

Abstract
Although the existence of repressor protein(s) involved in the regulation of highly inducible metallothionein-I (MT-I) gene expression has been postulated, none has been identified to date. We considered nuclear factor I (NFI) protein as a potential repressor, as three half-sites for NFI binding are present on MT-I promoter and NFI is known to downregulate several cellular gene promoters. Overexpression of all four isoforms of mouse NFI protein (NFI-A, -B, -C, and -X) suppressed both constitutive and heavy metal-induced activation of the MT-I promoter in HepG2 cells. However, unlike other target genes of NFI, direct interaction of NFI with MT-I promoter is not necessary to mediate its repression. Point mutation of the NFI binding sites within the MT-I promoter that abrogates NFI binding in vitro could not alleviate the repression. Similarly, NFI proteins also repress activity of minimal MT-I promoter deficient in the NFI binding sites. Further, an NFI-C deletion mutant lacking the DNA binding domain continued to repress MT-I promoter. Overexpression of MTF-1, the key trans-acting factor involved in MT-I gene transcription, surmounted NFI-mediated repression of the basal and zinc-induced MT-I promoter activity. These data demonstrate that NFI is a repressor of MT-I expression, where its DNA binding activity is not essential to downregulate the MT-I promoter. Interaction of NFI with another protein(s), probably MTF-1, may be involved in this repression.
Keywords: Nuclear factor I (NFI), Metallothionein Metal, transcription factor-1 (MTF-1)
METALLOTHIONEINS are a group of highly conserved, ubiquitously expressed, cysteine-rich metal binding proteins that are implicated in maintaining metal ion homeostasis and scavenging free radicals [for recent reviews, see (16,29,37)]. Four isoforms of metallothionein (MT) have been identified in mammals that are arranged in tandem in mouse chromosome 8 and human chromosome 16 (30,46). Unlike rodents, humans have several variants of MT-I genes (42,50). Among the four isoforms, the expression of murine MT-I and MT-II is coordinately regulated in all tissues (54), whereas MT-III expression is restricted to the glutaminergic neurons of the brain (43) and MT-IV is expressed in stratified squamous epithelium of skin and tongue (46). Unlike the constitutively expressed MT-III and MT-IV isoforms, MT-I and MT-II are highly inducible, and the positive regulators include heavy metal ions, reactive oxygen species, and UV radiation as well as different physiological and pathological stimuli such as infection, inflammation, and stress (24,27,29,42). Significant induction of MT-I and MT-II has also been reported in the liver of Cu, Zn superoxide dismutase (SOD) knockout mice (24,27,29,42). In most cell lines and tissues, the constitutive expression of MT proteins is usually low, which increases dramatically in response to various inducers by transcriptional activation of the respective gene (22,29). The molecular mechanism of induction of MT-I and MT-II genes has been explored in great detail (4,42,50,53). The metal response elements (MREs) through which the metal ions exert their effects consist of multiple copies of 13–15-bp imperfect repeats in either orientation. The MRE consensus sequence is CTCTGCRCBCSGCCC, where the residues represented in bold letters are absolutely required for heavy metal response, R is a purine, B is any base other than A, and S represents G or C (42,53). Several copies of MREs on MT promoters act cooperatively for maximal induction. Metal transcription factor-1 (MTF-1) is a well-characterized MT-I transcription factor that is essential for the basal and induced expression of MT-I/MT-II genes (13,21,23,47). Although this protein binds to all MREs, its affinity is highest for MRE-d, which is also the most potent cis element to mediate the heavy metal response (3). In vivo genomic footprinting studies have shown that MTF-1 occupies the MREs in response to heavy metals as well as free radical generators (13,33,38). Apart from MTF-1, Sp-1 and MLTF/USF also stimulate the basal expression of MT-I by binding to GC box and MLTF/ARE (adeno major late transcription factor/antioxidant response element) sequences, respectively.
In the course of our investigation to identify a repressor of MT-I gene expression in animal cells, we recently discovered that different Rat 1 cell-derived clonal cells that express the large subunit (p80) of the autoantigen Ku are unable to induce MT-I/MT-II genes in response to heavy metals. The wild-type cells or the cells overexpressing the small subunit (p70) continue to induce MT (17). Subsequent study showed that the major molecular mechanism for the suppression of MT-I expression in these cells is due to hypermethylation of CpG islands within its promoter. Similar inactivation of MT-I promoter activity by methylation was also observed in mouse lymphosarcoma cells (as opposed to the parental mouse thymocytes) and a rat hepatoma (as opposed to the parental liver tissue) (18,34). The methylation-induced suppression of MT promoter appears to be mediated by a repressor complex consisting of a methyl C binding protein (MeCP) and co-repressors such as histone deacetylase (7).
In the present study we explored the possibility that NFI may function as another repressor of MT expression. NFI belongs to a protein family ubiquitously expressed in higher eukaryotes, and is known for its ability to downregulate the promoter activity of some genes in a cell type-specific manner. Four highly conserved but distinct genes encoding NFI (NFI-A, NFI-B, NFI-C, and NFI-X) have been identified (35,45,49,51). Multiple products are generated from each of these genes by alternative splicing, with predicted molecular masses that range from 27 to 62 kDa (25,262,28). All NFI isoforms bind as homo- or heterodimers to the consensus sequence TTGGCN5G CCAA (40). The DNA binding domain resides in the N-terminal 220 amino acids of the protein and is highly conserved among the different isoforms. The trans-activation property is dictated by the C-terminal end, which shows greater extent of sequence variation (20,36). Human NFI-C was initially purified from HeLa nuclear extract as a factor that is necessary for efficient initiation of adenovirus replication in vitro (41). Subsequently, different isoforms of NFI have been implicated as both positive and negative modulators of different cellular gene promoters.
Upon analysis of MT-I promoter, we observed the presence of three half-sites for NFI binding. It is logical to postulate that NFI protein interacts with any one or all of the three NFI sites on MT-I promoter and modulates MT expression. We report that all four isofoms of mouse NFI as well as NFI-L cloned from rat liver downregulate both constitutive and heavy metal-inducible activity of the MT-I promoter. Unlike all other target genes of NFI, direct interaction of NFI protein with the cognate binding site on the promoter is not required to downregulate MT-I promoter activity.
MATERIALS AND METHODS
Plasmid Construction and Mutagenesis
The plasmid pMT-Luc was constructed by ligation of SmaI-BglII fragment (−292 to +64) of the plasmid pMT-IΔi (19) to the SmaI-BglII site of pGL2-basic vector containing firefly luciferase as reporter gene (Promega). We also generated a minimal mouse MT-I promoter/Luciferase reporter vector (MMP/Luc), by ligating −33 to +18 bp of the mouse MT-I promoter to the BglII-HindIII site of the pGL2-basic vector. The minimal MT-I promoter is BamHI-HindIII fragment of the plasmid 5′-delta33 (12). The reporter plasmids p(MRE-c′)4 and p(MRE-d)5 were generated by ligating four copies of MRE-c′ oligo and five copies of MRE-d oligo in tandem, respectively (see below), upstream of the MT-I minimal promoter in MMP/Luc. Similarly, p(MRE-c′/d) was generated by ligating (MRE-d)5 upstream of MRE-c′ oligomer of p(MRE-c′)4.
MRE-c′ oligo: 5′-GATCGAAAAGTGCGCTCGGCTCT GCCAAGG-3′
MRE-d oligo: 5′-GATCCGAGGGAGCTCTGCACTCCG CCCGAAAAGTG-3′
The reporter plasmid pMRE-c′mut was generated by site-directed mutation of MRE-c′ sequence of the pMT-Luc plasmid by multiple rounds of PCR using appropriate primers with altered bases (2). Similarly, the pNFImut plasmid was generated using appropriate oligos to mutate NFI binding half-sites of the plasmid pMRE-c′mut. The primer pairs used to make pMRE-c′mut plasmid were:
-
Pair 1: MTI-BglII: 5′-CCAACAGTACCGGAATGCCAAG-3′
MRE-c′-mut1: 5′-GAAAAGTGCGCTCGGCTCTTAAAAGG-3′
-
Pair 2: MTI-SmaI: 5′-GGTACTGTAACTGAGCTAACATAACCCGGG-3′
MRE-c′-mut2: 5′-CCTTTTAAGAGCCGAGCGCACTTTTC-3′
The mutated bases are in bold and underlined.
The primer pairs used to create the pNFImut plasmid were:
-
Pair 1: MTI-BglII: 5′-CCAACAGTACCGGAATGCCAAG-3′
MRE-c′-mut4: 5′-GGAGCAAAATCCTGCTGGGTGCAAAAAT TTTGC-3′
-
Pair 2: MTI-SmaI: 5′-GGTACTGTAACTGAGCTAACATAACCCGGG-3′
MRE-c′-mut3: 5′-CGAAAATTTTTGCACCCAGCAGG-3′
The mutated bases are in bold and underlined.
The construction of eukaryotic expression vectors pCHmNFI-A2, pCHmNFI-C, pCHmNFI-B, and pCHmNFI-X expressing the mouse NFI-A, -C, -B, and-X proteins, respectively, and pCHmNFI-C-160, pCHmNFI-C-240 expressing truncated NFI-C proteins were described previously (8). The plasmid RSV-NFL21 expressing the spliced variant of the rat liver NFI isoform was a generous gift from Dr. R. Cortese (IRBM, Rome, Italy) (45).
Cell Culture and Transfection Assays
HepG2 cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS). For transfection assay, 1.0 × 105 cells were plated onto 35-mm dishes 24 h prior to transfection and then transfected using DOTAP (Boehringer Mannheim) following the manufacturer’s protocol. Normally, each transfection cocktail contains a maximum of 3 μg of DNA that includes the reporter plasmid, pRLTK (renila luciferase reporter driven by HSV-tk promoter, Promega), as internal control (1/10th the amount of the reporter plasmid) and eukaryotic expression vector harboring the gene of interest in DOTAP (described in the respective figure legends). The cells were allowed to incubate in the presence of the transfection mixture in complete medium (DMEM + 10% FBS) for 16 h in a 37°C incubator with 5% CO2, followed by replacement with fresh medium. After 24 h in the fresh medium, the cells were treated with heavy metals for 6 h or as indicated, harvested in lysis buffer (Promega), and luciferase activity was measured using Dual luciferase assay kit (Promega) in a Luminometer (Lumat LB 9507; EG&G Berthold, Oak Ridge, TN).
Electrophoretic Mobility Shift Assay
Whole cell extracts used for the DNA binding activities of NFI proteins were prepared as described previously (17), incubated with [γ-32P]ATP-labeled MRE-c′ duplex probe, and resolved on a 6% nondenaturing polyacrylamide gel (5). The NFI antibody used for supershift experiment was a generous gift from Dr. N. Tanese (New York University, New York, NY) (52). The wild-type and mutated MRE-c′ oligonucleotides used for competition assay are as described in the previous section for generating reporter plasmids. The NFI consensus and the mutated oligos are from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). For measuring DNA binding activity of Spl and MTF-1 [α-32P]GMP-labeled MRE-d oligonucleotide was used as probe.
RESULTS
NFI Family of Proteins Can Repress MT-I Promoter Activity That Harbors Half-Site for NFI Binding
EMSA and Antibody Supershift Shows That NFI Family of Proteins Can Interact With MRE-c′ Element on MT Promoter
Previous study in this laboratory established the functional significance of a 26 base pair sequence MRE-c′ located on the mouse MT-I promoter, which encompasses the metal regulatory element MRE-c and additional 5′ and 3′ sequences (14). Analysis of the MRE-c′ element revealed that this sequence contains a half-site for NFI binding (Fig. 1A). Computer-aided analysis of MT-I promoter proximal to the +1 site also shows the presence of two additional half-sites for NFI binding around the same region. One of these sites overlaps with the MRE-b element and the other lies between the MLTF-ARE and MRE-b sites (Fig. 1A, B).
Figure 1.
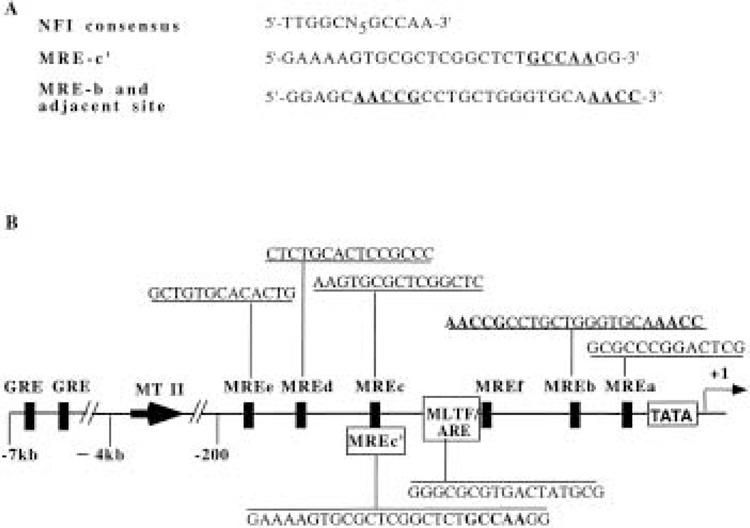
MT-I promoter harbors three half-sites for NFI binding. (A) Comparison of nucleotide sequence of MRE-c′ and MRE-b with NFI consensus element. Common nucleotides among the cis elements are shown in bold letters. (B) Schematic diagram of mouse MT-I promoter, depicting the cis elements. NFI half-sites are shown in bold letters and underlined.
The whole cell extracts or nuclear extracts from different cell lines that can induce MT-I gene formed an array of DNA/protein complexes upon incubation with 32P-labeled MRE-c′ oligo. The multiple complexes formed between whole cell extract from HepG2 cells and MRE-c′ oligo are presented in Figure 2. To determine the potential interaction of NFI proteins with MRE-c′ element, the competition of the MRE-c′ binding complexes with NFI consensus oligo was performed. A 50-fold molar excess of unlabeled NFI oligo was more potent in competing out the complexes than a 100-200-fold molar excess of cold MRE-c′ oligo (Fig. 2A, compare lanes 2, 3 with 6, 7). Both NFI mutant and MRE-c′ mutant oligos failed to disrupt the formation of DNA/protein complexes, which confirmed the specificity of the reaction between MRE-c′ oligo and NFI (Fig. 2A, lanes 4, 5 and 8, 9). This conclusion was further substantiated by supershift of the DNA/protein complexes attained after preincubation with anti-NFI antisera (Fig. 2B). When anti-NFI antiserum was added to the binding reaction, a fraction of the complexes formed with MRE-c′ oligo were supershifted whereas addition of normal rabbit serum had no such effect. This observation further indicates that the protein(s) binding to MRE-c′ oligo belongs to the NFI family. The binding of NFI proteins to MRE-c′ coupled with the existence of several half-sites for NFI binding on the MT-I promoter suggested that NFI proteins may modulate MT-I gene expression.
Figure 2.
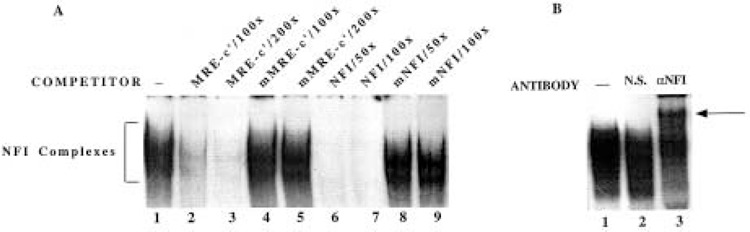
Factors forming complex with MRE-c′ oligo belong to NFI family of proteins. Whole cell extract (10 μg) from HepG2 cells (lane 1) was incubated with 0.1–0.2 ng of 32P-labeled MRE-c′ oligo at 4°C under optimum binding conditions and subjected to electrophoresis on polyacrylamide (6% acrylamide) gel. (A) To identify the proteins forming the complexes, the extract was preincubated with either of the following unlabeled oligos: (a) 100- or 200-fold molar excess of MRE-c′ (lanes 2 and 3), (b) MRE-c′ mutant (lanes 4 and 5), (c) 50- or 100-fold molar excess of NFI consensus oligo (lanes 6 and 7), or (d) NFI mutant (lanes 8 and 9) along with 0.5 μg of poly(dI-dC) as nonspecific competitor. (B) The extract was also preincubated separately with l μl of normal rabbit serum (lane 2) or anti-NFI antisera (lane 3) for 30 min on ice prior to addition of the labeled probe. The arrow indicates the position of the supershifted complex.
NFI Overexpression Downregulates MT-I Promoter Activity in HepG2 Cells
The presence of NFI proteins in the cell and their ability to bind MT-I promoter prompted us to investigate further their role in MT-I gene transcription. To study the effect of NFI proteins on the MT-I promoter activity, we performed a series of transient transfection assays using a promoter/reporter construct containing MT-I promoterdriven luciferase reporter gene (pMT-Luc) and eukaryotic vector expressing NFI proteins. When HepG2 cells were transiently transfected with pMT-Luc and treated with 10 μM cadmium sulfate, the MT-I promoter was activated nearly sixfold. In contrast, when the NFI-C expression vector was cotransfected with pMT-Luc, both the basal and cadmium-induced activities were inhibited by 90% and 89%, respectively (Fig. 3A). The empty vector for NFI expression plasmid had no such effect (data not shown). The inhibition of MT-I promoter was observed with all four isoforms of NFI (NFI-A, NFI-B, NFI-C, and NFI-X), although the extent of inhibition varied with the isoform (Fig. 3A, B). These four NFI variants, encoded by different genes, contain nearly identical N-terminal DNA binding domains but different C-terminal domains (20,36). The expression levels of all four isoforms of NFI upon transfection were comparable by Western blot analysis of the HA-tagged proteins (data not shown). The different degree of inhibition by different isoforms may be due to the sequence variation at the carboxy-terminal end that dictates the trans-activating/repressing property or difference in their capability to interact with co-repressor protein(s). The isoform expressed in rat liver, NFI-L2i (an NFI-A gene product), also showed the inhibitory effect on both constitutive and heavy metal-inducible activity of MT-I promoter. The extent of inhibition by this isoform, however, was less than that achieved with the other isoforms (Fig. 3B). This study was further extended to determine the effect on zinc-stimulated MT-I promoter activity by NFI-B and NFI-L. The zinc-induced and the basal MT-I promoter activity were inhibited by these NFI isoforms (Fig. 3B). These data indicate that all NFI isoforms that are highly conserved from human to chicken inhibit both basal and heavy metal-induced MT-I promoter activity in HepG2 cells.
Figure 3.
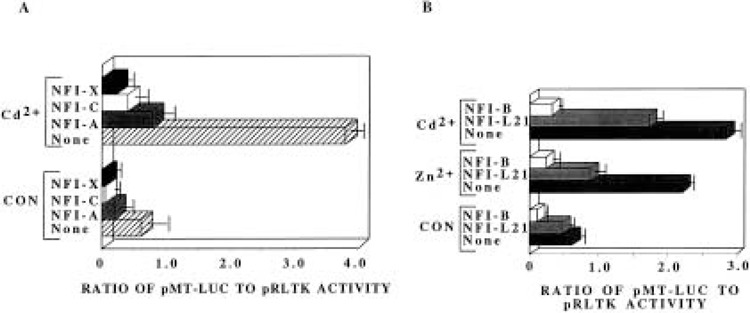
Both constitutive and heavy metal-induced activities of the MT-I promoter are downregulated by NFI family of proteins. (A) HepG2 cells were transfected with the reporter plasmid pMT-Luc (1.5 μg), the internal control pRLTK (0.15 μg), CMV control vector (1.5 μg), or CMV vectors expressing NFI-A, -C, and -X proteins (1.5 μg). Transfected cells were incubated with or without 10 μM cadmium sulfate for 6 h prior to harvest. Luciferase activity was determined by dual luciferase assay kit where renila luciferase activity (pRLTK) was used to normalize the transfection efficiency. The bars represent the mean and range of data from triplicate assays of three independent transfections. (B) HepG2 cells were transiently transfected (as stated in A) except that NFI-B and NFI-L expression vectors were cotransfected here. Transfected cells were either left untreated or treated with 100 μM zinc sulfate or 10 μM cadmium sulfate for 6 h prior to harvest.
The NFI-Mediated Repression of MT-I Promoter Activity Does Not Require NFI-DNA Interaction
Site-Directed Mutation of MRE-c′ Element and Other Putative NFI Binding Sites on MT-I Promoter Has No Effect on the NFI-Mediated Repression
To address the contribution of DNA binding activity of NFI in mediating MT-I suppression, the MRE-c′ element of the pMT-Luc plasmid was subjected to site-directed mutation that abrogated binding of NFI proteins to MRE-c′ element (pMRE-c′mut) (see Materials and Methods). As shown by in vitro binding assay (Fig. 2A, lanes 4 and 5), 100- or 200-fold molar excess of the mutant MRE-c′ oligo failed to compete out the formation of DNA/protein complex between HepG2 whole cell extract and wild-type MRE-c′ oligo. Further, HepG2 whole cell extract did not form a specific complex with 32P-labeled mutant MRE-c′ oligo (data not shown). When HepG2 cells were transfected with pMRE-c′mut plasmid, the basal promoter activity was comparable to that of cells transfected with pMT-Luc (Fig. 4, compare lanes 1 and 5). This is possible only if binding of NFI to MRE- c′ is not essential for the suppression of the basal promoter activity. When these cells were treated with 10 μM cadmium sulfate for 6 h, both pMT-Luc and pMRE-c′mut promoters were activated to the same level (4.5-fold, Fig. 4, lanes 2 and 6). Cotransfection with NFI-C expression vector inhibited both pMT-Luc and the pMRE-c′mut promoter to similar extent (68–72% of the basal, Fig. 4, compare lanes 1, 3 with 5, 7; 75-78% of the cadmium-induced activity, compare lanes 2, 4 with 6, 8). Although inhibition of NFI binding to MRE-c′ could not abrogate the repression observed upon NFI overexpression, it was conceivable that NFI proteins bind to the other half-site(s) present on the MT-I promoter. To eliminate this possibility both NFI sites of the plasmid pMRE-c′mut were mutated to generate pNFImut. HepG2 cells were transiently transfected with pNFImut and cotransfected with or without NFI overexpression vector. The extent of inhibition of the constitutive and metal-induced activity of the plasmid pNFImut by NFI-C was comparable to that of the wild-type pMT-Luc and the pMRE-c′mut promoters (Fig. 4). These data indicate that although there are potential binding sites for NFI protein on the MT-I promoter, the repression of the promoter is not mediated by conventional DNA-protein interaction.
Figure 4.
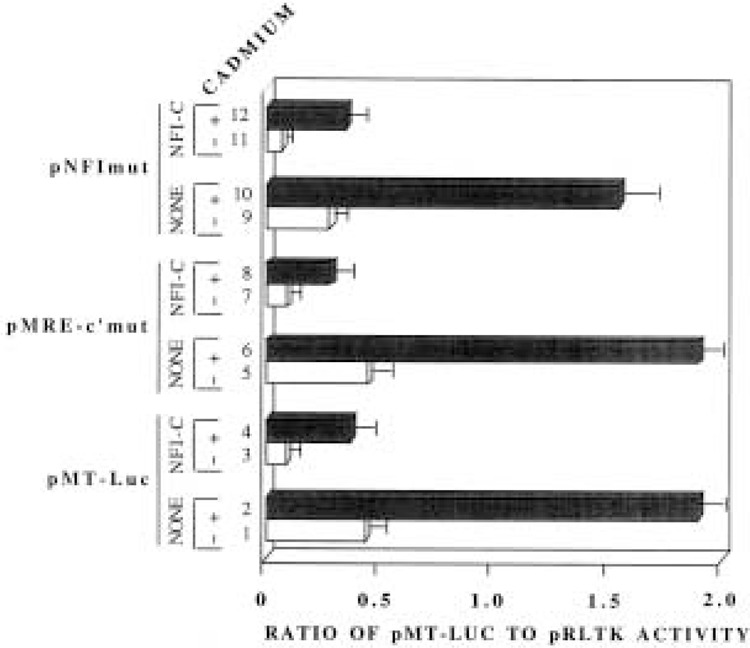
Mutation of NFI binding site on the MT-I promoter that abrogates binding of NFI protein does not abolish the repressor action of NFI proteins on the promoter. HepG2 cells were transfected with either pMT-Luc, pMRE-c′mut, or pNFImut (1.5 μg), pRLTK (0.15 μg), CMV control vector (not shown), or CMV vector expressing NFI-C (1.5 μg). Reporter plasmids are described in Materials and Methods. Transfected cells were either treated with or without 10 μM cadmium sulfate 6 h before harvesting. Ratio of reporter gene activity to that of the internal control is presented in the bar diagram. This is representative of two independent experiments with triplicate assays.
Synthetic MT-I Promoter Lacking NFI Binding Site Is Also Downregulated by NFI Proteins
To prove further that DNA binding activity of NFI is not required for suppression of MT-I promoter, we constructed plasmids containing synthetic promoters lacking NFI DNA binding site. These plasmids consist of minimal mouse MT-I promoter (–33 to + 18) tagged to the luciferase reporter vector, with a) four copies of MRE-c′ oligo [p(MRE-c′)4], b) five copies of MRE-d oligo [p(MRE-d)5], and c) combination of both MRE-c′ and MRE-d [p(MRE-c′/d)], ligated upstream of the minimal promoter (Fig. 5A). When HepG2 cells were transfected with these plasmids, p(MRE-c′)4 showed very low constitutive and heavy metal (both Zn2+, Fig. 5B, lanes 1 and 2, and Cd2+, data not shown) inducible activity that was further inhibited upon NFI overexpression (Fig. 5B, compare lanes 1, 2 with 3, 4, respectively). Whether or not NFI binds to the MRE-c′ multimer in vivo, this result can be anticipated. Transfection of cells with p(MRE-d)5 showed a much higher level (fivefold) of constitutive and heavy metal-inducible activity (12-fold, ZnSO4, Fig. 5B, compare lanes 1, 2 with 5, 6; eightfold CdSO4, data not shown) when compared to promoter activity with transfected p(MRE-c′)4. This observation is expected, as MRE-d is the most potent cis element that imparts heavy metal response to the basal promoter (42) and it does not contain an NFI binding site. Overexpression of NFI-C, however, resulted in inhibition of the promoter activity (49% basal; 60% Zn2+ induced) using this plasmid, despite the absence of a cis element capable of NFI binding (Fig. 5B, compare lanes 5, 6 with 7, 8, respectively). The difference in percentage inhibition by NFI-C of pMT-Luc (wild-type promoter) and p(MRE-d)5 (synthetic promoter) can be attributed to the difference in nature of the promoter. Nevertheless, this result confirms the earlier data (see Fig. 4) indicating that NFI-promoter interaction is not essential for NFI-mediated repression of MT promoter activity. A much higher constitutive and metal-induced activity was observed when HepG2 cells were transfected with the plasmid p(MRE-c′/d), as multiple MREs were incorporated into the promoter. These activities were again downregulated to the same extent as the other plasmids when NFI-C was overexpressed (Fig. 5B, compare lanes 9, 10 with 11, 12, respectively).
Figure 5.
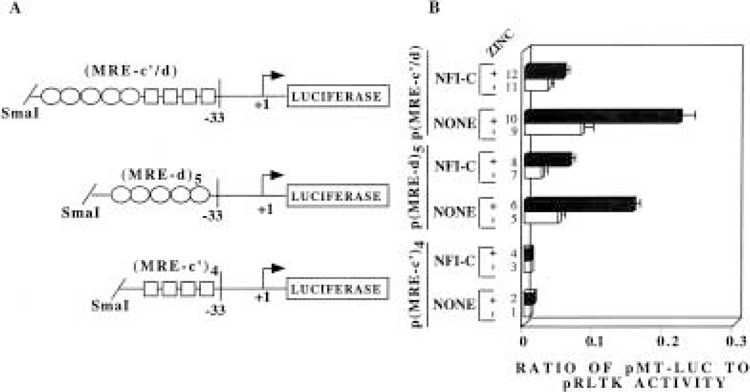
NFI proteins can downregulate both the constitutive and heavy metal-induced activity of the MT-I promoter irrespective of the presence of cognate cis element. (A) Schematic diagram of the promoter/reporter plasmids constructed (described in Materials and Methods). The boxes represent MRE-c′ sequences and the ovals represents MRE-d sequence. (B) Transient transfection of HepG2 cells and subsequent zinc treatment of the transfected cells are as described in the previous experiment (Fig. 3). The promoter/reporter plasmids transfected in HepG2 cells are either p(MRE-c′)4, p(MRE-d)s, or p(MRE-c′/d) (1.5 μg). The data are representative of three independent experiments.
An NFI-C Deletion Mutant Lacking DNA Binding Domain Can Still Repress MT-I Promoter
We used an NFI-C deletion mutant lacking the DNA binding domain to validate the notion that, unlike other genes, NFI-mediated suppression of MT-I does not involve DNA-protein interaction. The constructs expressing N-terminal 160 amino acids (NFI-C-160) and 240 amino acids (NFI-C-240) of the NFI-C protein were transfected into HepG2 cells along with pMT-Luc. The NFI-C-160 plasmid expresses truncated NFI-C protein that lacks the ability to bind to the cognate cis element whereas the protein expressed from NFI-C-240 plasmid can bind DNA (8). As expected, both basal and zinc-induced activities of the MT-I promoter in HepG2 cells were repressed when full-length NFI-C protein was expressed. The MT-I promoter was suppressed to a comparable extent when HepG2 cells were cotransfected with either NFI-C-240 or NFI-C-160 expression vectors (Fig. 6). The expression of the truncated proteins was verified by Western blot analysis of whole cell extracts prepared from the transfected cells (data not shown). Despite the absence of the DNA binding domain in NFI-C-160, it can repress both basal and heavy metal (zinc as well as cadmium, data not shown) induced MT-I promoter activity. This result further substantiates our conclusion that the DNA binding activity of NFI protein is not essential for the downregulation of MT-I promoter in HepG2 cells.
Figure 6.
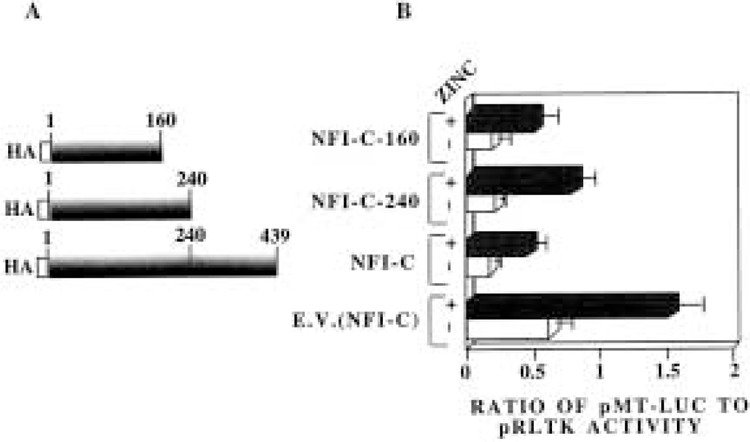
NFI-C truncated proteins lacking DNA binding domain can still exert its repressor action on the constitutive and heavy metal-induced MT-I promoter activity. (A) Schematic diagrams of HA-tagged NFI-C carboxy-terminal deletion constructs. (B) HepG2 cells were transfected with pMT-Luc reporter plasmid (1.5 μg), pRLTK (0.15 μg), and CMV vector expressing full-length NFI-C (pCHmNFI-C), or N-terminal 240 amino acids of NFI-C (pCHmNFI-C-240) or N-terminal 160 amino acids of NFI-C (pCHmNFI-C-160) (1.5 μg). Transfected cells were treated for 6 h with or without zinc sulfate (100 μM). The bars represent mean and range of data from three independent transfections with duplicate assays.
NFI Overexpression May Inhibit MT-I Promoter Activity by Compromising MTF-1 Function
Overexpression of MTF-1 Can Overcome the Inhibitory Effect of NFI Protein on MT-I Promoter
MTF-I is the key trans-acting factor required for the basal and induced expression of MT-I in response to heavy metals and reactive oxygen species (21). Genetic (47) and biochemical (3) data suggested the existence of a putative repressor that regulates MT-I expression. The observation that NFI proteins can repress both the basal and metal-induced MT-I promoter activity, irrespective of its DNA binding activity, led us to postulate that overexpression of MTF-1 (that is known to interact with all known MREs) may relieve NFI-mediated suppression of MT-I promoter. To test this possibility, we cotransfected HepG2 cells with pMT-Luc as well as NFI-C and MTF-1 expression vectors, and studied the effect on basal, cadmium-induced (Fig. 7A), and zinc-induced promoter activities (Fig. 7B). Following overexpression of NFI-C, the basal MT-I promoter activity was down-regulated by 72%, cadmium-induced expression by 77% (Fig. 7A, compare lanes 5, 6 with 1, 2, respectively), and zinc-induced activity by 83% (Fig. 7B, lanes 2 and 4). The control vector for NFI-C exhibited negligible effect on the basal or heavy metal-induced activity of the MT-I promoter (Fig. 7A, lanes 1, 2 and 3, 4). When the expression vectors harboring both mouse MTF-1 and NFI-C were introduced into HepG2 cells in equal amounts, the basal (Fig. 7B, lanes 1, 3, and 11) and zinc-induced (Fig. 7B, lanes 2, 4, and 12) MT-I promoter activities could be restored almost to their normal level even in the presence of NFI-C. However, there was only partial recovery of cadmium-induced MT-I promoter activity from NFI repression when MTF-1 was overexpressed (Fig. 7A, lanes 2, 6, and 14). Cotransfection of empty vector for MTF-1 could not abrogate the NFI-C-mediated inhibition of the promoter (Fig. 7A, compare lanes 5, 6 with 11, 12, respectively, and Fig. 7B, lanes 3, 4 with 9, 10, respectively). This result suggests that MTF-1 can overcome the inhibitory effect of NFI proteins on the constitutive and zinc-induced MT-I transcription. The inability of MTF-1 in restoring the cadmium-induced MT-I promoter activity can be attributed to the notion that an additional yet unidentified signaling mechanism is involved in MT-I activation by cadmium (21) that might be compromised by NFI overexpression. MTF-1 overexpression alone will not, therefore, restore the cadmium-induced promoter activity.
Figure 7.
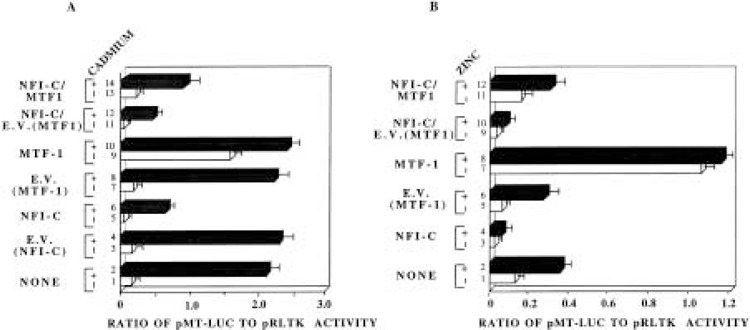
Overexpression of MTF-1 protein can recover the basal and zinc-induced MT-I promoter activity from NFI-mediated repression. HepG2 cells were transfected with pMT-Luc (1.0 μg), pRLTK (0.1 μg), and either of the following vectors (1.0 μg): (a) CMV control vector (E.V. NFI-C), (b) CMV vector expressing NFI-C, (c) control vector for MTF-1 (E.V. MTF-1), (d) MTF-1 expression vector or a combination of (b) and (c) or (b) and (d) (1.0 μg each), as indicated in the figure. Transfected cells were either left untreated or treated with 10 μM cadmium sulfate (A) or 100 μM zinc sulfate (B) 6 h prior to harvest. The data are representative of three independent experiments.
The expression of MTF-1 alone stimulated the constitutive MT-I promoter activity as much as 10fold, but no further significant stimulation was observed in the presence of cadmium (Fig. 7A, compare lanes 1, 2 with 9, 10, respectively) or zinc (Fig. 7B, compare lanes 1, 2 with 7, 8, respectively). This observation is consistent with the significant increase in the basal MT-I promoter activity but not metal-dependent MT expression observed after MTF-1 overexpression (47,48). This is most likely due to saturation of all the cognate binding sites on the promoter by the overexpressed MTF-1.
Overexpression of NFI Has No Effect on the DNA Binding Activity of MTF-1
It is well established that the key trans-activator of MT-I gene is MTF-1. Because overexpression of MTF-1 in transient transfection studies can alleviate the NFI-mediated repression of the MT-I promoter activity, it is possible that NFI overexpression might downregulate MTF-1 expression, consequently affecting its DNA binding activity. To address this issue, we prepared whole cell extract from HepG2 cells transfected with different NFI isoforms (A–X) and performed EMSA with radiolabled MRE-d oligonucleotide as probe. As can be seen in Figure 8, the DNA binding activity of MTF-1 in cells overexpressing NFI isoforms was comparable to that from cells transfected with the empty vector. The Sp1 DNA binding activity also remained unaltered upon overexpression of NFI proteins. These data suggest that NFI has no direct effect on the DNA binding activity of MTF-1.
Figure 8.
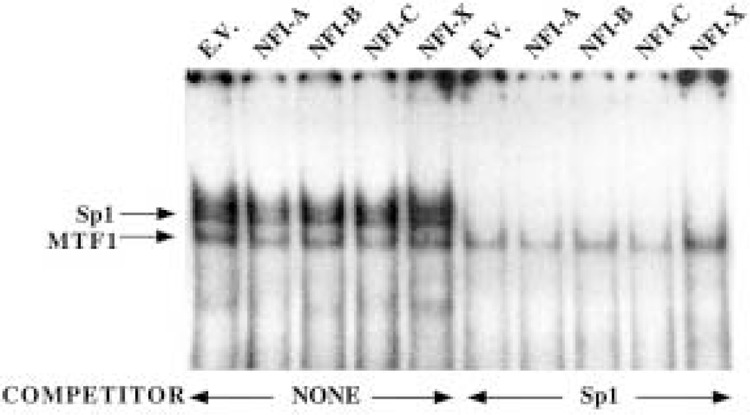
DNA binding activity of MTF-1 is not affected by NFI overexpression. Whole cell extract (10 μg) from HepG2 cells transfected with CMV control vector or different isoforms of NFI protein (NFI-A, -B, -C, or -X) was incubated with 0.1–0.2 ng of 32P-labeled MRE-d oligo at 4°C under optimum binding conditions and subjected to electrophoresis on polyacrylamide (6% acrylamide) gel. The extracts were preincubated with or without unlabeled Sp1 oligo as indicated to disrupt the Sp1 complex, so that specific MTF-1/MRE-d complex is distinctly visible.
DISCUSSION
The NFI family of proteins has been implicated in regulating the expression of many cellular genes (15). The 20 different spliced variants coded by four different genes isolated so far modulate positively or negatively both constitutive and inducible activity of different promoters. In all these cases, the binding of NFI protein to the promoter has been shown to be absolutely essential for the modulatory effect. The consensus sequence for NFI binding has been shown to be TTGGCN5GCCAA. Although most of the NFI target promoters like GLUT4 (10), PEPCK (phosphoenol pyruvate carboxy kinase) (11,31), peripherin (1), or α1B-adrenergic receptor (15) harbor a complete cis element for NFI binding, some genes like MT-I contain only half-site(s) for NFI binding (6,32). Irrespective of the presence of half-site or a complete cis element on the promoter, the DNA binding is crucial for NFI action.
The present study has demonstrated that the NFI family of proteins negatively regulates MT-I gene expression. The unique finding is that unlike the NFI-mediated suppression of other genes, the negative regulation of MT-I promoter does not involve direct NFI-promoter interaction. To our knowledge, this is also the first report of a specific protein that down-regulates MT promoter under the basal or heavy metal-induced conditions. The data presented in this study do not suggest binding of NFI proteins to a negative regulatory element on MT promoter. Rather, it is likely that protein–protein interaction is responsible for the suppression of MT-I promoter activity. First, the integrity of the NFI binding site on the MT-I promoter is not necessary for the suppression of the promoter. Second, the activity of a minimal MT-I promoter containing only MTF-1 binding sites can also be impeded by NFI. Finally, deletion mutants of NFI protein lacking functional DNA binding domain can repress the MT-I promoter activation. Because NFI overexpression does not affect the DNA binding activity of MTF-1 in vitro, NFI could either block the interaction of MTF-1 with a coactivator or a basal transcription factor by suppressing its trans-activation domain or prevent its binding to MREs by masking its DNA binding domain in vivo. Alternatively, NFI may function independently of MTF-1 by interacting with a component of the basal transcription machinery. Here again, overexpression of MTF-1 could surmount the suppressor action of NFI.
Interestingly, different isoforms of NFI can either repress or activate the same gene, an effect most likely due to distinct C-terminal domain on the different isoforms (36). For example, NFI-A and NFI-B stimulate the basal transcription of PEPCK gene promoter, but it is inhibited moderately by NFI-C and NFI-X in HepG2 cells (11). NFI-C and NFI-X, but not NFI-A or NFI-B, inhibit glucocorticoid-induced MMTV promoter activity in HeLa cells (9). A specific NFI isoform can either activate or repress the same promoter in a cell-specific manner. For example, NFI-C and NFI-X can repress the glucocorticoid-induced MMTV promoter activity in HeLa and COS-1 cells, whereas this repression is not observed in JEG-3 choriocarcinoma cells or 293 cells (9). Similarly, NFI/L (rat liver NFI-A isoform) or NFI/RedI (hamster liver NFI-B isoform) can activate au-adrenergic receptor gene promoter in primary rat hepatocytes, but acts as repressor in DDT1 MF-2 smooth muscle cells (15). This differential effect of NFI is most likely mediated through its interaction with a cell-specific coactivator/repressor. Alternatively, the cell type-specific accessibility of NFI to the promoter may determine whether NFI will act as an activator or a repressor (39). In the present study with HepG2 cells, we observed consistent inhibition of constitutive and metal-induced activity of the MT-I promoter by all four mouse isoforms as well as the rat isoform NFI-L21. This observation indicates that the repressor domain of the NFI isoforms is retained within the conserved region of these molecules. The continued repression of MT-I promoter by NFI-C deletion mutants (NFI-C-160 and NFI-C-240) lacking the C-terminal domain further supports the notion that the nonconserved C-terminal domain may not play any role in MT-I repression.
Despite the constitutive presence of the positive transcription factor MTF-1 in cells, the basal expression of MT-I is usually low in most cells/tissues, suggesting the existence of a repressor that blocks MTF-1 function. Such a notion is consistent with the increased accumulation of MT-I mRNA observed after inhibition of protein synthesis by cycloheximide (3,44). The MT induction by this agent is observed even when minimal MT-I promoter is transcribed by multiple copies of MRE-d, implying that the labile repressor inhibits MTF-1 function (44). However, no such protein has been identified. Alternatively, the repressor may indirectly affect MTF-1 function by competing for similar or overlapping binding sites on the promoter. This does not appear to be the mechanism for the downregulation of MT-I promoter as in vivo footprinting studies did not reveal occupancy of MREs by a repressor in unstimulated cells (18,33). Although our study suggests potential modification of MTF-1 by NFI, it is unlikely that NFI is related to the cycloheximide-sensitive repressor because of its relative stability (Majumder and Jacob, unpublished data). It is, therefore, logical to conclude that more than one mechanism operates to repress MT promoter.
ACKNOWLEDGMENTS
We are grateful to Dr. Walter Shaffner for MTF-1 expression vector, Dr. Richard Palmiter for mouse MT-I minigene, Dr. Naoko Tanase for anti-NFI antibody and Dr. Riccardo Cortese for NFL21 expression vector. This research was supported, in part, by a grant ES10874 (S.T.J.) for the National Institute of Environmental Health Sciences, and by grant HD-34908 (R.M.G.) from the National Institute of Health.
REFERENCES
- 1. Adams A. D.; Choate D. M.; Thompson M. A. NF1-L is the DNA-binding component of the protein complex at the peripherin negative regulatory element. J. Biol. Chem. 270:6975–6983; 1995. [Erratum appears in J. Biol. Chem. 270:19668; 1995] [DOI] [PubMed] [Google Scholar]
- 2. Aiyar A.; Leis J. Modification of the megaprimer method of PCR metagenesis: Improved amplification of the final product. Biotechniques 14:366–369; 1993. [PubMed] [Google Scholar]
- 3. Alam J.; Smith A. Heme-hemopexin-mediated induction of metallothionein gene expression. J. Biol. Chem. 267:16379–16384; 1992. [PubMed] [Google Scholar]
- 4. Andrews G. K. Regulation of metallothionein gene expression by oxidative stress and metal ions. Biochem. Pharmacol. 59:95–104; 2000. [DOI] [PubMed] [Google Scholar]
- 5. Aniskovitch L.; Jacob S. T. Purification and characterization of a rat liver protein that recognizes CCAAT-homologous sequences of the metallothionein promoter and trans-activates this promoter. Arch. Biochem. Biophys. 341:337–346; 1997. [DOI] [PubMed] [Google Scholar]
- 6. Barchurski C. J.; Kelly S. E.; Glasser S. W.; Currier T. A. Nuclear factor I family members regulate the transcription of surfactant protein-C. J. Biol. Chem. 272:32759–32766; 1997. [DOI] [PubMed] [Google Scholar]
- 7. Bird A. P.; Wolffe A. P. Methylation-induced repression—belts, braces, and chromatin. Cell 99:451–454; 1999. [DOI] [PubMed] [Google Scholar]
- 8. Chaudhry A. Z.; Vitulo A. D.; Gronostajski R. M. Nuclear factor I (NFI) isoforms differentially activate simple versus complex NFI-responsive promoters. J. Biol. Chem. 273:18538–18546; 1998. [DOI] [PubMed] [Google Scholar]
- 9. Chaudhry A. Z.; Vitullo A. D.; Gronostajski R. M. Nuclear factor I-mediated repression of the mouse mammary tumor virus promoter is abrogated by the coactivators p300/CBP and SRC-1. J. Biol. Chem. 274:7072–7081; 1999. [DOI] [PubMed] [Google Scholar]
- 10. Cooke D. W.; Lane M. D. The transcription factor nuclear factor I mediates repression of the GLUT4 promoter by insulin. J. Biol. Chem. 274:12917–12924; 1999. [DOI] [PubMed] [Google Scholar]
- 11. Crawford D. R.; Leahy P.; Hu C. Y.; Chaudhry A.; Gronostajski R.; Grossman G.; Woods J.; Hakimi P.; Roesler W. J.; Hanson R. W. Nuclear factor I regulates expression of the gene for phosphoenolpyruvate carboxykinase (GTP). J. Biol. Chem. 273:13387–13390; 1998. [DOI] [PubMed] [Google Scholar]
- 12. Culotta V. C.; Hamer D. H. Fine mapping of a mouse metallothionein gene metal response element. Mol. Cell. Biol. 9:1376–1380; 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dalton T. P.; Li Q.; Bittel D.; Liang L.; Andrews G. K. Oxidative stress activates metal-responsive transcription factor-1 binding activity. Occupancy in vivo of metal response elements in the metallothionein-I gene promoter. J. Biol. Chem. 271:26233–26241; 1996. [DOI] [PubMed] [Google Scholar]
- 14. Datta P. K.; Jacob S. T. Identification of a sequence within the mouse metallothionein-I gene promoter mediating its basal transcription and of a protein interacting with this event. Cell. Mol. Biol. Res. 39:439–449; 1993. [PubMed] [Google Scholar]
- 15. Gao B.; Kunos G. Cell type-specific transcriptional activation and suppression of the alpha1B adrenergic receptor gene middle promoter by nuclear factor 1. J. Biol. Chem. 273:2277–2286; 1998. [DOI] [PubMed] [Google Scholar]
- 16. Ghoshal K.; Jacob S. T. Regulation of metallothionein gene expression. Prog. Nucleic Acids Res. Mol. Biol. 66:357–384; 2000. [DOI] [PubMed] [Google Scholar]
- 17. Ghoshal K.; Li Z.; Jacob S. T. Overexpression of the large subunit of the protein Ku suppresses metallothionein-I induction by heavy metals. Proc. Natl. Acad. Sci. USA 95:10390–10395; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ghoshal K.; Majumder S.; Li Z.; Dong Z.; Jacob S. T. Suppression of metallothionein gene expression in a rat hepatoma because of promoter-specific DNA methylation. J. Biol. Chem. 275:539–547; 2000. [DOI] [PubMed] [Google Scholar]
- 19. Glanville N.; Duram D. M.; Palmiter R. D. Structure of mouse metallothionein-I gene and its mRNA. Nature 292:267–269; 1981. [DOI] [PubMed] [Google Scholar]
- 20. Gounari F.; De Francesco R.; Schmitt J.; van der Vliet P.; Cortese R.; Stunnenberg H. Amino-terminal domain of NF1 binds to DNA as a dimer and activates adenovirus DNA replication. EMBO J. 9:559–566; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gunes C.; Heuchel R.; Georgiev O.; Muller K. H.; Lichtlen P.; Bluthmann H.; Marino S.; Aguzzi A.; Schaffner W. Embryonic lethality and liver degeneration in mice lacking the metal-responsive transcriptional activator MTF-1. EMBO J. 17:2846–2854; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hamer D. H. Metallothionein. Annu. Rev. Biochem. 55:913–915; 1986. [DOI] [PubMed] [Google Scholar]
- 23. Heuchel R.; Radtke F.; Georgiev O.; Stark G.; Aguet M.; Schaffner W. The transcription factor MTF-1 is essential for basal and heavy. EMBO J. 13:2870–2875; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hidalgo J.; Gasull T.; Giralt M.; Armario A. Brain metallothionein in stress. Biol. Signals 3:198–210; 1994. [DOI] [PubMed] [Google Scholar]
- 25. Jackson S. P.; Tjian R. O-glycosylation of eukaryotic transcription factors: Implications for mechanisms of transcriptional regulation. Cell 55:125–133; 1988. [DOI] [PubMed] [Google Scholar]
- 26. Jackson S. P.; MacDonald J. J.; Lees-Miller S.; Tjian R. GC box binding induces phosphorylation of Sp1 by a DNA-dependent protein kinase. Cell 63:155–165; 1990. [DOI] [PubMed] [Google Scholar]
- 27. Jacob S. T. Ghoshal K.; Sheridan J. F. Inductinon of metallothionein by stress and its molecular mechanisms. Gene Expr. 7:301–310; 1999. [PMC free article] [PubMed] [Google Scholar]
- 28. Jones K. A.; Kadonaga J. T.; Rosenfled P. J.; Kelly T. J.; Tjian R. A cellular DNA-binding protein that activates eukaryotic transcription and DNA replication. Cell 63:155–165; 1990. [DOI] [PubMed] [Google Scholar]
- 29. Kagi J. A. An overview of metallothionein. Methods Enzymol. 205:613–626; 1991. [DOI] [PubMed] [Google Scholar]
- 30. Karin M.; Eddy R. L.; Henry W. M.; Haley L. L.; Byers M. G.; Shows T. B. Human metallothionein genes are clustered on chromosome 16. Proc. Natl. Acad. Sci. USA 81:5494–5498; 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leahy P.; Crawford D. R.; Grossman G.; Cronostajski R. M.; Hanson R. W. CREB binding protein coordinates the function of multiple transcription factors including nuclear factor I to regulate phosphoenolpyruvate carboxykinase (GTP) gene transcription. J. Biol. Chem. 274:8813–8822; 1999. [DOI] [PubMed] [Google Scholar]
- 32. Li S.; Rosen J. M. Nuclear factor I and mammary gland factor (STAT5) play a critical role in regulating rat whey acidic protein gene expression in transgenic mice. Mol. Cell. Biol. 15:2063–2070; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Majumder S.; Ghoshal K.; Li Z.; Jacob S. T. Hypermethylation of metallothionein-I promoter and suppression of its induction in cell lines overexpressing the large subunit of Ku protein. J. Biol. Chem. 274:28584–28589; 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Majumder S.; Ghoshal K.; Li Z.; Bo Y.; Jacob S. T. Suppression of metallothionein gene expression in a rat hepatoma because of promoter-specific DNA methylation. J. Biol. Chem. 275:539–547; 2000. [DOI] [PubMed] [Google Scholar]
- 35. Meisterernst M.; Rogge L.; Foeckler R.; Karaghiosoff M.; Winnacker E. L. Structural and functional organization of a porcine gene coding for nuclear factor I. Biochemistry (Mosc) 28:8191–8200; 1989. [DOI] [PubMed] [Google Scholar]
- 36. Mermod N.; O’Neill E. A.; Kelly T. J.; Tjian R. The proline-rich transcriptional activator of CTF/FN-I is distinct from the replication and DNA binding domain. Cell 58:741–753; 1989. [DOI] [PubMed] [Google Scholar]
- 37. Moffatt P.; Denizeau F. Metallothionein in physiological and physiopathological processes. Drug Metab. Rev. 29:261–307; 1997. [DOI] [PubMed] [Google Scholar]
- 38. Mueller P. R.; Salser S. J.; Wold B. Constitutive and metal-inducible protein:DNA interactions at the mouse metallothionein I promoter examined by in vivo and in vitro footprinting. Genes Dev. 2:412–427; 1988. [DOI] [PubMed] [Google Scholar]
- 39. Mymryak J. S.; Berard D.; Hager G. L.; Archer T. K. Mouse mammary tumor virus chromatin in human breast cancer cells is constitutively hypersensitive and exhibits steriod hormone-dependent loading of transcription factors in vivo. Mol. Cell. Biol. 15:26–34; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nagata K.; Guggenheimer R. A.; Hurwitz J. Specific binding of a cellular DNA replication protein to the origin of replication of adenovirus DNA. Proc. Natl. Acad. Sci. USA 80:6177–6181; 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nagata K.; Guggenheimer R. A.; Enomoto T.; Lichy J. H.; Hurwitz J. Adenovirus DNA replication in vitro: Identification of a host factor that stimulates synthesis of the preterminal protein-dCMP complex. Proc. Natl. Acad. Sci. USA 79:6438–6442; 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Palmiter R. D. Molecular biology of metallothionein gene expression. EXS 52:63–80; 1987. [DOI] [PubMed] [Google Scholar]
- 43. Palmiter R. D.; Findlye S. D.; Whitmore T. E.; Duram D. M. MT-III, a brain-specific member of the metallothionein gene family. Proc. Natl. Acad. Sci. USA 89:6333–6337; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Palmiter R. D. Regulation of metallothionein genes by heavy metals appears to be mediated by a zinc-sensitive inhibitor that interacts with a constitutively active transcription factor, MTF-1. Proc. Natl. Acad. Sci. USA 91:1219–1223; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Paonessa G.; Gounari F.; Frank R.; Cortese R. Purification of a NF1-like DNA-binding protein from rat liver and cloning of the corresponding cDNA. EMBO J. 7:3115–3123; 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Quaife C. J.; Findley S. D.; Erickson J. C.; Froelic G. J.; Kelly E. J.; Zambrowica B. P.; Palmiter R. D. Induction of a new metallothionein isoform (MT-IV) occurs during differentiation of stratified squamous epithelia. Biochemistry (Mosc) 33:7250–7259; 1994. [DOI] [PubMed] [Google Scholar]
- 47. Radtke F.; Heuchel R.; Georgiev O.; Hergersberg M.; Gariglio M.; Dembic Z.; Schaffner W. Cloned transcription factor MTF-1 activates the mouse metal-lothionein I promoter. EMBO J. 12:1355–1362; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Radtke F.; Georgiev O.; Muller H. P.; Brugnera E.; Schaffner W. Functional domains of the heavy metal-responsive transcription regulator MTF-1. Nucleic Acids Res. 23:2277–2286; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rupp R. A.; Kruse U.; Multhaup G.; Gobel U.; Beyreuther K.; Sippel A. E. Chicken NFI/TGGCA proteins are encoded by at least three independent genes: NFI-a, NFI-b and NFI-c with homologues in mammalian genomes. Nucleic Acids Res. 18:2607–2616; 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Samson S. L.; Gedamu L. Molecular analyses of metallothionein gene regulation. Prog. Nucleic Acid Res. Mol. Biol. 59:257–288; 1988. [DOI] [PubMed] [Google Scholar]
- 51. Santoro C.; Mermod N.; Andrews P. C.; Tjian R. A family of human CCAAT-box-binding proteins active in transcription and DNA replication: Cloning and expression of multiple cDNAs. Nature 334:218–224; 1988. [DOI] [PubMed] [Google Scholar]
- 52. Tanese N.; Pugh B. F.; Tjian R. Coactivators for a proline-rich activator purified from the multisubunit human TFIDD complex. Genes Dev. 5:2212–2224; 1991. [DOI] [PubMed] [Google Scholar]
- 53. Thiele D. J. Metal regulated transcription in eukaryotes. Nucleic Acid Res. 20:1183–1191; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yagle M. K.; Palmiter R. D. Coordinate regulation of mouse metallothionein I and II genes by heavy metals and glucocorticoids. Mol. Cell. Biol. 5:291–294; 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]