

Abstract
Mitochondrial dysfunction is thought to play a significant role in neurodegeneration observed in Parkinson’s disease (PD), yet the mechanisms underlying this pathology remain unclear. Here, we demonstrate that loss of mitoNEET (CISD1), an iron–sulfur containing protein that regulates mitochondrial bioenergetics, results in mitochondrial dysfunction and loss of striatal dopamine and tyrosine hydroxylase. Mitochondria isolated from mice lacking mitoNEET were dysfunctional as revealed by elevated reactive oxygen species (ROS) and reduced capacity to produce ATP. Gait analysis revealed a shortened stride length and decreased rotarod performance in knockout mice, consistent with the loss of striatal dopamine. Together, these data suggest that mitoNEET KO mice exhibit many of the characteristics of early neurodegeneration in PD and may provide a novel drug discovery platform to evaluate compounds for enhancing mitochondrial function in neurodegenerative disorders.
Keywords: mitoNEET, mitochondrial dysfunction, aging, drug discovery
Graphical abstract
INTRODUCTION
Parkinson’s disease (PD) is a chronic and progressive neurodegenerative disorder, which affects more than a million people in the United States and approximately seven million worldwide.1 PD is the second most prevalent neurodegenerative disease after Alzheimer’s disease and is characterized by resting tremors, rigidity, and bradykinesia. These symptoms greatly affect the quality of life of patients and place a heavy burden on the family and health care systems. The clinical presentation is a result of the loss of dopaminergic neurons in substantia nigra, pars compacta, and a concomitant loss of striatal dopamine levels.2 No treatments are available to slow or stop the disease progression leading to a quest for identifying new drug targets that may modify disease outcomes.3
The etiology of PD remains to be fully elucidated, but current understanding points to both genetic and environmental components.4 Irrespective of the origin, mitochondrial dysfunction has been implicated to play a central role in the neurodegeneration of dopaminergic cells.5–8 Mitochondria play a critical role in maintaining the energy balance of neurons via oxidative phosphorylation. Elevated oxidative stress from reactive oxygen species (ROS) generated by dysfunctional mitochondria has been linked to neuron loss and abnormal mitophagy.8 Dysfunctional mitochondria that are not cleared may promote the ROS environment and potentiate neuronal damage.4
MitoNEET (CISD1) is an iron–sulfur (Fe–S) protein that was identified as an integral component of the outer mitochondrial membrane.9 Studies suggest mitoNEET may act as a redox-sensor for mitochondria.10 Additional functions proposed include maintenance of the oxidative capacity of mitochondria isolated from cardiac cells and roles in obesity and type 2 diabetes.11–13 This study was initiated to evaluate the relationships between mitochondrial dysfunction, dopaminergic markers, and PD as a result of loss of mitochondrial mitoNEET.
RESULTS AND DISCUSSION
Characterization of mitoNEET Knockout Mice
Immunohistochemical evaluation of mitoNEET revealed decreased immunoreactivity in mitoNEET KO mice (Figure 1B) as compared to wild-type (WT) mice (Figure 1A). Densitometric analysis of immunoreactivity revealed a significant decrease in mitoNEET staining as a result of gene deletion (Figure 1C, p < 0.05). Follow-up with Western blot shows that the antibody has low cross-reactivity with CISD2 (Naf-1/miner1), which can be seen in Figure 1D.14 This corroborates previously published data on these mice in a recent study focused on the liver.15
Figure 1.
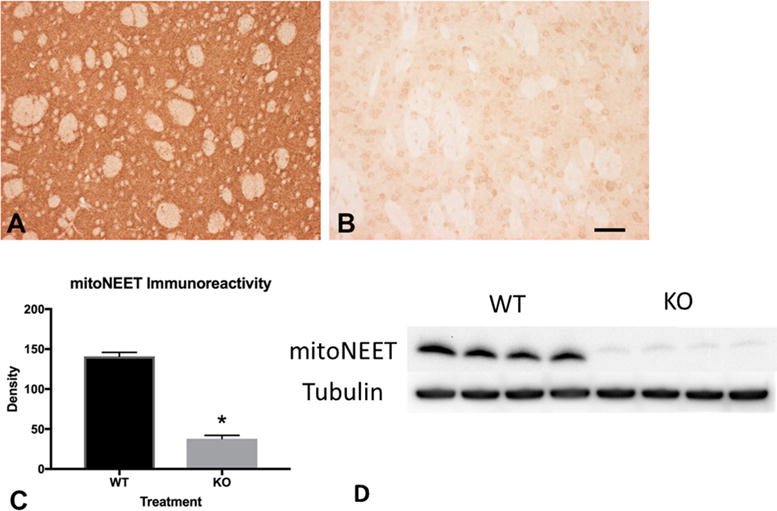
Immunohistochemical evaluation of mitoNEET deletion. Immunolocalization of mitoNEET revealed a near complete absence in knockout mice (B) compared to wild-type control animals (A). Quantitative analysis of immunoreactivity revealed a significant decrease as a result of mitoNEET deletion (C, p < 0.05). Scale bar = 50 μm. (D) Western Blot showing depletion of mitoNEET in KO mice. Abbreviations are wild-type (WT); mitoNEET knockout (KO).
Loss of mitoNEET Leads to Mitochondrial Dysfunction in Brain of KO Mice
We evaluated the effects of loss of mitochondrial mitoNEET from striatal mitochondria under normal conditions, where the respiration buffer was supplemented with pyruvate. Mitochondria isolated from mitoNEET KO mice showed an increased production of reactive oxygen species (Figure 2A), as well as superoxide (Figure 2B), compared to wild-type animals. Elevated superoxide production in knockout mice was visualized by DHE staining (Figure 2C). Deletion of mitoNEET decreased the ability of striatal mitochondria to produce ATP by approximately 50% (Figure 2D). State III respiration was decreased in mitochondria isolated from knockout mice by approximately 30% (Figure 2E) when compared to the control mitochondria, and the respiratory control ratio (RCR) was decreased accordingly in knockout mice (Figure 2F).
Figure 2.
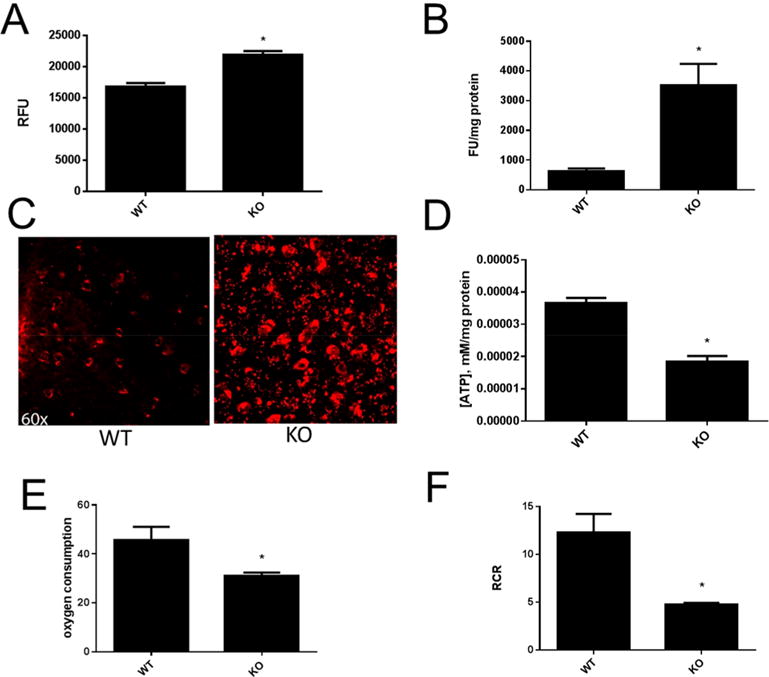
Loss of mitochondrial mitoNEET leads to mitochondrial dysfunction. Mitochondria from mitoNEET KO mice showed increased production of ROS as measured by Amplex Red (A). Superoxide levels measured by DHE were higher in mitochondria isolated from mitoNEET KO mice (B). In vivo staining of the striatum with DHE showed increased superoxide levels (C). Mitochondria isolated from knockout mice showed decreased ATP production compared to wild-type control animals (D). State III respiration was decreased in mitochondria isolated from mitoNEET KO mice (E) and RCR was reduced (F). Data are mean ± SEM where N = 5–7 mice. *Statistical significance P < 0.05.
mitoNEET KO Mice Show a Decrease in Striatal Tyrosine Hydroxylase (TH) and Dopamine Levels
We evaluated dopaminergic terminal integrity by analyzing the density of immunostaining for tyrosine hydroxylase in the striatum of wild-type and knock out mice (Figure 3). At six months, striatal TH levels were significantly decreased compared to control animals (p < 0.05). Deletion of mitoNEET resulted in an approximate 16% decrease in striatal TH staining (Figure 3C). Decreased striatal TH immunostaining observed at six months was accompanied by decreased tyrosine hydroxylase levels as evaluated by Western blotting (Figure 3D). Levels of the housekeeping protein tubulin did not vary across animals. Quantitation of blot data revealed an approximate 25% decrease in TH level following mitoNEET knockout (Figure 3E).
Figure 3.
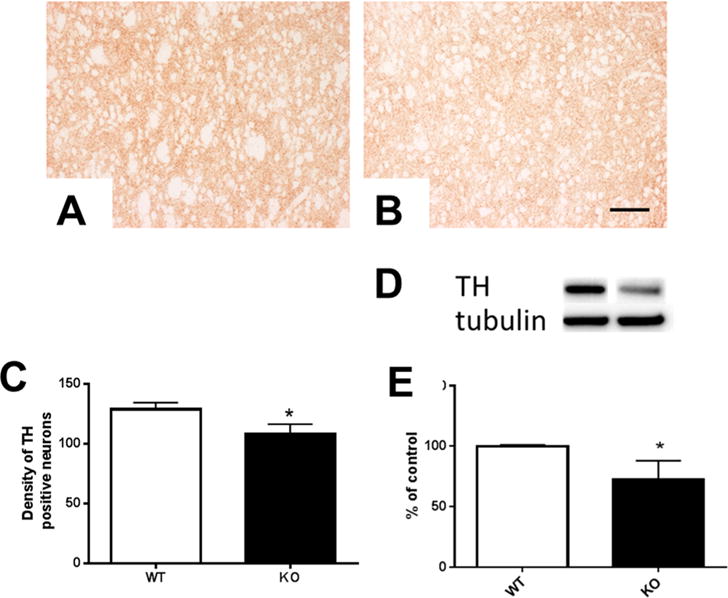
Tyrosine hydroxylase is reduced in mitoNEET KO striatum. mitoNEET deletion caused a temporal reduction in immunostaining for tyrosine hydroxylase in the striata of mice. At six months of age, knockout mice showed reduced TH immunostaining in the striatum (WT mice (A) and mitoNEET KO mice (B)). TH densitometric analysis of immunostaining revealed a significant reduction (p < 0.05) in levels of TH immunoreactivity (C). Scale bar = 60 μm. Striatal tyrosine hydroxylase levels were reduced in mitoNEET KO mice (D) compared to WT mice when TH was normalized to the housekeeping gene tubulin. Quantitative analysis of Western blot data revealed an approximate 25% decrease in TH of knockout mice (E). Data are mean ± SEM where n = 5–7 mice. *Statistical significance P < 0.05. Abbreviations are wild-type (WT); mitoNEET knockout (KO).
Deletion of mitoNEET Causes Iron Accumulation in Striatum
Histological evaluation of iron staining revealed increased iron content in the striatum of mitoNEET knockout mice at six months compared to wild-type animals (Figure 4). Levels of iron in knockout animals at one month did not differ from controls (data not shown). Most of the iron was present in fibers of passage coursing through the striatum. Iron-containing oligodendrocytes were observed among the fibers in both genotypes. These cells were round or slightly oval and contained more iron at one end of the cell. Iron-containing processes were observed throughout the neuropil and could be seen connecting to fiber bundles.
Figure 4.
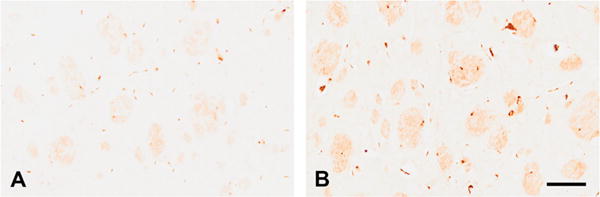
Iron accumulates following deletion of mitoNEET. Evaluation of iron staining in striatum revealed an accumulation of iron in mitoNEET knockout mice at six months of age (B) compared to control animals (A). Iron staining was observed in the fibers of passage and oligodendrocytes that are located at the borders of the fascicles. Iron-containing cells accumulate the metal at one pole of the cell. Little to no staining was observed in the matrix of either genotype but processes can be observed coursing from iron-positive cells to the fiber bundles. Scale bar = 50 μm.
mitoNEET KO Mice Show Motor Deficits
We evaluated gait and locomotor activity in mitoNEET KO and wild-type mice. Stride length was significantly decreased by nearly two centimeters in the mitoNEET KO mice as compared to wild-type controls (Figure 5A). mitoNEET KO mice fell from the rotarod in approximately 20 s, as compared to wild-type control animals that remained on the rod for nearly 95 s (Figure 5B). HPLC analysis of striatal homogenates revealed a significant decrease in levels of dopamine (Figure 5C) at six months following deletion of mitoNEET (p < 0.05).
Figure 5.
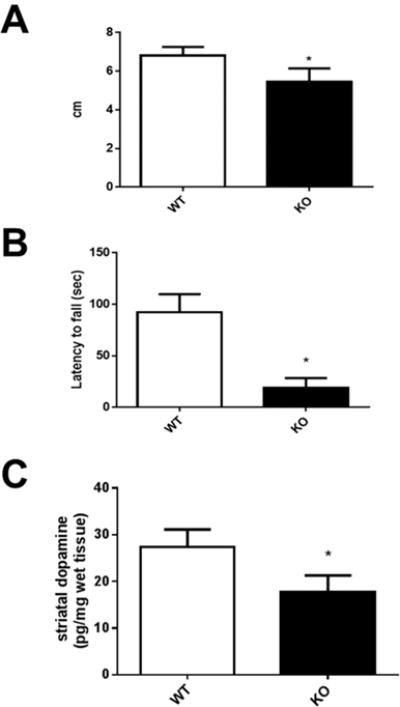
Behavior and neurochemical analysis of mitoNEET knockout mice. (A) The stride length of mice was decreased significantly following the deletion of mitoNEET. (B) Transgenic mice were less able to maintain balance on the rotarod and fell from the rod quicker than wild-type control animals. (C) HPLC analysis of striatal homogenates revealed a significant decrease in levels of dopamine at six months following deletion of mitoNEET. Data are mean ± SEM where N = 5–7 mice. *Statistical significance P < 0.05. Abbreviations are wild-type (WT); mitoNEET knockout (KO).
CONCLUSION
We evaluated the effects of loss of the mitochondrial protein, mitoNEET, in mice and report a progressive mitochondrial dysfunction and loss of immunoreactivity for TH in striatum at six months of age. Deletion of mitoNEET caused an increase in ROS and superoxide levels concomitant with decreased respiration and ATP production. Striatal dopamine levels were decreased with an increase in age, and this was accompanied by behavioral deficits associated with decreased dopamine levels. Thus, deletion of mitoNEET produces a neurochemical and behavioral phenotype consistent with that observed with dopaminergic neurodegeneration.
MitoNEET was initially discovered as a ligand of the type-2 diabetes drug, pioglitazone. The NEET family of proteins consists of three members, CISD1–3 (mitoNEET, Miner1, and Miner2), which share the CDGSH domain that contains the Fe–S cluster characteristic of this family. Recent characterization of NEET family function suggests these proteins are involved in cluster transfer or electron transfer.16 Protonation of histidine residues can cause release and transfer of an Fe–S cluster to an apo-acceptor protein; however, further research is required to understand the physiological functions of these transfers.
Mitochondrial dysfunction has been implicated in the loss of striatal dopamine and is thought to contribute to the pathology associated with Parkinson’s disease.8 Previous studies in cardiac myocytes and adipocytes revealed mitochondrial dysfunction following the loss of mitoNEET.12 Conversely, overexpression of mitoNEET in adipocytes of ob/ob mice resulted in increased uptake and storage of lipids and reduction of mitochondrial iron, ROS, and inflammatory markers.13 These results suggest that mitoNEET plays an important role in regulating the bioenergetics of mitochondria.
Deletion of mitoNEET was associated with a reduction in stride length and balance on the rotarod suggesting that the loss of striatal dopamine is reflected in a dysfunction of motor control in transgenic mice. The reduction in latency to fall was particularly striking, approximately 10 s in knockout mice, suggesting balance is severely affected in these animals. Only one other study evaluated rotarod performance as a function of mitoNEET deletion, and our data are consistent with latency times reported.17 Since PD is a movement disorder, our results suggest that loss of mitoNEET interferes with the normal activity of these neurons. Future studies will determine whether ligands of mitoNEET are able to modulate behavioral parameters.
Mitochondrial dysfunction is implicated in the pathogenesis of PD.18 Our work supports the hypothesis that mitoNEET is important to the proper function of mitochondria and that loss of mitochondrial integrity is associated with increased ROS, decreased striatal TH immunoreactivity and dopamine levels, and behavioral deficits. We observed that the deletion of mitoNEET caused a temporal accumulation of iron in the striatum of knockout mice compared to wild-type control animals. It seems likely that the increased iron is responsible for the elevated ROS and superoxide production, which drives the pathology associated with this manipulation. Iron is localized to the fibers of passage, which is the same physical location as the nerve terminals that display immunoreactivity for tyrosine hydroxylase. Perhaps the increased ROS and superoxide production produces a hostile environment in the striatum that facilitates damage to the dopaminergic nerve terminals and eventually causes their destruction and disappearance.
Ligands of mitoNEET have shown promise in the modulation of mechanisms involved in pathological processes.19 Known ligands belong to the thiazolidinedione (TZD) family; however, this binding motif is not absolutely necessary for binding to mitoNEET.19 Our laboratory has evaluated the ligand NL-1 and found it to be protective to cardiac stem cells in an oxidative environment.20 Other studies investigating the ligand TT01001 have shown protection from hyperglycemia, hyperlipidemia, and glucose intolerance in an animal model of diabetes.21 Future studies aimed at discovering ligands of mitoNEET may result in therapeutic interventions that are useful in the treatment of cancer, diabetes, Parkinson’s disease, or other pathologies that feature ROS as a component of the mitochondrial dysfunction.
METHODS
Animals
Mice were housed at Northeast Ohio Medical University (NEOMED). MitoNEET knockout (KO) mice were obtained from Taconic Farms, were backcrossed to N9 on C57BL6/J mice and bred as heterozygous mice. All animal experiments were done in accordance with the NEOMED Institutional Animal Care and Use Committee (IACUC), protocol number 12-021. For each of the wild-type (WT) and mitoNEET KO (homozygous mice), the groups consisted of five to seven mice to satisfy power analysis requirements and reflect an acceptable standard error. Animals were sacrificed by decapitation for biochemical experiments and overdose/perfusion for immunohistochemical experiments, at the age of six months of age, and all the current mice were male.
Mitochondrial Respiration
Mitochondrial respiration studies were performed as described previously.22 The brains were removed, and the striata were dissected on a glass plate chilled by ice. Striata from two mice were pooled and homogenized in isolation buffer (215 mM mannitol, 75 mM sucrose, 20 mM 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), 1 mM ethylene glycol-bis(β-amino-ethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) and 0.1% bovine serum albumin (BSA), pH 7.2). Tissue homogenate was centrifuged for 3 min at 1300g, 4 °C, and the supernatant was transferred to new centrifuge tube. The pellet was resuspended and recentrifuged, and the supernatant was combined with the first collection. Pooled supernatants were centrifuged for 10 min at 13000g, 4 °C. The pellet was resuspended in isolation buffer without EGTA, layered onto a discontinuous Ficoll gradient (7.5%, 10%), and centrifuged at 50000g for 30 min, 4 °C. The pellet was washed by resuspension in isolation buffer without EGTA, and the solution was centrifuged for 10 min at 10000g to obtain the final product. The pellet was resuspended in 50 μL of isolation buffer without EGTA and was used in the respiration studies. Mitochondrial activity was measured in an Oxytherm Clark-type electrode (Hansatech Instruments, UK). Respiration buffer consisted of 125 mM KCl, 20 mM HEPES, 2 mM MgCl2 and 2.5 mM KH2PO4, pH 7.2. The rate of oxygen consumption was measured by the addition of 2.5 μL of 2.5 mM pyruvate and 5 mM malate and the addition of ADP, 1.25 μL of 150 μM (State III), followed by 1 μL of 1 μM oligomycin (State IV). The respiratory control ratio (RCR) was determined by dividing State III by State IV.
Mitochondrial Reactive Oxygen Species and Superoxide and ATP Detection
Mitochondria were freshly isolated and purified for the ROS and superoxide experiments from the striata of both WT and mitoNEET KO mice. The mitochondria were resuspended in respiration buffer, and 50 μL samples were added to a 96-well black opaque bottom plate. Baseline ROS activity was determined using an Amplex Red kit (A22188, Invitrogen, Eugene, OR) and the protocol for H2O2 assay, including positive and negative controls. Briefly, 50 μL of the working solution was added to each well, and the plate was incubated for 30 min at room temperature in the dark. The fluorescence intensity was recorded in a Cytation 3 plate reader (CYT3F, BioTek, Winooski, VT) with excitation at 550 nm and emission detection at 590 nm. To detect baseline superoxide, 50 μL of 30 μM dihydroethidium (DHE) (D1168, Molecular Probes, Thermo-Fisher, Waltham, MA) was added to the wells, and the plate was incubated for 30 min at 37 °C. The fluorescence intensity was measured in a Cytation 3 plate reader (CYT3F, BioTek, Winooski, VT) with excitation at 400 nm and emission detection at 560 nm.23 For ATP, the same method was used to isolate mitochondria, and the 50 mL samples were added to white opaque bottom plates. To this was added ATP Glo (Promega, Madison WI), and the luminescence was read in the Cytation 3 plate reader (CYT3F, BioTek, Winooski, VT).
Superoxide Detection in Vivo
To evaluate the presence of superoxide in the brain, mice received an intraperitoneal injection of DHE (1.0 mg/kg in DMSO). Following a 4 h recovery, the brains were collected and prepared for immunohistochemistry as described below.
Brains from both WT and mitoNEET KO mice injected with DHE were cryoprotected overnight in 20% sucrose in Dulbecco’s phosphate-buffered saline (DPBS), sectioned at 50 μm on a sliding microtome (AO860, American Optical), and mounted onto microscope slides (ColorFrost+, Fisher Scientific, Pittsburgh, PA) unstained. The slides were covered with a coverslip with VectaShield (Vector Laboratories, Burlingame, CA) and examined by fluorescence microscopy.
Tissue Collection and Processing for Immunohistochemistry
Mice were overdosed with sodium pentobarbital (ip injection) and perfused with 25 mL of 0.9% saline followed by 50 mL of 4% paraformaldehyde at 4 °C using a perfusion pump set to 5.0 mL/min. The brains were removed from the skull from both the WT and mitoNEET KO mice, postfixed in 4% paraformaldehyde for 24 h, and changed into DPBS. Brains selected for paraffin processing were transferred into DPBS and prepared for shipping to AML Laboratories (St. Augustine, FL). At AML, brains were processed, embedded in paraffin, and sectioned at 15 μm using standard histological procedures.
Immunohistochemistry: Paraffin Sections
Slides were processed through a standard deparaffination to water and incubated in an antigen retrieval solution (10 mM citric acid, pH = 6.0) at 100 °C for 10 min. The solution was allowed to cool, and the slides were rinsed in DPBS. The sections were outlined with a hydrophobic pen (Fisher Scientific, Pittsburgh, PA) and processed for immunohistochemistry using a modified ABC procedure (Vector Laboratories, Burlingame, CA). Slides were transferred to a humidity chamber and treated with 10% methanol and 10% hydrogen peroxide in Dulbecco’s modified phosphate buffered saline (136 mM NaCl, 8 mM Na2HPO4, 2.6 mM KCl, 1.5 mM KH2PO4) for 15 min to quench endogenous peroxidase. Following three rinses in DPBS for 5 min each, sections were incubated in a permeabilizing solution (1.8% L-lysine, 4% normal horse serum, 0.2% Triton X-100 in DPBS) for 30 min at room temperature. The solution was drained from the slide and replaced with primary antibody solution in DPBS + 4% normal horse serum, and the slides were incubated overnight in the humidity chamber at room temperature. The following day, slides were rinsed three times in DPBS for 5 min each in a staining dish, then transferred back to the chamber for incubation with secondary antibody solution in DPBS + 4% normal horse serum for 2 h at room temperature. Following three rinses in DPBS, sections were incubated in Avidin D-HRP (1:1000 in DPBS, Vector Laboratories, Burlingame, CA) in the chamber for 1 h at room temperature, rinsed three times in DPBS, and incubated with chromogen solution (3–3′ diaminobenzidine, 50 mg in 100 mL of DPBS + 50 μL of 30% hydrogen peroxide (Electron Microscopy Sciences, Hatfield, PA) for 5 min. The slides were rinsed in DPBS for 5 min, dehydrated through a standard histological series, and covered with a coverslip with Permount (Fisher Scientific, Pittsburgh, PA). Antibodies investigated are listed in Table 1.
Table 1.
Antibodies and Dilutions Utilized
| antibody | company | catalog no. | location | primary | secondary |
|---|---|---|---|---|---|
| TH | Pel-Freeze | P40101-150 | Rogers, AR | 1:5000 | 1:1000 |
| mitoNEET | ProteinTech | 16006-1-AP | Rosemont, IL | 1:100 | 1:500 |
Histochemical Detection of Iron
Iron levels were determined in brain sections of wild-type and knockout mice using a modification of the Perl’s Prussian Blue stain with DAB enhancement.24 Briefly, slides were deparaffinized to water, and were incubated in fresh Perl’s solution (1% potassium ferrocyanide, pH = 1, 5% polyvinylpyrrolidone (PVP)) for 60 min with shaking. The slides were rinsed in PBS three times for 5 min each and transferred to methanol containing 0.01 M sodium azide and 0.3% hydrogen peroxide for 60 min with shaking. Following three rinses, slides were incubated in DAB solution (0.025% DAB, 0.12% hydrogen peroxide in 0.01 M Tris, pH = 7.4) for 30 min with shaking. The slides were rinsed three times as described, dehydrated through a standard histological series, and covered with a coverslip with Permount (Fisher Scientific, Pittsburgh, PA).
Microscopy
Slides were viewed on a Zeiss Axio Imager.Z2 microscope using AxioVision 4.8.2 software. Images were captured with an AxioCam ICc1 at 20× and stored on a dedicated server. Independent images were obtained for left and right striatum from approximately the same location in the striatum.
Image Analysis
Images were viewed and analyzed in Photoshop CC2017.0.0 (Adobe Systems Inc., San Jose, CA). To evaluate the density of immunostaining in striatum, images were converted to gray scale and saved. A 0.5 × 0.5 cm2 square was drawn and dragged to ten random positions (in the fibers of passage) in the striatum. The mean intensity of the immunostain was recorded from the histogram at each of the ten positions. Intensity values were recorded in Excel (Microsoft Corp., Redmond, WA) and subtracted from 276 (the maximum bright level) to reconcile the inverse relationship between reduced staining and increased brightness. Left and right striata were measured and recorded independently for each brain but were combined at the analysis stage since the treatment (mitoNEET deletion) presumably affects both hemispheres equally. The average staining density was calculated for each animal and recorded in Excel.
Protein Determination and Western Blotting
Striata were isolated from both the WT and mitoNEET KO mice and homogenized in cold lysis buffer (10-fold wet weight) containing protease inhibitors and phenylmethanesulfonylfluoride (PMSF). The homogenate was centrifuged for 15 min (14000g, 4 °C), and the supernatant was removed to a new tube. Protein in the supernatant was quantified using a BCA protein assay kit (23225, Pierce, ThermoFisher Scientific, Waltham, MA). Ten micrograms of each sample was separated using a 4–12% mini-PROTEAN precast gel running at 200 V for 35 min (Bio-Rad, Hercules, CA). The protein was transferred to a poly(vinylidene fluoride) (PVDF) membrane at 100 V for 1 h. The membrane was blocked for 1 h with reconstituted 5% nonfat dry milk in 0.2% TBST (Tris-buffered saline, 0.1% Tween 20). The membrane was incubated with a primary antibody solution in TBST (rabbit anti-TH, 1:1000, Millipore, Billerica, MA) overnight at 4 °C, washed for 5 min in PBS at room temperature, and incubated with a secondary antibody in TBST (goat anti-rabbit IgG-HRP, 1:3000, Invitrogen, ThermoFisher Scientific, Waltham, MA) for 1 h at room temperature. The blot was exposed to Pierce ECL Western blotting substrate (32106, Pierce, ThermoFisher Scientific, Waltham, MA) and analyzed on a FluorChem M chemiluminescent detector (92-15312-00, ProteinSimple, San Jose, CA).
HPLC Determination of Striatal Dopamine (DA)
To determine levels of DA, striata were dissected out from both the WT and mitoNEET KO mice and were weighed and placed in cold HClO4 (0.1 N, 500 μL, 4 °C). Tissue samples were sonicated and centrifuged at 1200g for 5 min, and an aliquot was removed to measure DA and 3,4-dihydroxyphenylacetic acid (DOPAC) by electrochemical detection. Biogenic amines were separated on a Supelco column (Discovery C18, 10 cm × 3 mm × 5 μm) with a 20 μL injection loop. Samples were eluted with a degassed isogradient mobile phase (50 mM sodium acetate, 27.4 mM citric acid, 10 mM NaOH, 0.1 mM sodium octyl sulfate, 0.1 mM EDTA, and 5% MeOH in filtered deionized water, pH = 4.5, and filtered with a 0.45 μm Millipore filter). Standards were diluted in 0.1 N perchloric acid in increments of 3.1, 6.2, 12.5, 25, 50, 100, 200, and 400 pg/20 μL. Chromatographs were analyzed using the ESA 501 program. The assay sensitivity (6.2–12.5 pg/20 μL) was determined by the presence of detectable peaks above baseline noise.
Stride Length Determination
The consequences of mitoNEET deletion on motor function were evaluated by gait and locomotor testing as performed previously.17,25 To analyze gait differences between the two groups of mice, both the WT and mitoNEET KO mice were placed in a walkway that was 4.5 cm wide, 12 cm tall, and 40 cm long and allowed to walk toward a dark home box. The front paws were dipped in orange ink and the back paws in black ink, and a white sheet of paper was placed along the walkway. Each mouse was allowed to walk in the walkway separately. The distances between steps were measured and recorded in Excel (Microsoft Corporation, Redmond, WA). The ability of animals to maintain balance and motor coordination was evaluated using the rotarod performance test.17 Time and speed parameters were obtained from a dose–response of saline-injected wild-type mice and were at a point prior to normal fatigue (150 s at 15 rpm). Mice were trained for 10 min at 5 rpm. The latency to fall (number of seconds on the rod) was recorded in Excel, and average latencies were calculated for each treatment group.
Statistical Analysis
Group means and standard errors were calculated with Prism 7.0c (GraphPad Software Inc., La Jolla, CA). Variance between means (WT and mitoNEET KO) was evaluated by Student t test. Group means were considered different when p < 0.05.
Acknowledgments
Funding
This study was funded in part by the Richard Nicely and Glenn and Karen Leppo Parkinson’s Disease Research Funds, the Stark Community Foundation Canton, Ohio, USA, and a grant from the Michael J. Fox Foundation (W.J.G.). The project was also supported by NIH Grants U54GM104942 and P20 GM109098 (W.J.G. and C.M.B.), K01NS081014 (C.M.B.), and R01ES021800 (J.R.R.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or any other funder.
Footnotes
ORCID
Werner J. Geldenhuys: 0000-0002-2405-376X
Author Contributions
W.J.G, S.A.B., L.L., H.M.Y., and A.S.D. designed and performed the research; W.J.G. and S.A.B. analyzed the data; W.J.G., S.D.C., P.G.S., C.M.B., and J.R.R. wrote the paper.
Notes
The authors declare no competing financial interest.
References
- 1.Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology. 2007;68:384–6. doi: 10.1212/01.wnl.0000247740.47667.03. [DOI] [PubMed] [Google Scholar]
- 2.Lees AJ, Hardy J, Revesz T. Parkinson’s disease. Lancet. 2009;373:2055–66. doi: 10.1016/S0140-6736(09)60492-X. [DOI] [PubMed] [Google Scholar]
- 3.Lee A, Gilbert RM. Epidemiology of Parkinson Disease. Neurol Clin. 2016;34:955–965. doi: 10.1016/j.ncl.2016.06.012. [DOI] [PubMed] [Google Scholar]
- 4.Nakamura T, Lipton SA. Redox regulation of mitochondrial fission, protein misfolding, synaptic damage, and neuronal cell death: potential implications for Alzheimer’s and Parkinson’s diseases. Apoptosis. 2010;15:1354–63. doi: 10.1007/s10495-010-0476-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson’s disease. Nat Rev Neurosci. 2006;7:207–19. doi: 10.1038/nrn1868. [DOI] [PubMed] [Google Scholar]
- 6.Arduino DM, Esteves AR, Cortes L, Silva DF, Patel B, Grazina M, Swerdlow RH, Oliveira CR, Cardoso SM. Mitochondrial metabolism in Parkinson’s disease impairs quality control autophagy by hampering microtubule-dependent traffic. Hum Mol Genet. 2012;21:4680–702. doi: 10.1093/hmg/dds309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blesa J, Trigo-Damas I, Quiroga-Varela A, Jackson-Lewis VR. Oxidative stress and Parkinson’s disease. Front Neuroanat. 2015;9:91. doi: 10.3389/fnana.2015.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moon HE, Paek SH. Mitochondrial Dysfunction in Parkinson’s Disease. Exp Neurobiol. 2015;24:103–16. doi: 10.5607/en.2015.24.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colca JR, McDonald WG, Waldon DJ, Leone JW, Lull JM, Bannow CA, Lund ET, Mathews WR. Identification of a novel mitochondrial protein (″mitoNEET″) cross-linked specifically by a thiazolidinedione photoprobe. Am J Physiol Endocrinol Metab. 2004;286:E252–60. doi: 10.1152/ajpendo.00424.2003. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Landry AP, Ding H. The mitochondrial outer membrane protein mitoNEET is a redox enzyme catalyzing electron transfer from FMNH2 to oxygen or ubiquinone. J Biol Chem. 2017;292:10061. doi: 10.1074/jbc.M117.789800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paddock ML, Wiley SE, Axelrod HL, Cohen AE, Roy M, Abresch EC, Capraro D, Murphy AN, Nechushtai R, Dixon JE, Jennings PA. MitoNEET is a uniquely folded 2Fe 2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc Natl Acad Sci U S A. 2007;104:14342–7. doi: 10.1073/pnas.0707189104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiley SE, Murphy AN, Ross SA, van der Geer P, Dixon JE. MitoNEET is an iron-containing outer mitochondrial membrane protein that regulates oxidative capacity. Proc Natl Acad Sci U S A. 2007;104:5318–23. doi: 10.1073/pnas.0701078104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kusminski CM, Holland WL, Sun K, Park J, Spurgin SB, Lin Y, Askew GR, Simcox JA, McClain DA, Li C, Scherer PE. MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat Med. 2012;18:1539–49. doi: 10.1038/nm.2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiley SE, Andreyev AY, Divakaruni AS, Karisch R, Perkins G, Wall EA, van der Geer P, Chen YF, Tsai TF, Simon MI, Neel BG, Dixon JE, Murphy AN. Wolfram Syndrome protein, Miner1, regulates sulphydryl redox status, the unfolded protein response, and Ca2+ homeostasis. EMBO Mol Med. 2013;5:904–18. doi: 10.1002/emmm.201201429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu X, Jogasuria A, Wang J, Kim C, Han Y, Shen H, Wu J, You M. MitoNEET Deficiency Alleviates Experimental Alcoholic Steatohepatitis in Mice by Stimulating Endocrine Adiponectin-Fgf15 Axis. J Biol Chem. 2016;291:22482–22495. doi: 10.1074/jbc.M116.737015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tamir S, Paddock ML, Darash-Yahana-Baram M, Holt SH, Sohn YS, Agranat L, Michaeli D, Stofleth JT, Lipper CH, Morcos F, Cabantchik IZ, Onuchic JN, Jennings PA, Mittler R, Nechushtai R. Structure-function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease. Biochim Biophys Acta, Mol Cell Res. 2015;1853:1294–315. doi: 10.1016/j.bbamcr.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 17.Antzoulatos E, Jakowec MW, Petzinger GM, Wood RI. Sex differences in motor behavior in the MPTP mouse model of Parkinson’s disease. Pharmacol, Biochem Behav. 2010;95:466–72. doi: 10.1016/j.pbb.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scott L, Dawson VL, Dawson TM. Trumping neurodegeneration: Targeting common pathways regulated by autosomal recessive Parkinson’s disease genes. Exp Neurol. 2017 doi: 10.1016/j.expneurol.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geldenhuys WJ, Yonutas HM, Morris DL, Sullivan PG, Darvesh AS, Leeper TC. Identification of small molecules that bind to the mitochondrial protein mitoNEET. Bioorg Med Chem Lett. 2016;26:5350–5353. doi: 10.1016/j.bmcl.2016.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Logan SJ, Yin L, Geldenhuys WJ, Enrick MK, Stevanov KM, Carroll RT, Ohanyan VA, Kolz CL, Chilian WM. Novel thiazolidinedione mitoNEET ligand-1 acutely improves cardiac stem cell survival under oxidative stress. Basic Res Cardiol. 2015;110:19. doi: 10.1007/s00395-015-0471-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi T, Yamamoto M, Amikura K, Kato K, Serizawa T, Serizawa K, Akazawa D, Aoki T, Kawai K, Ogasawara E, Hayashi J, Nakada K, Kainoh M. A novel MitoNEET ligand, TT01001, improves diabetes and ameliorates mitochondrial function in db/db mice. J Pharmacol Exp Ther. 2015;352:338–45. doi: 10.1124/jpet.114.220673. [DOI] [PubMed] [Google Scholar]
- 22.Geldenhuys WJ, Funk MO, Barnes KF, Carroll RT. Structure-based design of a thiazolidinedione which targets the mitochondrial protein mitoNEET. Bioorg Med Chem Lett. 2010;20:819–23. doi: 10.1016/j.bmcl.2009.12.088. [DOI] [PubMed] [Google Scholar]
- 23.Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci U S A. 2006;103:15038–43. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sands SA, Leung-Toung R, Wang Y, Connelly J, LeVine SM. Enhanced Histochemical Detection of Iron in Paraffin Sections of Mouse Central Nervous System Tissue: Application in the APP/PS1Mouse Model of Alzheimer’s Disease. ASN Neuro. 2016;8:175909141667097. doi: 10.1177/1759091416670978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geldenhuys WJ, Guseman TL, Pienaar IS, Dluzen DE, Young JW. A novel biomechanical analysis of gait changes in the MPTP mouse model of Parkinson’s disease. PeerJ. 2015;3:e1175. doi: 10.7717/peerj.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]