

Abstract
Prions are infectious aggregation-prone isoforms of the normal proteins, supposedly able to seed aggregation of the normal cellular counterparts. In vitro, prion proteins form amyloid fibers, resembling cytoskeletal structures. Yeast prion [PSI], which is a cytoplasmically inherited aggregated isoform of the translation termination factor Sup35p (eRF3), serves as a useful model for studying mechanisms of prion diseases and other amyloidoses. The previously described interaction between Sup35p and cytoskeletal assembly protein Slalp points to the possible relationships between prions and cytoskeletal networks. Although the Sup35PSI+ aggregates do not colocalize with actin patches, we have shown that yeast cells are efficiently cured of the [PSI] prion by prolonged incubation with latrunculin A, a drug disrupting the actin cytoskeleton. On the other hand, treatments with sodium azide or cycloheximide, agents blocking yeast protein synthesis and cell proliferation but not disrupting the cytoskeleton, do not cause a significant loss of [PSI]. Moreover, simultaneous treatment with sodium azide or cycloheximide blocks [PSI] curing by latrunculin A, indicating that prion loss in the presence of latrunculin A requires a continuation of protein synthesis during cytoskeleton disruption. The sodium azide treatment also decreases the toxic effect of latrunculin A. Latrunculin A influences neither the levels of total cellular Sup35p nor the levels of chaperone proteins, such as Hspl04 and Hsp70, which were previously shown to affect [PSI]. This makes an indirect effect of latrunculin A on [PSI] via induction of Hsps unlikely. Fluorescence microscopy detects changes in the structure and/or localization of the Sup35PSI+ aggregates in latrunculin A-treated cells. We conclude that the stable maintenance of the [PSI] prion aggregates in the protein-synthesizing yeast cells partly depends on an intact actin cytoskeleton, suggesting that anticytoskeletal treatments could be used to counteract some aggregation-related disorders.
Keywords: Actin, Sup35p, Release factor, [PSI], Protein aggregation, Sodium azide, Cycloheximide
PRIONS are infectious proteins that cause neurodegenerative diseases in mammals, including humans [see (35) for review], and determine certain cytoplasmically inherited traits in yeast [see (43) for review]. A prion isoform may have the same amino acid sequence as the normal isoform of the same protein, but it is folded in an alternative conformation. Moreover, prion conformation is reproducible due to the ability of the prion protein to convert normal protein into a prion shape. Several unrelated proteins of different functions demonstrate prion behavior. Examples of prions include the mammalian membrane-associated protein PrP, yeast regulatory protein in the nitrogen metabolism pathway Ure2p, and the yeast counterpart of a eukaryotic release (i.e., translation termination) factor eRF3 (Sup35p) [see (35,43) for review]. This suggests that prion phenomena are widespread in nature. Prion diseases in mammals and humans are fatal and incurable, and some of them, such as “mad cow disease,” gained significant publicity recently due to the possibility of prion transmission from animals to humans [see (35) for review].
One of the models suggests that prions are aggregated proteins reproduced by the process of “seeded” or “nucleated” polymerization, so that preexisting oligomeric prion “seeds” facilitate polymerization of the normal soluble protein [see (28,29) for review]. Both mammalian and yeast prions produce aggregates in vivo (11,18,33,34) and amyloid-like fibers in vitro (20,25,36,37,40), which supports a nucleated polymerization model. This also provides a parallel between prion diseases and other amyloidoses (such as Alzheimer’s disease, Parkinson’s disease, etc.) caused by nucleated polymerization of abnormal proteins [see (26) for review]. This makes prions a useful model for studying general mechanisms of formation and propagation of the stable protein aggregates in the cell. Factors preventing aggregate propagation could be considered as potential antiamyloidosis treatments.
Nucleated polymerization of prions and other amyloid-like aggregates resembles formation of the cy-toskeletal networks. Indeed, we have shown that the cytoskeletal assembly protein Slalp physically interacts to the prion-forming domain of the Saccharomyces cerevisiae release factor Sup35p and influences formation and propagation of [PSI], a polymerized prion form of Sup35p (4). The Slalp protein has previously been implicated in forming “nuclei,” which seed polymerization of the cortical actin microfilaments (22). This points to the possible connection between actin and prion polymerization.
Here, we investigate the effects of latrunculin A, a drug disrupting actin microfilaments, on propagation of the [PSI] prion. Latrunculin A is a structurally unique marine toxin isolated from the Red Sea sponge Latrunculia magnifica. It binds near the nucleotide binding pocket of actin monomers and forms an assembly-incompetent complex with monomeric actin (10). As a result, latrunculin A sequesters free actin monomers, preventing actin filament assembly while allowing disassembly (2,10,30). Therefore, actin polymers, normally undergoing rapid cycles of assembly and disassembly in yeast, are disassembled in the presence of latrunculin A. Disruption of both actin cables and cortical actin patches by latrunculin A was observed in yeast (3). We have found that latrunculin A treatment leads to structural alterations of prion aggregates and interferes with [PSI] maintenance. These data establish a functional connection between the actin cytoskeleton and [PSI] prion, and suggest a new approach to potential antiprion treatments.
MATERIALS AND METHODS
Yeast Strains and Growth Conditions
Isogenic yeast strains OT55 (“weak” [PSI +]), OT56 (“strong” [PSI +]), OT60 ([psi − PIN +]) and GT17 ([psi − pin −]), described previously (4,13,14), are derivatives of 74-D694 (MAT a adel-14 his3 leu2 trpl ura3). The [psi − pin −] strain GT234 (MATα adel-14 his3 leu2 lys2 trpl ura3) is a derivative of GT81-1D (8,9) obtained by guanidine-HCl treatment as described previously (14). The UGA mutation adel-14, which causes adenine auxotrophy and accumulation of a red pigment, is suppressed by [PSI] due to readthrough of the UGA codon, caused by the malfunction of the aggregated Sup35p. Therefore, [psi −] colonies are red and Ade−, while [PSI +] colonies are light pink and Ade+. Standard yeast media and cultivation conditions were used (23). Yeast cultures were grown at 30°C.
Plasmids
The episomal 2μ. DNA plasmids YEpl3 (5), which bears a LEU2 marker, and pSTR7 (41), which is an YEpl3 derivative bearing the S. cerevisiae SUP35 gene under its own promoter, were described previously. The plasmids pHGPD-sGFP, pHGPD-NMsGFP, and pmCUP-NMsGFP, kindly provided by S. Lindquist, are centromeric (single-copy) yeast vectors bearing the HIS3 and URA3 yeast markers, respectively. The plasmid pHGPD-sGFP, used as a formal control, contains the gene for green fluorescent protein (GFP) under the yeast constitutive promoter for the glycerophosphate dehydrogenase gene (GPD). This GPD-GFP construct produces the soluble GFP protein in yeast. The plasmid pHGPD-NMsGFP contains the Sup35p N-proximal region (SUP35NM) under the GPD promoter, which is fused to a GFP ORF (GPD-SUP35NM::GFP). This plasmid produces a chimeric Sup35NM-GFP protein, which forms fluorescent clumps in the [PSI +] cells but not in the [psi −] cells (33). The plasmid pmCUP-NMsGFP contains the same SUP35NM-GFP construct under the control of copper-inducible CUP1 promoter. The centromeric plasmid pLSpSUP35NM-GFP has been constructed by E. Lewitin in Y. Chernoff’s lab by inserting the smaller BamHI-SacI fragment of pmCUPNM-GFP, bearing the SUP35NM-GFP ORF, into the BamHI-SacI digested plasmid pRS315-Sp-SUP35HA, kindly provided by J. Weissman. In this way, the SUP35 ORF is substituted by the chimeric SUP35NM-GFP ORF. The resulting construct contains the SUP35NM-GFP fused gene under the control of the SUP35 own promoter. This construct also produces the [PSI]-dependent fluorescent clumps, although of significantly smaller size, if compared with the highly expressed GPD-promoted construct.
Latrunculin A Treatments
Latrunculin A was purchased from the University of California at Santa Cruz. The 10 mM stock solution in dimethyl sulfoxide (DMSO) was prepared and added to the exponential yeast cells (OD600 = 0.6) in the volume needed to reach the required final concentration. An equal volume of DMSO without latrun-culin A was added to another sample of cells used as a control. After specified periods of time, cells were washed three times in liquid YPD medium and plated onto solid YPD medium. After 2 or 3 days of incubation at 30°C and 2 subsequent days at 4°C, red ([psi −]) and white-pink ([PSI +]) colonies were counted. To confirm that red colonies have lost [PSI], the representative red and white-pink colonies were velveteen replica plated onto the synthetic medium lacking adenine and incubated for 7–10 days at 30°C. For each concentration of latrunculin A and each time point (shown in Table 1) at least four independent cultures were analyzed. Results were generally homogenous among the cultures. Total numbers are included in Table 1. In cases when sodium azide or cycloheximide were used (Table 4), these compounds were added 15 min prior to latrunculin A treatment.
TABLE 1.
[PSI] LOSS IN THE PRESENCE OF LATRUNCULIN A IN THE STRAIN OT55
| Time of Incubation (h) | Concentration of Latrunculin A | Number of Colonies | |
|---|---|---|---|
| [psi −] | Total | ||
| 1 | none | 0 | 2913 |
| 1 | 40 μM | 5 (0.75%) | 665 |
| 1 | 200 μM | 9 (1.4%) | 624 |
| 4 | none | 4 (0.1%) | 4049 |
| 4 | 40 μM | 127 (5.7%) | 2226 |
| 4 | 200 μM | 417 (12.8%) | 3253 |
| 18–22 | none | 13 (0.08%) | 15484 |
| 18–22 | 40 μM | 236 (53.9%) | 438 |
| 18–22 | 200 μM | 354 (61.1%) | 579 |
TABLE 4.
[PSI] STABILITY IN THE PRESENCE OF PROTEIN SYNTHESIS INHIBITORS IN THE STRAIN OT55
| Time of Incubation (h) | Concentrations | Number of Colonies | |||
|---|---|---|---|---|---|
| Na3N (mM) | Cyc (μg/ml) | Lat A (μM) | [psi −] | Total | |
| 0 | — | — | — | 2 (0.1%) | 2418 |
| 4 | 10 | — | — | 5 (0.8%) | 622 |
| 4 | — | 100 | — | 1 (0.8%) | 130 |
| 4 | 10 | — | 40 | 0 | 141 |
| 4 | 10 | — | 200 | 1 (0.8%) | 119 |
| 4 | — | 100 | 40 | 1 (0.7%) | 138 |
| 4 | — | 100 | 200 | 4 (0.3%) | 1367 |
| 22 | 10 | — | — | 4 (0.5%) | 789 |
| 22 | — | 100 | — | 0 | 197 |
| 22 | 10 | — | 40 | 0 | 225 |
| 22 | 10 | — | 200 | 1 (0.6%) | 163 |
| 22 | — | 100 | 40 | 1 (0.5%) | 210 |
| 22 | — | 100 | 200 | 1 (1.4%) | 73 |
Detection of [PIN] Factor
To score for the [PIN] factor, the red [psi −] colonies obtained as described above were patched on YPD medium and mated by velveteen replica plating onto the lawn of the GT234 [psi − pin −] strain transformed with the 2μ plasmid pSTR7, which contains the SUP35 gene. Mating to the same strain transformed with the control plasmid YEpl3 was used as a negative control. Diploids were selected on the synthetic medium lacking lysine and leucine, and subsequently velveteen replica plated onto the synthetic medium lacking lysine, leucine, and adenine. The [psi − pin −] diploids are unable to grow on this medium, while [psi − PIN +] diploids containing the pSTR7 plasmid exhibit intense papillation after 7–10 days of incubation due to [PIN]-dependent induction of [PSI] by the multicopy SUP35 gene, which results in suppression of the adel-14 mutation [see (14)]. The isogenic diploids obtained by mating the same GT234 transformants to the [psi − pin −] strain GT17 and [psi − PIN +] strain OT60 were used as controls.
Fluorescent Detection of Sup35NM-GFP
Yeast cells, carrying pHGPD-sGFP, pHGPD-NMsGFP, or pLSpSUP35NM-GFP, were grown to early exponential phase, spun down, resuspended in DABCO buffer (2.45% of 1,4-diazabicyclo-octane, Sigma, in l× PBS and 7.5% glycerol) or in phenylenediamine mounting solution (1 mg/ml p-phenylenediamine, Sigma, in l× PBS and 90% glycerol) and pipetted onto a poly-lysine (Sigma)-coated slide. The slide was covered with a glass coverslip (#1.5, Sigma) and sealed with nail polish. Fluorescence was examined with a Nikon Diaphot fluorescence microscope with an HB100 W/Z high-pressure mercury lamp and a Nikon 100× Plan-Neofluar oil immersion objective with phase optics. The microscope’s parameters were set at: excitation 460-490 nm, beamsplitter 505 nm, and emission 515–550 nm. Photographs were taken using a Nikon 2000 camera and Kodak TMAX 400 film.
Alternatively, samples were scanned using a Zeiss LSM510 UV confocal laser scanning microscope (Carl Zeiss Inc., Thornwood, NY) with a Zeiss 63× Plan-Neofluar oil immersion lens. The excitation wavelength for the argon laser (GFP fluorescence) was at 488 nm. Pictures were generated by the Carl Zeiss Laser Scanning System LSM510 and analyzed using the Zeiss LSM Image Browser (Carl Zeiss, Jena, Germany).
Fluorescence Staining of Cellular Actin
In order to visualize actin patches, filamentous actin was stained with rhodamine, which was conjugated to phalloidin, a drug that is known to stabilize actin polymers (31). The procedure of Adams and Pringle (1) was used in modification of Karpova et al. (24). Exponential cells at OD600 = 0.3–0.6 were fixed in synthetic medium containing 4% formaldehyde for 15 min to 1 h at 30°C, and incubated with 5% of a 3.3 μM solution of rhodamine phalloidin (Molecular Probes, Eugene, Oregon) for 1 h at 4°C in the dark. After three washes in l× PBS, fixed stained cells were resuspended in DABCO buffer or phenylenediamine mounting buffer and analyzed by fluorescence microscopy. When the confocal microscope was used, the excitation wavelength for the helium neon laser (rhodamine-phalloidin fluorescence) was 543 nm.
Molecular Biology Techniques and Reagents
Standard techniques were used for DNA isolation and analysis, yeast and bacterial transformation, protein isolation, and Western blotting. To identify the insoluble (prion) aggregates of Sup35p, protein extracts were fractionated by centrifugation as described earlier (32). Antibodies to Sup35p, Hspl04, Hsp82, and Hsp26 were kindly provided by S. Lindquist. Antibodies to Hsp70 were kindly provided by E. Craig. Amounts of protein loaded were controlled by Coomassie staining. Western blotting was performed according to Amersham protocols. The chem-iluminescent ECL detection kit from Amersham was used for visualization of the protein bands.
RESULTS
Latrunculin A Causes Loss of [PSI]
The yeast [PSI +] strains, obtained in the identical genetic backgrounds, differ from each other by both stringency of suppression and mitotic stability of [PSI] [see (13)]. These characteristics are reproduced upon [PSI] propagation and represent the specific patterns of the individual “strains” of a prion. For this study, we have chosen the “weak” [PSI +] strain OT55 (32). This strain is characterized by less efficient suppression and lower level of stability, compared with the “strong” [PSI +] strains of the same genotype. However, suppression of adel-14 remains easily detectable in OT55 by both color and growth, and the level of reproduction of [PSI +] remains sufficient (usually about 99.9% or higher) to achieve stable propagation of [PSI] in control experiments. On the other hand, the [PSI] curing treatments described previously, such as guanidine-HCl (17) or overex-pressed Hspl04 protein (7), cause much more efficient elimination of [PSI] in the strain OT55, compared with the “stronger” [PSI +] strains of the same genotypic background. This makes OT55 a very useful experimental model potentially capable of identifying even relatively weak prion-curing agents.
To detect the effects of latrunculin A on [PSI] maintenance, the exponential culture of OT55 was incubated with and without latrunculin A for various time periods. Cells were plated on YPD, and the [PSI +] and [psi −] colonies were identified, based on color and ability to grow on -Ade medium as described above (see Material and Methods). As expected, latrunculin A inhibited growth of yeast cultures, while overnight incubation with latrunculin A caused high cell death (Fig. 1A). While several minutes of incubation with latrunculin A were previously shown to be sufficient for the actin cytoskeleton disruption (3), loss of [PSI] required longer periods. Detectable [PSI] loss was observed beginning from 1 h of incubation with latrunculin A (Table 1). After 4 h with latrunculin A, 6–13% of the cells became [psi −] (depending on concentration of the drug), while after overnight incubation (18–22 h) up to 61% of viable cells became [psi −] (Table 1, Fig. 1A, see below). Quite remarkably, the vast majority of colonies obtained after latrunculin A treatment were either complete [PSI +] or complete [psi −], while the mosaic (sectored) colonies composed of [PSI +] and [psi −] cells were rare (Fig. 2A, see below). This shows that [PSI] was cured instantly by latrunculin A treatment, rather than destabilized and lost in the subsequent cell divisions.
Figure 1.
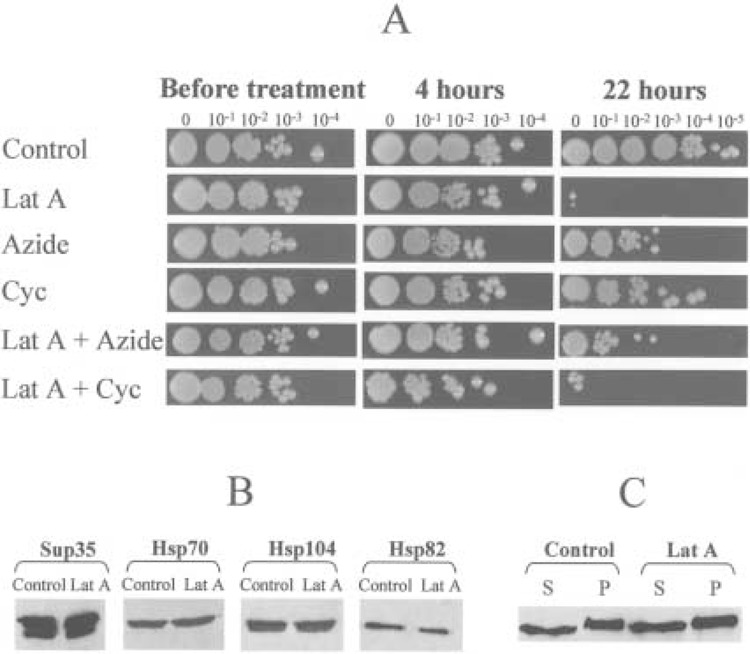
Effects of latrunculin A on yeast viability and protein levels. (A) Viability of yeast cells in the control culture and cultures treated with latrunculin A (Lat A), sodium azide (Azide), and cycloheximide (Cyc) in various combinations. Cultures were grown in YPD at 30°C as described in Materials and Methods. At each time point, serial decimal dilutions were made, and 3-μ1 aliquots of each dilution were spotted onto YPD plates. Plates have been photographed after 3 days of incubation at 30°C. Concentrations: latrunculin A, 200 μM; sodium azide, 10 mM; cycloheximide, 100 μg/ml. (B) Levels of the Sup35p and Hsps in the control (Control) and latrunculin A-treated (Lat A) cultures of the strain OT55, as determined by Western blotting. Latrunculin A treatment was for 4 h. Equal amounts of total cellular protein were loaded in each lane, as verified by Coomassie staining (not shown). (C) Distribution of Sup35p between soluble (S, supernatant) and insoluble (P, pellet) fractions is not affected by latrunculin A. Yeast extracts were prepared and fractionated as described (32). Latrunculin A treatment was for 4 h.
Figure 2.
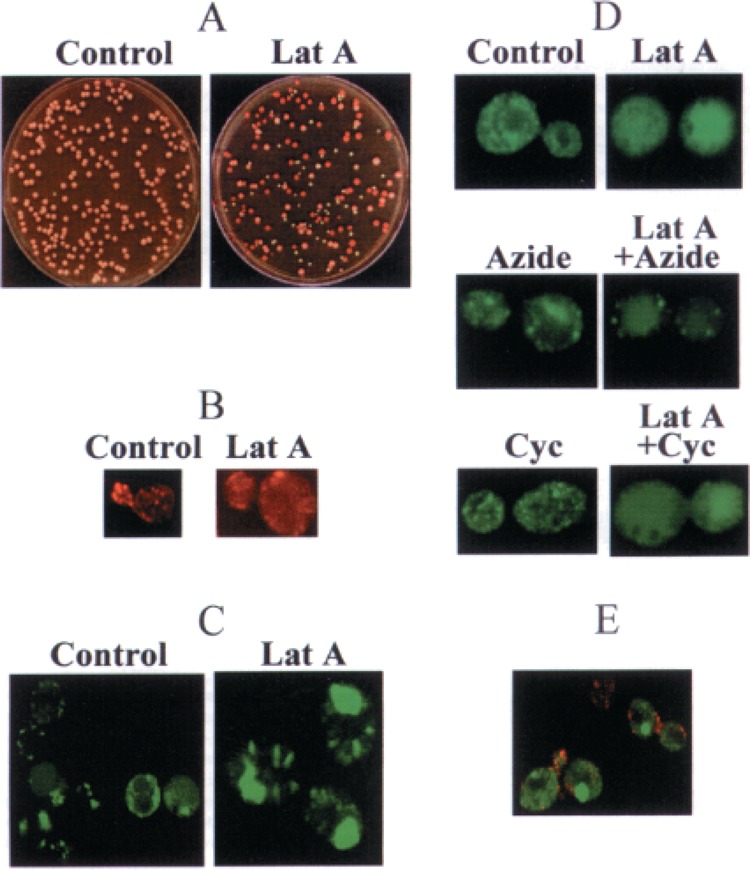
[PSI] loss and patterns of the Sup35p aggregates in the control and latrunculin A-treated cells. (A) Latrunculin A-induced loss of [PSI] as visualized by color on YPD plates. The cultures of the [PSI +] strain OT55 were plated onto YPD medium after 18 h of treatment with latrunculin A (Lat A, right) or without such a treatment (Control, left). Red colonies are [psi −], while light pink colonies are [PSI +]. (B) Latrunculin A causes dissociation of the actin patches. Actin patches were visualized by rhodamine-phalloidin staining (red). Patches are clearly seen in the control cells of the strain OT55 and preferentially concentrate in the area of new bud and bud neck (left). Most patches disappear after latrunculin A treatment; red background indicates that actin becomes distributed evenly in the cytoplasm (right). Latrunculin A treatment was for 4 h. (C) Visualization of the Sup35pPSI+ aggregates in the yeast strain transformed with the plasmid pHGPD-NMsGFP, containing the chimeric SUP35NM-GFP gene under the control of strong GPD promoter. Most of the Sup35NM-GFP aggregates (green) are relatively small and compact in the control cells of the [PSI +] strain OT55 (left). Four-hour treatment with 200 μM latrunculin A leads to diffusion of the aggregates (right). Images of the control and latrunculin A-treated cells were taken at the same scale. Difference in size is due to uncontrolled cell growth in the conditions when cell division is blocked for 4 h by latrunculin A treatment. (D) Visualization of the Sup35pPSI+ aggregates in the yeast strain transformed with the plasmid pLSpSUP35NM-GFP, containing the chimeric SUP35NM-GFP gene under the control of the SUP35 own promoter, which is moderately expressed. Due to lower levels of the Sup35-GFP production, the prion aggregates in the control cells are smaller in size, compared with (C). Sodium azide blocks the Sup35-GFP diffusion caused by latrunculin A more efficiently than cycloheximide. See also comments in the text. All treatments were for 1 h. Similar results were observed after 4-h treatments (not shown). All images were taken at the same scale. Concentrations: latrunculin A, 200 μM; sodium azide, 10 mM; cycloheximide, 100 μg/ml. (E) Actin patches (red) usually do not colocalize with the Sup35NM-GFP aggregates (green) in the cells of [PSI +] strain OT55. The actin patches were visualized by rhodamine-phalloidin staining as in (B). The fluorescent Sup35-GFP protein was produced by the plasmid pHGPD-NMsGFP, as in (C). Images used for (B)–(E) were obtained by using a confocal microscope, as described in Materials and Methods.
Presence of [PSI] Does Not Affect Sensitivity of the Yeast Cells to Latrunculin A
One could suggest that spontaneously appearing [psi −] cells are less sensitive to latrunculin A and therefore gain a selective advantage upon prolonged incubation. To check this, we have compared latrunculin A effects on viabilities of the [PSI +] strain OT55 and isogenic [psi −] strain GT17. No difference in sensitivity to latrunculin A was observed between these strains (Table 2).
TABLE 2.
EFFECT OF LATRUNCULIN A ON VIABILITY OF THE [PSI +] AND [psi −] CELLS
| Strain | Conditions | Colony Forming Units (cfu) per 1 ml at | ||
|---|---|---|---|---|
| Start | 4 h | 18 h | ||
| OT55 [PSI +] | control | 4.0 × 106 | 1.1 × 107 | 9.6 × 107 |
| latranculin A (200 μM) | 4.0 × 106 | 4.1 × 106 | 2.7 × 104 | |
| GT17 [psi −] | control | 4.0 × 106 | 7.7 × 106 | 1.2 × 108 |
| latranculin A (200 μM) | 4.0 × 106 | 5.7 × 106 | 2.9 × 104 | |
Latrunculin A Does Not Cure Yeast of the [PIN] Factor
The [PIN] factor is a yeast non-Mendelian element that greatly increases ability of yeast cells to generate [PSI] de novo upon Sup35p overproduction (14). Based on genetic evidence, it has been suggested that [PIN] is a prion that is distinct from [PSI] (14,15). Some [PSI] curing treatments (e.g., guanidine-HCl or inactivation of Hspl04) also cure yeast cells of [PIN], while other [PSI] curing treatments (e.g., excess Hspl04) do not affect [PIN] (14). We have checked whether [psi −] colonies generated by latrunculin A treatment have also lost [PIN]. Our data (Table 3) show that all these colonies retained [PIN +], indicating that in contrast to [PSI], [PIN] is not sensitive to latrunculin A treatment.
TABLE 3.
RETENTION OF THE [PIN] FACTOR IN LATRUNCULIN A-CURED [psi −] COLONIES
| Time of Incubation (h) | Concentration of Latrunculin A (μM) | Number of [psi −] Colonies | ||
|---|---|---|---|---|
| [PIN +] | [pin −] | Total | ||
| 1 | 40 | 3 | 0 | 3 |
| 1 | 200 | 4 | 0 | 4 |
| 4 | 40 | 3 | 0 | 3 |
| 4 | 200 | 12 | 0 | 12 |
| 18–22 | 40 | 40 | 0 | 40 |
| 18–22 | 200 | 11 | 0 | 11 |
| Total | 73 | 0 | 73 | |
Blocking of Cell Division by Treatments Other Than Latrunculin A Does Not Necessarily Cure Yeast Cells of [PSI]
Because [PSI] curing has not immediately followed cytoskeleton disruption and required prolonged incubation in the presence of latrunculin A, one could suggest that [PSI] loss in the strain OT55, which is a relatively “weak” [PSI +] strain (13), occurs as a result of continuous cell division blockage, rather than in direct response to the cytoskeleton disruption by latrunculin A. To address this question, we have studied effects of the other proliferation-blocking agents on [PSI] maintenance. For this purpose, the yeast cells were incubated in the presence of either 10 mM sodium azide or 100 μg/ml cyclo heximide. Cycloheximide is a protein synthesis inhibitor [see (6) for review], while sodium azide is an uncoupling agent and ATPase inhibitor with wide spectrum of action [for examples, see (21,27)], inhibiting protein synthesis [for instance, see (16)] and other cellular processes. Cycloheximide was not reported to specifically affect cytoskeleton, while sodium azide blocks movements of the actin patches but does not disrupt the actin cytoskeleton (42). We have confirmed that the concentrations of the sodium azide or cycloheximide used in these experiments in hibit growth of the yeast cells (Fig. 1A) and block Hsp induction in the strain OT55 during heat shock (G. P. Newnam and Y. O. Chernoff, unpublished data). However, [PSI] maintenance was not significantly affected by sodium azide or cycloheximide in the strain OT55 (Table 4), confirming that block of cell division itself does not cure yeast cells of [PSI].
[PSI] Curing by Latrunculin A Depends on Protein Synthesis
To check whether [PSI] curing in the presence of latrunculin A depends on protein synthesis, we have treated yeast cells of the strain OT55 with latrunculin A in conditions when protein synthesis was inhibited by sodium azide or cycloheximide. Our results show that sodium azide or cycloheximide treatments almost completely inhibit the [PSI] curing (Table 4), but not antiactin (not shown) effect of latrunculin A. These data indicate that [PSI] loss requires continuation of the metabolic processes, in particular of protein synthesis, during latrunculin A treatment. The sodium azide treatment has also increased viability of the yeast cells upon prolonged incubation with latrunculin A, although such an effect was not observed for cycloheximide and thus could not be attributed solely to inhibition of the protein synthesis (Fig. 1A).
Latrunculin A Treatment Does Not Affect Levels of Sup35p and Heat Shock Proteins
The interplay between stress-inducible proteins of Hsp100 and Hsp70 families modulates [PSI] maintenance in yeast (7,8,32). Because latrunculin A-induced loss of [PSI] depends on protein synthesis, one could suggest that latrunculin A affects [PSI] indirectly by inducing heat shock proteins, in particular Hspl04, excess of which was previously shown to cure yeast cells of [PSI] (7). It was difficult to analyze levels of Hsps in the yeast cells, which have been incubated overnight in the presence of latrunculin A, because the vast majority of the culture was dead by that time. However, we have checked Hsp levels in the cells treated with latrunculin A for 4 h, conditions that cause significant loss of [PSI] but do not yet result in the considerable cell death. For this purpose, we have isolated total cellular proteins from the latrunculin A-treated cells, separated them by SDS-PAGE, and reacted them to Hsp-specific antibodies. Our results failed to detect any visible change in the levels of three different stressed-induced proteins, Hspl04, Hsp70 (Ssa), and Hsp82, after 4-h incubation with latrunculin A (Fig. 1B). Neither control nor latrunculin A-treated cells contained detectable levels of Hsp26, which is usually not expressed in the exponential phase (not shown). These data confirm that the latrunculin A treatment does not induce general stress response in yeast. Quite remarkably, the levels of Sup35p itself were not affected by latrunculin A treatment (Fig. 1B).
Visualization of the Latrunculin A Effect on Sup35p Aggregates
To check whether latrunculin A treatment affects the physical characteristics of prion aggregates, both centrifugation assay and fluorescence microscopy were used. The distribution of Sup35p between soluble and insoluble (prion) fractions in the [PSI +] strain remained unaffected by latrunculin A (Fig. 1C). As described previously (2,3), cortical actin patches, as visualized by rhodamine-phalloidin staining, essentially disappeared in the latrunculin A-treated cells (Fig. 2B). By using the chimeric protein Sup35NM-GFP (33), which originated from the fusion between the Sup35p prion-forming region (Sup35NM) and green fluorescent protein (GFP), we have shown that the latrunculin A treatment also affects properties of the Sup35pPSI+ aggregates. Instead of the relatively small and compact fluorescent “seeds” observed in the untreated [PSI +] cells, the latrunculin A-treated cells contained large amorphous spots of the fluorescent material diffused over the significant portions of the cytoplasm (Fig. 2C, D). One-hour incubation with latrunculin A was sufficient to induce the aggregate diffusion (Fig. 2D). In contrast, the cycloheximide and especially sodium azide treatments made the Sup35-GFP “seeds” more sharp and clearly seen, compared with the control untreated cultures (Fig. 2D). One possible explanation is that block of protein synthesis in the presence of sodium azide or cycloheximide prevents accumulation of the newly synthesized Sup35p and Sup35-GFP, leading to concentration of most of the Sup35-GFP in the aggregated structures. It is also likely that sodium azide may “sharpen” the prion clumps due to prevention of production and/or inhibition of activity of the proteins involved in “seeding” of the Sup35pPSI+ aggregates (i.e., in dissociation of larger aggregates into the smaller oligomers, which initiate the next round of prion “replication”), for instance Hspl04 [according to some models; see (28,43) for review]. When the plasmid overexpressing the SUP35-GFP under the strong constitutive promoter was employed, some control (untreated) cells accumulated large agglomerates of the Sup35-GFP instead or in addition to the smaller seeds. Such agglomerates were also observed in a fraction of the cycloheximide-treated cells expressing the SUP35-GFP under the moderate SUP35 promoter (not shown). While reasons for the formation of such large agglomerates are unknown, they appeared different in size and location from the diffused spots induced by latrunculin A. In parallel with the [PSI] curing results, the sodium azide treatment suppressed the latrunculin A-mediated Sup35-GFP aggregate diffusion (Fig. 2D). However, the latrunculin A treatment still caused some diffusion of the Sup35-GFP aggregates in the presence of cycloheximide (Fig. 2D), despite the lack of the [PSI] curing effect (Table 4, see above).
Prion Aggregates and Actin Patches Do Not Colocalize
Because our data suggest that latrunculin A affects the actin cytoskeleton and prion aggregates in similar ways, one could suggest that these cellular components are physically connected to each other. However, we have not observed colocalization of the fluorescent Sup35NM-GFP aggregates with cortical actin patches, visualized by the rhodamine-phalloidin staining and found preferentially near the bud neck (Fig. 2E). This suggests that at least relatively large, visually detectable prion aggregates are not necessarily associated with actin patches.
DISCUSSION
Latrunculin A is an agent that sequesters mono-meric actin. It has been shown to inhibit actin polymerization in vitro (38) and to disrupt microfilament organization in vivo in invertebrates (39) and in yeast (3). The rapidity with which actin filaments are disassembled in latrunculin A-treated cells indicates that, although yeast are nonmotile, actin filaments’ turnover is very fast in vivo, and that living yeast cells undergo rapid cycles of assembly and disassembly (3). The effect of latrunculin A on actin cytoskeleton is fully reversible in yeast cells, requiring about 60 min to regain their normal polarized actin cytoskeleton (3).
Our new data show that in addition to the reversible disruption of actin cytoskeleton, latrunculin A treatment results in [PSI] loss that is not reversed by removing latrunculin A (Table 1, Fig. 2A). Presence of [PSI] had no effect on cell viability during latrunculin A treatment (Table 2). Therefore, the [PSI] loss observed could not be explained by differential survival rates of the [psi −] and [PSI +] cells in the presence of latrunculin A. The loss of [PSI] also could not be interpreted as a nonspecific prion instability in the cells maintained in marginal conditions. First, efficient [PSI] loss was detected after 4-h treatment with latrunculin A (Table 1), which did not cause significant cell death (Fig. 1A, Table 2). Second, neither other agents blocking cell proliferation, such as sodium azide and cycloheximide (Table 4, see above), nor prolonged incubation of nondividing yeast cells at low temperature (Y. O. Chernoff and G. Newnam, unpublished data) or in the stationary phase (Y. O. Chernoff, G. Newnam, and J. Kumar, unpublished data) lead to a significant loss of [PSI].
The efficient [PSI]-curing chemical agents described previously, such as guanidine-HCl, acted on proliferating cells and caused proliferation-dependent loss of [PSI] (17). However, yeast cells do not proliferate in the presence of latrunculin A due to the disruption of the actin cytoskeleton. There were very few mosaics (sectored [PSI +]/[psi −] colonies) among the [psi −] colonies observed after latrunculin A treatment (Fig. 2A). This suggests that latrunculin A treatment either destroys prion aggregates or makes them unable to reproduce themselves. Some antiprion treatments characterized previously, such as an excess Hsp104 (7), were accompanied by solubilization of the Sup35pPSI+ aggregates (33,34). Moreover, reversible Sup35p solubilization preceded the [PSI] loss: in the original [PSI +] yeast culture, which over-expressed Hspl04 and contained primarily soluble Sup35p, only 2% of cells formed [psi −] colonies (34). However, our data show that latrunculin A treatment does not shift a balance between soluble and insoluble Sup35p even in the conditions when 6–13% of the cells form [psi −] colonies (Fig. 1C, Table 1). This confirms that solubilization of the Sup35pPSI+ aggregates does not represent a primary effect of the latrunculin A treatment. However, the fluorescence microscopy results (Fig. 2C) show that structural patterns of the Sup35p aggregates are altered dramatically in the presence of latrunculin A. The latrunculin A treatment converts the compact Sup35p aggregates into more diffused and amorphous agglomerates. Apparently, intracellular localization of aggregates is also altered. Most likely, the ordered structure of prion polymers is disrupted in the latrunculin A-treated cells. Although most of Sup35p remains in the aggregated form, large amorphous agglomerates probably reduce their ability to reproduce the prion state by “seeding” new cycles of the Sup35p polymerization. One reason for this could be a decrease of surface available for interaction with the new Sup35p molecules, due to a dramatic increase of the average size of the aggregate. It is also possible that the conformation of the Sup35p molecules within the large amorphous agglomerates is changed, compared with the small compact aggregates. This might make Sup35p less active in prion conversion. It should be noted that observable changes in the structural patterns of aggregates are not sufficient to explain a defect in prion maintenance. The cycloheximide treatment protected [PSI] from curing by latrunculin A (Table 4), despite the fact that it did not completely prevent latrunculin A from altering the aggregate structure (Fig. 2D). This means that “loose” latrunculin A-affected aggregates are able to recover in case protein synthesis did not occur during the latrunculin A treatment. Possibly, the “loose” unstructured aggregates become easily accessible for proteases or refolding chaperones, which eventually destroy them unless production of these proteases and chaperones is inhibited due to block of protein synthesis. This agrees with the notion that diffusion of the Sup35pPSI+ aggregates is almost complete after 1-h treatment with latrunculin A (Fig. 2C), while curing of [PSI] needs longer time to become efficient (Table 1).
It is not likely that latrunculin A acts on prion aggregates directly. The only known direct effect of latrunculin A is sequestration of actin. It is more probable that while a prion isoform of the Sup35p is able to survive for certain periods of time in the absence of an actin cytoskeleton, it cannot maintain its structure and/or ability to propagate indefinitely if the integrity of the cytoskeleton is not restored. One possibility is that actin microfilaments provide a network that assists in either breaking the larger aggregates down into the smaller “seeds” or in transporting these “seeds” to new locations where they can initiate new rounds of prion replication. This would keep the prion aggregates in the compacted and highly ordered form, which is resistant to the proteolytic and/or refolding systems of the cell. Indeed, we have previously detected physical and functional interactions between the Sup35p prion-forming domain and Slalp, a cytoskeletal assembly protein involved in actin nucleation (4). Moreover, Slalp is dispensable for the regular propagation of [PSI] aggregates, but it becomes crucial for [PSI] recovery in marginal conditions (4). This suggests that interactions with the cytoskeletal networks are not necessary for the normal proliferation of the [PSI] prion, but could become important in the situation when [PSI] ability to proliferate is impaired.
It is worth mentioning that various [PSI +] strains, even of isogenic origin, differ from each other with regard to their sensitivity to the [PSI] curing effect of latrunculin A. For example, latrunculin A caused much more efficient loss of prion in the “weak” [PSI +] strain than in the isogenic “strong” [PSI +] strain OT56 (P. A. Bailleul-Winslett and Y. O. Chernoff, unpublished data). It is possible that the “strong” [PSI] isoforms are more resistant to the cellular “antiprion” systems, compared with the “weak” [PSI] isoforms. This enables the “strong” [PSI]s to survive for longer periods of time in the absence of actin cytoskeleton. This supports a notion that while cytoskeletal structures assist in maintaining [PSI], they are not essential for [PSI] maintenance.
The easiest explanation for the Slalp and latrunculin A effects would be direct association between the prion aggregates and cytoskeletal structures. However, we have not observed a colocalization of Sup35NM-GFP aggregates and actin patches (Fig. 2E). It is possible that the interactions between the actin networks and prion “seeds” are of a transient nature and cannot be caught by co-staining techniques. On the other hand, it remains to be seen whether relatively large, visible aggregates of the Sup35NM-GFP fusion protein contribute to the next cycle of prion formation. It is still possible that such visually detected “seeds” (even those observed in the cells not treated with latrunculin A) represent the “dead ends” of the prion propagation pathway and are not able to propagate anymore. In support of this model, we have observed that certain [PSI] curing treatments, such as partial inactivation of Hspl04, increase rather than decrease the average size of the Sup35-GFP aggregates in [PSI +] cells (R. D. Wegrzyn, K. Bapat, and Y. O. Chernoff, in preparation). It cannot be ruled out that prion transmission occurs primarily via relatively small Sup35p oligomers, which are not readily visualized by fluorescence microscopy. The effects of latrunculin A and Slalp suggest that propagation of these oligomers could be assisted by the cytoskeletal networks.
An alternative explanation for the latrunculin A effect could be that latrunculin A cures yeast cells of [PSI] indirectly by inducing the stress response. Indeed, the [PSI] curing effect of latrunculin A is blocked by sodium azide or cycloheximide, the agents that inhibit protein synthesis (Table 4). However, there is only one heat shock protein, Hspl04, that is proven to cure yeast cells of [PSI] upon overproduction (7). We have not detected any increase of Hspl04 levels in the presence of latrunculin A (Fig. 1B). While slight variations in Hspl04 levels (10–15%) in response to latrunculin A could not be ruled out by the experimental approach used, such variations would not be sufficient to explain [PSI] loss. Previoulsy, we observed at least twofold or higher increase in Hspl04 levels in the strain OT55 in conditions when [PSI] was cured (7,32). Thus, our data show that latrunculin A-induced [PSI] loss does not occur by Hspl04 induction. Levels of other heat shock proteins tested also remain unaffected in latrunculin A-treated cells (Fig. 1B). It is also unlikely that latrunculin A causes [PSI] loss by inactivating Hspl04, as previously proposed for guanidine-HCl (17,19). Hspl04 inactivation results in the loss of both [PSI] and [PIN] (14) while latrunculin A treatment cures [PSI] but does not cure [PIN] (Table 3).
It is still possible that latrunculin A treatment induces an unknown protein other than Hspl04, which causes loss of [PSI]. Identification of such a protein would be of great interest because Hspl04 remains the only protein thus far shown to cause [PSI] loss in yeast. However, the protection of [PSI] in the conditions when protein synthesis is inhibited could also have a simpler explanation. First, the unregulated growth of the Sup35p aggregates, leading to their structural alterations in the absence of the actin cytoskeleton network, could be facilitated by a continuous supply of the newly synthesized Sup35p. Second, the newly synthesized Sup35p is also needed in order to produce the soluble (Sup35psi−) population of the Sup35p molecules once the structure and prion-forming abilities of the aggregates are impaired. Third, continuous supply of the proteases and/or refolding chaperones (not necessarily those specifically induced by latrunculin A) could be needed for destruction of the prion aggregates altered in the presence of latrunculin A. Once the protein synthesis is blocked, supply of these proteins becomes insufficient.
It should be noted that the sodium azide treatment not only interfered with the latrunculin A-induced [PSI] loss (Table 4) but also protected a compact structure of the Sup35-GFP aggregates during the latrunculin A treatment (Fig. 2D). It is possible that, in addition to blocking protein synthesis, sodium azide is also influencing the interactions between the prion aggregates and unidentified cellular factors causing dissociation of aggregates in the conditions when the cytoskeleton is disrupted. This agrees with previous observation that the sodium azide treatment is “freezing” cortical actin patches and preventing them from moving (42). Interestingly, the sodium azide treatment also increased viability of the yeast cells after prolonged incubation with latrunculin A (Fig. 1A). This indicates that the death of the cytoskeleton-depleted cells includes a sodium azide-sensitive component. It is unlikely that the protecting effect of azide on viability is due to the block of protein synthesis, because inhibition of protein synthesis by cycloheximide did not rescue yeast cells from the latrunculin A-induced cell death (Fig. 1A).
It remains to be seen whether latrunculin A affects prions other than [PSI]. We observed that latrunculin A does not cure yeast cells of [PIN] (Table 3), which modulates the frequency of [PSI] appearance (14). However, the molecular basis of [PIN] remains unknown. While mammalian PrP is an extracellular protein and is unlikely to be directly affected by actin cytoskeleton, some other amyloidoses (e.g., Huntington’s disease) involve intracellular proteins. Similarities were previously noted between Sup35p and the aggregation-prone (poly-Gln expanded) derivative of huntingtin in regard to both sequence organization (12) and protein interaction patterns (4). “Instant” curing of [PSI] by latrunculin A paves the way for potential antiaggregation treatments in animal systems. Our new data point to anticytoskeletal drugs as a potential tool in the fight against aggregation-related disorders.
ACKNOWLEDGMENTS
We are grateful to Tatiana S. Karpova for the useful technical comments, rhodamine-phalloidin staining protocol, and critical reading of the manuscript. We thank E. Lewitin for constructing the plasmid pLSpSup35-GFP, T. Serio for guidance in the GFP assays, S. Woodard for the confocal microscopy training, E. Craig, S. Lindquist, and J. Weissman for plasmids and antibodies. This work was supported by grant R01GM58763 from the National Institutes of Health to Y.O.C., and by the GAANN (Graduate Assistance in the Areas of National Needs) Fellowship from the U.S. Department of Education to P.A.B.-W.
REFERENCES
- 1. Adams A. E. M.; Pringle J. Relationship of actin and tubulin distribution to bud growth in wild-type and morphogenetic mutant of Saccharomyces cerevisiae . J. Cell. Biol. 98:934–945; 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ayscough K. Use of latrunculin-A, an actin monomer-binding drug. Methods Enzymol. 298:18–25; 1998. [DOI] [PubMed] [Google Scholar]
- 3. Ayscough K. R.; Stryker J.; Pokala N.; Sanders M.; Crews P.; Drubin D. G. High rates of actin filament turnover in budding yeast and roles for actin in establishment and maintenance of cell polarity revealed using the actin inhibitor latrunculin-A. J. Cell Biol. 137: 399–416; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bailleul P. A.; Newnam G. P.; Steenbergen J. N.; Chernoff Y. O. Genetic study of interactions between the cytoskeletal assembly protein Slal and prion-forming domain of the release factor Sup35 (eRF3) in Saccharomyces cerevisiae . Genetics 153:81–94; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Broach J. R.; Strathern J. N.; Hicks J. B. Transformation of yeast: Development of the hybrid cloning vector and isolation of the CAN1 gene. Gene 8:121–133; 1979. [DOI] [PubMed] [Google Scholar]
- 6. Carrasco L.; Fernandez-Puentes C; Vazquez D. Antibiotics and compounds affecting translation by eukaryotic ribosomes. Specific enhancement of aminoacyl-tRNA binding by methylaxnthines. Mol. Cell. Biochem. 10:97–122; 1976. [DOI] [PubMed] [Google Scholar]
- 7. Chernoff Y. O.; Lindquist S. L.; Ono B.; Inge-Vechtomov S. G.; Liebman S. W. Role of the chaperone protein Hspl04 in propagation of the yeast prion-like factor [pst]. Science 268:880–884; 1995. [DOI] [PubMed] [Google Scholar]
- 8. Chernoff Y. O.; Newnam G P.; Kumar J.; Allen K.; Zink A. Evidence for a “protein imitator” in yeast: Role of the Hsp70-related chaperone Ssb in formation, stability and toxicity of the [PSI] prion. Mol. Cell. Biol. 19:8103–8112; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chernoff Y. O.; Galkin A. P.; Lewitin E.; Chernova T. A.; Newnam G. P.; Belenkiy S. M. Evolutionary conservation of prion-forming abilities of the yeast Sup35 protein. Mol. Microbiol. 35:865–876; 2000. [DOI] [PubMed] [Google Scholar]
- 10. Coue M.; Brenner S. L.; Spector I.; Korn E. D. Inhibition of actin polymerization by latrunculin A. FEBS Lett. 213:316–318; 1987. [DOI] [PubMed] [Google Scholar]
- 11. DeArmond S. J.; McKinley M. P.; Barry R. A.; Braunfeld M. B.; McColloch J. R.; Prusiner S. B. Identification of prion amyloid filaments in scrapie-infected brain. Cell 41:221–235; 1985. [DOI] [PubMed] [Google Scholar]
- 12. De Pace A. H.; Santoso A.; Hillner P.; Weissman J. S. A critical role for amino-terminal glutamine/asparagine repeats in the formation and propagation of a yeast prion. Cell 93:1241–1252; 1998. [DOI] [PubMed] [Google Scholar]
- 13. Derkatch I. L.; Chernoff Y. O.; Kushnirov V. V.; Inge-Vechtomov S. G.; Liebman S. W. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae . Genetics 144:1375–1386; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Derkatch I. L.; Bradley M.; Zhou P.; Chernoff Y. O.; Liebman S. W. Genetic and environmental factors affecting the de novo appearance of the [PSI +] prion in Saccharomyces cerevisiae . Genetics 147:507–519; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Derkatch I. L.; Bradley M. E.; Masse S. V.; Zadorsky S. P.; Polozkov G. V.; Inge-Vechtomov S. G.; Liebman S. W. Dependence and independence of [PSI +] and [PIN]: A two-prion system in yeast? EMBO J. 19:1942–1952; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Duncan R. F.; Hershey J. W. Initiation factor protein modifications and inhibition of protein synthesis. Mol. Cell. Biol. 7:1293–1295; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eaglestone S. S.; Ruddock L. W.; Cox B. S.; Tuite M. F. Guanidine hydrochloride blocks a critical step in the propagation of the prion-like determinant [PSI+] of Saccharomyces cerevisiae . Proc. Natl. Acad. Sci. USA 97:240–244; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Edskes H. K.; Gray V. T.; Wickner R. B. The [URE3] priori is an aggregated form of Ure2p that can be cured by overexpression of Ure2p fragments. Proc. Natl. Acad. Sci. USA 96:1498–1503; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Glover J. R.; Lindquist S. Hspl04, Hsp70 and Hsp40: A novel chaperone system that rescues previously aggregated proteins. Cell 94:1–20; 1998. [DOI] [PubMed] [Google Scholar]
- 20. Glover J. R.; Kowal A. S.; Schirmer E. C.; Patino M. M.; Liu J. J.; Lindquist S. Self-seeded fibers formed by Sup35, the protein determinant of [PSI +], a heritable prion-like factor of Saccharomyces cerevisiae . Cell 89:811–819; 1997. [DOI] [PubMed] [Google Scholar]
- 21. Harris D. A. Azide as a probe of co-operative interactions in the mitochondrial Fl-ATPase. Biochim. Biophys. Acta 974:156–162; 1989. [DOI] [PubMed] [Google Scholar]
- 22. Holtzman D. A.; Yang S.; Drubin D. G Synthetic-lethal interactions identify two novel genes, SLA1 and SLA2, that control membrane cytoskeleton assembly in Saccharomyces cerevisiae . J. Cell Biol. 122:635–644; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kaiser C; Michaelis S.; Mitchell A. Methods in yeast genetics. A Cold Spring Harbor Laboratory course manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- 24. Karpova T. S.; McNally J. G.; Moltz S. L.; Cooper J. A. Assembly and function of the actin cytoskeleton in yeast: Relationships between cables and patches. J. Cell Biol. 142:1501–1517; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. King C. Y.; Tittmann P.; Gross H.; Gebert R.; Aebi M.; Wuthrich K. Prion-inducing domain 2-114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc. Natl. Acad. Sci. USA 94:6618–6622; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koo E. H.; Lansbury P. T.; Kelly J. W. Amyloid diseases: Abnormal protein aggregation in neurodegeneration. Proc. Natl. Acad. Sci. USA 96:9989–9990; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kubak B. M.; Yotis W. W. Staphylococcus aureus adenosine triphosphatase: Inhibitor sensitivity and release from membrane. J. Bacteriol. 146:385–390; 1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kushnirov V. V.; Ter-Avanesyan M. D. Structure and replication of yeast prions. Cell 94:13–16; 1998. [DOI] [PubMed] [Google Scholar]
- 29. Lansbury P. T.; Caughey B. The chemistry of scrapie reaction: The “ice 9” metaphore. Chem. Biol. 2:1–5; 1995. [DOI] [PubMed] [Google Scholar]
- 30. Lappalainen P.; Drubin D. G. Cofilin promotes rapid actin filament turnover in vivo . Nature 288:78–82; 1997. [DOI] [PubMed] [Google Scholar]
- 31. Li R.; Zheng Y.; Drubin D. G. Regulation of cortical actin cytoskeleton assembly during polarized cell growth in budding yeast. J. Cell Biol. 128:599–615; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Newnam G. P.; Wegrzyn R. D.; Lindquist S. L.; Chernoff Y. O. Antagonistic interactions between yeast chaperones Hspl04 and Hsp70 in prion curing. Mol. Cell. Biol. 19:1325–1333; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Patino M. M.; Liu J. J.; Glover J. R.; Lindquist S. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science 273:622–626; 1996. [DOI] [PubMed] [Google Scholar]
- 34. Paushkin S. V.; Kushnirov V. V.; Smirnov V. N.; Ter-Avanesyan M. D. Propagation of the yeast prion-like [psi +] determinant is mediated by oligomerization of the SUP35-encoded polypeptide chain release factor. EMBO J. 15:3127–3134; 1996. [PMC free article] [PubMed] [Google Scholar]
- 35. Prusiner S. B. Prions. Proc. Natl. Acad. Sci. USA 95: 13363–13383; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Prusiner S. B.; McKinley M. P., Bowman K. A.; Bolton D. C.; Bendheim P. E.; Groth D. F.; Glenner G. G. Scrapie prions aggregate to form amyloid-like birefringent rods. Cell 35:349–358; 1983. [DOI] [PubMed] [Google Scholar]
- 37. Schlumpberger M.; Wille H.; Baldwin M. A.; Butler D. A.; Herskowitz I.; Prusiner S. B. The prion domain of yeast Ure2p induces autocatalytic formation of amyloid fibers by a recombinant fusion protein. Protein Sci. 9:440–451; 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Spector I.; Shochet N. R.; Kashman Y.; Groweiss A. Latrunculins: Novel marine toxins that disrupt micro-filament organization in cultured cells. Science 219: 493–495; 1983. [DOI] [PubMed] [Google Scholar]
- 39. Spector I.; Shochet N. R.; Blasberger D.; Kashman Y. Latrunculins—novel marine macrolides that disrupt microfilament organization and affect cell growth: I. Comparison with cytochalasin D. Cell. Motil. Cytoskel. 13:127–144; 1989. [DOI] [PubMed] [Google Scholar]
- 40. Taylor K. L.; Cheng N.; Williams R. W.; Steven A. C.; Wickner R. B. Prion domain initiation of amyloid formation in vitro from native Ure2p. Science 283: 1339–1343; 1999. [DOI] [PubMed] [Google Scholar]
- 41. Telckov M. V.; Surguchov A. P.; Dagkesamanskaya A. R.; Ter-Avanesyan M. D. Isolation of a chromosomal DNA fragment containing SUP2 gene of the yeast Saccharomyces cerevisiae . Genetika (Russ.) 22: 17–25; 1986. [Google Scholar]
- 42. Waddle J. A.; Karpova T. S.; Waterston R. H.; Cooper J. A. Movement of cortical actin patches in yeast. J. Cell Biol. 132:861–870; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wickner R. B.; Chernoff Y. O. Prions of fungi: [URE3], [PSI], and [Het-s] discovered as heritable traits. In: Prusiner S. B., ed. Prion biology and diseases. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999:229–272. [Google Scholar]