

Abstract
Purpose of review:
Leukodystrophies are genetic disorders primarily and predominantly affecting CNS white matter. They are associated with connotations such as “much unknown,” “progressive myelin loss,” and “nothing can be done.” Recent technological progress is reversing this picture.
Recent findings:
Next-generation sequencing has created the revolution of whole-exome/genome sequencing, allowing disease definition and gene identification for numerous ultra-rare disorders by focusing on very small groups and individual patients. Knowledge of many new “white matter proteins” is transforming our understanding of white matter physiology and pathophysiology. Regarding therapy, especially stem cell and gene therapy are evolving rapidly, aiming at personalized therapy for a specific patient with a specific disease. Multimodal approaches targeting multiple aspects of the disease hold the highest promise.
Summary:
Technological developments are revolutionizing the leukodystrophy field. Unknown becomes known and untreatable becomes treatable. New insight is that not all leukodystrophies are irreversible and that some improve spontaneously.
Leukodystrophies can be defined as genetic disorders primarily and predominantly affecting the white matter of the CNS. There are many different leukodystrophies, most of which are rare or exceedingly rare, hampering diagnosis and therapy development.
Patients with the same leukodystrophy share distinct patterns of abnormalities on MRI, different from the patterns seen in other leukodystrophies. This is the basis of MRI pattern recognition, which greatly facilitates the diagnostic process and has allowed definition of “novel” disorders among the numerous patients with an unclassified leukodystrophy. The genes mutated in these newly defined disorders were initially mainly found through genetic linkage studies. These strategies require numbers, both at disease definition and gene hunting stages. They worked well for more common leukodystrophies, but not for very rare disorders. The required new technology came with next-generation sequencing, which allowed whole-exome sequencing and whole-genome sequencing in small groups of patients, single families, or even single patients. Using this technology, a rapidly increasing number of new leukodystrophies and their underlying genetic defects has been and is being defined.
Definition of new diseases and identification of the related genes have provided important information to patients and families particularly regarding family planning, but advances in treatment have been elusive. Very few leukodystrophies are treatable in the earliest disease stages by IV “hematopoietic stem cell (HSC) transplantation” (HSCT), with considerable morbidity and mortality and uncertain outcome. Gene therapy strategies were a failure in the 1990s and neural stem cell transplantation was hampered by issues of immune rejection and ethical debates about the source of the stem cells. New technologies were also needed to address treatment.
New genes, pathomechanisms, and leukodystrophy concepts
The traditional view on leukodystrophies is that they are diseases in which not enough myelin is formed (hypomyelinating disorders) or in which myelin is lost (demyelinating disorders) on the basis of a genetic defect.1 Myelin vacuolization never entered the official leukodystrophy definition, although it was already clear in 1996 that the pathologic basis of megalencephalic leukoencephalopathy with subcortical cysts (MLC), well accepted as a leukodystrophy,2 is myelin vacuolization and not myelin loss.3
Only recently has molecular insight shed light on mechanisms underlying intramyelinic edema. MLC is caused by mutations in 2 genes: MLC1 and GLIALCAM.3 Defects in both genes lead to loss of MLC1 function, which concerns volume regulation by astrocytes.3 Whole-exome sequencing revealed that defects in chloride channel 2 (ClC-2), encoded by CLCN2, and connexin32, encoded by GJB1, may also lead to brain myelin vacuolization.4 Important additional information is that mutant mice with loss of ClC-2 function are blind due to a severe retinopathy and have myelin vacuolization throughout the CNS, except the optic nerve, where no action potentials occur because of the blindness. Together, these pieces of information indicate that the process disrupted in MLC and ClC2-related disease concerns compensation of intramyelinic water generated by action potentials (cartoon in reference 4) and that MLC1, ClC2, and connexins are important proteins in this process.
The MRI features of myelin vacuolization are not uniform. Diffusion parameters are determined by the size of vacuoles and extracellular spaces. In most cases of acute myelin vacuolization, intramyelinic vacuoles are small and extracellular spaces are compressed by swollen myelin sheaths (myelin microvacuolization), resulting in restricted diffusion.4 By contrast, intramyelinic vacuoles and extracellular spaces are large in MLC (myelin macrovacuolization), resulting in increased diffusion (figure 1).3
Figure 1. Myelin vacuolization: MRI features and reversibility.

In megalencephalic leukoencephalopathy with subcortical cysts (A–C), T2-weighted images (A) show diffuse cerebral white matter abnormalities, which have increased diffusion leading to low signal on diffusion-weighted images (B) and high signal on apparent diffusion coefficient maps (C), indicating large water spaces. In the early stages of leukoencephalopathy with thalamus and brainstem abnormalities and lactate elevation (D–F), MRI shows extensive cerebral white matter abnormalities and involvement of the thalamus (D). Diffusion is markedly decreased in the abnormal areas with high signal on diffusion-weighted images (E) and low signal on apparent diffusion coefficient maps (F), indicating compressed water spaces. Two years later (G–I), most signal abnormalities have disappeared (G) and the diffusion has normalized (H and I).
The word “leukodystrophy” is associated with the connotation of progression.1 Myelin vacuolization may, however, be reversible. First, defects in GJB1 cause peripheral neuropathy, but may be complicated by transient episodes of encephalopathy with acute intramyelinic edema.5 Second, MLC caused by dominant GLIALCAM mutations starts as classic MLC, but from the second year, improvement occurs and the leukoencephalopathy disappears.3 Third, the classic form of LTBL (“leukoencephalopathy with thalamus and brainstem abnormalities and lactate elevation”) is characterized by neurologic deterioration in infancy with, on MRI, a severe leukoencephalopathy and diffusion restriction, suggesting myelin microvacuolization.6 This dramatic onset is followed by clinical and MRI improvement, with disappearance of most abnormalities (figure 1).6 Reversibility in the context of leukodystrophies seems like a new concept, but in fact it is not. It is known that neonatal maple syrup urine disease is characterized by acute myelin microvacuolization of areas that contain myelin in a neonate and that the abnormalities are reversible with adequate treatment.7
Hidden mutations, a new phenotype, and novel pathomechanism
PLP1, encoded by PLP1, is the major myelin protein. Together with its smaller isoform DM20, they constitute more than half of the CNS myelin protein mass. Expression of both isoforms is tightly regulated in space and time. PLP1 alterations, mostly duplications, cause Pelizaeus-Merzbacher disease (PMD). Milder mutations are associated with X-linked spastic paraplegia.
Recently, a new hypomyelinating disorder was defined on the basis of an unusual pattern of myelin deficit that is more prominent in early myelinating structures than in later myelinating ones, in contrast to other hypomyelinating entities (figure 2). The disease, called “hypomyelination of early myelinating structures” (HEMS) has an X-linked inheritance.8 It defied all efforts to find the mutated gene for quite some time, even using whole-exome sequencing. Eventually, well-hidden noncoding mutations deep in intron 3 of PLP1 were discovered in several patients.9 In the other patients, synonymous exonic mutations, missense mutations, a small deletion, and a mutation near a splice-donor site were found. All variants are located either in intron 3 or in exon 3B, the latter spliced out in isoform DM20. Besides the deletion, which leads to truncation of PLP1 but not of DM20, all other mutations affect correct splicing of PLP1, including the deep intronic changes, which are clustered within 2 long-distance interacting sites. Computational tools showed that HEMS mutations cause a disturbed secondary messenger RNA structure. In vitro, PLP1 expression was demonstrated to be much reduced compared to DM20.10 DM20 is preferably expressed in the developing CNS, but PLP1 during and after the period of myelination. A disturbed PLP1/DM20 ratio is therefore likely to influence the normal sequence of myelination.
Figure 2. Abnormal sequence of myelination in HEMS.

Silver staining of a sagittal neonatal brain slice demonstrates presence of myelin in the brainstem and also already in the subcortical white matter of the precentral gyrus (A). Silver staining of an axial slice of a 4-month-old infant shows myelin in the external optic radiation (red arrow, D). Axial T2-weighted images of 2 patients with HEMS (B and E) demonstrate lack of normal myelin signal in the brainstem (B) and the optic radiation (red arrow, E). The corresponding images from a child with typical Pelizaeus-Merzbacher disease (C and F) show a much better myelinated brainstem (C) and optic radiation (red arrow, F). HEMS = hypomyelination of early myelinating structures. Panels A and D are used with permission from Thieme Publishers, Georg Thieme Verlag KG. Flechsig P. Anatomie des menschlichen Gehirns und Rückenmarks auf myelogenetischer Grundlage. Leipzig: Thieme; 1920.
These findings illustrate several points. First, MRI pattern recognition remains a powerful tool, even presently when many diagnoses are made using nonselective genetic approaches such as gene panel analysis or whole-exome sequencing. For HEMS, definition of the MRI phenotype allowed identification of a homogeneous group of patients sufficiently large to detect the unusual mutations. Second, of note, even deep intronic mutations may be disease-causing. Third, MRI findings may be useful in concept formation regarding disease mechanisms. On the basis of the fact that MRI demonstrated preferential hypomyelination of structures that normally myelinate early, the name HEMS was coined, already suggesting a defect affecting the normal temporospatial regulation of myelination.8 With HEMS, a new phenotype with a new pathomechanism has been added to the PMD spectrum.
Stem cell therapy
Since the 1980s, the potential of stem cells in the context of leukodystrophy therapy has been considered with hope and expectation. The first stem cells used were allogenic HSCs harvested from bone marrow. Mortality of IV allogenic HSCT was high and treatment success unclear. At present, the success of HSCT in patients with early stages of the childhood cerebral form of X-linked adrenoleukodystrophy (X-ALD) is undisputed, although therapy-related morbidity and mortality remain considerable and HSCT does not prevent myelopathy.11 The efficacy of HSCT in adults with the cerebral form of X-ALD is less clear, and treatment-related complications are more frequent.12 For metachromatic leukodystrophy (MLD), allogenic HSCT may halt the disease in patients with clinically presymptomatic juvenile or adult disease, even with already considerable white matter abnormalities on MRI, but HSCT is unsuccessful in patients with late-infantile MLD, also in the presymptomatic stage.13 Krabbe disease mostly presents as an infantile variant with rapid deterioration, and HSCT is not effective in stopping this downhill course.14 With newborn screening, HSCT can be applied in presymptomatic infants and it mitigates the fast natural disease course, but medium-term outcome is disappointing, as all children develop important neurologic impairment.15 Important recent news is that in contrast to the well-established idea that IV HSCT can at best stabilize a disease, improvement of white matter abnormalities on MRI has been observed after HSCT in several patients with X-ALD16 and MLD,17 suggesting the potential of white matter recovery.
HSCT is applicable in only a few leukodystrophies, leaving all other leukodystrophies unaddressed. Neural stem cell therapy is a promising new treatment option. Although leukodystrophies have many different causes, they share lack of proper function of astrocytes and oligodendrocytes (macroglia or in short glia),2 which makes glia replacement therapy an excellent option. In 2004, the Goldman lab showed that administration of intracerebral human glial precursor cells could rescue myelin-deficient shiverer mice.18 Subsequent studies confirmed the regenerative capacity of human glia after transplantation in rodent models of hypomyelination. Based on these outcomes, the first clinical study involving 4 patients with PMD was initiated.19 This phase I trial aimed at testing the safety of intracerebral injection of clinical-grade human CNS stem cells and at detecting (re)myelination. One year after transplantation, no adverse effects had occurred and some signs of myelin formation were seen. This study strengthens the belief that neural stem cell–based therapies will be part of future treatment strategies for leukodystrophies.
The discovery that somatic cells can be reprogrammed into embryonic stem cell–like cells, so-called induced pluripotent stem cells (iPSC),20 opened possibilities for personalized medicine, including cell replacement. iPSC technology offers the possibility of generating large numbers of autologous cells and eliminates chances of immune rejection as well as ethical concerns around the use of embryonic stem cells. Further improved iPSC generation and differentiation procedures are needed, which are efficient and nonintegrative, and can be quality-controlled at all stages. With the speed current stem cell technologies are evolving, the first clinical studies using iPSC derivatives are not far away.
It is still challenging to determine what cell type(s) needs to be replaced in which leukodystrophy and in which disease stage. In different leukodystrophies, distinct cell populations are primarily or secondarily involved. For only a few disorders, the primarily affected cell type is known. Examples are PMD, in which the myelin protein PLP1 expressed by oligodendrocytes is mutated, and Alexander disease, which is caused by mutations in GFAP predominantly expressed in astrocytes. However, for most leukodystrophies, sufficient insight into the underlying disease mechanism is lacking and further studies are necessary to determine which cell populations are primarily affected. It is expected that for many leukodystrophies, neural stem cell replacement should target more than myelinating oligodendrocytes alone.
Next to residing cell populations, the white matter microenvironment, including the extracellular matrix, determines the success of cell replacement. A recent cell therapy study for mice with Krabbe disease failed, because accumulation of galactosyl-ceramide and its toxic derivative psychosine killed the maturing oligodendrocytes after transplantation.21 Our studies in patients with vanishing white matter disease showed that hyaluronan is increased in the brain extracellular matrix,22 which impedes maturation of oligodendrocyte progenitors and may also affect transplanted oligodendrocyte progenitors.
Gene therapy
Current gene therapy strategies either involve HSCs in combination with lentiviral vectors to target microglia precursors or intracerebral delivery of adeno-associated virus (AAV) vectors.
Lentiviral-based gene therapies of HSCs have shown successful outcomes in X-ALD and MLD.23,24 This strategy overcomes the problem of finding a matched donor and avoids the morbidity associated with allogenic HSCT. In a trial of 2 boys with X-ALD, IV transplantation of lentivirally transduced autologous HSCs stopped progressive demyelination.23 In 3 presymptomatic patients with MLD, IV administration of autologous HSCs after lentiviral-based gene therapy halted disease progression.24 Phase II/III clinical trials are currently ongoing for X-ALD and are in preparation for MLD. The development of more efficient lentiviral vectors may further improve clinical outcomes.
Another strategy relies on direct intracerebral administration of AAV vectors, which has the potential of more rapid and higher expression in the brain than transplantation of lentivirally transduced HSCs. Results of a phase I/II clinical trial testing intraparenchymal gene delivery with a recombinant AAV serotype 2 (AAV2) vector in Canavan disease look promising.25 Treated children showed decreased brain N-acetylaspartic acid concentrations and MRI suggested more normal myelin.25 AAV transduction methods, however, need improvement. Intraparenchymal infusion is not suitable for widespread CNS transduction because of localized delivery and limited diffusion capacity of AAV vectors. The recent discovery of new AAV serotypes that can cross the blood–brain barrier was therefore welcomed with great enthusiasm, as IV injection allows broad dispersion and is less invasive than intracerebral delivery methods.26 Mice with Canavan disease could be cured with a single IV recombinant AAV injection.27 None of the current AAV vectors efficiently and selectively targets oligodendrocytes. Newer serotypes that cross the blood–brain barrier and are more efficient and more specific for different neural cell types are needed. In addition, the half-life of the transduced cells and the time point of treatment in disease progression need further study.
The recently developed technology CRISPR (clustered regularly interspaced short palindromic repeat)/Cas9 (CRISPR-associated nuclease 9) system provides a simple and efficient method to precisely manipulate the genome. Because of the ease and speed of designing CRISPR-guided nucleases, the CRISPR/Cas9 system has quickly evolved to a widely used DNA editing tool. It repairs disease-causing alleles by changing the DNA sequence at the exact location on the chromosome. In 2014, the first use of CRISPR/Cas9 to fix a mutation in a living animal was shown.28 Although the efficiency in this study was very low, it showed the potential of this technology for human gene therapy in the future.
Multimodal therapy
Multimodal therapy approaches have the highest potential not only of halting but also repairing the complex and multifactorial pathology of leukodystrophies (figure 3). Modalities can be used as add-on to enhance the efficacy of another modality. Gene editing of HSCs allows transplantation of genetically corrected autologous cells.23,24 This strategy not only overcomes problems such as finding a matched donor and complications of allogenic HSCT, it also allows further manipulations of stem cells. Transplanted autologous HSCs after lentiviral-based gene therapy, designed to overexpress the normal enzyme, increased enzyme activity in the CNS and halted disease progression in MLD.24 Combinations of individual modalities are probably even more efficacious. Gene therapy targeting neural cells is a fast-working strategy to combat the primary defect, while cell replacement and modulations of the microenvironment have high promise to accelerate repair of already acquired damage. This point is illustrated by a mouse model for Krabbe disease that was treated by either neural stem cells or AAV2/5-mediated gene therapy, followed by HSCT.29 Each approach gave clinical improvements, but a combination of procedures dramatically increased treatment efficacy. Also, HSCT plus substrate reduction and plus gene therapy, or HSCT plus enzyme replacement, were superior to single therapies in Krabbe mice.30
Figure 3. Multimodal therapy of leukodystrophies.
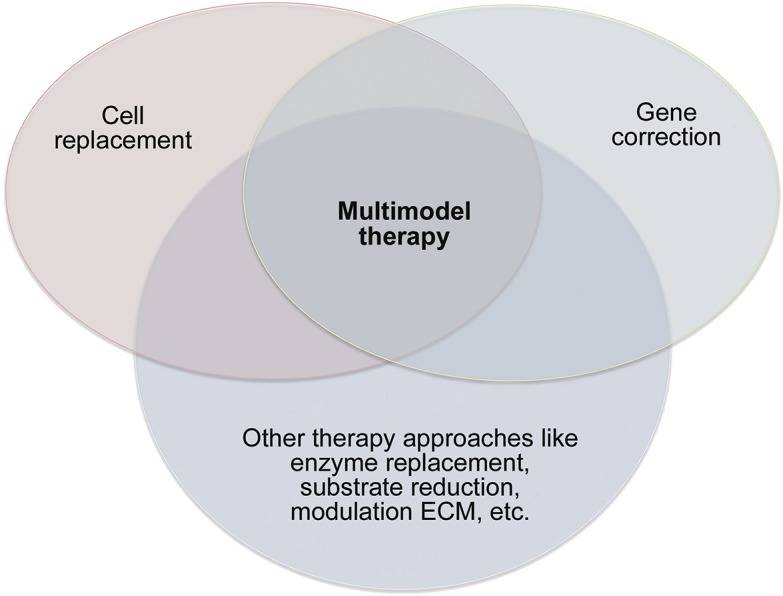
ECM = extracellular matrix.
As treatment options become better and presymptomatic or early symptomatic patients profit most from any treatment, early diagnosis is an urgent issue. Testing of clinically unaffected siblings, even when older than the index patient, and newborn screening are important in this context.
Leukodystrophies: Five new things
New genetic techniques have allowed the definition and elucidation of numerous new rare and ultra-rare leukodystrophies, providing unprecedented insight into brain white matter physiology and pathophysiology, also showing that not all leukodystrophies are irreversible and that some improve spontaneously.
MRI and genetic findings came together in HEMS, a novel disease phenotype within the PMD spectrum with disturbed developmental regulation of myelination.
The stem cell transplantation field benefits greatly from technological developments, allowing the use of different patient-derived cell types, improving the prospects for cell replacement therapy for leukodystrophies.
Gene therapy is a rapidly advancing method for personalized medicine involving correction of autologous cell transplants and intracerebral gene correction.
Multimodal therapy approaches are applied with increasing success and are superior to unimodal approaches in halting the disease process and repairing the multifactorial complex pathology of leukodystrophies.
AUTHOR CONTRIBUTIONS
All authors contributed to conceptualizing, drafting, and editing the manuscript.
STUDY FUNDING
No targeted funding reported.
DISCLOSURES
M.S. van der Knaap serves on the editorial board of Neuropediatrics, and receives research support from ZonMw, E-Rare, NutsOhra Foundation, Optimix Foundation, and Hersenstichting Nederland. N.I. Wolf serves on scientific advisory boards for the European Leukodystrophy Association (ELA) and Mission Massimo Foundation, has received funding for travel from ELA, serves as editor for Neuropediatrics, communicating editor for Journal of Inherited Metabolic Disease, and on the editorial board of Neurology®, receives research support from Hersenstichting Nederland, Metakids, Stichting Stofwisselkracht and M.O. Knip Stichting, and holds stock/options in Aer Beatha. V.M. Heine receives research support from ZonMw, ELA and E-Rare. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.

REFERENCES
- 1.Morell P, Wiesmann U. A correlative synopsis of the leukodystrophies. Neuropediatrics 1984;15(suppl):62–65. [DOI] [PubMed] [Google Scholar]
- 2.Vanderver A, Prust M, Tonduti D, et al. Case definition and classification of leukodystrophies and leukoencephalopathies. Mol Genet Metab 2015;114:494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Knaap MS, Boor I, Estévez R. Megalencephalic leukoencephalopathy with subcortical cysts: chronic white matter oedema due to a defect in brain ion and water homoeostasis. Lancet Neurol 2012;11:973–985. [DOI] [PubMed] [Google Scholar]
- 4.Depienne C, Bugiani M, Dupuits C, et al. Brain white matter oedema due to ClC-2 chloride channel deficiency: an observational analytical study. Lancet Neurol 2013;12:659–668. [DOI] [PubMed] [Google Scholar]
- 5.Paulson HL, Garbern JY, Hoban TF, et al. Transient central nervous system white matter abnormality in X-linked Charcot-Marie-Tooth disease. Ann Neurol 2002;52:429–434. [DOI] [PubMed] [Google Scholar]
- 6.Steenweg ME, Ghezzi D, Haack T, et al. Leukoencephalopathy with thalamus and brainstem involvement and high lactate “LTBL” caused by EARS2 mutations. Brain 2012;135:1387–1394. [DOI] [PubMed] [Google Scholar]
- 7.Jan W, Zimmerman RA, Wang ZJ, Berry GT, Kaplan PB, Kaye EM. MR diffusion imaging and MR spectroscopy of maple syrup urine disease during acute metabolic decompensation. Neuroradiology 2003;45:393–399. [DOI] [PubMed] [Google Scholar]
- 8.Steenweg ME, Wolf NI, Schieving JH, et al. Novel hypomyelinating leukoencephalopathy affecting early myelinating structures. Arch Neurol 2012;69:125–128. [DOI] [PubMed] [Google Scholar]
- 9.Kevelam SH, Taube JR, van Spaendonk RM, et al. Altered PLP1 splicing causes hypomyelination of early myelinating structures. Ann Clin Transl Neurol 2015;2:648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taube J, Sperle K, Banser L, et al. PMD patient mutations reveal a long-distance intronic interaction that regulates PLP1/DM20 alternative splicing. Hum Mol Genet 2014;23:5464–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Geel BMM, Poll-The BT, Verrips A, Boelens JJ, Kemp S, Engelen M. Hematopoietic cell transplantation does not prevent myelopathy in X-linked adrenoleukodystrophy: a retrospective study. J Inherit Metab Dis 2015;38:359–361. [DOI] [PubMed] [Google Scholar]
- 12.Köhler W, Kühl JS. Hematopoietic stem cell transplantation for adult cerebral X-linked adrenoleukodystrophy. Neurology 2014;82:SP5.174. [Google Scholar]
- 13.Boucher AA, Miller W, Shanley R, et al. Long-term outcomes after allogeneic hematopoietic stem cell transplantation for metachromatic leukodystrophy: the largest single-institution cohort report. Orphanet J Rare Dis 2015;10:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duffner P, Caggana M, Orsini J, et al. Newborn screening for Krabbe disease: the New York state model. Pediatr Neurol 2009;40:245–252. discussion 253–255. [DOI] [PubMed] [Google Scholar]
- 15.Escolar M, Poe M, Provenzale J, et al. Transplantation of umbilical-cord blood in babies with infantile Krabbe's disease. N Engl J Med 2005;352:2069–2081. [DOI] [PubMed] [Google Scholar]
- 16.Shapiro E, Krivit W, Lockman L, et al. Long-term effect of bone-marrow transplantation for childhood-onset cerebral X-linked adrenoleukodystrophy. Lancet 2000;356:713–718. [DOI] [PubMed] [Google Scholar]
- 17.van Egmond ME, Pouwels PJ, Boelens JJ, et al. Improvement of white matter changes on neuroimaging modalities after stem cell transplant in metachromatic leukodystrophy. JAMA Neurol 2013;70:779–782. [DOI] [PubMed] [Google Scholar]
- 18.Windrem MS, Nunes MC, Rashbaum WK, et al. Fetal and adult human oligodendrocyte progenitor cell isolates myelinate the congenitally dysmyelinated brain. Nat Med 2004;10:93–97. [DOI] [PubMed] [Google Scholar]
- 19.Gupta N, Henry RG, Strober J, et al. Neural stem cell engraftment and myelination in the human brain. Sci Transl Med 2012;4:155ra137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007;131:861–872. [DOI] [PubMed] [Google Scholar]
- 21.Kuai XL, Ni RZ, Zhou GX, et al. Transplantation of mouse embryonic stem cell derived oligodendrocytes in the murine model of globoid cell leukodystrophy. Stem Cell Res Ther 2015;6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bugiani M, Postma N, Polder E, et al. Hyaluronan accumulation and arrested oligodendrocyte progenitor maturation in vanishing white matter disease. Brain 2013;136:209–222. [DOI] [PubMed] [Google Scholar]
- 23.Cartier N, Hacein-Bey-Abina S, Bartholomae CC, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009;326:818–823. [DOI] [PubMed] [Google Scholar]
- 24.Biffi A, Montini E, Lorioli L, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science 2013;341:1233158. [DOI] [PubMed] [Google Scholar]
- 25.Leone P, Shera D, McPhee SW, et al. Long-term follow-up after gene therapy for Canavan disease. Sci Transl Med 2012;4:165ra163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang B, Li S, Wang H, et al. Global CNS transduction of adult mice by intravenously delivered rAAVrh.8 and rAAVrh.10 and nonhuman primates by rAAVrh.10. Mol Ther 2014;22:1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahmed SS, Li H, Cao C, et al. A single intravenous rAAV injection as late as P20 achieves efficacious and sustained CNS gene therapy in Canavan mice. Mol Ther 2013;21:2136–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yin H, Xue W, Chen S, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol 2014;32:551–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ricca A, Rufo N, Ungari S, et al. Combined gene/cell therapies provide long-term and pervasive rescue of multiple pathological symptoms in a murine model of globoid cell leukodystrophy. Hum Mol Genet 2015;24:3372–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hawkins-Salsbury JA, Shea L, Jiang X, et al. Mechanism-based combination treatment dramatically increases therapeutic efficacy in murine globoid cell leukodystrophy. J Neurosci 2015;35:6495–6505. [DOI] [PMC free article] [PubMed] [Google Scholar]