

Abstract
The growth and development of some of the male sex accessory organs such as the prostate requires the conversion of testosterone to dihydrotestosterone (DHT) by 5α-reductase. To provide insights into the role of testosterone versus DHT in the prostate, we studied the impact of finasteride, a potent and specific inhibitor of 5α-reductase, on the expression of prostatic androgen-response genes in testis-intact rats and in 7-day castrated rats. Finasteride inhibition of the conversion of testosterone to DHT was confirmed by measuring serum and intraprostatic androgens. As expected, finasteride treatment caused a reduction in the wet weight of the prostate in the testis-intact rats and inhibited the testosterone-stimulated prostatic regrowth in the 7-day castrated rats. Although finasteride treatment had little or no effect on the expression of the surveyed androgen-response genes in testis-intact rats, its administration enhanced the expression of many androgen-response genes during the testosterone-stimulated regrowth of the regressed prostate in castrated rats. These observations suggest that testosterone is more potent than DHT in stimulating the expression of many androgen-response genes in the regressed prostate. The expression of androgen-response genes is mainly prostate specific and thus is likely to be associated with androgen-dependent prostatic differentiation. Therefore, testosterone is more potent than DHT in inducing differentiation and weaker in stimulating proliferation during prostate regrowth. The fact that testosterone is a strong inducer of prostatic differentiation has potential clinical implications.
Keywords: 5α-Reductase, Androgen-response genes, Testosterone, Dihydrotestosterone
THE most biologically active androgens are testosterone and dihydrotestosterone (DHT) (12). Testosterone is synthesized in the testis and secreted into bloodstream circulation. Testosterone is converted into DHT by 5α-reductase in target organs, including the prostate (5,6). From previous studies by others, it is clear that DHT is the active form of androgens in many male sex accessory tissues. Inactivation of 5α-reductase in humans leads to pseudohermaphroditism, a disease characterized by undervirilization of the external genitalia and prostate (14,31). Attempts have been made to determine the functional differences between testosterone and DHT, despite that both testosterone and DHT bind to the same androgen receptor (AR). DHT has a higher binding affinity to the AR relative to testosterone (15,19,34). This observation suggested that the only difference between testosterone and DHT is relative potency (11,15). Excessive testosterone can compensate for the lack of DHT, and the conversion of testosterone to DHT is merely a metabolic amplification step that enhances androgen action. This is further supported by the observation that the maximum activation of the MMTV-CAT reporter gene by testosterone and DHT was the same, although the kinetics of activation by testosterone was slower than that by DHT (9). Other studies suggest that DHT and testosterone play different roles in androgen action. DHT is required for prostatic cell proliferation (22,25). Testosterone is incapable of stimulating proliferation, although it can attenuate apoptosis (26,36). Testosterone and DHT have been shown to differentially regulate the expression of some genes, including 5α-reductase, TDD5, and other novel genes (2,13,20). The functional differences between testosterone and DHT could be due to different conformations of the liganded AR.
Finasteride is a potent 5α-reductase inhibitor developed for the treatment of benign prostatic hyperplasia (BPH) (24,29). Because of its clinical significance, the effect of finasteride has been studied extensively in animal models. Inhibition of 5α-reductase by finasteride can reduce both serum and intraprostatic DHT to castration levels (10,11,23,25). Thus, finasteride decreases the size of the prostate in the rat (16,23,25–27,37).
Because the androgen receptor is a ligand-dependent transcription factor that regulates the expression of androgen-response genes, the impact of both testosterone and DHT on the prostate is mediated through the expression of androgen-response genes, either directly or indirectly (21,30,33,38). If functional differences between testosterone and DHT exist, they are likely to be mediated through differential regulation of androgen-response genes. Using a PCR-based cDNA subtraction method, we have identified more than 20 androgen-response genes on the basis of their induction by androgens in the regressed rat ventral prostate (32,33). These genes were induced in the presence of both testosterone and DHT because the injected testosterone propionate can be converted to DHT by endogenous 5α-reductase in the rat prostate. This gene collection permitted us to test whether testosterone and DHT differentially regulate the expression of androgen-response genes in the rat ventral prostate.
MATERIALS AND METHODS
Materials
DSL-4000 ACTIVE Testosterone (T) and DSL-9600 Dihydrotestosterone (DHT) Radioimmunoassay Kits were from Diagnostic Systems Laboratories, Inc. (Webster, TX). Leupeptin, pepstatin, phenylmethyl-sulfonyl fluoride (PMSF), antifoam B, and crystalline testosterone propionate and DHT were from Sigma Chemical Co. (St. Louis, MO). Phosphate-buffered saline (PBS) was from Gibco. Ethyl alcohol and n-hexane were from Fisher Scientific (Pittsburgh, PA). Methoxyflurane (Metofane) was from Schering-Plough Animal Health. Finasteride was generously provided by Merck Research Laboratories.
Experimental Animals
Adult Sprage-Dawley male rats, weighing 250–300 g each, were purchased from Harlan Laboratories (Indianapolis, IN) and fed with normal chow (Teklad 1521-1512, Harlan Teklad) ad libitum. Castration of the rat was performed through a single scrotal incision while the animals were under methoxyflurane anesthesia. Androgen replacement of 7-day castrated rats was conducted by implanting the animals with silastic tubes filled with crystalline testosterone or DHT. These implants were made by filling testosterone or DHT into silastic tubing with inner diameter of 0.07 in. and outer diameter of 0.125 in. (Dow-Corning, Midland, MI) for various lengths and plugging the ends with wood sticks and adhesive clear silicone (Dow Corning) (8). Finasteride treatment was carried by daily subcutaneous injection of 1 ml vehicle (10% ethanol in sesame oil) supplemented with finasteride at a dose of 40 mg/kg or 12 mg/rat for appropriate times depending upon the experiment (25). For the 7-day castrated rats, a daily dose of carrier alone or finasteride was administrated beginning 1 day prior to the androgen implantation. The sacrifice of the anesthetized rats was carried out by thoracotomy followed by withdrawing blood via cardiac puncture for measuring serum testosterone and DHT levels. Dissection of seminal vesicles, ventral, lateral, and dorsal prostate lobes was performed as previously described (17). The dissected tissues from all animal groups were weighed and immediately frozen in liquid nitrogen for RNA isolation and measurement of testosterone and DHT levels in the tissues.
RNA Preparation and Northern Blot Analysis
Total RNA was isolated from the right lobe of the ventral prostate using the guanidinium/CsCl gradient method (7). Purified RNA samples were fractionated on a 1% agarose-formaldehyde gel. Ten micrograms of total RNA sample was loaded in each lane, electrophoresed, and transferred to nylon membrane for Northern blot hybridization. The probes for Northern blot were prepared using the cDNAs of various prostatic androgen-response genes from the rat ventral prostate (33). The intensity of Northern blot hybridization bands was determined by either phosphoimaging or by Kodak Digital Science 1D image analysis.
Serum and Prostate Extract Preparation
The blood samples collected by cardiac puncture were centrifuged at 2500 rpm for 5 min to obtain serum. Serum samples from two to three animals were pooled for androgen measurement. The left ventral prostate lobes were homogenized in a homogenate buffer (HB) consisting of 1× PBS, 100 mM EDTA, 100 μM PMSF, 10 μM leupeptin, 1 μM pepstatin, and antifoam B emulsion (150 μl per 50 ml of HB) using the tissue homogenizer (Ultra-Turrax T25) at top speed. The homogenized samples were centrifuged at 3700 rpm for 20 min at 4°C and the supernatants collected. Equal amounts of the ventral prostate lobes from two to three animals were pooled to achieve an 80 mg/ml tissue extract concentration for androgen measurement.
Measurement of Testosterone Concentration
To measure testosterone concentration in serum or tissue extract, testosterone standards and controls (provided in DSL-4000 kit) were reconstituted individually in 0.5 ml deionized water. Fifty microliters of serum, tissue extract, control, or standard was pipetted to the bottom of each anti-testosterone-coated tube (provided in DSL-4000 kit) in duplicate. Immediately, 500 μl testosterone (I-125) reagent was added to each blue tube. The contents were mixed by gentle vortexing at low speed. All tubes were incubated at 37°C for 60–70 min. After incubation, the contents were decanted into a radioactive waste receptacle, and the tubes were left to drain on an absorbent paper towel for 2 min. All tubes were measured for bound testosterone (I-125) using a gamma counter for 1 min per tube. Because every testosterone measurement was done in duplicates, the results were expressed as average bound radioactive counts per minute (cpm). A testosterone standard curve was constructed for every experiment to determine the testosterone concentration in serum or in tissue extract. The coefficiency of intra-assay variation is 7.9–9.6% and the coefficiency of interassay variation is 8.4–9.1%.
Measurement of DHT Concentration
The first step to measure DHT concentration in serum or tissue extract is the organic extraction of DHT. DHT standards and controls (provided in DSL-9600 kit) were reconstituted separately in 1.0 ml deionized water. In duplicates, 200 μl of unknown sample or control was transferred to a 15 ml polypropylene tube and 250 μl of the oxidation solution (provided by the kit) was added to each tube. The contents were thoroughly vortexed and incubated at room temperature (∼25°C) for 15 min. Each oxidized sample was extracted by adding 2 ml n-hexane (98% hexane: 2% ethanol) and vortexing for 1 min. Twenty-five microliters of DHT sample buffer (provided by the kit) was added to vortexed samples and mixed gently by inverting capped tubes 3–4 times. The mixed contents were centrifuged at 2500 rpm at 2–8°C for 15 min. A volume of 1.25 ml upper organic layer was transferred to an Eppendorf tube. The samples were completely dried in a speed-vac assembly with heating. Prior to DHT detection with RIA, the dried material was reconstituted with 125 μl DHT sample diluent and vortexed for 1 min.
To measure DHT concentration in the extracted and reconstituted samples, 100 μl reconstituted material, control, or DHT standard was pipetted to the bottom of each anti-DHT-coated tube (provided in DSL-9600 kit) in duplicate. Immediately, 500 μl DHT (I-125) reagent was added to the bottom of each tube. The contents were mixed by gentle vortexing on low setting. All tubes were covered and incubated at room temperature (∼25°C) with shaking at 180 rpm for 2 h. After incubation, the contents were decanted into a radioactive waste receptacle and the tubes were left to drain on an absorbent paper towel for 2 min. All tubes were measured for bound DHT (I-125) using a gamma counter for 1 min per tube. Because every DHT measurement was done in duplicate, the results were expressed as average bound radioactive counts per minute (cpm). A DHT standard curve was constructed for every experiment to determine the DHT concentration in tissue extract. The coefficiency of intra-assay variation is 3.1–6.2% and the coefficiency of interassay variation is 2.3–8.5%.
Calculations and Statistical Analysis
The concentration of free androgen in serum or tissue extract samples was calculated using GraphPad Prism by GraphPad Software Incorporated. Results were verified by two different investigators.
The statistical significance of differences in results of prostate weights was evaluated with Kruskal Wallis nonparamentric test, followed by Mann Whitney tests when significant differences were detected (SPSS statistic software package). All data were expressed as the means ± SEM of the samples examined, and values of p < 0.05 were considered significant.
RESULTS
Finasteride Inhibits the Regrowth of the Prostate in the 7-Day Castrated Rats
To explore the role of testosterone versus DHT on the proliferation of prostatic cells, we studied the impact of finasteride on the regrowth of the ventral prostate in 7-day castrated rats. The regrowth of the regressed prostate was induced by implanting the rats 7 days after castration with silastic tubing filled with 0.375, 0.75, or 1.5 cm of testosterone propionate. The results in Figure 1 show that the wet weight of the prostate 7 days after the implantation of testosterone was significantly increased and the extent of the wet weight increase was greater with longer silastic tubing. Finasteride treatment caused a significant reduction in the wet weight of the regrown ventral prostate in the castrated rats treated with testosterone. In the animals implanted with 0.375 cm testosterone tubing, the wet weight of the ventral prostate in the absence of finasteride was 285 ± 19 mg and was reduced to 185 ± 10 mg by the administration of finasteride. The difference was statistically significant (p = 0.021). In animals implanted with 0.75 cm testosterone tubing, the wet weight of the ventral prostate in the absence of finasteride was 323 ± 21 mg and was reduced to 193 ± 14 mg in the presence of finasteride. The difference was statistically significant (p = 0.021). In animals with 1.5-cm testosterone tubing, the wet weight of the ventral prostate in the absence of finasteride was 331 ± 26 mg and was reduced to 250 ± 19 mg in the presence of finasteride. However, the difference was not statistically significant.
Figure 1.
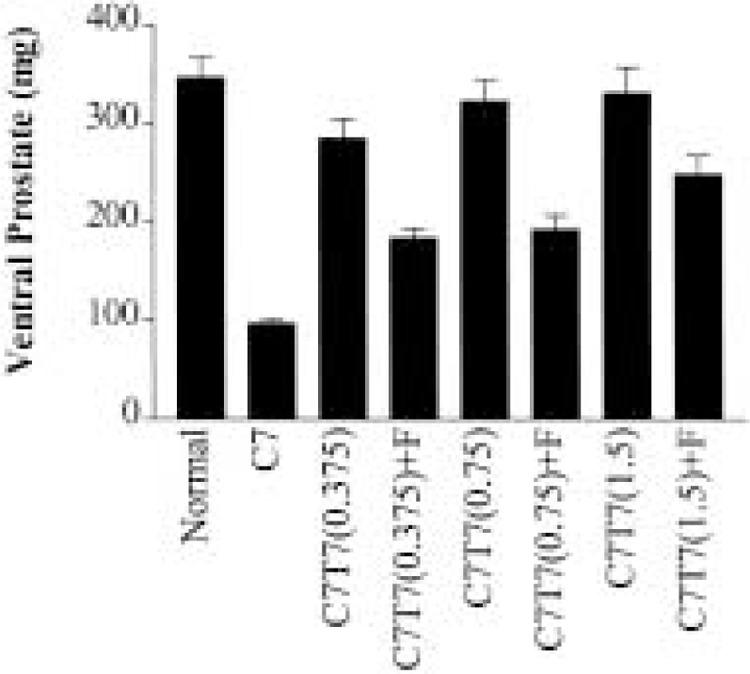
The effect of finasteride on the wet weight of the ventral rat prostate. The ventral prostate was dissected from the following groups of rats: Normal = testis intact; C7 = 7-day castrated; C7T7(0.375) = 7-day castrated followed by additional 7-day implantation with silastic tubing containing 0.375-cm length of testosterone with vehicle; C7T7(0.375)+F = 7-day castrated followed by additional 7-day implantation with silastic tubing containing 0.375-cm length of testosterone and treatment with finasteride; C7T7(0.75) = 7-day castrated followed by additional 7-day implantation with silastic tubing containing 0.75-cm length of testosterone with vehicle; C7T7(0.75)+F = 7-day castrated followed by additional 7-day implantation with silastic tubing containing 0.75cm length of testosterone and treatment with finasteride; C7T7(1.5) = 7-day castrated followed by additional 7-day implantation with silastic tubing containing 1.5-cm length of testosterone with vehicle; C7T7(1.5)+F = 7-day castrated followed by additional 7-day implantation with silastic tubing containing 1.5-cm length of testosterone and treatment with finasteride. The finasteride treatment was initiated 1 day prior to the implantation of the silastic tubing.
We further tested the inhibitory effect of finasteride on prostate regrowth by implanting the 7-day castrated rats with 0.75-cm testosterone tubing for 2 or 7 days either in the presence or absence of finasteride. The 0.75-cm testosterone tubing was chosen in this study because the wet weight of the ventral prostate exhibited the biggest reduction in the finasteride-treated rats relative to the controls in animals implanted with the 0.75-cm tubing (Fig. 1). As expected, the results in Figure 2 show that the wet weight of the ventral prostate was reduced by finasteride relative to the controls at both 2 and 7 days after the testosterone implantation. The wet weight of the ventral prostate at day 2 in the absence of finasteride was 90 ± 5 mg and was reduced to 80 ± 3 mg in the presence of finasteride. This reduction was not statistically significant. At day 7 after the implantation, the wet weight of the ventral prostate was 240 ± 16 mg in the absence of finasteride and 168 ± 10 mg in the presence of finasteride. This reduction was dramatic and was statistically significant (p = 0.04). The finasteride inhibition on the lateral and dorsal lobes of the prostate is virtually identical to that of the ventral prostate. The impact of finasteride on the seminal vesicles was found to be similar to that of the prostate (results not shown).
Figure 2.
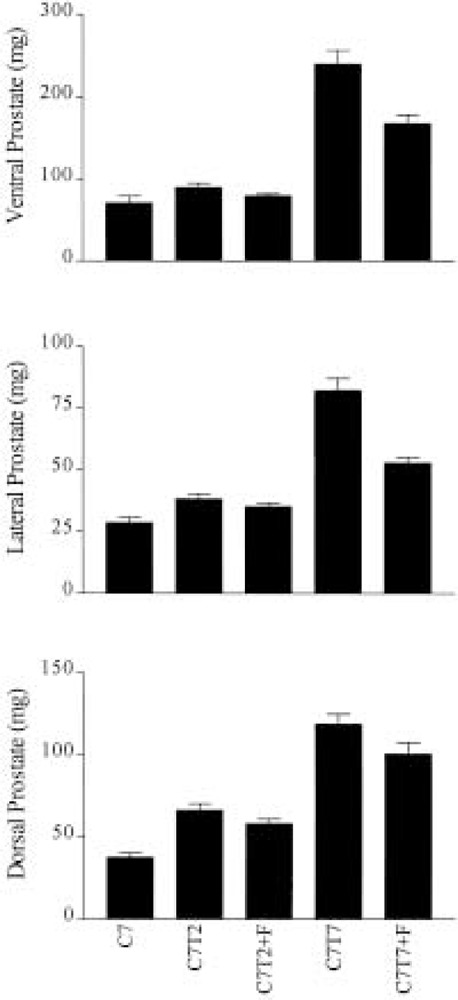
The effect of finasteride on the regrowth of the ventral, lateral, and dorsal rat prostate. The ventral, lateral, and dorsal prostate were dissected from the following groups of rats: C7 = 7-day castrated; C7T2 = 7-day castrated followed by additional 2-day implantation with silastic tubing containing 0.75-cm length of testosterone and treatment with vehicle without finasteride; C7T2+F = 7-day castrated followed by additional 2-day implantation with silastic tubing containing 0.75-cm length of testosterone and treatment with finasteride; C7T7 = 7-day castrated followed by additional 7-day implantation with silastic tubing containing 0.75-cm length of testosterone and treatment with vehicle without finasteride; C7T7+F = 7-day castrated followed by additional 7-day implantation with silastic tubing containing 0.75-cm length of testosterone and treatment with finasteride. The finasteride treatment was initiated 1 day prior to the implantation of the silastic tubing.
Although the regrowth of the castrated prostate was significantly inhibited by finasteride treatment, the wet weight of the ventral, lateral, and dorsal prostate in the animals treated with both testosterone and finasteride was substantially higher relative to the castrated prostate at day 7 after testosterone implantation. The difference was statistically very significant for both the ventral and lateral prostate (p = 0.001) and significant for dorsal prostate (p = 0.01), suggesting that testosterone can support the regrowth of the regressed prostate. However, we cannot rule out the potential proliferative effect of very low levels of prostatic DHT in the presence of finasteride.
Finasteride Inhibits the Conversion of Exogenous Testosterone to DHT in Rats
Serum testosterone and DHT were measured using the commercially available kits as described in Materials and Methods. To explore the applicability of the kits for measuring testosterone and DHT in prostatic tissue, we first determined whether prostatic extracts interfered with the assay for the detection of testosterone and/or DHT. The protein homogenates were prepared from the normal prostate or regressed prostate as described in Materials and Methods. Our experiments showed that the presence of various amounts of prostatic tissue homogenates did not interfere with the measurement of testosterone and DHT standards, respectively (results not shown). We then measured both intraprostatic and serum androgens and the results are shown in Figure 3. The intraprostatic testosterone and DHT were determined in the left lobes of the ventral prostate.
Figure 3.
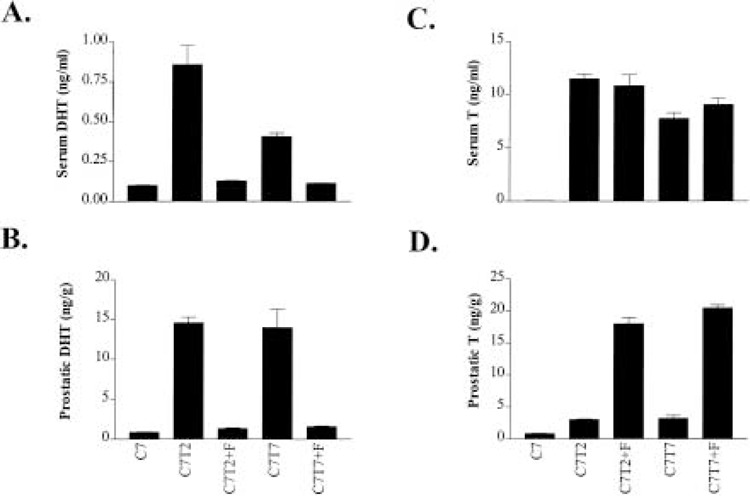
Effect of finasteride on DHT in serum (A), DHT in prostatic tissue (B), testosterone in serum (C), and testosterone in prostatic tissue (D). The measurement of serum and intraprostatic DHT and testosterone was described in Materials and Methods. The animals used in the study are from the same groups of rats illustrated in Figure 2.
Finasteride treatment caused a reduction in serum DHT levels (Fig. 3A). In 7-day castrated rats implanted with testosterone, finasteride reduced the circulating DHT from 0.86 ± 0.12 to 0.13 ± 0.003 ng/ml and from 0.41 ± 0.02 to 0.11 ± 0.001 ng/ml at 2 and 7 days after the testosterone implantation, respectively. The serum DHT levels in the presence of finasteride were very similar or identical to the castration serum DHT level, which was 0.10 ± 0.03 ng/ml. Finasteride also caused a marked reduction in intraprostatic DHT levels (Fig. 3B). In 7-day castrated rats implanted with testosterone, finasteride reduced the intraprostatic DHT from 14.55 ± 0.72 to 1.35 ± 0.09 ng/g and from 13.95 ± 2.33 to 1.58 ± 0.09 ng/g at 2 and 7 days after the testosterone implantation, respectively. The intraprostatic DHT levels in the presence of finasteride were very similar to the intraprostatic DHT level in the castrated rats, which was 0.87 ± 0.03 ng/g.
In the castrated rats implanted with testosterone, finasteride treatment had little or no detectable effect on the circulating testosterone (Fig. 3C). However, finasteride treatment caused a marked elevation in intraprostatic testosterone levels (Fig. 3D). In 7-day castrated rats implanted with testosterone, finasteride elevated the intraprostatic testosterone from 3.06 ± 0.09 to 18.02 ± 1.00 ng/g and from 3.25 ± 0.42 to 20.48 ± 0.45 ng/g at 2 and 7 days after the testosterone implantation, respectively.
The above results indicated that the inhibition of 5α-reductase by finasteride treatment was highly effective in castrated rats. In the presence of finasteride, both serum and intraprostatic DHT levels in testis-intact rats or castrated rats with testosterone implants are very similar, if not identical, to that in the castrated rats. Our results on serum and intraprostatic testosterone and DHT are very similar to previously reported results by others (11,25,26).
Finasteride Enhances the Induction of Some Androgen-Response Genes in the Regressed Prostate by Testosterone Replacement
To investigate whether testosterone and DHT differentially regulate the expression of androgen-response genes in the prostate, we extracted the total RNA from the right lobes of the ventral prostate for Northern blot analysis. Examples of the Northern blots are shown in Figure 4. Interestingly, finasteride caused further upregulation of some androgen-response genes during the testosterone-stimulated regrowth. Figure 4 shows that finasteride reproducibly caused further upregulation of prostatein C3, calreticulin, adrenomedullin, spermidine synthase, and U17 during the androgen-stimulated regrowth of the castrated prostate (33). The extent of finasteride upregulation varies for different genes. The most significant reproducible upregulation, about two- to threefold, was observed for prostatein C3 and U17. Not all the androgen-response genes were further upregulated by finasteride during the regrowth of the prostate. Some of the androgen-response genes, such as U16 and U23, were not influenced by finasteride (results not shown) whereas others, such as U25 and FPP synthase, were slightly downregulated by finasteride (Fig. 4). U16, U17, U23, and U25 are novel genes upregulated by androgen in the prostate (33).
Figure 4.
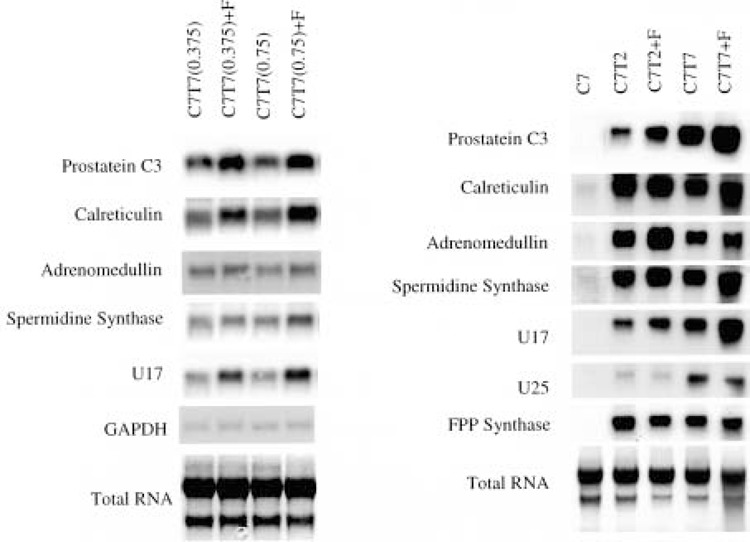
The effect of finasteride on the expression of androgen-response genes in the rat ventral prostate. Northern blot analysis was used to detect the expression of androgen-response genes. In the left panel, total RNA was extracted from the prostate samples described in Figure 1. In the right panel, total RNA was extracted from the prostate samples described in Figure 2. The expression of indicated androgen-response genes is shown. U16, U17, U23, and U25 are novel androgen-response genes (33). The total RNA loaded in the gels was visualized by staining the transferred membrane with methylene blue. The expression of GAPDH was also included in the left panel as a control.
As a control experiment, we tested the effect of finasteride on DHT-induced gene expression in the prostate. The expression of androgen-response genes was not influenced at day 2 after the implantation of DHT in 7-day castrated rats. Figure 5 shows several examples of finasteride’s effect on DHT-induced androgen-response gene expression. This result implies that the impact of finasteride on the expression of androgen-response genes induced by the implantation of testosterone is mediated through the inhibition of the conversion from testosterone to DHT by 5α-reductase.
Figure 5.
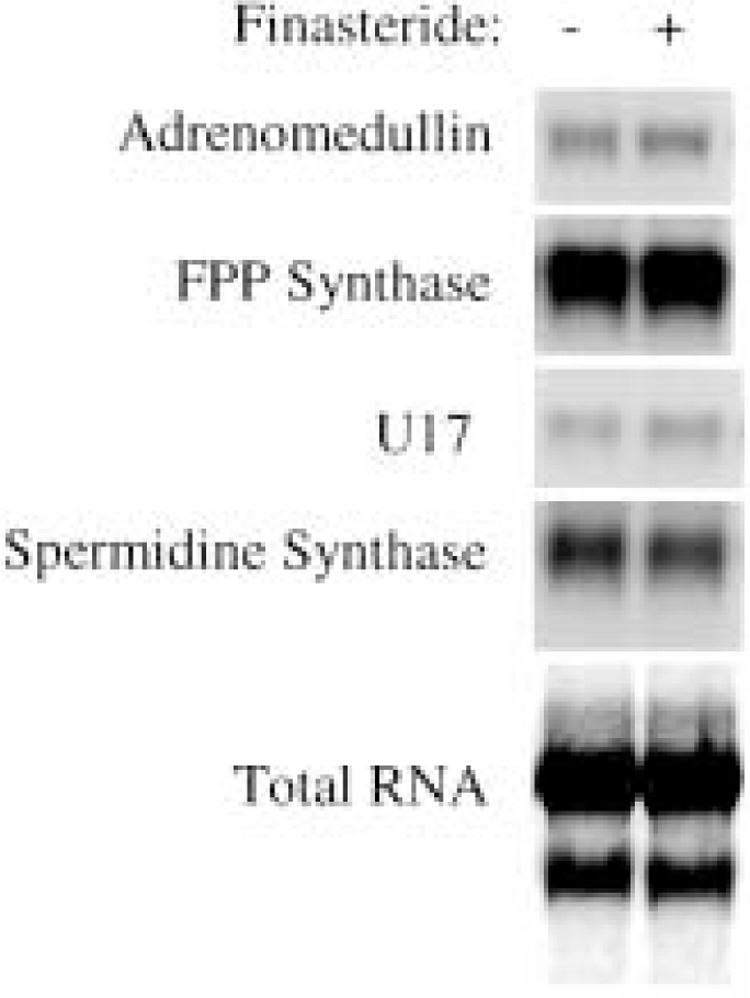
The effect of finasteride on the expression of androgen-response genes in the DHT-stimulated ventral prostate regrowth in 7-day castrated rats. A total of eight castrated rats were implanted with silastic tubing containing 3-cm DHT powder for 2 days. Four of the rats were treated with finasteride daily beginning 1 day prior to the implantation, whereas the other four were treated with vehicle as controls. Northern blot analysis was used to detect the expression of indicated androgen-response genes, including adrenomedullin, FPP synthase, spermidine synthase, and U17 novel gene (33). The total RNA loaded in the gels was visualized by staining the transferred membrane with methylene blue.
The Effect of Finasteride on the Prostate in the Testis-Intact Rats
We also studied the effect of finasteride administration on the normal prostate of testis-intact rats. The results in Table 1 showed that finasteride treatment for 96 h results in a reduction in the wet weight of the ventral prostate in testis-intact rats. The wet weight of the prostate in the untreated animals was 347 ± 20 mg. The wet weight of the ventral prostate treated with finasteride for 96 h was 283 ± 46 mg, which represents an 18% reduction relative to the normal prostate, and the reduction was statistically significant (p = 0.03).
TABLE 1.
SUMMARY OF THE EFFECT OF FINASTERIDE ON THE WET WEIGHT OF THE VENTRAL PROSTATE, SERUM TESTOSTERONE (T) AND DHT, AND INTRAPROSTATIC T AND DHT IN TESTIS-INTACT RATS
| Serum | Prostate | ||||
|---|---|---|---|---|---|
| T (ng/ml) | DHT (ng/ml) | T (ng/g) | DHT (ng/g) | Weight (mg) | |
| Normal | 2.63 ± 0.76 | 0.22 ± 0.02 | 3.01 ± 0.07 | 6.54 ± 1.73 | 347 ± 20 |
| Finasteride | 4.60 ± 0.41 | 0.11 ± 0.003 | 17.56 ± 1.48 | 1.02 ± 0.01 | 283 ± 46 |
| Castrated | 0.04 ± 0.002 | 0.10 ± 0.004 | 0.86 ± 0.04 | 0.87 ± 0.06 | 97 ± 4.4 |
The rats were treated with finasteride for 96 h. The 7-day castrated rats were included to provide a reference.
In testis-intact rats, finasteride reduced the circulating serum DHT from 0.22 ± 0.02 to 0.11 ± 0.003 ng/ml and the intraprostatic DHT from 6.54 ± 1.73 to 1.02 ± 0.01 ng/g at 96 h after the treatment in these animals. The levels of serum and intraprostatic DHT in finasteride-treated animals were similar to that observed in the castrated rats. On the other hand, finasteride treatment elevated the serum testosterone levels in testis-intact animals (Table 1) from 2.63 ± 0.76 to 4.60 ± 0.41 ng/ml and the intraprostatic testosterone from 3.01 ± 0.07 to 17.56 ± 1.48 ng/g at 96 h after the treatment. These observations are consistent with the result from other laboratories that significant serum testosterone elevation is induced by finasteride (11,25).
Although finasteride had significant impact on the wet weight of the prostate and inhibited the conversion of testosterone to DHT in testis-intact rats, no significant alterations in the expression of various androgen-response genes was observed by finasteride administration (Fig. 6). Only the expression of calreticulin, adrenomedullin, and U25 appears to be slightly inhibited by finasteride in the testis-intact animals. In our experiment, finasteride did not affect the expression of prostatein C3 in testis-intact rats, which is consistent with previous finding by Rittmaster et al. (25). The influence of finasteride on androgen-response gene expression in fully grown prostate appears to be different from that in regressed prostate.
Figure 6.
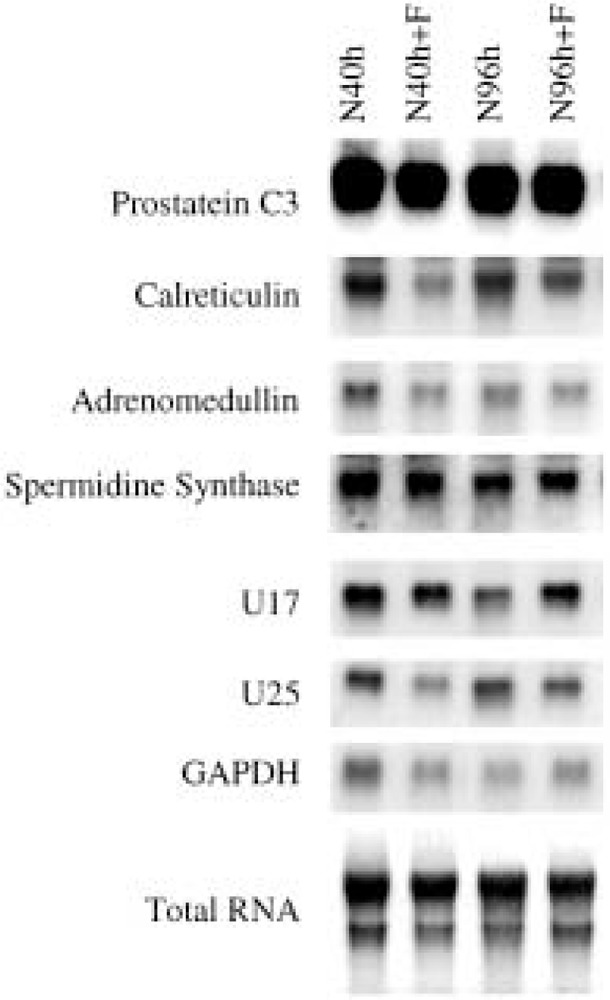
The effect of finasteride on the expression of androgen-response genes in the ventral prostate of testis-intact rats. Four experimental groups of rats were following: N40h = treated with vehicle for 40 h; N40h+F = treated with finasteride for 40 h; N96h = treated with vehicle for 96 h; and N96h+F = treated with finasteride for 96 h. Four animals were used in each group. Northern blot was used to detect the expression of various androgen-response genes and the patterns of representative genes were shown. The total RNA loaded in the gels was visualized by staining the transferred membrane with methylene blue. The expression of GAPDH gene was also included as a control.
DISCUSSION
The present article addresses one of the critical questions in androgen action: whether or not testosterone and DHT differentially regulate the expression of androgen-response genes in the prostate. Our experiments showed that finasteride treatment in testis-intact rats induced moderate regression of the prostate and inhibited the conversion of testosterone to DHT (Table 1). However, finasteride had little or no influence on the expression of androgen-response genes (Fig. 6). These findings are consistent with the previous findings by others (25,36) that inhibition of 5α-reductase by finasteride reduced the wet weight of the prostate in testis-intact animals without influencing the expression of androgen-response genes, including prostatein C3.
We also studied the impact of finasteride on the regrowth of the prostate in 7-day castrated rats. Our studies showed that finasteride inhibited the conversion of exogenous testosterone to DHT very effectively and significantly reduced the wet weight of the regrown prostate, which is consistent with previously reported findings (27,35). Wright et al. (35) reported that the wet weight changes correlated with DNA content changes during the regrowth of the regressed prostate either in the presence or absence of finasteride. Although the inhibition of the regrowth by finasteride is obvious, prostatic regrowth did occur at a slower rate in the presence of finasteride (Figs. 1 and 2) (35). These results suggest that testosterone is mitogenic in the 7-day castrated ventral prostate. The dorsal and lateral lobes of the prostate responded to finasteride treatment in the same way as was seen in the ventral prostate (Fig. 2). The extent of the wet weight reduction for the dorsal and lateral prostate resembles that of the ventral prostate during their regrowth from a regressed state in the castrated animals. Also, the response of the wet weight of the seminal vesicles to finasteride treatment is very similar to that of the prostate (results not shown). Therefore, the differential action of testosterone and DHT is involved in regulating the regrowth of various lobes of the prostate and seminal vesicles in adult rats.
The observation that elevated testosterone is not mitogenic for the prostate in the testis-intact animals but is mitogenic for the regressed prostate can be reconciled by considering the differences between the regressed prostate and the fully grown prostate. Androgens do not stimulate the proliferation of a fully grown prostate, although they are required for the maintenance of the structural and functional integrity of the prostate (4). In contrast, androgens stimulate rapid and massive proliferation of a regressed prostate. Cells in the regressed prostate of a 7-day castrated rat respond to androgens very differently from cells in the fully grown prostate of a testis-intact rat.
The impact of finasteride on the expression of androgen-response genes during the regrowth of the prostate in 7-day castrated rats is different from its impact in the testis-intact animals (Figs. 4 and 6). Although the expression of various androgen-response genes was not influenced by finasteride in the testis-intact rats, the expression of some of these genes was upregulated by finasteride during the regrowth of the regressed prostate. It is also interesting that one of these genes, U25, was downregulated by finasteride during prostate regrowth. The differential response of androgen-response genes to finasteride treatment between testis-intact rats and 7-day castrated rats suggests that differentiation status of the prostate could influence the effects of testosterone and DHT on androgen-response genes. Abundant evidences exist in literature for the differences in differentiation status between the fully grown prostate and regressed prostate. Morphologically, the epithelial cells in regressed prostate are cuboidal or low-columnar, whereas the epithelial cells in fully grown prostate are tall-columnar in shape (18). Their responsiveness to androgen manipulation is also different. Androgen replacement only stimulates proliferation in regressed prostate but not in fully grown prostate (4). Finally, some prostate-specific genes, such as prostatein C3, are considered differentiation markers and only expressed in the fully grown prostate but not in the castrated prostate (Fig. 4, right panel) (28).
The androgen-response genes that are upregulated by finasteride require testosterone for their full activation. Elevated levels of testosterone in the finasteride-treated animals enhanced their expression, suggesting that these genes may be more responsive to testosterone relative to their response to DHT. In contrast, the genes that are downregulated by finasteride are likely to require DHT for their full activation. Testosterone alone can activate their expression but not to the maximum levels. For androgen-response genes that are not influenced by finasteride, their maximum activation can be stimulated by either testosterone or DHT.
At the level of androgen-response gene expression, our results suggested that testosterone and DHT have overlapping but different biological functions during prostate regrowth. The mechanism of differential activation of some androgen-response genes by testosterone and DHT is not clear. One possible explanation is that the interactions between AR and some AR associated factors may be influenced by the bound ligand, which in turn can affect the transcription of various androgen-response genes. This possibility is consistent with the fact that some of the androgen-response genes that are influenced by finasteride, prostatein C3 and calreticulin, for example, are direct androgen-response genes (28,39,40). However, our study cannot rule out the possibility that finasteride may influence the posttranscriptional regulation of some androgen-response genes. Although our data did not address the mechanism by which finasteride influences the expression of androgen-response genes, the results showed that finasteride can enhance the expression of these genes at the mRNA level. This finding is important because gene expression is commonly regulated at the level of transcription initiation.
It is novel and unexpected that inhibition of the conversion from testosterone to DHT can lead to increased expression of androgen-response genes in vivo during the regrowth of the regressed prostate. Although the affinity of testosterone to AR is lower than that of DHT, testosterone appears to be more potent than DHT in stimulating the expression of these genes. The androgen-dependent expression of these genes appears to be prostate specific and could serve as markers for androgen-dependent prostate differentiation (33). The association of the higher level expression of many androgen-response genes with the inhibition of the regrowth in the presence of finasteride also suggests that these androgen-response genes (i.e., prostatein C3) are associated with differentiation rather than proliferation during the regrowth. Thus, our studies suggest that testosterone, compared with DHT, is a more potent inducer of differentiation with a weaker influence on proliferation.
The measurement of intraprostatic testosterone and DHT showed that finasteride inhibited the conversion of testosterone to DHT and had little effect on the total amount of androgens in the regrowing prostate of castrated rats (Fig. 3). The concentration of androgens in the finasteride-treated prostate was about 19–22 ng/g, with testosterone at about 18–20 ng/g and DHT at 1.3–1.5 ng/g. The concentration of androgens in the absence of finasteride was about 17.5 ng/g, with testosterone at 3.0–3.5 ng/g and DHT at 14.0–14.5 ng/g. This result further suggests that testosterones is more potent than DHT, at similar molar concentrations, in the induction of many androgen-response genes in castrated prostate.
The concept that testosterone is a potent inducer of differentiation relative to DHT during prostate regrowth may have a potential clinical application for enhancing the intermittent androgen ablation therapy of prostate cancer. The cycling of androgens during such therapy is thought to stimulate differentiation of prostate cancer cells, which is beneficial to patients in terms of life quality and/or survival (1). According to the present study, it would be advantageous if the cycling androgen is testosterone, without DHT, resulting in strong differentiation and weak proliferation responses. Elevated levels of testosterone can be achieved by treating patients with 5α-reductase inhibitor during the off-treatment part of the therapy cycles when testicular function is recovering. The fact that the finasteride may prolong the off-treatment interval in some patients undergoing intermittent androgen ablation therapy encourages further exploration of this approach in conjunction with finasteride administration (3). Also, the differential action of testosterone and DHT on gene expression raises the possibility of developing novel androgen analogues that stimulate efficient differentiation with little or no mitogenic activity.
In summary, our studies indicate that the expression of many androgen-response genes induced by testosterone is greater when compared with that induced by DHT during the regrowth of a regressed prostate, although testosterone is less potent than DHT in stimulating regrowth of prostate. The finasteride-induced upregulation of androgen-response genes appears to be associated with differentiation rather than proliferation of prostatic cells. The recognition that testosterone is a potent differentiation inducer has potential clinical significance.
ACKNOWLEDGMENT
We thank Chung Lee for help in the dissection of ventral, lateral, and dorsal rat prostate; Mahesh Alur, Jomol Cyriac, Feng Jiang, David Klumpp, Shane Oram, Wuhan Xiao, and Qiuheng Zhang for critical readings; Borko Jovanovic for consultation on statistical analysis; and Merck Pharmaceuticals for providing finasteride. We are especially grateful to Nicholas Bruchovsky and Wade Bushman for insightful discussion. This research was supported by a NIH grant R01 51193, a grant from Merck Pharmaceutical, and DOD prostate cancer program award DAMD17-98-18543.
REFERENCES
- 1. Akakura K.; Bruchovsky N.; Goldenberg S.; Rennie P.; Buckley A.; Sullivan L. Effects of intermittent androgen suppression on androgen-dependent tumors. Apoptosis and serum prostate-specific antigen. Cancer 71:2782–2790; 1993. [DOI] [PubMed] [Google Scholar]
- 2. Avila D.; Fuqua S.; George F.; McPhaul M. Identification of genes expressed in the rat prostate that are modulated differently by castration and finasteride treatment. J. Endocrinol. 159:403–411; 1998. [DOI] [PubMed] [Google Scholar]
- 3. Bruchovsky N.; Klotz L.; Sadar M.; Crook J.; Hoffart D.; Godwin L.; Warkentin M.; Gleave M.; Goldenberg S. Intermittent androgen suppression for prostate cancer: Canadian prospective trial and related observation. Mol. Urol. 4:191–200; 2000. [PubMed] [Google Scholar]
- 4. Bruchovsky N.; Lesser B.; Doorn E. V.; Craven S. Hormonal effects on cell proliferation in rat prostate. Vitam. Horm. 33:61–102; 1975. [DOI] [PubMed] [Google Scholar]
- 5. Bruchovsky N.; Wilson J. The conversion of testosterone to 5-alpha-androstan-17-beta-ol-3-one by rat prostate in vivo and in vitro. J. Biol. Chem. 243:20122021; 1968. [PubMed] [Google Scholar]
- 6. Bruchovsky N.; Wilson J. The intranuclear binding of testosterone and 5-alpha-androstan-17-beta-ol-3-one by rat prostate. J. Biol. Chem. 243:5953–5960; 1968. [PubMed] [Google Scholar]
- 7. Chirgwin J.; Przbyla A.; MacDonald R.; Rutter W. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry 18: 5294–5299; 1979. [DOI] [PubMed] [Google Scholar]
- 8. Darras F.; Lee C.; Huprikar S.; Rademaker A.; Grayhack J. Evidence for a non-androgenic role of testis and epididymis in androgen-supported growth of the rat ventral prostate. J. Urol. 148:432–440; 1992. [DOI] [PubMed] [Google Scholar]
- 9. Deslypere J.; Young M.; Wilson J.; McPhaul M. Testosterone and 5 alpha-dihydrotestosterone interact differently with the androgen receptor to enhance transcription of the MMTV-CAT reporter gene. Mol. Cell. Endocrinol. 88:15–22; 1992. [DOI] [PubMed] [Google Scholar]
- 10. di Salle E.; Giudici D.; Biagini L.; Cominato C.; Briatico G.; Panzeri A. Effects of 5 alpha-reductase inhibitors on intraprostatic androgens in the rat. J. Steroid Biochem. Mol. Biol. 53:381–385; 1995. [DOI] [PubMed] [Google Scholar]
- 11. George F. Androgen metabolism in the prostate of the finasteride-treated, adult rat: A possible explanation for the differential action of testosterone and 5 alpha-dihydrotestosterone during development of the male urogenital tract. Endocrinology 138:871–877; 1997. [DOI] [PubMed] [Google Scholar]
- 12. George F.; Wilson J. Sex determination and differentiation. New York: Raven Press; 1994. [Google Scholar]
- 13. George F. W.; Russell D. W.; Wilson J. D. Feedforward control of prostate growth: Dihydrotestosterone induces expression of its own biosynthetic enzyme, steroid 5 alpha-reductase. Proc. Natl. Acad. Sci. USA 88:8044–8047; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Griffin J.; McPhaul M.; Russell D.; Wilson J. The androgen resistance syndromes: Steroid 5a-reductase 2 deficiency, testicular feminization, and related disorders. New York: McGraw-Hill; 1995. [Google Scholar]
- 15. Grino P.; Griffin J.; Wilson J. Testosterone at high concentrations interacts with the human androgen receptor similarly to dihydrotestosterone. Endocrinology 126:1165–1172; 1990. [DOI] [PubMed] [Google Scholar]
- 16. Imperato-McGinley J.; Sanchez R.; Spencer J.; Yee B.; Vaughan E. Comparison of the effects of the 5 alpha-reductase inhibitor finasteride and the antiandrogen flutamide on prostate and genital differentiation: Dose-response studies. Endocrinology 131:1149–1156; 1992. [DOI] [PubMed] [Google Scholar]
- 17. Lee C. Gross dissection of three lobes of the rat prostate. Prog. Clin. Biol. Res. 239:577–582; 1987. [PubMed] [Google Scholar]
- 18. Lee C.; Sensibar J.; Dudek S.; Hiipakka R. A.; Liao S. Prostatic ductal system in rats: Regional variation in morphological and functional activities. Biol. Reprod. 43:1079–1086; 1990. [DOI] [PubMed] [Google Scholar]
- 19. Liao S.; Liang T.; Fang S.; Castaneda E.; Shao T. Steroid structure and androgenic activity. Specificities involved in the receptor binding and nuclear retention of various androgens. J. Biol. Chem. 248:6154–6162; 1973. [PubMed] [Google Scholar]
- 20. Lin T.; Chang C. Cloning and characterization of TDD5, an androgen target gene that is differentially repressed by testosterone and dihydrotestosterone. Proc. Natl. Acad. Sci. USA 94:4988–4993; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mainwaring W. The mechanism of action of androgen. New York: Springer Verlag; 1977. [Google Scholar]
- 22. Nakayama O.; Hirosumi J.; Chida N.; Takahashi S.; Sawada K.; Kojo H.; Notsu Y. FR146687, a novel steroid 5 alpha-reductase inhibitor: In vitro and in vivo effects on prostates. Prostate 31:241–249; 1997. [DOI] [PubMed] [Google Scholar]
- 23. Prahalada S.; Rhodes L.; Grossman S.; Heggan D.; Keenan K.; Cukierski M.; Hoe C.; Berman C.; van Zwieten M. Morphological and hormonal changes in the ventral and dorsolateral prostatic lobes of rats treated with finasteride, a 5-alpha reductase inhibitor. Prostate 35:157–164; 1998. [DOI] [PubMed] [Google Scholar]
- 24. Rhodes L.; Primka R.; Berman C.; Vergult G.; Gabriel M.; Pierre-Malice M.; Gibelin B. Comparison of finasteride (Proscar), a 5 alpha reductase inhibitor, and various commercial plant extracts in in vitro and in vivo 5 alpha reductase inhibition. Prostate 22:43–51; 1993. [DOI] [PubMed] [Google Scholar]
- 25. Rittmaster R.; Magor K.; Manning A.; Norman R.; Lazier C. Differential effect of 5 alpha-reductase inhibition and castration on androgen-regulated gene expression in rat prostate. Mol. Endocrinol. 5:1023–1029; 1991. [DOI] [PubMed] [Google Scholar]
- 26. Rittmaster R.; Manning A.; Wright A.; Thomas L.; Whitefield S.; Norman R.; Lazier C.; Rowden G. Evidence for atrophy and apoptosis in the ventral prostate of rats given the 5 alpha-reductase inhibitor finasteride. Endocrinology 136:741–748; 1995. [DOI] [PubMed] [Google Scholar]
- 27. Shao T.; Kong A.; Marafelia P.; Cunningham G. Effects of finasteride on the rat ventral prostate. J. Androl. 14:79–86; 1993. [PubMed] [Google Scholar]
- 28. Tan J.; Marschke K.; Ho K.; Perry S.; Wilson E.; French F. Response elements of the androgen-regulated C3 gene. J. Biol. Chem. 267:4456–4466; 1992. [PubMed] [Google Scholar]
- 29. Tempany C.; Partin A.; Zerhouni E; Zinreich S.; Walsh P. The influence of finasteride on the volume of the peripheral and periurethral zones of the prostate in men with benign prostatic hyperplasia. Prostate 22: 39–42; 1993. [DOI] [PubMed] [Google Scholar]
- 30. Tenniswood M. Role of epithelial-stromal interactions in the control of gene expression in the prostate: An hypothesis. Prostate 9:375–385; 1986. [DOI] [PubMed] [Google Scholar]
- 31. Walsh P.; Madden J.; Harrod M.; Goldstein J.; MacDonald P.; Wilson J. Familial incomplete male pseudohermaphroditism, type 2. Decreased dihydrotestosterone formation in pseudovaginal perineoscrotal hypospadias. N. Engl. J. Med. 291:944–949; 1974. [DOI] [PubMed] [Google Scholar]
- 32. Wang Z.; Brown D. A gene expression screen. Proc. Natl. Acad. Sci. USA 88:11505–11509; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Z.; Tufts R.; Haleem R.; Cai X. Genes regulated by androgen in the rat ventral prostate. Proc. Natl. Acad. Sci. USA 94:12999–13004; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wilson E.; French F. Binding properties of androgen receptors. Evidence for identical receptors in rat testis, epididymis, and prostate. J. Biol. Chem. 251:5620–5629; 1976. [PubMed] [Google Scholar]
- 35. Wright A.; Douglas R.; Thomas L.; Lazier C.; Rittmaster R. Androgen-induced regrowth in the castrated rat ventral prostate: Role of 5alpha-reductase. Endocrinology 140:4509–4515; 1999. [DOI] [PubMed] [Google Scholar]
- 36. Wright A.; Thomas L.; Douglas R., Lazier C.; Rittmaster R. Relative potency of testosterone and dihydrotestosterone in preventing atrophy and apoptosis in the prostate of the castrated rat. J. Clin. Invest. 98: 2558–2563; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yamashita A.; Hayashi N.; Sugimura Y.; Cunha G.; Kawamura J. Influence of diethylstilbestrol, leuprolelin (a luteinizing hormone-releasing hormone analog), finasteride (a 5 alpha-reductase inhibitor), and castration on the lobar subdivisions of the rat prostate. Prostate 29:1–14; 1996. [DOI] [PubMed] [Google Scholar]
- 38. Zhou Z.; Wong C.; Sar M.; Wilson E. The androgen receptor: An overview [Review]. Recent Prog. Horm. Res. 49:249–274; 1994. [DOI] [PubMed] [Google Scholar]
- 39. Zhu N.; Pewitt E. B.; Cai X.; Cohn E. B.; Lang S.; Chen R.; Wang Z. Calreticulin: An intracellular Ca++-binding protein abundantly expressed and regulated by androgen in prostatic epithelial cells. Endocrinology 139:4337–4344; 1998. [DOI] [PubMed] [Google Scholar]
- 40. Zhu N.; Wang Z. Calreticulin expression is associated with androgen regulation of the sensitivity to calcium ionophore-induced apoptosis in LNCaP prostate cancer cells. Cancer Res. 59:1896–1902; 1999. [PubMed] [Google Scholar]