, include those that encode genes responsible for L-fucose metabolism and a bacteriocin/antibiotic associated locus.
Pathogenic clade 2 is clustered closely by gene content to many non-pathogenic strains and non-chicken isolates. Therefore, it is hypothesised that pathogenic clade 2 strains were not originally pathogenic chicken strains, but have been introduced into chickens from other sources, and that the strains have subsequently inherited the virulence plasmids through horizontal gene transfer, resulting in the emergence of new strains capable of causing necrotic enteritis in chickens. In C. perfringens horizontal gene transfer in the laboratory has been demonstrated to convert strains to different toxin types and disease causing capability [7, 36, 37] so it is possible that this process can also occur in the environment or in the chicken gastrointestinal tract.
It is common to isolate netB negative strains of C. perfringens from chickens suffering necrotic enteritis [25, 38–41]. It is considered that these strains are either normal or transient flora or isolates in which the plasmid has been lost during initial recovery; seven such netB negative isolates are included in this analysis. Comparison of the chromosomes of these strains reveals a degree of similarity to pathogenic clade 2 strains, but with more individual variation. PFGE fingerprinting revealed that each strain has a unique fingerprint, providing evidence that they originate from different backgrounds or have a high level of genomic rearrangements. There is a relatively high degree of genomic variation within all the non-pathogenic chicken strains whether they were isolated from diseased birds or healthy birds, suggesting a high level of divergence or genetic drift, partly driven by the integration and mobilisation of mobile genetic elements that are prolific within the C. perfringens species.
Strains isolated from healthy birds with no signs of necrotic enteritis are believed to be commensal or transient C. perfringens strains. Both the sequence data and PFGE indicated that most of these strains did not contain large clostridial plasmids. These strains have no association with necrotic enteritis, and lack the netB gene. By comparative chromosome analysis, it was seen that several of these strains, including PBD1, PBD5 and PC5, have highly divergent chromosomes compared to other chicken strains. Strains SAF-1 and 11 are an exception; even though these strains lacked large plasmids, they clustered much closer to pathogenic clade 2 and sat within the group of strains that were isolated as netB negative from diseased birds. The genetic diversity of these healthy bird isolates suggests that further sampling and sequencing of different C. perfringens isolates from healthy individuals may further expand the known diversity of C. perfringens genomes. This may help to inform our understanding of strain evolution, conjugative transfer of toxin plasmids, pathogenesis, and normal flora-host relationships.
The ability of strains to adhere to and colonise the host is essential for virulence in many disease systems [23, 31, 42]. The presence of many different capsular biosynthesis and adhesion-associated regions in pathogenic chicken strains suggests that there may be various mechanisms by which strains can cause disease. For necrotic enteritis to occur, expression of the NetB toxin is required, however, a pathogenic strain also needs to compete with other microbes within the gut environment of the chicken host [43]. The presence of the VR-10B adhesion allele is strongly associated with necrotic enteritis [23, 30, 44], suggesting that this allele may provide a selective advantage for colonisation in chickens. The other major alternative allele, VR-10A, is highly mutated in many chicken pathogenic isolates, suggesting that this locus is not required for colonisation or disease in chickens.
In other Gram-positive bacteria capsules play an important role in the disease process [39–45] Three major capsule types were observed in pathogenic strains. Capsules are likely to be a virulence factor as capsule has been implicated in various diseases, with particular roles in colonisation and immune evasion [45–47]. The capsule loci encode proteins with varied UDP-sugar biosynthesis and glycosyltransferase functions, suggesting that very different capsule polysaccharide chains may be synthesised by different strains. Different polysaccharide capsules have been characterised by NMR in C. perfringens [48–51]. A recent study characterising the polysaccharide structures in ATCC13124 demonstrated distinct polymer structures containing β-mannosamine, rhamnose, glucosamine and phosphocholine linkage [52], this supports our characterisation of the capsule loci, as ATCC13124 contains the corresponding genes within the CpCAP. Using Streptococcus sp. as a model we predict that the conserved genes on both the 5′ and the 3′ end of the locus produce the minimum polysaccharide capsule [45]. Typically most of the enzymes necessary for polysaccharide synthesis are encoded within polysaccharide-specific loci, with many of the nucleotide diphospho-sugars (NDP-sugars) common to other cellular pathways and obtained from cellular pools without the need for the involvement of unique enzymes [46]. Therefore, some difficulty arises in estimating the structure of the polysaccharide capsule without physical examination, although initial studies into structure have been performed by NMR spectroscopy, and mechanical isolation with agglutination tests using immune sera [48–50, 53].
Allelic replacement of capsule regions within and between bacterial species could explain the dramatic amount of capsule and serotype variation observed in bacterial species such as Staphylococcus sp. and Streptococcus sp. [45, 54, 55] . It is likely that genetic exchange can occur between bacterial species that persist in the same environment as C. perfringens. Many other species co-exist in the gastrointestinal tract of animals and so it is potentially an environment of high genomic exchange, which could result in the transfer and exchange of CpCAP regions [45, 46, 56, 57].
It has been proposed [43] that bacteriocin production may be one of the factors responsible for the typical single strain dominance [58] in necrotic enteritis cases. It has been demonstrated that co-challenge with pathogenic and non-pathogenic C. perfringens strains resulted in the displacement of non-pathogenic strains from broilers, an important step in the pathogenesis of necrotic enteritis [32, 43, 59]. Therefore, bacteriocin production can be regarded as a potentially important virulence-associated factor. In the isolates in this study the bacteriocin perfrin (cpp) is encoded by an integrated phage regions and has similarities to other phage encoded inhibitory proteins [60].
Finally, the presence of a large variety of prophage elements is not only an important factor in establishing phylogenetic relationships between strains, but they can also be an important source of virulence factors [61]. In other A-T rich genomes, such as those of Staphylococcus aureus and Streptococcus pyogenes, phage integrations account for a substantial amount of the inter-strain genetic variations that are seen [62, 63]. As shown here, and in previous C perfringens genome papers, prophages account for a large proportion of the variation seen between C. perfringens genomes [14, 23]. In the clostridia, prophages have the potential to manipulate the regulation of genes through their insertion into genes involved in sporulation and metabolism [64]. Most of the prophages found to be associated with C. perfringens belong to the family Siphoviridae of the order Caudoviridales [64–67]. Several unique prophages have been found in previous sequenced strains such as ATCC13124, strain 13, ATCC3626, and SM101, as well as in many strains not investigated in this study [14, 64, 68, 69]. Some strains have a single prophage integration, while other strains have up to eight different integration events. These high levels of variation caused by prophage integrations are likely to have a strong influence on C. perfringens by potentially introducing new and varied virulence factors such as bacteriocin genes and/or affecting gene regulation. Bacterial immune systems were also characterised including type I and type II restriction modification systems and a CRISPR.
Representative non-chicken strain sequences, accessed from the public databases, were used to compare the variation observed within the necrotic enteritis-associated C. perfringens to that seen in the wider population of C. perfringens from other sources, including strains of different toxinotypes. Interestingly, a recent WGS study has concluded, based on the carriage of know virulence factors, that the pathogenesis of C. perfringens caused necrotic enteritis in turkeys is likely to be different to that seen in chickens [70]. The analysis of non-chicken derived C. perfringens isolates suggested that regardless of host, toxinotype, and plasmid content, the non-chicken strains are more closely related to each other and to the non-pathogenic clade 3 chicken isolates and pathogenic strains in clade 2 than to the strains of pathogenic clade 1. Since the toxinotyping system for classifying C. perfringens is dependent upon plasmid-determined toxin production [2], the removal of plasmids from typing studies would result in every C. perfringens strain being classified as toxinotype A. Therefore, the lack of diversity in these non-pathogenic strains may indicate that extra-chromosomal elements such as plasmids are a major source of genomic variation in non-chicken strains. Another possible explanation for the lack of diversity in non-chicken isolates may be that the strains that can cause disease in mammals share common traits for colonisation and pathogenesis, as mammals are likely more structurally and biologically like one another than to chickens and other poultry in terms of their digestive physiology [71]. Increasing the sampling and sequencing of isolates that are implicated in other diseases, together with the addition of non-pathogenic strains from other animals, will reveal more about the chromosome variation in isolates from other toxinotypes and diseases. Perhaps there is a greater diversity such as that observed in chicken isolates for each different animal host, a suggestion which can only be tested by investigation of much larger numbers of diverse isolates.
Conclusion
Overall, the current study has shown that necrotic enteritis-causing isolates of C. perfringens group into several pathogenic clades and subpopulations based on their chromosomal gene content. This study has demonstrated that pathogenicity does not correlate with core genome content, but rather is correlated with accessory gene content located on the chromosome and plasmids. This study has identified several variable genomic loci with potential roles in virulence that could contribute to alternative pathogenesis mechanisms in different isolates of NetB producing necrotic enteritis causing strains of C. perfringens. The variation in the capsule loci, adhesion-associated locus, and prophage elements accounts for most of the chromosomal variation between strains. The presence of virulence and antibiotic resistance plasmids was also shown to be variable between clades. It will be of interest to determine whether any of these loci provide increased fitness or provide a selective advantage of one pathogenic clade over another, as all subtypes appear to persist in the environment, temporally and spatially. In summary, these results suggest that netB-positive, necrotic enteritis-causing strains have evolved via several different mechanisms, including clonal expansion and horizontal gene transfer.
Methods
Strain collection
The collection of 36 C. perfringens strains selected for whole genome sequencing (WGS) included isolates from a range of international locations, isolation dates and host disease status. Their isolation and previous characterisations are detailed in the references listed in Table 1. Diverse strains were selected based on laboratory tests and virulence in a chicken model of necrotic enteritis induction as reported in previous published studies. The common finding is that only isolates that carry the netB gene can reproducibly produce disease. We have therefore classified strains that have not been directly tested in a disease induction model as either virulent or avirulent based on their carriage of netB. The dataset was supplemented with the addition of 20 WGS from published sequences available in GenBank (Table 1).
DNA extraction and genome sequencing
Strains were cultured from -80 °C glycerol stocks on fresh Tryptone-Sulphite-Cycloserine (TSC) plates (Oxoid). To prepare genomic DNA for Roche/454 and Illumina MiSeq library preparation single colonies were used to seed overnight cultures in Tryptone-Peptone-Glucose (TPG) broth (5% (w/v) Bacto-Tryptone (BD Biosciences), 0.5% (w/v) proteose peptone (BD Biosciences), 0.4% (w/v) glucose and 0.1% (w/v/) sodium thioglycolate). The overnight broth was used to seed a fresh TPG broth and incubated for 8 h. Cultures were then centrifuged at 14,000 rpm for 30 min and the supernatant was discarded. The pellets were then subjected to a phenol/chloroform DNA extraction. Sequencing libraries were prepared for Roche/454 sequencing using the NEBNext Quick DNA Library Prep Reagent Set (New England Biolabs) and for Illumina MiSeq using the Nextera XT DNA Library Preparation Kit (Illumina). Genomes were sequenced using Roche/454 (Titanium chemistry) and Illumina MiSeq (2 × 300 bp paired-end reads) methods. Roche/454 read data were assembled using Roche GS De Novo Assembler v2.6 with default settings. Illumina MiSeq read data were quality trimmed using EA-utils v1.1.2 fastq-mcf program [72]: sequence with an average quality score lower than 15 in a 5 bp sliding window was trimmed. Reads with lengths less than 50 bp or more than 70% low complexity were discarded. MiSeq reads were assembled using IDBA-UD v1.1.1 with default settings [73]. For PacBio sequencing, EHE-NE18 was grown overnight as previously described, 10 μl was subcultured into five 1.5 ml Heart Infusion broths (Oxoid) for 3 hours until mid-log phase. Each culture was subjected to the MagAttract HMW Kit (Qiagen) as per the manufacturers’ instructions. The quality of purified DNA was assessed using the Qubit 2.0 (ThermoFisher Scientific) and the High Sensitivity dsDNA kit as per the manufacturer’s instructions. 20 Kb size selection was done using BluePippin Size Selection (Sage Science) and DNA was prepared for sequencing using the SMRTbell Template Preparation Kit 1.0 (Pacific Biosciences) as per the manufactures’ instructions. The genome was sequenced on an RS2 instrument (PacificBioSciences). The RS2 read data were assembled using the Canu Assembler v1.3 with the following options: mhapMerSize = 21′, ‘genomeSize = 3.5 m’, ‘OvlMerSize = 21′, and ‘-pacbio-raw’ [74]. NCBI Blast was used to align the beginning and end of the molecule and matching sequence was detemined from the beginning of the molecule. Seqret (EMBOSS v6.6.0.0) was used to reformat the circulised file to appropriate FASTA format. The genome was then polished using blasr version 5.2.b99b47c (pitchfork release, 10/6/16 PacificBioSciences, 2015). The genome was then subjected to short read error correction using Pilon v1.18 using the MiSeq sequencing data [75]. The whole genome sequence data has been deposited at DDBJ/ENA/GeBank under BioProject PRJNA422745.
Bioinformatic analysis
The program, HARVEST tools, including Parsnp v1.2, was used to align the core genomes of C. perfringens strains and to identify SNPs [76]. This software was designed to identify regions of recombination and thus avoid mis-calling of SNPs; we tested program runs with and without this feature (option ‘-x’) and found no difference in results. Default program settings were used for all other options. To ensure accuracy, the program Snippy v.3.2 (Github.com/tseemann/snippy) was also used to establish core genome SNP phylogeny. Phylogenetic trees of maximum likelihood were created using the RAxML v 8.0 using the general time reversible model with gamma distribution and a 1000 bootstrap replicates [33].
For the pangenome analysis, coding DNA sequences (CDS) were identified in assembled genomes using Prokka v1.11 [77] set to ‘fast’ mode with no rRNA or tRNA detection. Plasmid CDS were removed from the data by first creating a database of C. perfringens plasmid sequences from the NCBI nucleotide database using NCBI E-utilities [78]. Command = “esearch -db nucleotide -query ‘(Clostridium perfringens plasmid complete sequence) AND “Clostridium perfringens”[porgn:__txid1502] NOT (chromosome)’ | efetch -format fasta > cp_ncbi_plasmids.fasta”. Plasmid CDS were then identified using Prokka as described for genomes and made into a BLAST protein database (BLAST v2.2.30). Genome CDS were searched against the plasmid database using blastp. All CDS with an alignment e-value of 1 × 10− 10 or less were removed from the genome’s CDS file using a python script (pullplasmids_cds.py, available on GitHub: https://github.com/tallnuttcsiro/pullplasmids_cds.py). The presence of CDS among strains was assigned using cluster analysis. All CDS protein sequences were placed in a single FASTA file and analysed using the UCLUST algorithm of the USEARCH program [29] with a 60% identity radius for clusters (i.e. proteins with 60% alignment identity or greater were clustered as a single CDS around a ‘centroid’ CDS). A full example of linux commands and the associated scripts used is available: https://github.com/tallnuttcsiro/pangenome_using_uclust). Note that the clustering threshold of 60% chosen here is arbitrarily used as a threshold below which we assume non-homologous CDS will not be scored as present among strains. Increasing this threshold gives a lower probability that non-homologous CDS will be amalgamated, whereas raising the threshold divides truly homologous CDS into spurious subdivisions. We found that 60% provided a reasonable compromise, giving a realistic number of pangenome CDS in the sequenced genomes. Our analysis was also confirmed using the Bacterial Pangenome Analysis Pipeline (BPGA) v1.3.0 [79], which also applies the USEARCH algorithms to determine evolutionary relationships. Using the same thresholds on the chromosomal sequences of our dataset provided the same evolutionary relationships. KEGG and COG analyses on the core, accessory and unique genes were performed using the BPGA software [79]. Maximum likelihood tree of the pangenome was created with RAxML v 8.0 using the binary pan matrix as the input and gamma model of rate heterogeneity with 1000 bootstrap replicates [33].
Prophage regions within the assembled chromosome of C. perfringens were characterised and located using PHAST (PHAge Search Tool) http://phast.wishartlab.com [80] and further expanded manually. Other variable regions were examined manually and blastn was utilised to examine the presence or absence of these loci within our strain database.
PFGE fingerprinting
C. perfringens strains were grown overnight in 6 ml of TPG broth. Bacterial pellets were collected by centrifugation at 14,000 rpm and the cells were resuspended in 1 ml of 1% PFGE-certified agarose and adjusted to an OD600 of 0.8. Agarose plugs were incubated for 1 h with gentle shaking at 37 °C in lysis buffer (0.5 M EDTA pH 8.0, 2.5% of 20% N-lauryl-sarkosyl, 0.25% lysozyme) and subsequently incubated in 2% proteinase buffer (0.5 M EDTA pH 8.0, 2.5% of 20% N-lauryl-sarkosyl 0.25% Proteinase K) overnight at 55 °C. For each isolate, a plug was equilibrated in 200 μL of CutSmart restriction buffer (NEB) at room temperature for 20 min and then digested with 20 U of SmaI at 25 °C overnight. Electrophoresis was performed in a 1% PFGE-certified agarose gel and separated with a CHEF-DR-III PFGE system (Bio-Rad Laboratories) in 0.5× Tris-borate-EDTA (TBE) buffer at 14 °C at 300 V for 20 h with a ramped pulse time of 4 to 38 s at an angle of 120°. Gels were stained with GelRed (Biotium) and visualized using UV light. Mid-range and Low-Range PFGE markers (New England Biolabs) were used as molecular DNA ladders. PFGE gels were analysed using GelCompare II software v 6.6 (Applied Maths). Band matching was performed using a 1% position tolerance and 0% optimization factor, and cluster analysis was performed using the Dice similarity coefficient and unweighted pair group method with arithmetic mean (UPGMA).
Acknowledgements
We thank Dr. Angela Hartman for supplying C. perfringens strains from the collection held by Zoetis, USA.
Funding
This research was conducted within the Poultry Cooperative Research Centre (CRC 1.1.2), established and supported under the Australian Government’s Cooperative Research Centres Program.
Availability of data and materials
The whole genome sequence data generated as part of this study is available from NCBI. The accession numbers are listed in Table 1.
Abbreviations
- CDS
Coding DNA sequence
- CGH
Comparative genome hybridization
- CpCap
Capsular polysaccharide synthesis locus
- CRISPR
Clustered regularly interspaced short palindromic repeats
- MLST
Multi-locus sequence type
- NCBI
National Centre for Biotechnology Information
- NMR
Nuclear magnetic resonance
- ORFs
Open reading frames
- PCA
Principal component analysis
- PFGE
Pulsed-field gel electrophoresis
- RMS
Restriction modification system
- SNP
Single nucleotide polymorphism
- TPG
Tryptone peptone glucose
- TSC
Tryptone sulphite cycloserine
- WGS
Whole genome sequencing
Authors contributions
RM, PJ and DL initiated and supervised the study, contributed to data interpretation and writing of the manuscript. JL, TA contributed in data gathering, data analysis, data interpretation, and writing of the manuscript. TS, XH, JR, BV, TH, HC contributed in data gathering. TS, VH, and BW contributed in data gathering and analysis. AK contributed to study design and data gathering. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable. All the work reported is laboratory based, not requiring human or animal ethics approvals or consents.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jake A. Lacey, Email: jake.lacey@monash.edu
Theodore R. Allnutt, Email: theo.allnutt@deakin.edu.au
Ben Vezina, Email: ben.vezina@rmit.edu.au.
Thi Thu Hao Van, Email: thithuhao.van@rmit.edu.au.
Thomas Stent, Email: thomas.stent@monash.edu.
Xiaoyan Han, Email: xiao.han@monash.edu.
Julian I. Rood, Email: julian.rood@monash.edu
Ben Wade, Email: christopher.wade@deakin.edu.au.
Anthony L. Keyburn, Email: anthony.keyburn@csiro.au
Torsten Seemann, Email: tseemann@unimelb.edu.au.
Honglei Chen, Email: Honglei.Chen@csiro.au.
Volker Haring, Email: volker.haring@gmail.com.
Priscilla A. Johanesen, Email: priscilla.johanesen@monash.edu
Dena Lyras, Email: dena.lyras@monash.edu.
Robert J. Moore, Email: rob.moore@rmit.edu.au
References
- 1.Uzal FA, Freedman JC, Shrestha A, Theoret JR, Garcia J, Awad MM, et al. Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol. 2014;9:361–377. doi: 10.2217/fmb.13.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li J, Adams V, Bannam TL, Miyamoto K, Garcia JP, Uzal FA, et al. Toxin plasmids of Clostridium perfringens. Microbiol Mol Biol Rev. 2013;77:208–233. doi: 10.1128/MMBR.00062-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parreira VR, Costa M, Eikmeyer F, Blom J, Prescott JF. Sequence of two plasmids from Clostridium perfringens chicken necrotic enteritis isolates and comparison with C. perfringens conjugative plasmids. PLoS One. 2012;7:e49753. doi: 10.1371/journal.pone.0049753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sayeed S, Li J, McClane BA. Virulence plasmid diversity in Clostridium perfringens type D isolates. Infect Immun. 2007;75:2391–2398. doi: 10.1128/IAI.02014-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bueschel DM, Jost BH, Billington SJ, Trinh HT, Songer JG. Prevalence of cpb2, encoding beta2 toxin, in Clostridium perfringens field isolates: correlation of genotype with phenotype. Vet Microbiol. 2003;94:121–129. doi: 10.1016/S0378-1135(03)00081-6. [DOI] [PubMed] [Google Scholar]
- 6.Garmory HS, Chanter N, French NP, Bueschel D, Songer JG, Titball RW. Occurrence of Clostridium perfringens beta2-toxin amongst animals, determined using genotyping and subtyping PCR assays. Epidemiol Infect. 2000;124:61–67. doi: 10.1017/S0950268899003295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bannam TL, Yan X-X, Harrison PF, Seemann T, Keyburn AL, Stubenrauch C, et al. Necrotic enteritis-derived Clostridium perfringens strain with three closely related independently conjugative toxin and antibiotic resistance plasmids. MBio. 2011;2:e00190-11. doi: 10.1128/mBio.00190-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adams V, Li J, Wisniewski JA, Uzal FA, Moore RJ, McClane BA, et al. Virulence plasmids of spore-forming bacteria. Microbiol Spectr. 2014;2:PLAS-0024-2014. [DOI] [PMC free article] [PubMed]
- 9.Freedman JC, Theoret JR, Wisniewski JA, Uzal FA, Rood JI, McClane BA. Clostridium perfringens type A–E toxin plasmids. Res Microbiol. 2015;166:264–279. doi: 10.1016/j.resmic.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keyburn AL, Boyce JD, Vaz P, Bannam TL, Ford ME, Parker D, et al. NetB, a new toxin that is associated with avian necrotic enteritis caused by Clostridium perfringens. PLoS Pathog. 2008;4:e26. doi: 10.1371/journal.ppat.0040026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nowell VJ, Kropinski AM, Songer JG, MacInnes JI, Parreira VR, Prescott JF. Genome sequencing and analysis of a type a Clostridium perfringens isolate from a case of bovine clostridial abomasitis. PLoS One. 2012;7:e32271. doi: 10.1371/journal.pone.0032271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong YM, Juan JC, Gan HM, Austin CM. Draft genome sequence of Clostridium perfringens strain JJC, a highly efficient hydrogen producer isolated from landfill leachate sludge. Genome Announc. 2014;2:e00064–14. [DOI] [PMC free article] [PubMed]
- 13.Johansson A, Aspan A, Bagge E, Båverud V, Engström BE, Johansson K-E. Genetic diversity of Clostridium perfringens type a isolates from animals, food poisoning outbreaks and sludge. BMC Microbiol. 2006;6:47. doi: 10.1186/1471-2180-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myers GSA, Rasko DA, Cheung JK, Ravel J, Seshadri R, DeBoy RT, et al. Skewed genomic variability in strains of the toxigenic bacterial pathogen, Clostridium perfringens. Genome Res. 2006;16:1031–1040. doi: 10.1101/gr.5238106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chalmers G, Bruce HL, Hunter DB, Parreira VR, Kulkarni RR, Jiang Y-F, et al. Multilocus sequence typing analysis of Clostridium perfringens isolates from necrotic enteritis outbreaks in broiler chicken populations. J Clin Microbiol. 2008;46:3957–3964. doi: 10.1128/JCM.01548-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chalmers G, Martin SW, Hunter DB, Prescott JF, Weber LJ, Boerlin P. Genetic diversity of Clostridium perfringens isolated from healthy broiler chickens at a commercial farm. Vet Microbiol. 2008;127:116–27. [DOI] [PubMed]
- 17.Nauerby B, Pedersen K, Madsen M. Analysis by pulsed-field gel electrophoresis of the genetic diversity among Clostridium perfringens isolates from chickens. Vet Microbiol. 2003;94:257–266. doi: 10.1016/S0378-1135(03)00118-4. [DOI] [PubMed] [Google Scholar]
- 18.Hassan KA, Elbourne LDH, Tetu SG, Melville SB, Rood JI, Paulsen IT. Genomic analyses of Clostridium perfringens isolates from five toxinotypes. Res Microbiol. 2015;166:255–263. doi: 10.1016/j.resmic.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 19.Parish WE. Necrotic enteritis in the fowl (Gallus Gallus Domesticus). I. Histopathology of the disease and isolation of a strain of Clostridium welchii. J Comp Pathol Ther. 1961;71:377–393. doi: 10.1016/S0368-1742(61)80043-X. [DOI] [PubMed] [Google Scholar]
- 20.Songer JG. Clostridial enteric diseases of domestic animals. Clin Microbiol Rev. 1996;9:216–234. doi: 10.1128/cmr.9.2.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lovland A, Kaldhusdal M. Severely impaired production performance in broiler flocks with high incidence of Clostridium perfringens-associated hepatitis. Avian Pathol. 2001;30:73–81. doi: 10.1080/03079450020023230. [DOI] [PubMed] [Google Scholar]
- 22.Lepp D, Roxas B, Parreira VR, Marri PR, Rosey EL, Gong J, et al. Identification of novel pathogenicity loci in Clostridium perfringens strains that cause avian necrotic enteritis. PLoS One. 2010;5:e10795. doi: 10.1371/journal.pone.0010795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lepp D, Gong J, Songer JG, Boerlin P, Parreira VR, Prescott JF. Identification of accessory genome regions in poultry Clostridium perfringens isolates carrying the netB plasmid. J Bacteriol. 2013;195:1152–1166. doi: 10.1128/JB.01032-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keyburn AL, Bannam TL, Moore RJ, Rood JI. NetB, a pore-forming toxin from necrotic enteritis strains of Clostridium perfringens. Toxins. 2010;2:1913–1927. doi: 10.3390/toxins2071913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keyburn AL, Yan X-X, Bannam TL, Van Immerseel F, Rood JI, Moore RJ. Association between avian necrotic enteritis and Clostridium perfringens strains expressing NetB toxin. Vet Res. 2010;41:21. doi: 10.1051/vetres/2009069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coursodon CF, Glock RD, Moore KL, Cooper KK, Songer JG. TpeL-producing strains of Clostridium perfringens type a are highly virulent for broiler chicks. Anaerobe. 2012;18:117–121. doi: 10.1016/j.anaerobe.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 27.Allaart JG, de BND, van AAJAM, Fabri THF, Gröne A. NetB-producing and beta2-producing Clostridium perfringens associated with subclinical necrotic enteritis in laying hens in the Netherlands. Avian Pathol. 2012;41(6):541. doi: 10.1080/03079457.2012.729809. [DOI] [PubMed] [Google Scholar]
- 28.Han X, Du X-D, Southey L, Bulach DM, Seemann T, Yan X-X, et al. Functional analysis of a bacitracin resistance determinant located on ICECp1, a novel Tn916-like element from a conjugative plasmid in Clostridium perfringens. Antimicrob Agents Chemother. 2015;59:6855–6865. doi: 10.1128/AAC.01643-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinforma. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 30.Wade B, Keyburn A, Haring V, Ford M, Rood J, Moore R. The adherent abilities of Clostridium perfringens strains are critical for the pathogenesis of avian necrotic enteritis. Vet Microbiol. 2016;197:53–61. doi: 10.1016/j.vetmic.2016.10.028. [DOI] [PubMed] [Google Scholar]
- 31.Wade B, Keyburn AL, Seemann T, Rood JI, Moore RJ. Binding of Clostridium perfringens to collagen correlates with the ability to cause necrotic enteritis in chickens. Vet Microbiol. 2015;180:299–303. doi: 10.1016/j.vetmic.2015.09.019. [DOI] [PubMed] [Google Scholar]
- 32.Timbermont L, De Smet L, Van Nieuwerburgh F, Parreira VR, Van Driessche G, Haesebrouck F, et al. Perfrin, a novel bacteriocin associated with netB positive Clostridium perfringens strains from broilers with necrotic enteritis. Vet Res. 2014;45:40. doi: 10.1186/1297-9716-45-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neumann AP, Rehberger TG. MLST analysis reveals a highly conserved core genome among poultry isolates of Clostridium septicum. Anaerobe. 2009;15:99–106. doi: 10.1016/j.anaerobe.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 35.Gholamiandekhordi AR, Ducatelle R, Heyndrickx M, Haesebrouck F, Van Immerseel F. Molecular and phenotypical characterization of Clostridium perfringens isolates from poultry flocks with different disease status. Vet Microbiol. 2006;113:143–152. doi: 10.1016/j.vetmic.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 36.Hughes ML, Poon R, Adams V, Sayeed S, Saputo J, Uzal FA, et al. Epsilon-toxin plasmids of Clostridium perfringens type D are conjugative. J Bacteriol. 2007;189:7531–7538. doi: 10.1128/JB.00767-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brynestad S, Sarker MR, McClane BA, Granum PE, Rood JI. Enterotoxin plasmid from Clostridium perfringens is conjugative. Infect Immun. 2001;69:3483–3487. doi: 10.1128/IAI.69.5.3483-3487.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crespo R, Fisher DJ, Shivaprasad HL, Fernández-Miyakawa ME, Uzal FA. Toxinotypes of Clostridium perfringens isolated from sick and healthy avian species. J Vet Diagn Investig. 2007;19:329–333. doi: 10.1177/104063870701900321. [DOI] [PubMed] [Google Scholar]
- 39.Brady J, Hernandez-Doria JD, Bennett C, Guenter W, House JD, Rodriguez-Lecompte JC. Toxinotyping of necrotic enteritis-producing and commensal isolates of Clostridium perfringens from chickens fed organic diets. Avian Pathol. 2010;39:475–481. doi: 10.1080/03079457.2010.521141. [DOI] [PubMed] [Google Scholar]
- 40.Park JY, Kim S, Oh JY, Kim HR, Jang I, Lee HS, et al. Characterization of Clostridium perfringens isolates obtained from 2010 to 2012 from chickens with necrotic enteritis in Korea. Poult Sci. 2015;94:1158–1164. doi: 10.3382/ps/pev037. [DOI] [PubMed] [Google Scholar]
- 41.Engström BE, Johansson A, Aspan A, Kaldhusdal M. Genetic relatedness and netB prevalence among environmental Clostridium perfringens strains associated with a broiler flock affected by mild necrotic enteritis. Vet Microbiol. 2012;159:260–264. doi: 10.1016/j.vetmic.2012.03.024. [DOI] [PubMed] [Google Scholar]
- 42.Martin TG, Smyth JA. The ability of disease and non-disease producing strains of Clostridium perfringens from chickens to adhere to extracellular matrix molecules and Caco-2 cells. Anaerobe. 2010;16:533–539. doi: 10.1016/j.anaerobe.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 43.Barbara AJ, Trinh HT, Glock RD, Glenn Songer J. Necrotic enteritis-producing strains of Clostridium perfringens displace non-necrotic enteritis strains from the gut of chicks. Vet Microbiol. 2008;126:377–382. doi: 10.1016/j.vetmic.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 44.Ronco T, Stegger M, Ng KL, Lilje B, Lyhs U, Andersen PS, et al. Genome analysis of Clostridium perfringens isolates from healthy and necrotic enteritis infected chickens and turkeys. BMC Res Notes. 2017;10:270. doi: 10.1186/s13104-017-2594-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yother J. Capsules of Streptococcus pneumoniae and other bacteria: paradigms for polysaccharide biosynthesis and regulation. Annu Rev Microbiol. 2011;65:563–581. doi: 10.1146/annurev.micro.62.081307.162944. [DOI] [PubMed] [Google Scholar]
- 46.Bentley SD, Aanensen DM, Mavroidi A, Saunders D, Rabbinowitsch E, Collins M, et al. Genetic analysis of the capsular biosynthetic locus from all 90 pneumococcal serotypes. PLoS Genet. 2006;2:e31. doi: 10.1371/journal.pgen.0020031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seo N, Tamai K, Nishida S. Criteria for identification of Clostridium perfringens. 5. Capsule formation and urease production. Igaku To Seibutsugaku. 1969;79:215–218. [PubMed] [Google Scholar]
- 48.Baine H, Cherniak R. Capsular polysaccharides of Clostridium perfringens Hobbs 5. Biochemistry (Mosc) 1971;10:2949–2952. [PubMed] [Google Scholar]
- 49.Cherniak R, Frederick HM. Capsular polysaccharide of Clostridium perfringens Hobbs 9. Infect Immun. 1977;15:765–771. doi: 10.1128/iai.15.3.765-771.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sheng S, Cherniak R. Structure of the capsular polysaccharide of Clostridium perfringens Hobbs 10 determined by NMR spectroscopy. Carbohydr Res. 1997;305:65–72. doi: 10.1016/S0008-6215(97)00280-2. [DOI] [PubMed] [Google Scholar]
- 51.Stringer MF, Turnbull PC, Hughes JA, Hobbs BC. An international serotyping system for Clostridium perfringens (welchii) type a in the near future. Dev Biol Stand. 1976;32:85–89. [PubMed] [Google Scholar]
- 52.Vinogradov E, Aubry A, Logan SM. Structural characterization of wall and lipidated polysaccharides from Clostridium perfringens ATCC 13124. Carbohydr Res. 2017;448:88–94. doi: 10.1016/j.carres.2017.06.003. [DOI] [PubMed] [Google Scholar]
- 53.Henderson DW. The somatic antigens of the Cl. welchii group of organisms. J Hyg. 1940;40:501–512. doi: 10.1017/S002217240002800X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chaguza C, Andam CP, Harris SR, Cornick JE, Yang M, Bricio-Moreno L, et al. Recombination in Streptococcus pneumoniae lineages increase with carriage duration and size of the polysaccharide capsule. MBio. 2016;7:e01053–16. [DOI] [PMC free article] [PubMed]
- 55.O’Riordan K, Lee JC. Staphylococcus aureus capsular polysaccharides. Clin Microbiol Rev. 2004;17:218–234. doi: 10.1128/CMR.17.1.218-234.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caimano MJ, Hardy GG, Yother J. Capsule genetics in Streptococcus pneumoniae and a possible role for transposition in the generation of the type 3 locus. Microb Drug Resist. 1998;4:11–23. doi: 10.1089/mdr.1998.4.11. [DOI] [PubMed] [Google Scholar]
- 57.Smith HE, Damman M, van der Velde J, Wagenaar F, Wisselink HJ, Stockhofe-Zurwieden N, et al. Identification and characterization of the cps locus of Streptococcus suis serotype 2: the capsule protects against phagocytosis and is an important virulence factor. Infect Immun. 1999;67:1750–1756. doi: 10.1128/iai.67.4.1750-1756.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Prescott JF, Parreira VR, Mehdizadeh Gohari I, Lepp D, Gong J. The pathogenesis of necrotic enteritis in chickens: what we know and what we need to know: a review. Avian Pathol. 2016;45:288–294. doi: 10.1080/03079457.2016.1139688. [DOI] [PubMed] [Google Scholar]
- 59.Timbermont L, Lanckriet A, Pasmans F, Haesebrouck F, Ducatelle R, Van Immerseel F. Intra-species growth-inhibition by Clostridium perfringens is a possible virulence trait in necrotic enteritis in broilers. Vet Microbiol. 2009;137:388–391. doi: 10.1016/j.vetmic.2009.01.017. [DOI] [PubMed] [Google Scholar]
- 60.Simmons M, Donovan DM, Siragusa GR, Seal BS. Recombinant expression of two bacteriophage proteins that lyse Clostridium perfringens and share identical sequences in the C-terminal cell wall binding domain of the molecules but are dissimilar in their N-terminal active domains. J Agric Food Chem. 2010;58:10330–10337. doi: 10.1021/jf101387v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boyd EF, Brüssow H. Common themes among bacteriophage-encoded virulence factors and diversity among the bacteriophages involved. Trends Microbiol. 2002;10:521–529. doi: 10.1016/S0966-842X(02)02459-9. [DOI] [PubMed] [Google Scholar]
- 62.Baba T, Takeuchi F, Kuroda M, Yuzawa H, Aoki K, Oguchi A, et al. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet. 2002;359:1819–1827. doi: 10.1016/S0140-6736(02)08713-5. [DOI] [PubMed] [Google Scholar]
- 63.Smoot LM, McCormick JK, Smoot JC, Hoe NP, Strickland I, Cole RL, et al. Characterization of two novel pyrogenic toxin superantigens made by an acute rheumatic fever clone of Streptococcus pyogenes associated with multiple disease outbreaks. Infect Immun. 2002;70:7095–7104. doi: 10.1128/IAI.70.12.7095-7104.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim K-P, Born Y, Lurz R, Eichenseher F, Zimmer M, Loessner MJ, et al. Inducible Clostridium perfringens bacteriophages ΦS9 and ΦS63. Bacteriophage. 2012;2:89–97. doi: 10.4161/bact.21363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zimmer M, Scherer S, Loessner MJ. Genomic analysis of Clostridium perfringens bacteriophage φ3626, which integrates into guaA and possibly affects sporulation. J Bacteriol. 2002;184:4359–4368. doi: 10.1128/JB.184.16.4359-4368.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shimizu T, Ohtani K, Hirakawa H, Ohshima K, Yamashita A, Shiba T, et al. Complete genome sequence of Clostridium perfringens, an anaerobic flesh-eater. Proc Natl Acad Sci. 2002;99:996–1001. doi: 10.1073/pnas.022493799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lucchini S, Desiere F, Brüssow H. Similarly organized lysogeny modules in temperate Siphoviridae from low GC content gram-positive bacteria. Virology. 1999;263:427–435. doi: 10.1006/viro.1999.9959. [DOI] [PubMed] [Google Scholar]
- 68.Gervasi T, Curto RL, Narbad A, Mayer MJ. Complete genome sequence of ΦCP51, a temperate bacteriophage of Clostridium perfringens. Arch Virol. 2013;158:2015–2017. doi: 10.1007/s00705-013-1647-1. [DOI] [PubMed] [Google Scholar]
- 69.Morales CA, Oakley BB, Garrish JK, Siragusa GR, Ard MB, Seal BS. Complete genome sequence of the podoviral bacteriophage ΦCP24R, which is virulent for Clostridium perfringens. Arch Virol. 2012;157:769–772. doi: 10.1007/s00705-011-1218-2. [DOI] [PubMed] [Google Scholar]
- 70.Ronco T, Stegger M, Ng KL, Lilje B, Lyhs U, Andersen PS, Pedersen K. Genome analysis of Clostridium perfringens isolates from healthy and necrotic enteritis infected chickens and turkeys. BMC Res Notes. 2017;10:270. doi: 10.1186/s13104-017-2594-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Karasov WH, Douglas AE. Comparative digestive physiology. Compr Physiol. 2013;3:741–783. doi: 10.1002/cphy.c110054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aronesty E. Comparison of sequencing utlitity programs. Open Bioinform J. 2013;7:1–8. doi: 10.2174/1875036201307010001. [DOI] [Google Scholar]
- 73.Peng Y, Leung HCM, Yiu SM, Chin FYL. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinforma. 2012;28:1420–1428. doi: 10.1093/bioinformatics/bts174. [DOI] [PubMed] [Google Scholar]
- 74.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Canu PAM. Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Treangen TJ, Ondov BD, Koren S, Phillippy AM. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014;15:524. doi: 10.1186/s13059-014-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinforma. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 78.Sayers E. Entrez Programming Utilities Help. 2010. [Google Scholar]
- 79.Chaudhari NM, Gupta VK, Dutta C. BPGA- an ultra-fast pan-genome analysis pipeline. Sci Rep. 2016;6:24373. doi: 10.1038/srep24373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. PHAST: a fast phage search tool. Nucleic Acids Res. 2011;39:W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Roach DJ, Burton JN, Lee C, Stackhouse B, Butler-Wu SM, Cookson BT, et al. A year of infection in the intensive care unit: prospective whole genome sequencing of bacterial clinical isolates reveals cryptic transmissions and novel microbiota. PLoS Genet. 2015;11:e1005413. doi: 10.1371/journal.pgen.1005413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sheedy SA, Ingham AB, Rood JI, Moore RJ. Highly conserved alpha-toxin sequences of avian isolates of Clostridium perfringens. J Clin Microbiol. 2004;42:1345–1347. doi: 10.1128/JCM.42.3.1345-1347.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mehdizadeh Gohari I, Kropinski AM, Weese SJ, Parreira VR, Whitehead AE, Boerlin P, et al. Plasmid characterization and chromosome analysis of two netF+ Clostridium perfringens isolates associated with foal and canine necrotizing enteritis. PLoS One. 2016;11:e0148344. doi: 10.1371/journal.pone.0148344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stanley D, Keyburn AL, Denman SE, Moore RJ. Changes in the caecal microflora of chickens following Clostridium perfringens challenge to induce necrotic enteritis. Vet Microbiol. 2012;159:155–162. doi: 10.1016/j.vetmic.2012.03.032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The whole genome sequence data generated as part of this study is available from NCBI. The accession numbers are listed in Table 1.