

Abstract
Signaling mediated by growth factors receptors has long been suggested as one of the key factors responsible for failure of endocrine treatment in breast cancer (BCa). Herein we present that in the presence of tamoxifen, FGFs (Fibroblast Growth Factors) promote BCa cell growth with the strongest effect being produced by FGF7. FGFR2 was identified as a mediator of FGF7 action and the FGFR2-induced signaling was found to underlie cancer-associated fibroblasts-dependent resistance to tamoxifen. FGF7/FGFR2-triggered pathway was shown to induce ER phosphorylation, ubiquitination and subsequent ER proteasomal degradation which counteracted tamoxifen-promoted ER stabilization. We also identified activation of PI3K/AKT signaling targeting ER-Ser167 and regulation of Bcl-2 expression as a mediator of FGFR2-promoted resistance to tamoxifen. Analysis of tissue samples from patients with invasive ductal carcinoma revealed an inversed correlation between expression of FGFR2 and ER, thus supporting our in vitro data. These results unveil the complexity of ER regulation by FGFR2-mediated signaling likely to be associated with BCa resistance to endocrine therapy.
Abbreviations: BCa, breast cancer; CAFs, cancer associated fibroblasts; ER, estrogen receptor α; FGF, fibroblast growth factor; OHT, 4-hydroxytamoxifen; PR, progesterone receptor
Introduction
Histologically, breast cancer (BCa) is a heterogeneous disease characterized by specific morphological patterns and distinct biological properties. [1]. The most frequent are hormone-dependent subtypes, with luminal A (ER+, PR+) and luminal B (ER+, PR-) representing 50–60% and 15–20% of all BCa cases, respectively [2]. The treatment of patients with ER-positive BCa is based on selective ER modulators (SERM) (e.g. tamoxifen) and/or aromatase inhibitors. Since its approval in 1972 in the UK, tamoxifen has become a drug of first choice in patients with luminal BCa (now being replaced by aromatase inhibitors).
It is estimated that approximately 45% of women do not respond to tamoxifen (de novo resistance) [3], whereas acquired resistance to the drug develops ultimately in all tamoxifen-receiving patients. Around 33% of responders are reported to relapse within 15 years after 5-years of adjuvant tamoxifen-based therapy [4]. Thus, although tamoxifen was hailed as a major breakthrough in the management of patients with hormone-dependent BCa, development of resistance to the treatment poses serious clinical problem. The molecular mechanisms underlying cell sensitivity to the drug are still poorly understood and require further studies.
ER can be activated in two pathways: (i) hormone-dependent – associated with binding of estradiol to the AF2 domain, and (ii) hormone-independent – triggered, in the absence of its cognate ligand, by activation of AF1 domain by various cellular kinases [5]. GFRs-mediated ER activation results in various post-translational ER modifications such as phosphorylation or ubiquitination, that in favorable conditions (domination of co-activators) promote transcriptional function of ER, followed by receptor degradation [6]. There are at least 6 serines in ER's AF-1 domain that can be phosphorylated [5]. It was shown that activation of MAPK downstream of EGF (epidermal growth factor) and IGF (insulin-like growth factor) receptors specifically phosphorylates Ser118, which then promotes ER-transcriptional activity [7], [8], [9], [10]. On the other hand, phosphorylation of Ser167 by AKT1 [11], [12], [13], RSK [10], Src [11] and MAPK [13] was proved to stabilize ER interaction with ER-dependent promoters and lead to estrogen-regulated genes expression [10], [11], [12].
There is growing evidence to suggest that, besides up-regulation of estrogen-regulated genes (cyclin D1, Bcl-2) [14], activity of GFRs, e.g. HER2 [15], [16], EGFR (epidermal growth factor receptor) [7], [8], [15], [17], IGFR (insulin-like growth factor receptor) [7], [8], VEGF/VEGFR (vascular endothelial growth factor/receptor) [18] and FGFRs (fibroblast growth factor receptors; FGFR1–4) [19], is the main cause of failure of tamoxifen-based therapy. GFRs-triggered signaling leads to activation of MAPK and/or PI3K (phosphatidylinositol-3-kinase)/AKT pathways, which mediates ER phosphorylation at Ser167 [9] and Ser118 [7], [9], known to be associated with cell resistance to tamoxifen [6]. In addition, GFRs enhance cell proliferation (increased cyclin D1 expression) [17], [20] and anti-apoptotic signals (up-regulation of Bcl-2) [16] that overcome cytotoxic effect of tamoxifen.
It is generally acknowledged that tumor microenvironment can play an important role in resistance to tamoxifen. For instance, MCF7 and T47D breast cancer cells exposed to growth factors secreted by cancer-associated fibroblasts (CAFs) developed resistance to tamoxifen-induced apoptosis [21]. In the murine model of estradiol-dependent BCa, CAFs protected cancer cells from tamoxifen-induced cell death, acting through AKT and MAPK pathways, which led to ER phosphorylation at Ser118 [22].
Herein an impact of a crosstalk between fibroblast growth factor receptor 2 (FGFR2) and ER on development of resistance to tamoxifen was studied in two luminal BCa cell lines (MCF7 and T47D). We found that activation of the FGF7/FGFR2 axis abolished growth-inhibitory effect of tamoxifen, which was confirmed in cells exposed to CAFs-conditioned medium. FGF7/FGFR2-signaling was shown to phosphorylate ER, promote ER proteasomal degradation and reverse tamoxifen-driven ER stabilization. Activation of PI3K/AKT pathway targeting ER-Ser167 and regulation of Bcl-2 expression in response to FGF7 treatment were identified to underlie FGFR2-dependent resistance to tamoxifen. Analysis of tissue samples from patients with invasive ductal breast carcinoma revealed an inversed correlation between expression of FGFR2 and ER, thus supporting our in vitro data.
Materials and Methods
Cell Lines, Antibodies, and Reagents
Luminal (ER+) BCa cell lines: MCF7 and T47D were obtained from ATCC. Fibroblasts derived from BCa tumors (CAFs) were from the Cell Bank of the Department of Medical Biotechnology, Medical University of Gdansk (isolated as previously described [23], [24]). Cells were grown in DMEM or phenol red-free DMEM (CAFs) supplemented with 10% FBS and penicillin/streptomycin (100 U/ml/100 μg/ml) at 37°C in a humidified atmosphere of 5% CO2. All media and their supplements were purchased from Sigma-Aldrich, HyClone or Biowest. Anti-Ub (sc-8017) antibodies were from Santa Cruz. Antibody against β-actin (A5316) was obtained from Sigma-Aldrich, anti-Bcl-2 (clone 124) and anti-ERα (clone 1D5) were from Dako, anti-ERα-Ser167 was from Invitrogen (PA5–37570). The remaining antibodies including anti-FGFR2 (#23328), anti-Akt (#9272), anti-Akt-Ser473 (#4060), anti-Bax (#5023), anti-ERα-Ser118 (#2511), anti-ERK1/2 (#4695), anti-ERK1/2-Thr202/Tyr204 (#9101) were from Cell Signaling Technology. Immunohistochemistry for FGFR2 was done with mouse anti-human antibodies (1:600; Abnova #H00002263-M01). All fibroblasts growth factors (FGFs) were purchased from PeproTech, 4-hydroxytamoxifen (OHT), fulvestrant, heparin sodium salt, PD173074 and MG132 were from Sigma-Aldrich, LY294002 and ABT-199 were from Selleckchem. Growth factors-reduced matrigel matrix was obtained from BD Biosciences.
Western Blotting
Cells were scraped in cold PBS and lysed with Laemmli buffer (2× concentrated) with 2 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 5 mM EGTA, 1 mM EDTA, 2 mM Na4P2O7, 5 mM NaF and 5 mM Na3VO4. An equal amount of protein from each sample was loaded per well, resolved in SDS–PAGE and then transferred onto nitrocellulose membrane. After 1 h of blocking in 5% skimmed milk in TBST, membranes were incubated overnight with specific primary antibodies at 4°C. Appropriate secondary antibodies conjugated with horseradish peroxidase (Sigma-Aldrich) and Western Lightning Plus-ECL (PerkinElmer) were used to visualize specific protein bands.
Immunoprecipitation
Cells were lysed in 1% Triton X-100 in PBS containing 2 mM PMSF, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 5 mM EGTA, 1 mM EDTA, 2 mM Na4P2O7, 5 mM NaF and 5 mM Na3VO4. Supernatant was incubated with 1 μg of anti-ERα antibody (Dako) overnight at 4°C. The samples were mixed with anti-mouse IgG conjugated with agarose beads (Sigma Aldrich, A6531) according to manufacturer's instructions. Immunocomplexes were eluted from the beads with Laemmli buffer and analyzed by western blotting.
FGFR2 Knock-Down and Overexpression of FGFR2
T47D shFGFR2 cell line was generated with lentiviral system based on pLKO.1-TRC vector (Addgene, #10878) [25] with cloned shRNA designed on the basis of the following siRNA sequence of FGFR2 5′- GAG AUU UGG UAU UUG GUU GGU GGC - 3′ [26], [27]. In all experiments with FGFR2-negative variants of cell lines as a control we used cells transfected with backbone pLKO.1 plasmid. T47D FGFR2 cells were established with retroviral vector pBp-FGFR2b-WT (Addgene, #45698) [28].
Signaling Analyses, Stimulation With Growth Factors
For analysis of signaling triggered by growth factors, cells were starved overnight in serum-free media followed by stimulation with FGF7 (10 ng/ml) and/or OHT (1 μM) for indicated periods of time. In all experiments, FGF7 was used together with heparin sulfate (10 ng/ml) which is critical for the formation of an active FGFs/FGFRs signaling complex [29]. PD173074 (100 nM) and MG132 (0.05 μM) were applied for inhibition of FGFR and proteasomal degradation, respectively. LY294002 (2 μM) was used to inhibit PI3K/AKT signaling, ABT-199 (5 μM) was applied to abolish Bcl-2 activity (BH3 mimetic).
Culturing Cells in Three-Dimensional Matrigel
Cell culturing in three-dimensional matrigel was carried out as previously described [30]. Cells were cultured in regular medium and, when appropriate, supplemented with FGF7 (10 ng/ml) together with heparin sulfate (10 ng/ml) and/or OHT (1 μM). Media were replaced every third day. To evaluate cell growth, colonies were measured after 14 days of culture (at least 50 colonies for each condition) using ZEISS PrimoVert microscope and ImageJ software.
Quantitative PCR
RNA was isolated with TriPURE reagent (Roche) according to the manufacturer's protocol. Reverse transcription with random hexamer primers was performed with Transcriptor cDNA First Strand Synthesis Kit (Roche). Gene expression analysis was carried out for ESR1 gene (forward primer: 5′-AAGAAAGAACAACATCAGCAGTAAAGTC-3′, reverse primer: 5′-GGGCTATGGCTTGGTTAAACAT-3′) and reference genes: ACTB (forward primer: 5′-TGACGTGGACATCCGCAAAG-3′, reverse primer: 5′-CTGGAAGGTGGACAGCGAGG-3′) and GAPDH (forward primer: 5′-GACAGTCAGCCGCATCTTCT-3′, reverse primer: 5′-TTAAAAGCAGCCCTGGTGAC-3′). Twenty-microliter reactions were realized using Maxima SYBR Green qPCR Master Mix (Thermo Scientific) on 96-well plates in CFX96 cycler (Bio-Rad, Hercules). For analysis of for MYO1B and PSME1 expression TaqMan probes Hs00362654_m1 and Hs00389210_g1 and TaqMan Universal PCR Master Mix (Applied Biosystem) were used. Reactions were done in duplicates. Each plate contained an inter-run calibrator, a set of non-template controls and controls for gDNA contamination. Gene expression was calculated using a modified ΔΔC approach.
Soft Agarose Assay for Anchorage-Independent Growth (Supplement)
Anchorage-independent growth was evaluated as previously described [31]. Briefly, cells (5 × 104 per well) were suspended in 3 ml of 0.4% low gelling temperature agarose (Sigma Aldrich) prepared in DMEM containing 10% FBS and overlaid on 3 ml of solidified 0.5% agarose made up in the same medium. The top layer was covered with 3 ml DMEM medium and, when appropriate, supplemented with FGF7 (10 ng/ml) and/or OHT (1 μM). Medium was replaced every 3–4 days. After 21 days of culture, colonies were counted using ZEISS PrimoVert microscope and ImageJ software.
Clinical Data, Patient Selection, and Samples
Specimens of primary invasive ductal carcinoma were obtained from 166 women treated at the Oncology Department of Copernicus Memorial Hospital in Łódź between 1997 and 2001 according to the local ethical regulations. All patients had undergone a radical mastectomy with axillary lymph node dissection. Adjuvant therapy based on tamoxifen was received by 109 [ER+ (N = 52) and ER- (N = 57)] patients. Samples were histologically graded using the Nottingham criteria and the disease was staged according to the TNM system. ER/PR/HER2 status was determined by routine histopathological assessment. The characteristics of the study population are summarized in Table 1.
Table 1.
Patient Characteristics.
| Number of Patients (N) | 166 |
|---|---|
| Age (years) | |
| < 50 | 52 |
| ≥ 50 | 114 |
| Disease stage | |
| I | 39 |
| II | 84 |
| III | 43 |
| T status | |
| T1 | 55 |
| T2 | 103 |
| T3 | 1 |
| T4 | 7 |
| Grade | |
| 1–2 | 95 |
| 3 | 71 |
| Nodal status | |
| Negative | 82 |
| Positive | 84 |
| Estrogen receptor status | |
| Negative | 73 |
| Positive | 93 |
| HER2 status | |
| Negative | 138 |
| Positive | 28 |
Immunohistochemistry
Serial 5 μm paraffin sections of formalin-fixed blocks were processed for immunohistochemistry for FGFR2 (mouse anti-human; 1:600; Abnova #H00002263-M01), using protocols recommended by the manufacturers. As a negative control for immunostaining, primary antibodies were replaced by non-immune sera. Scoring of immunostaining was carried out as follows: (i) 0 – no reactivity or only faint cytoplasmic/membranous reactivity in <10% of tumor cells; (ii) 1+ − faint cytoplasmic/membranous reactivity in ≥10% of tumor cells; (iii) 2+ − weak to moderate cytoplasmic/membranous reactivity in ≥10% of tumor cells; (iv) 3+ − strong cytoplasmic/membranous reactivity in ≥10% of the tumor cells. Immunohistochemical staining was evaluated and scored independently by two observers (HMR and RK). The agreement on staining intensity was >90%. Where there was disagreement, intensity was determined by consensus. Dichotomisation of final scores into: (a) ‘negative/low’ for 0–1 and (b) ‘positive/high’ for 2–3 was guided by intensity of immunostaining in positive controls (human stomach carcinoma) recommended by the manufacturer.
Statistical Analysis
Pearson's χ2 test or Fisher's exact test were used to assess the associations between expression of FGFR2 and clinicopathological variables. The results were considered statistically significant when two-sided P was less than .05. The analyses were performed using the StatsDirect (StatsDirect Ltd., Altrincham, UK) and Statistica 9.1 (StatSoft Inc. Tulsa, OK, USA) software. Colonies size in 3D cultures was measured with ImageJ. Data are expressed as means ± SD from at least three independent experiments. Comparative data were analyzed with the unpaired Student's t-test using the STATISTICA software (version 10, StatSoft). Two-sided P < .05 was considered as significant.
Results
FGFs/FGFR Action Impairs Tamoxifen-Dependent Growth Inhibition of BCa Cells
To analyze an impact of FGFs/FGFR activation on tamoxifen-induced biological effect, T47D luminal BCa cells grown in 3D matrigel were treated with FGF1, FGF2, FGF4, FGF7 or FGF9 and/or OHT (4-hydroxytamoxifen – active metabolite of tamoxifen, used in all further experiments). We found that all tested FGFs promoted cell proliferation (reflected by colony size) (Figure 1A, left panel) whereas incubation with OHT resulted in significant reduction of cell growth (Figure 1A, right panel). OHT action was partially abolished by FGFs with the strongest effect being produced by FGF2 and FGF7. The latter was used in further studies. This choice was additionally justified by numerous reports strongly implicating FGF7 in both physiology and pathophysiology of the mammary gland [32], [33], [34]. The observed FGF7 effect on growth of OHT-treated cells was confirmed in the MCF7 cell line (Supplementary data, Figure 1). As in T47D cells, FGF7 promoted MCF7 cells growth and protected the cells from inhibitory influence of tamoxifen. For both T47D and MCF7 cell lines, the FGF7-induced resistance to OHT was observed also in cells cultivated in adhesion-independent conditions (Supplementary data, Figure 2).
Figure 1.

FGFs/FGFR action promotes 3D growth of BCa cells and impairs tamoxifen/OHT action. (A) T47D cells were grown in 3D matrigel for 14 days in the presence of FGF1, 2, 4, 6, 7, 9 (10 ng/ml) and/or tamoxifen (1 μM), B) with/without PD173074-FGFR inhibitor (100 nM). Representative pictures were taken, colonies measured and statistically analyzed with ImageJ. Scale bar represents 100 μm, *P ≤ .05, **P ≤ .001, n = 3.
1Ratios to control/non-treated cells.
Supplementary data, Figure 1.

FGF7-induced protective effect in OHT-treated BCa cells. MCF7 cells were grown in 3D matrigel and treated with FGF7 and/or tamoxifen. Representative pictures were taken after 14 days of culture. The experiment was done in duplicates. Scale represents 100 μm.
Supplementary data, Figure 2.

FGF7 promotes adhesion-independent growth of BCa cells and impairs tamoxifen/OHT action. MCF7 and T47D cells were seeded and grown in soft agarose as described in materials and methods section. Cultures were treated with FGF7 and/or tamoxifen, after 21 days colonies were counted, *P ≤ .05, **P ≤ .001, n = 3.
To verify an engagement of FGF receptors in FGF-induced resistance to OHT, T47D cells were grown with PD173074 (a well characterized, selective FGFR inhibitor [35], [36]) and/or FGF7. We found that PD173074 nearly completely abolished both FGF7-mediated promotion of T47D cell growth (Figure 1B, left panel) and FGF7 protective effect from OHT (Figure 1B, right panel). These results indicate that FGF/FGFR-driven signaling is involved in mediation of tamoxifen resistance in BCa cells.
FGF7-Dependent Protective Effect in OHT-Treated Cells is Mediated by FGFR2
Since it is well documented that FGF7 binds with the highest affinity to FGFR2 [37], [38], stable knock-down and overexpression of FGFR2 gene were generated in T47D cells (as described previously [27]) to confirm FGFR2 involvement in FGF7-triggered resistance to tamoxifen (Figure 2A). Lack of any off-target effects of applied shRNA has been confirmed in previous studies [26], [27]. Results showed that FGFR2 silencing and overexpression impaired and enhanced T47D cell growth in 3D, respectively (Figure 2B). On the other hand, FGF7-dependent promotion of cell growth was nearly completely abolished following FGFR2 knock-down or enhanced upon FGFR2 overexpression (Figure 2B). As shown in Figure 2C, knock-down of FGFR2 abrogated, whereas overexpression of FGFR2 promoted FGF7-stimulated growth of OHT-treated cells. This suggests that protective effect of FGF7 against tamoxifen-induced growth inhibition is mediated by FGFR2.
Figure 2.

FGFR2 mediates FGF7-dependent protective effect in OHT-treated BCa cells. A) T47D cell lines with knock-down or overexpression of FGFR2 were established as described in materials and methods section. B-C) Their response to FGF7 (10 ng/ml) and/or tamoxifen (1 μM) was evaluated in 3D matrigel. Representative pictures were taken after 14 days of growth; colonies were measured and statistically analyzed with ImageJ. Scale bar represents 100 μm, *P ≤ .05, n = 3.
1Ratios to control/non-treated cells.
FGFR2 is Involved in Protective Effect of Breast Cancer Associated Fibroblasts from OHT
To assess an influence of tumor microenvironment on pharmacological effectiveness of tamoxifen, T47D cells were incubated with CAFs-conditioned medium (CAFs-CM, rich in growth factors/cytokines secreted by fibroblasts) and/or tamoxifen (Figure 3, A–B). We found that CAFs-CM promoted cell growth both: a) under control conditions, i.e. untreated cells (Figure 3A, 2.16× bigger colonies) and, b) subjected to treatment with tamoxifen (Figure 3B, 4.9× bigger colonies). As shown in Figure 3, A and B, application of PD173074 (an inhibitor of FGFRs) strongly diminished promoting effects of CAFs-CM, which suggests that FGFs/FGFRs signaling is involved in the observed phenomenon (Figure 3, A and B). Analyses of T47D vs T47D shFGFR2 revealed functional participation of FGFR2 in CAFs-induced growth and protection from tamoxifen (Figure 3, C and D). These has been additionally confirmed in MCF7 cells (Supplementary data, Figure 3).
Figure 3.

Cancer Associated Fibroblasts (CAFs) promote BCa cells growth and resistance to tamoxifen through FGFR2. A) T47D cells were grown for 14 days in 3D matrigel in the presence of CAFs-conditioned medium with/without FGFR inhibitor (PD173074, 100 nM), B-D) FGFR2 involvement in CAFs-mediated cell growth and resistance to tamoxifen was evaluated. Scale represents 100 μm, *P ≤ .05, **P ≤ .001, n = 3.
1Ratios to control/non-treated cells.
Supplementary data, Figure 3.

FGFR2 mediates CAF-driven MCF7 cells growth and resistance to tamoxifen. MCF7 and MCF7 shFGFR2 cells were grown in 3D matrigel and treated with FGF7 and/or tamoxifen. Representative pictures were taken after 14 days of culture. The experiment was done in duplicates. Scale represents 100 μm.
FGF7/FGFR2 Signaling Regulates Degradation of ER
To determine whether FGF7-triggered signaling affects ER expression and interferes with tamoxifen action, T47D and MCF7 cells were serum-starved and incubated for indicated periods of time (24, 48, 72 h) with FGF7 and/or tamoxifen (Figure 4A). We found that incubation of BCa cells with FGF7 gradually decreased expression of ER in both analyzed luminal BCa cell lines (Figure 4A). On the other hand, as previously shown [39], exposure to tamoxifen strongly increased level of ER, which was restored to the steady-state by FGF7 (Figure 4A). Moreover, CAF-CM triggered exactly the same effect as FGF7, i.e., counteracted tamoxifen-dependent increase in ER expression (Supplementary data, Figure 4). FGFR2 was proved to play a key role in both FGF7- and CAF-CM-dependent regulation of ER expression (Figure 4B and Supplementary data, Figure 4). In order to confirm that the observed FGF7/FGFR2-induced down-regulation of ER (Figure 4, A and B) was caused by receptor's degradation, T47D and MCF7 cells were treated with FGF7 and/or tamoxifen in the presence of MG132 (a well-characterized, specific 26S proteasome inhibitor [40]). As anticipated, lack of proteasomal activity abolished FGF7-triggered down-regulation of ER (Figure 4C, Supplementary data, Figure 5). We then confirmed an involvement of FGF7/FGFR2 in ER degradation/turnover and showed that FGF7 promoted, whereas tamoxifen impaired ubiquitination of ER (Figure 4D, left panel). In cells exposed to both FGF7 and tamoxifen, ER was ubiquitinated to the same extent as in control/non-treated cells (immunoprecipitated samples were normalized to ER level). As expected, knock-down of FGFR2 abolished FGF7-regulated ER ubiquitination (Figure 4D, right panel). FGFR2 role in ubiquitination of ER induced by CAF-CM was also proved (Supplementary data, Figure 6). In addition, as shown by qPCR analyses, in both cell lines, neither tamoxifen nor FGF7 significantly affected ER mRNA level (ESR1 gene product) (Figure 4E). This set of experiments clearly demonstrates that FGF7/FGFR2 signaling overrides an effect of tamoxifen on ER stability in luminal BCa cells. In order to confirm specificity of FGF7/FGFR2–tamoxifen interaction, in control experiment we incubated cells with fulvestrant, a first approved ER down-regulator, known to trigger ER degradation [41]. As expected, FGF7 treatment did not interfere with Fulvestrant-dependent growth inhibition (Supplementary data, Figure 7) as their action towards ER is based on a similar mechanism (enhanced receptor's degradation).
Figure 4.

FGF7/FGFR2 signaling regulates proteasomal degradation of ER in BCa cells. (A) T47D and MCF7 cells were grown in the presence of FGF7 and/or tamoxifen for 24, 48, and 72 h. ER expression was evaluated by western blotting. Densitometry was done with ImageJ software. (B) FGF7-dependent effect on ER expression in T47D cells is mediated by FGFR2 and (C) involves proteasomal activities (48 h incubation with MG132). D) FGF7/FGFR2-signaling triggers ER ubiquitination. T47D vs T47D shFGFR2 cells were serum starved and incubated with FGF7 (10 ng/ml) and/or or tamoxifen (1 μM) for 24 h. Cell lysis and immunoprecipitation of ER was done in 1% Triton-X. Ubiquitination level was evaluated by western blot analyses. Protein amount in both lysate and immunoprecipitated fraction was normalized. E) qPCR analyses of ESR1 (coding ER) expression upon FGF7 and/or tamoxifen exposure. A, D, E experiments were done in triplicates; B and C experiments were done in duplicates.
Supplementary data, Figure 4.
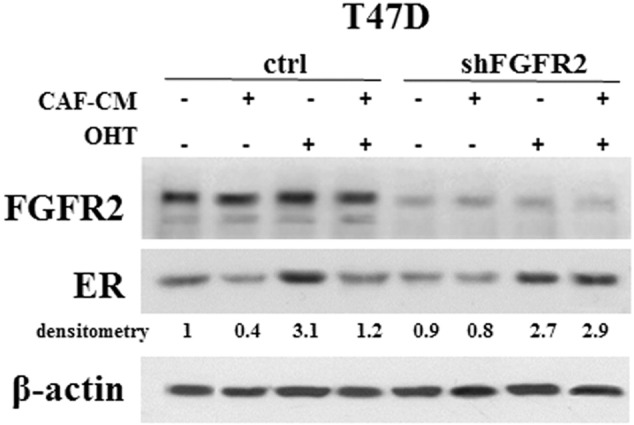
FGFR2 mediates CAF-CM-dependent regulation of ER level. T47D cells were grown in CAF-CM with/without tamoxifen for 72 h. ER expression was analyzed by western blotting. The experiment was done in duplicates.
Supplementary data, Figure 5.
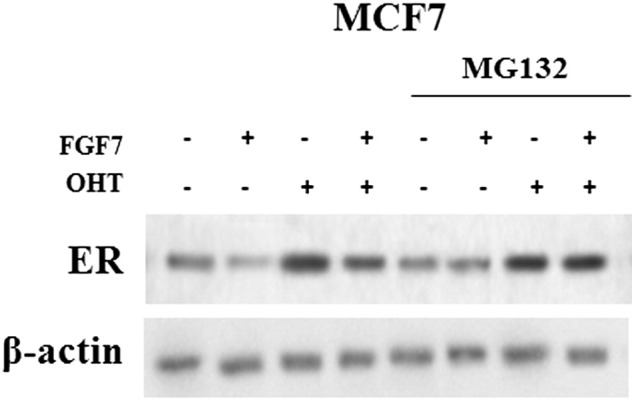
FGF7 promotes proteasomal degradation of ER and abrogates tamoxifen-dependent ER stabilization. MCF7 cells were treated with FGF7 and/or tamoxifen in the presence of MG132-proteasomal inhibitor (0.05 μM). The experiment was done in duplicates.
Supplementary data, Figure 6.
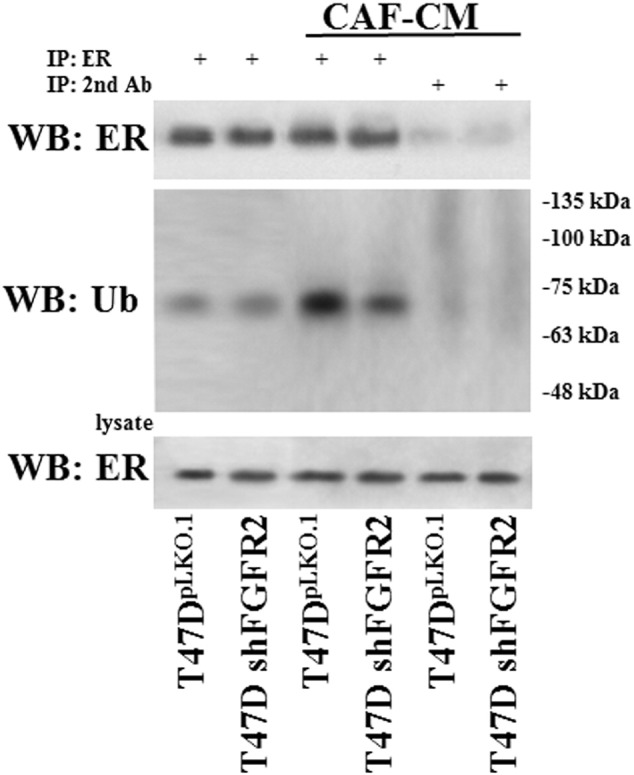
CAF-CM-induced ubiquitination of ER is mediated by FGFR2. T47D vs T47D shFGFR2 cells were serum starved and incubated with CAF-CM for 24 h. Cell lysis and immunoprecipitation of ER was done in 1% Triton-X. Ubiquitination level was evaluated by western blot analyses. Protein amount in both lysate and immunoprecipitated fraction was normalized. The experiment was done in duplicates.
Supplementary data, Figure 7.
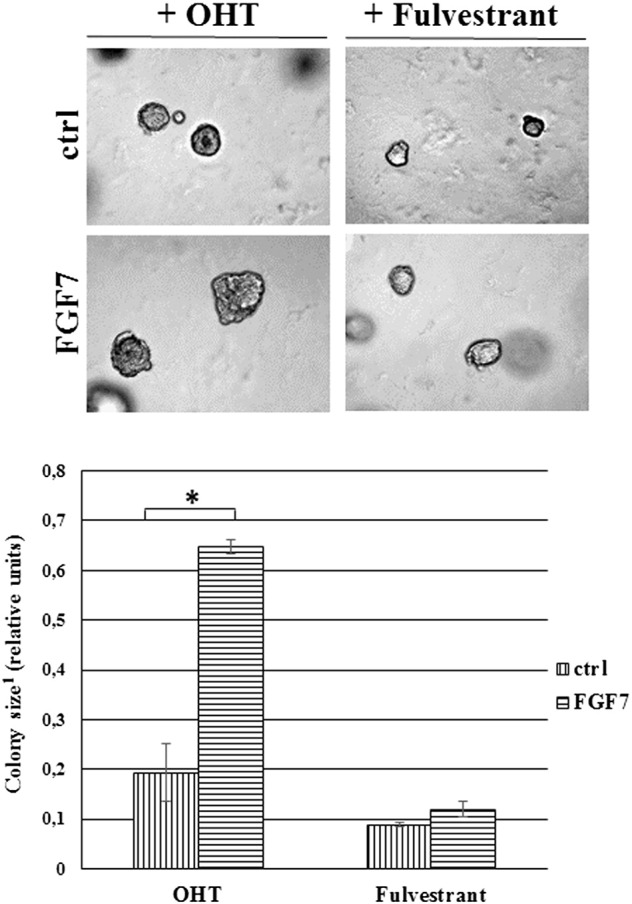
FGF7 does not prevent fulvestrant-mediated growth inhibition. Cells were grown in 3D matrigel and treated with tamoxifen (1 μM) or fulvestrant (100 nM) ± FGF7 (10 ng/ml). Representative pictures were taken after 14 days of culture. *P ≤ .05, n = 3
1Ratios to control/non-treated cells
PI3K/AKT and Bcl-2 Mediate FGF7/FGFR2-Driven Resistance to Tamoxifen
In confirmation of a well-documented activation of ER by growth factors-triggered signaling [5], we demonstrated that in both analyzed cell lines, FGF7 induced phosphorylation of ER at Ser118 and Ser167 (Figure 5A). Surprisingly, treatment with tamoxifen resulted in activation of ER-Ser118 while having a very modest impact on ER-Ser167. In addition, FGF7 conferred an additive effect, as exposure of cells to both agents resulted in phosphorylation of both Ser118 and Ser167 (Figure 5A). These differences between phosphorylation of Ser118 and Ser167 are in agreement with observed activation pattern of ERK (extracellular signal-regulated kinase) and AKT kinases, previously shown to phosphorylate ER-Ser118 and ER-Ser167, respectively [7], [8], [12]. As demonstrated in Figure 5B, FGFR2 was involved in mediation of FGF7-induced activation of ER. Knock-down of FGFR2 nearly completely abolished phosphorylation of ER as well as activation of upstream kinases (ERK and AKT) in response to FGF7. FGF7/FGFR2 role in enhancing ER activity has been confirmed by analysis of ER-dependent genes (MYO1B and PSME1, previously shown to be up-regulated and down-regulated in tamoxifen resistance, respectively [42], [43]) (Supplementary data, Figure 8). Specificity of AKT towards phosphorylation of ER at Ser167 induced by FGF7/FGFR2 signaling was confirmed with the PI3K/AKT inhibitor (LY294002) (Figure 5C). AKT activity was also shown to be involved in turnover of ER following treatment with FGF7 (Figure 5D). Inhibition of PI3K/AKT signaling strongly impaired FGF7-triggered ER degradation. Moreover, AKT mediated FGF7-dependent both promotion of cell growth and protection from tamoxifen (Figure 6, A and B).
Figure 5.

FGF7/FGFR2-dependent phosphorylation of ER-Ser167 by PI3K/AKT mediates tamoxifen resistance. (A) MCF7 and T47D cells were treated with FGF7 (10 ng/ml) and/or tamoxifen (1 μM) for 10–60 minutes and analyzed for activation of ER, ERK and AKT. (B) FGF7/FGFR2 signaling targets ER phosphorylation. (C) Inhibition of PI3K/AKT signaling abolishes FGF7-driven activation of ER-Ser167. (D) PI3K/AKT activity is involved in FGF7-dependent regulation of ER expression.
Supplementary data, Figure 8.

FGF7 promotes ER transcriptional activity. T47D cells were serum starved and treated with estradiol (E2, 10 nM) and/or FGF7 (10 ng/ml) for 12 h. qPCR analysis was done as described in material and methods section, *P ≤ 0.05, n = 3.
Figure 6.

PI3K/AKT activity is indispensable for FGF7-triggered resistance to tamoxifen. T47D cells were grown in matrigel and treated with FGF7 (10 ng/ml) and/or tamoxifen (1 μM) with/without PI3K/AKT inhibitor (LY294002, 2 μM). Representative pictures were taken after 14 days of growth; colonies were measured and statistically analyzed with ImageJ. Scale represents 100 μm, **P ≤ .001, n = 3.
1Ratios to control/non-treated cells.
We also demonstrated that FGF7/FGFR2 signaling regulated Bcl-2 expression (Figure 7A). This anti-apoptotic protein was previously identified as an ER target gene [44], [45]. We showed that tamoxifen down-regulated Bcl-2 level was abrogated by FGF7 treatment. Surprisingly, we found that knock-down of FGFR2 led also to decrease of Bcl-2 expression, which might suggest a potentially prominent role of FGFR2 in control of apoptosis. Our results indicate as well that Bcl-2 might act as one of candidate mediators of FGF7-driven resistance to tamoxifen. Inhibition of Bcl-2 by ABT-199 (a potent and selective inhibitor [46]) completely abrogated FGF7-triggered resistance to tamoxifen (Figure 7, B and C).
Figure 7.

Bcl-2 is involved in FGF7/FGFR2-dependent resistance of BCa cells to tamoxifen. (A) T47D vs T47D shFGFR2 cells were treated with FGF7 and or tamoxifen for 72 h and analyzed for Bcl-2 expression. The experiment was done in triplicates. Densitometry was done with ImageJ software. (B) Inhibition of Bcl-2 activity abrogates FGF7-mediated resistance to tamoxifen. Cells were grown in 3D matrigel for 14 days in the presence of FGF7 and/or tamoxifen, with/without ABT-199 (5 μM). Colonies were measured and statistically analyzed with ImageJ. Scale bar represents 100 μm, **P ≤ 0.001, n = 3.
1Ratios to control/non-treated cells.
Expression of FGFR2 Inversely Correlates with ER in Breast Cancer
To examine a potential translational value of the demonstrated molecular link between FGFR2 and ER, we analyzed a relationship between expression of these proteins in tissue samples from patients with invasive ductal carcinoma (IDC) (Table 1, patients characteristic). FGFR2-positivity was seen in 32% of ER-positive (30/93) and 48% (35/73) of ER-negative tumors. Examples of high and low levels of FGFR2 expression are presented in Figure 8. Inverse FGFR2/ER relationship was seen in 63/166 (38%) and 35/166 (21%) cases for FGFR2−/ER+ and FGFR2+/ER-, respectively. FGFR2 was significantly associated only with lack of estrogen receptor (P = .04) (Table 2) and there was a trend towards statistical significance of the relationship between FGFR2 and tumor grade (P = .095). As ER itself is a strong determinant of the degree of cell dedifferentiation (ER versus grade P < .0001), a possible correlation between inverse FGFR2/ER expression and tumor grade was not further assessed. Lack of stringent criteria for patients selection for tamoxifen therapy precluded the analysis of an impact of FGFR2 alone or FGFR2/ER interdependence on patients' survival.
Figure 8.

Expression of ER and FGFR2 in BCa tissue samples. Examples of low (A and C) and high (B and D) immunoreactivity for ER and FGFR2, respectively. Scale bar 500 μm.
Table 2.
Relationship Between Clinicopathological Features and Expression of FGFR2 and ER.
| Feature | FGFR2+ (N = 65) vs Rest (N = 101) |
ER+ (N = 93) vs Rest (N = 73) |
|---|---|---|
| Tumor size | 0.131 | 0.136 |
| Nodal Status | 0.724 | 0.515 |
| Stage | 0.510 | 0.391 |
| Grade | 0.095 | <0.0001 |
| HER2 | 0.197 | <0.0001 |
| ER- | 0.040 |
Discussion
Tamoxifen has been used for over 40 years for luminal breast cancer treatment. Despite its undisputed therapeutic benefits, a significant number of patients recur [4]. Tumor microenvironment-originating growth factors may initiate molecular mechanisms that affect cell response to tamoxifen and are indeed implicated in the development of resistance to the drug [47], [48]. In our study we focused on FGFs that are abundantly secreted by breast cancer-associated fibroblasts [49], [50], [51]. We present the most comprehensive so far analysis of an impact of FGFs (FGF1, FGF2, FGF4, FGF6, FGF7 and FGF9) on resistance to tamoxifen and corroborate an involvement of CAFs in this process. We showed for the first time that FGFR2 is a mediator of signals derived from tumor microenvironment, responsive for development of resistance to the drug.
Although a role of FGFs/FGFRs in cell response to tamoxifen was studied before [20], [52], [53], [54], the underlying mechanism still remains unexplained. The most in-depth analysis undertaken by Turner et al. [20] showed that stimulation with FGF2 resulted in resistance to tamoxifen in cells with elevated expression of FGFR1. FGF2/FGFR1 signaling was suggested to overcome tamoxifen-induced growth arrest and apoptosis, which has been linked with high MAPK and AKT baseline activity as well as increased level of cyclin D1 [20].
Herein we focused on a crosstalk between FGFR2-triggered signaling and ER activity in relation to the growth inhibitory effectiveness of tamoxifen. We found a clear effect of FGF7 resulting in activation of AKT/ER-Ser167 but not ERK/ER-Ser118 in cells treated with the drug. Therefore we conclude that FGF7/FGFR2 signaling drives resistance to tamoxifen through PI3K/AKT (shown before to specifically target ER-Ser167 [12]) rather than MAPK-pathway (proved to activate ER-118 [7]). This is in agreement with data presented by Kirkegaard which demonstrate a clinical value of a correlation between phosphorylated AKT and ER-Ser167 status [55]. However, some reports put a question mark over prognostic significance of ER phosphorylation sites [56], [57], [58], [59]. This might be due to tissue-specific dominance of different signaling pathways leading to ER phosphorylation [60], likely to be associated with molecular diversity of analyzed cohorts of patients. Interestingly, there is also a contradictory aspect of PI3K/AKT-ER axis regulation as inhibition of PI3K was reported to activate ER-dependent transcription. In addition, suppression of ER activity can sensitize tumors to PI3K inhibitors [61]. Although PI3K/AKT-ER interdependence seems to be complex, it is clear that combined therapy involving PI3K/AKT and ER inhibitors is a promising approach for ER-positive BCa patients.
The molecular mechanism of tamoxifen action, which includes stabilization of ER based on impaired receptor's degradation/turnover, is very complex [39]. In our analyses of FGFR2-mediated cell resistance to tamoxifen, we found that FGF7 as well as CAF-CM stimulation led to ER loss in MCF7 and T47D BCa cell lines. Elevated expression of ER resulting from tamoxifen treatment was restored to steady-state level (observed in non-treated cells) by FGF7 (and CAF-CM). Effect of FGF7/CAF-CM towards regulation of ER level was proved to be mediated by FGFR2. As reduced expression of ER was shown to be associated with failure of endocrine therapy [62], we conclude that loss of ER caused by microenvironmental stimuli and mediated FGF7/FGFR2 could be one of the underlying mechanisms of recurrence.
Regulation of ER expression by FGF7/FGFR2-triggered signaling was previously reported in MCF7 cells and shown to down-regulate ER expression at transcription level [63]. We demonstrated that FGF7/FGFR2 and CAF-CM/FGFR2 signaling promoted ER ubiquitination and cell incubation with MG132 (a well described 26S proteasome inhibitor [40]) abolished FGFR2-dependent ER down-regulation. This was shown to overcome tamoxifen-driven increase in ER level, which was dependent on impaired receptor's degradation reflected by diminished ER ubiquitination. Further analysis revealed a modest impact of FGF7 stimulation on regulation of ESR1 expression (down-regulation by 5% and 14% in MCF7 and T47D cells, respectively - similar range as reported by Chang et al. [63]). Taken together, these data suggest that degradation/turnover of ER protein, rather than regulation of ESR1 expression, is the key mechanism whereby FGF7/FGFR2-initiated signaling axis interferes with ER function and is responsible for alteration of cell response to tamoxifen. This is in agreement with a study involving in vitro and clinical analyses (228 BCa patients) demonstrating that CUEDC2 (a ubiquitin-binding motif-containing protein which regulates ER degradation) is a key factor in resistance to tamoxifen-based therapy [64].
Inconsistency in therapeutic regimen applied to the patients of the studied cohort precluded an analysis of an impact of FGFR2 on both response to tamoxifen and patients survival. However, an assessment of the relationship between FGFR2, ER and clinicopathological features demonstrated an inverse association between the receptors and indicated the FGFR2 presence is likely to be linked to histologically more advanced tumors, which gives support to the in vitro data.
Our study demonstrated for the first time that the FGF7/FGFR2-dependent signaling pathways regulate ER function and affect response of breast cancer cells to tamoxifen. FGFR2 may be an important mediator of resistance to endocrine therapy driven by microenvironmental stimuli (CAFs), which might lead to progression to hormone-independence.
The following are the supplementary data related to this article.
Funding
This work was supported by National Science Centre: UMO-2012/06/M/NZ3/00023 (to RS), UMO-2013/09/B/NZ4/02512 (to HMR) and UMO-2015/17/B/NZ4/02157 (to RK) grants.
Acknowledgments
Acknowledgements
pLKO.1-TRC cloning vector was a gift from David Root (Broad Institute). pBp-FGFR2b-WT was a gift from Matthew Meyerson (Dana-Farber Cancer Institute).
Ethical Approval and Consent to Participate
The study was conducted in accordance with the Declaration of Helsinki and approved by the Local Research Ethics Committee of the Medical University of Łódź (project license #217/2014/TEBZN). Patient consent was not required.
Competing Interests
The authors declare that they have no competing interests.
Contributor Information
Hanna M. Romanska, Email: hanna.romanska@gmail.com.
Rafal Sadej, Email: rsadej@gumed.edu.pl.
References
- 1.Reis-Filho JS, Pusztai L. Gene expression profiling in breast cancer: classification, prognostication, and prediction. Lancet. 2011;378:1812–1823. doi: 10.1016/S0140-6736(11)61539-0. [DOI] [PubMed] [Google Scholar]
- 2.Yersal O, Barutca S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J Clin Oncol. 2014;5:412–424. doi: 10.5306/wjco.v5.i3.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clarke R, Liu MC, Bouker KB, Gu Z, Lee RY, Zhu Y, Skaar TC, Gomez B, O'Brien K, Wang Y. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene. 2003;22:7316–7339. doi: 10.1038/sj.onc.1206937. [DOI] [PubMed] [Google Scholar]
- 4.Davies C, Godwin J, Gray R, Clarke M, Cutter D, Darby S, McGale P, Pan HC, Taylor C, Wang YC. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 2011;378:771–784. doi: 10.1016/S0140-6736(11)60993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maggi A. Liganded and unliganded activation of estrogen receptor and hormone replacement therapies. Biochim Biophys Acta. 2011;1812:1054–1060. doi: 10.1016/j.bbadis.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou W, Slingerland JM. Links between oestrogen receptor activation and proteolysis: relevance to hormone-regulated cancer therapy. Nat Rev Cancer. 2014;14:26–38. doi: 10.1038/nrc3622. [DOI] [PubMed] [Google Scholar]
- 7.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 8.Bunone G, Briand PA, Miksicek RJ, Picard D. Activation of the unliganded estrogen receptor by EGF involves the MAP kinase pathway and direct phosphorylation. EMBO J. 1996;15:2174–2183. [PMC free article] [PubMed] [Google Scholar]
- 9.Joel PB, Traish AM, Lannigan DA. Estradiol-induced phosphorylation of serine 118 in the estrogen receptor is independent of p42/p44 mitogen-activated protein kinase. J Biol Chem. 1998;273:13317–13323. doi: 10.1074/jbc.273.21.13317. [DOI] [PubMed] [Google Scholar]
- 10.Joel PB, Smith J, Sturgill TW, Fisher TL, Blenis J, Lannigan DA. pp90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Mol Cell Biol. 1998;18:1978–1984. doi: 10.1128/mcb.18.4.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shah YM, Rowan BG. The Src kinase pathway promotes tamoxifen agonist action in Ishikawa endometrial cells through phosphorylation-dependent stabilization of estrogen receptor (alpha) promoter interaction and elevated steroid receptor coactivator 1 activity. Mol Endocrinol. 2005;19:732–748. doi: 10.1210/me.2004-0298. [DOI] [PubMed] [Google Scholar]
- 12.Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. Phosphatidylinositol 3-kinase/AKT-mediated activation of estrogen receptor alpha: a new model for anti-estrogen resistance. J Biol Chem. 2001;276:9817–9824. doi: 10.1074/jbc.M010840200. [DOI] [PubMed] [Google Scholar]
- 13.Sun M, Paciga JE, Feldman RI, Yuan Z, Coppola D, Lu YY, Shelley SA, Nicosia SV, Cheng JQ. Phosphatidylinositol-3-OH Kinase (PI3K)/AKT2, activated in breast cancer, regulates and is induced by estrogen receptor alpha (ERalpha) via interaction between ERalpha and PI3K. Cancer Res. 2001;61:5985–5991. [PubMed] [Google Scholar]
- 14.Merino D, Lok SW, Visvader JE, Lindeman GJ. Targeting BCL-2 to enhance vulnerability to therapy in estrogen receptor-positive breast cancer. Oncogene. 2016;35:1877–1887. doi: 10.1038/onc.2015.287. [DOI] [PubMed] [Google Scholar]
- 15.Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, Schiff R. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004;96:926–935. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 16.Kumar R, Mandal M, Lipton A, Harvey H, Thompson CB. Overexpression of HER2 modulates bcl-2, bcl-XL, and tamoxifen-induced apoptosis in human MCF-7 breast cancer cells. Clin Cancer Res. 1996;2:1215–1219. [PubMed] [Google Scholar]
- 17.Moerkens M, Zhang Y, Wester L, van de Water B, Meerman JH. Epidermal growth factor receptor signalling in human breast cancer cells operates parallel to estrogen receptor α signalling and results in tamoxifen insensitive proliferation. BMC Cancer. 2014;14:283. doi: 10.1186/1471-2407-14-283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aesoy R, Sanchez BC, Norum JH, Lewensohn R, Viktorsson K, Linderholm B. An autocrine VEGF/VEGFR2 and p38 signaling loop confers resistance to 4-hydroxytamoxifen in MCF-7 breast cancer cells. Mol Cancer Res. 2008;6:1630–1638. doi: 10.1158/1541-7786.MCR-07-2172. [DOI] [PubMed] [Google Scholar]
- 19.Meijer D, Sieuwerts AM, Look MP, van Agthoven T, Foekens JA, Dorssers LC. Fibroblast growth factor receptor 4 predicts failure on tamoxifen therapy in patients with recurrent breast cancer. Endocr Relat Cancer. 2008;15:101–111. doi: 10.1677/ERC-07-0080. [DOI] [PubMed] [Google Scholar]
- 20.Turner N, Pearson A, Sharpe R, Lambros M, Geyer F, Lopez-Garcia MA, Natrajan R, Marchio C, Iorns E, Mackay A. FGFR1 amplification drives endocrine therapy resistance and is a therapeutic target in breast cancer. Cancer Res. 2010;70:2085–2094. doi: 10.1158/0008-5472.CAN-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martinez-Outschoorn UE, Goldberg A, Lin Z, Ko YH, Flomenberg N, Wang C, Pavlides S, Pestell RG, Howell A, Sotgia F. Anti-estrogen resistance in breast cancer is induced by the tumor microenvironment and can be overcome by inhibiting mitochondrial function in epithelial cancer cells. Cancer Biol Ther. 2011;12:924–938. doi: 10.4161/cbt.12.10.17780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pontiggia O, Sampayo R, Raffo D, Motter A, Xu R, Bissell MJ, Joffé EB, Simian M. The tumor microenvironment modulates tamoxifen resistance in breast cancer: a role for soluble stromal factors and fibronectin through β1 integrin. Breast Cancer Res Treat. 2012;133:459–471. doi: 10.1007/s10549-011-1766-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sadej R, Romanska H, Baldwin G, Gkirtzimanaki K, Novitskaya V, Filer AD, Krcova Z, Kusinska R, Ehrmann J, Buckley CD. CD151 regulates tumorigenesis by modulating the communication between tumor cells and endothelium. Mol Cancer Res. 2009;7:787–798. doi: 10.1158/1541-7786.MCR-08-0574. [DOI] [PubMed] [Google Scholar]
- 24.Pontiggia O, Rodriguez V, Fabris V, Raffo D, Bumaschny V, Fiszman G, de Kier Joffé EB, Simian M. Establishment of an in vitro estrogen-dependent mouse mammary tumor model: a new tool to understand estrogen responsiveness and development of tamoxifen resistance in the context of stromal-epithelial interactions. Breast Cancer Res Treat. 2009;116:247–255. doi: 10.1007/s10549-008-0113-3. [DOI] [PubMed] [Google Scholar]
- 25.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 26.Dutt A, Salvesen HB, Chen TH, Ramos AH, Onofrio RC, Hatton C, Nicoletti R, Winckler W, Grewal R, Hanna M. Drug-sensitive FGFR2 mutations in endometrial carcinoma. Proc Natl Acad Sci U S A. 2008;105:8713–8717. doi: 10.1073/pnas.0803379105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piasecka D, Kitowska K, Czaplinska D, Mieczkowski K, Mieszkowska M, Turczyk L, Skladanowski AC, Zaczek AJ, Biernat W, Kordek R. Fibroblast growth factor signalling induces loss of progesterone receptor in breast cancer cells. Oncotarget. 2016 doi: 10.18632/oncotarget.13322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liao RG, Jung J, Tchaicha J, Wilkerson MD, Sivachenko A, Beauchamp EM, Liu Q, Pugh TJ, Pedamallu CS, Hayes DN. Inhibitor-sensitive FGFR2 and FGFR3 mutations in lung squamous cell carcinoma. Cancer Res. 2013;73:5195–5205. doi: 10.1158/0008-5472.CAN-12-3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ornitz DM. FGFs, heparan sulfate and FGFRs: complex interactions essential for development. Bioessays. 2000;22:108–112. doi: 10.1002/(SICI)1521-1878(200002)22:2<108::AID-BIES2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 30.Sadej R, Romanska H, Kavanagh D, Baldwin G, Takahashi T, Kalia N, Berditchevski F. Tetraspanin CD151 regulates transforming growth factor beta signaling: implication in tumor metastasis. Cancer Res. 2010;70:6059–6070. doi: 10.1158/0008-5472.CAN-09-3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Czaplinska D, Turczyk L, Grudowska A, Mieszkowska M, Lipinska AD, Skladanowski AC, Zaczek AJ, Romanska HM, Sadej R. Phosphorylation of RSK2 at Tyr529 by FGFR2-p38 enhances human mammary epithelial cells migration. Biochim Biophys Acta. 2014;1843:2461–2470. doi: 10.1016/j.bbamcr.2014.06.022. [DOI] [PubMed] [Google Scholar]
- 32.Imagawa W, Cunha GR, Young P, Nandi S. Keratinocyte growth factor and acidic fibroblast growth factor are mitogens for primary cultures of mammary epithelium. Biochem Biophys Res Commun. 1994;204:1165–1169. doi: 10.1006/bbrc.1994.2585. [DOI] [PubMed] [Google Scholar]
- 33.Palmieri C, Roberts-Clark D, Assadi-Sabet A, Coope RC, O'Hare M, Sunters A, Hanby A, Slade MJ, Gomm JJ, Lam EW. Fibroblast growth factor 7, secreted by breast fibroblasts, is an interleukin-1beta-induced paracrine growth factor for human breast cells. J Endocrinol. 2003;177:65–81. doi: 10.1677/joe.0.1770065. [DOI] [PubMed] [Google Scholar]
- 34.Ulich TR, Yi ES, Cardiff R, Yin S, Bikhazi N, Biltz R, Morris CF, Pierce GF. Keratinocyte growth factor is a growth factor for mammary epithelium in vivo. The mammary epithelium of lactating rats is resistant to the proliferative action of keratinocyte growth factor. Am J Pathol. 1994;144:862–868. [PMC free article] [PubMed] [Google Scholar]
- 35.Bansal R, Magge S, Winkler S. Specific inhibitor of FGF receptor signaling: FGF-2-mediated effects on proliferation, differentiation, and MAPK activation are inhibited by PD173074 in oligodendrocyte-lineage cells. J Neurosci Res. 2003;74:486–493. doi: 10.1002/jnr.10773. [DOI] [PubMed] [Google Scholar]
- 36.Pardo OE, Latigo J, Jeffery RE, Nye E, Poulsom R, Spencer-Dene B, Lemoine NR, Stamp GW, Aboagye EO, Seckl MJ. The fibroblast growth factor receptor inhibitor PD173074 blocks small cell lung cancer growth in vitro and in vivo. Cancer Res. 2009;69:8645–8651. doi: 10.1158/0008-5472.CAN-09-1576. [DOI] [PubMed] [Google Scholar]
- 37.Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their receptors in cancer. Biochem J. 2011;437:199–213. doi: 10.1042/BJ20101603. [DOI] [PubMed] [Google Scholar]
- 38.Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281:15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wijayaratne AL, McDonnell DP. The human estrogen receptor-alpha is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J Biol Chem. 2001;276:35684–35692. doi: 10.1074/jbc.M101097200. [DOI] [PubMed] [Google Scholar]
- 40.Han YH, Moon HJ, You BR, Park WH. The effect of MG132, a proteasome inhibitor on HeLa cells in relation to cell growth, reactive oxygen species and GSH. Oncol Rep. 2009;22:215–221. [PubMed] [Google Scholar]
- 41.Robertson JF. ICI 182,780 (Fulvestrant)--the first oestrogen receptor down-regulator--current clinical data. Br J Cancer. 2001;85(Suppl. 2):11–14. doi: 10.1054/bjoc.2001.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jansen MP, Foekens JA, van Staveren IL, Dirkzwager-Kiel MM, Ritstier K, Look MP, Meijer-van Gelder ME, Sieuwerts AM, Portengen H, Dorssers LC. Molecular classification of tamoxifen-resistant breast carcinomas by gene expression profiling. J Clin Oncol. 2005;23:732–740. doi: 10.1200/JCO.2005.05.145. [DOI] [PubMed] [Google Scholar]
- 43.Brechbuhl HM, Finlay-Schultz J, Yamamoto TM, Gillen AE, Cittelly DM, Tan AC, Sams SB, Pillai MM, Elias AD, Robinson WA. Fibroblast Subtypes Regulate Responsiveness of Luminal Breast Cancer to Estrogen. Clin Cancer Res. 2017;23:1710–1721. doi: 10.1158/1078-0432.CCR-15-2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang Y, Ray S, Reed JC, Ibrado AM, Tang C, Nawabi A, Bhalla K. Estrogen increases intracellular p26Bcl-2 to p21Bax ratios and inhibits taxol-induced apoptosis of human breast cancer MCF-7 cells. Breast Cancer Res Treat. 1997;42:73–81. doi: 10.1023/a:1005777219997. [DOI] [PubMed] [Google Scholar]
- 45.Perillo B, Sasso A, Abbondanza C, Palumbo G. 17beta-estradiol inhibits apoptosis in MCF-7 cells, inducing bcl-2 expression via two estrogen-responsive elements present in the coding sequence. Mol Cell Biol. 2000;20:2890–2901. doi: 10.1128/mcb.20.8.2890-2901.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19:202–208. doi: 10.1038/nm.3048. [DOI] [PubMed] [Google Scholar]
- 47.García-Becerra R., Santos N., Díaz L., Camacho J. Mechanisms of resistance to endocrine therapy in breast cancer: focus on signaling pathways, miRNAs and genetically based resistance. Int J Mol Sci. 2012;14:108–145. doi: 10.3390/ijms14010108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631–643. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 49.Zhang X, Nie D, Chakrabarty S. Growth factors in tumor microenvironment. Front Biosci (Landmark Ed) 2010;15:151–165. doi: 10.2741/3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Katoh M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review) Int J Mol Med. 2016;38:3–15. doi: 10.3892/ijmm.2016.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giulianelli S, Cerliani JP, Lamb CA, Fabris VT, Bottino MC, Gorostiaga MA, Novaro V, Gongora A, Baldi A, Molinolo A. Carcinoma-associated fibroblasts activate progesterone receptors and induce hormone independent mammary tumor growth: A role for the FGF-2/FGFR-2 axis. Int J Cancer. 2008;123:2518–2531. doi: 10.1002/ijc.23802. [DOI] [PubMed] [Google Scholar]
- 52.Tomlinson DC, Knowles MA, Speirs V. Mechanisms of FGFR3 actions in endocrine resistant breast cancer. Int J Cancer. 2012;130:2857–2866. doi: 10.1002/ijc.26304. [DOI] [PubMed] [Google Scholar]
- 53.Sikora MJ, Cooper KL, Bahreini A, Luthra S, Wang G, Chandran UR, Davidson NE, Dabbs DJ, Welm AL, Oesterreich S. Invasive lobular carcinoma cell lines are characterized by unique estrogen-mediated gene expression patterns and altered tamoxifen response. Cancer Res. 2014;74:1463–1474. doi: 10.1158/0008-5472.CAN-13-2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McLeskey SW, Zhang L, El-Ashry D, Trock BJ, Lopez CA, Kharbanda S, Tobias CA, Lorant LA, Hannum RS, Dickson RB. Tamoxifen-resistant fibroblast growth factor-transfected MCF-7 cells are cross-resistant in vivo to the antiestrogen ICI 182,780 and two aromatase inhibitors. Clin Cancer Res. 1998;4:697–711. [PubMed] [Google Scholar]
- 55.Kirkegaard T, Witton CJ, McGlynn LM, Tovey SM, Dunne B, Lyon A, Bartlett JM. AKT activation predicts outcome in breast cancer patients treated with tamoxifen. J Pathol. 2005;207:139–146. doi: 10.1002/path.1829. [DOI] [PubMed] [Google Scholar]
- 56.Yamashita H, Nishio M, Toyama T, Sugiura H, Kondo N, Kobayashi S, Fujii Y, Iwase H. Low phosphorylation of estrogen receptor alpha (ERalpha) serine 118 and high phosphorylation of ERalpha serine 167 improve survival in ER-positive breast cancer. Endocr Relat Cancer. 2008;15:755–763. doi: 10.1677/ERC-08-0078. [DOI] [PubMed] [Google Scholar]
- 57.Jiang J, Sarwar N, Peston D, Kulinskaya E, Shousha S, Coombes RC, Ali S. Phosphorylation of estrogen receptor-alpha at Ser167 is indicative of longer disease-free and overall survival in breast cancer patients. Clin Cancer Res. 2007;13:5769–5776. doi: 10.1158/1078-0432.CCR-07-0822. [DOI] [PubMed] [Google Scholar]
- 58.Weitsman GE, Li L, Skliris GP, Davie JR, Ung K, Niu Y, Curtis-Snell L, Tomes L, Watson PH, Murphy LC. Estrogen receptor-alpha phosphorylated at Ser118 is present at the promoters of estrogen-regulated genes and is not altered due to HER-2 overexpression. Cancer Res. 2006;66:10162–10170. doi: 10.1158/0008-5472.CAN-05-4111. [DOI] [PubMed] [Google Scholar]
- 59.Kok M, Holm-Wigerup C, Hauptmann M, Michalides R, Stål O, Linn S, Landberg G. Estrogen receptor-alpha phosphorylation at serine-118 and tamoxifen response in breast cancer. J Natl Cancer Inst. 2009;101:1725–1729. doi: 10.1093/jnci/djp412. [DOI] [PubMed] [Google Scholar]
- 60.de Leeuw R, Neefjes J, Michalides R. A role for estrogen receptor phosphorylation in the resistance to tamoxifen. Int J Breast Cancer. 2011;2011:232435. doi: 10.4061/2011/232435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bosch A, Li Z, Bergamaschi A, Ellis H, Toska E, Prat A, Tao JJ, Spratt DE, Viola-Villegas NT, Castel P. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci Transl Med. 2015;7:283ra251. doi: 10.1126/scitranslmed.aaa4442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnston SR, Saccani-Jotti G, Smith IE, Salter J, Newby J, Coppen M, Ebbs SR, Dowsett M. Changes in estrogen receptor, progesterone receptor, and pS2 expression in tamoxifen-resistant human breast cancer. Cancer Res. 1995;55:3331–3338. [PubMed] [Google Scholar]
- 63.Chang HL, Sugimoto Y, Liu S, Ye W, Wang LS, Huang YW, Lin YC. Keratinocyte growth factor (KGF) induces tamoxifen (Tam) resistance in human breast cancer MCF-7 cells. Anticancer Res. 2006;26:1773–1784. [PubMed] [Google Scholar]
- 64.Pan X, Zhou T, Tai YH, Wang C, Zhao J, Cao Y, Chen Y, Zhang PJ, Yu M, Zhen C. Elevated expression of CUEDC2 protein confers endocrine resistance in breast cancer. Nat Med. 2011;17:708–714. doi: 10.1038/nm.2369. [DOI] [PubMed] [Google Scholar]