

Abstract
The Notch signaling pathway is a highly conserved pathway involved in cell fate determination in embryonic development and also functions in the regulation of physiological processes in several systems. It plays an especially important role in vascular development and physiology by influencing angiogenesis, vessel patterning, arterial/venous specification, and vascular smooth muscle biology. Aberrant or dysregulated Notch signaling is the cause of or a contributing factor to many vascular disorders, including inherited vascular diseases, such as cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy, associated with degeneration of the smooth muscle layer in cerebral arteries. Like most signaling pathways, the Notch signaling axis is influenced by complex interactions with mediators of other signaling pathways. This complexity is also compounded by different members of the Notch family having both overlapping and unique functions. Thus, it is vital to fully understand the roles and interactions of each Notch family member in order to effectively and specifically target their exact contributions to vascular disease. In this chapter, we will review the Notch signaling pathway in vascular smooth muscle cells as it relates to vascular development and human disease.
1. INTRODUCTION
The Notch signaling pathway is an evolutionarily conserved pathway that plays a critical role in cell fate decisions for numerous systems in vertebrates (Artavanis-Tsakonas, Rand, & Lake, 1999; Ehebauer, Hayward, & Arias, 2006). Over the past ~15 years, many studies have demonstrated the importance of Notch signaling in regulating vascular development and physiology, through control of endothelial cells and smooth muscle cells and their interactions with each other. The overall contribution of Notch signaling to the vasculature and especially the role of endothelial cell-derived Notch signaling has been well studied and previously covered in several exceptional reviews (Fouillade, Monet-Lepretre, Baron-Menguy, & Joutel, 2012; Gridley, 2007, 2010; Iso, Hamamori, & Kedes, 2003; Phng & Gerhardt, 2009; Roca & Adams, 2007; Rostama, Peterson, Vary, & Liaw, 2014). In this review, we will focus on the role of Notch signaling in vascular smooth muscle cells (VSMCs) and how it regulates the development and homeostasis of the vasculature, and how when deficient or perturbed can contribute to disease states.
Originally discovered in Drosophila (and named for the “notched” wings that resulted from its mutation) (Morgan, 1917), the canonical Notch signaling pathway is relatively straightforward, consisting of a membrane-bound ligand and receptor engagement on neighboring cells leading to cleavage of the Notch intracellular domain (NICD) of the receptor, which shuttles to the nucleus and acts a transcriptional cofactor. This series of events allows for a signal to be passed directly from receptor activation to transcriptional modulation while bypassing the signaling intermediates and kinase cascades necessary for many other signaling pathways. This linear system coupled with its inherent feedback mechanisms allows for the creation of signal-sending: signal-receiving binary cell fate determinations in early developmental processes, such as those seen in the lateral specification of neural and epidermal cells in Drosophila (Heitzler & Simpson, 1991). However, beyond its role initially described in lower species, this basic signaling pathway has been adapted and utilized by nearly every cell type and organ system to enact a myriad of functions.
The cardiovascular system is the first organ system to develop in embryogenesis and is made up of the heart and an expansive network of vessels of different types and sizes (Patel-Hett & D’Amore, 2011). Early in development, angioblasts differentiate into endothelial cells and aggregate into a nascent vascular network in a process called vasculogenesis. This network is extended, pruned, and remodeled over the course of development to meet the needs of the developing embryo. To support the growing network of endothelial cell tubes, mural cells are recruited and differentiate into VSMCs or pericytes. These mural cells are vital to the maturation of vessels into functional arteries, veins, and capillaries. VSMCs and pericytes play countless roles in these vessels, from controlling blood flow to regulating permeability, and those roles shift depending upon the state of the vessel, embryological origin, and the environmental context. VSMCs are not “terminally differentiated” cells and are capable of adapting their activities in response to molecular and physiological cues. This ability of VSMCs to radically alter their functions in different contexts is known as VSMC phenotypic switching or plasticity. This concept has been well studied and reviewed (Alexander & Owens, 2012; Gomez & Owens, 2012; Owens, Kumar, & Wamhoff, 2004) and attributed to many of the activities of VSMCs in vascular diseases.
In VSMCs, the Notch receptors Notch2 and Notch3 and the ligand Jagged1 predominate (High et al., 2007; Tang et al., 2010). Notch1 and Notch4 are more highly expressed in endothelial cells, although Notch1 has been shown to play a role in VSMCs in the context of vascular injury (Li, Takeshita, et al., 2009) and in pericytes (Kofler, Cuervo, Uh, Murtomaki, & Kitajewski, 2015). Over the past few decades, extensive work has been performed to ascertain the role of these receptors and Notch signaling as a whole in VSMCs. While significant progress has been made and strong connections have linked the Notch pathway to vascular development, VSMC differentiation and phenotype, and vascular disease, there is still much that is not understood about the precise mechanisms involved (Fig. 1).
Fig. 1.
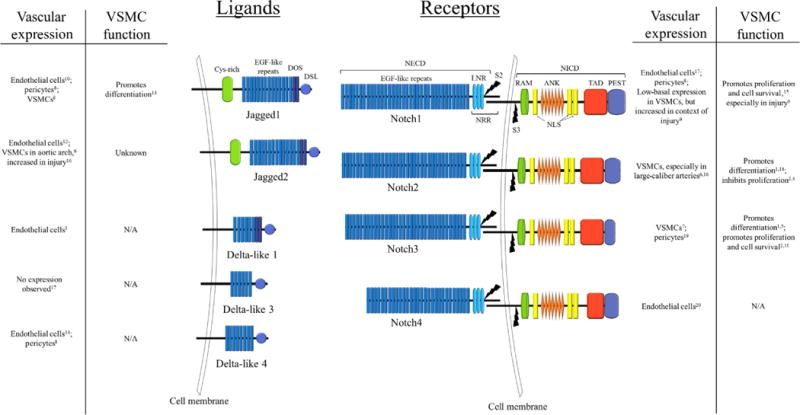
Notch family of ligands and receptors. Abbreviations: ANK, ankyrin repeats; Cys-rich, cysteine-rich region; DOS, Delta and OSM11-like proteins motif; DSL, Delta/Serrate/Lag2 motif; LNR, Lin12-Notch repeats; NECD, Notch extracellular domain; NICD, Notch intracellular domain; NLS, nuclear localization signal; NRR, negative regulatory region; PEST, proline/glutamic acid/serine/threonine-rich motifs; RAM, RBPJassociation module; S2, cleavage site 2; S3, cleavage site 3; TAD, transactivation domain. References: 1, Baeten, Jackson, McHugh, and Lilly (2015); 2, Baeten and Lilly (2015); 3, Beckers, Clark, Wunsch, Hrabe De Angelis, and Gossler (1999); 4, Boucher, Harrington, Rostama, Lindner, and Liaw (2013); 5, Domenga et al. (2004); 6, High et al. (2007); 7, Joutel et al. (2000); 8, Kofler et al. (2015); 9, Li, Takeshita, et al. (2009); 10, Lindner et al. (2001); 11, Liu, Zhang, Kennard, Caldwell, and Lilly (2010); 12, Luo, Aster, Hasserjian, Kuo, and Sklar (1997); 13, Manderfield et al. (2012); 14, Shutter et al. (2000); 15, Sweeney et al. (2004); 16, Varadkar et al. (2008); 17, Villa et al. (2001); 18, Wang, Zhao, Kennard, and Lilly (2012); 19, Wang, Pan, Moens, and Appel (2014); 20, Wu et al. (2005).
2. THE NOTCH SIGNALING PATHWAY
2.1 The Canonical Notch Signaling Pathway
In mammals, there are four Notch receptors (Notch1–4) and five ligands (Jagged1 and 2, Delta-like 1, 3, and 4) which are all Type I transmembrane proteins. Both the ligands and receptors have long extracellular domains (ECDs) made up of epidermal growth factor-like (EGF) repeats which serve as the binding platform for the receptor–ligand interaction. The receptor arrives at the cell membrane as a heterodimer after it is cleaved by furin at the site 1 (S1). After a receptor is bound by ligand, the negative regulatory region (NRR) is displaced, exposing the site 2 (S2) for cleavage by one of the ADAM metalloproteases (a disintegrin and metalloproteinase domain-containing protein), and the site 3 (S3) for cleavage by the γ-secretase complex, releasing the NICD fragment. After cleavage at the S3 site, the NICD’s nuclear localization signal triggers translocation to the nucleus. The NICD acts as a transcriptional cofactor by forming a complex with the CSL (CBF1/SuH/Lag1) transcription factor named for its homologs in mammals, Drosophila, and Caenorhabditis elegans, though it is now designated RBPJ (recombination signal-binding protein for immunoglobulin kappa J region) in mammals. This complex binds to its target DNA sequence and to one of the three mastermind-like (MAML) proteins, which act as transcriptional activators. The formation of this complex displaces transcriptional repressors including SKIP (Ski-interacting protein), CIR (CBF1-interacting corepressor), KyoT2, and SHARP (SMRT- and HDAC-associated repressor) and histone deacetylases (HDACs) from RBPJ, leading to expression of Notch target genes (Borggrefe & Oswald, 2009). Among the most highly activated genes are a family of basic helix–loop–helix (bHLH) proteins hairy/enhancer of split (HES) and HES-related proteins (HEY/HRT/HERP) which act as transcriptional repressors.
Though the transduction of the Notch signal via NICD cleavage and nuclear translocation is well established, there are many opportunities along its path for regulation to occur that introduces signaling diversity. One such mechanism is the Fringe family of glycosyltransferases, Lunatic Fringe, Manic Fringe, and Radical Fringe (Moloney et al., 2000). Both Notch ligands and receptors have attached sugar moieties on the EGF repeats of the ECDs, and Fringe glycosyltransferases are able to elongate N- and O-linked glycans at certain positions during posttranslational processing in the Golgi. The length and position of these glycans can determine whether ligands and receptors interact between neighboring cells (trans activation) or within the same cell (cis inhibition) (LeBon, Lee, Sprinzak, Jafar-Nejad, & Elowitz, 2014). The glycosylation state can also influence the binding affinities for the different receptor–ligand pairs (Hicks et al., 2000; Stanley & Okajima, 2010). This influence on which receptor and ligand interact can then control ultimate effect of Notch signaling, as the different Notch ligands and Notch receptors are capable of producing different signaling outcomes. For instance, the ligands JAG1 and DLL4 have opposing effects on angiogenesis and their activity is regulated by their glycosylation (Benedito et al., 2009), and DLL4, but not DLL1, can induce pericyte differentiation in myoblasts (Cappellari et al., 2013). Also, the NICDs of the different Notch receptors can activate transcription of their target genes with varying efficacy depending on the orientation and number of RBPJ-binding sites in the promoter (Ong et al., 2006). Another potential mechanism of Notch signal diversity is the control of the intensity and duration of the Notch signal. It has been demonstrated that some Notch target genes can be activated by low levels of active NICD, while others require high levels of activity (Ong et al., 2006). The number of NICD molecules present in the nucleus is dependent on both the influx of NICD from receptor activation and the turnover or degradation of NICD. The degradation of NICDs is regulated by the E3 ubiquitin ligases (Lai, 2002) which ubiquitinate NICDs in their PEST domain (rich in proline [P], glutamic acid [E], serine [S], and threonine [T]) and target them for degradation by the proteasome (Fig. 2).
Fig. 2.
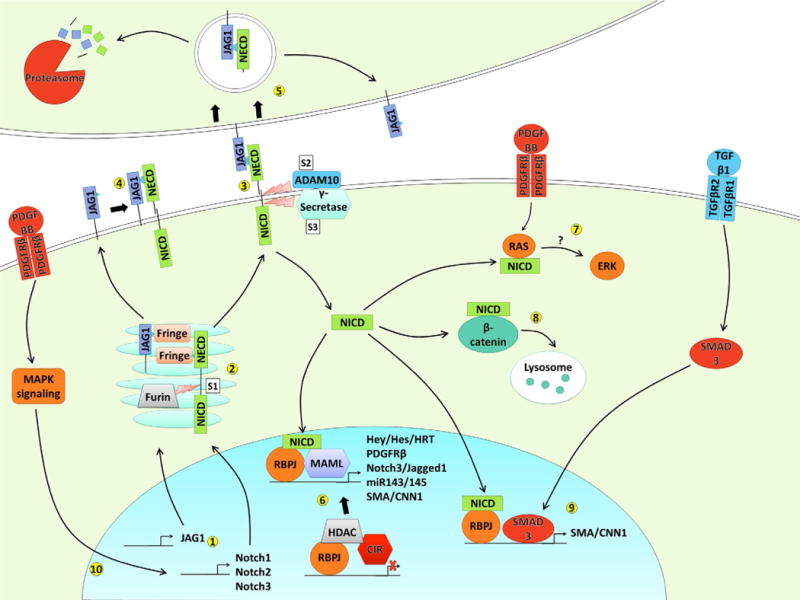
Notch signaling pathway. (1) Notch receptor and ligand genes are transcribed and translated into protein. (2) During posttranslational processing in the Golgi, Notch receptors are cleaved by furin to create heterodimers; both ligands and receptors are glycosylated on their EGF-like repeats by Fringe family enzymes. (3) trans activation of Notch receptor by Notch ligand from a neighboring cell, triggering cleavage by ADAM10 at S2 and γ-secretase at S3, releasing the NICD. (4) cis inhibition of Notch receptor by Notch ligand on the same cell, preventing activation. (5) Endocytosis of Notch ligand with associated NECD can be degraded by the proteasome or recycled back to the cell surface. (6) NICD is shuttled to the nucleus where it binds to RBPJ, displacing HDACs and corepressors such as CIR and recruiting MAML to activate transcription of Notch target genes. (7) NICD can both directly interact with Ras and promote ERK phosphorylation, though the mechanism is not yet known. (8) NICD can bind to the Wnt signaling mediator β-catenin and target it for degradation in the lysosome. (9) Transcription of some genes, including smooth muscle contractile genes, is coregulated by NICD/RBPJ and the TGFβ transcription factor SMAD3. (10) Many signaling pathways can regulate expression of Notch components, including PDGFRβ signaling via the MAPK pathway.
2.2 Interaction with Other Signaling Pathways and Noncanonical Signaling
In addition to the long understood canonical Notch pathway, there is increasing evidence that the Notch pathway can exert effects outside of its prominent role as a transcriptional cofactor (Andersen, Uosaki, Shenje, & Kwon, 2012; Ayaz & Osborne, 2014; Martinez Arias, Zecchini, & Brennan, 2002; Sanalkumar, Dhanesh, & James, 2010). An example of this is the ability of the NICD to bind active β-catenin, leading to its degradation by the lysosome (Kwon et al., 2011). Though non-canonical activities of Notch have been shown in many different cell types and systems, evidence for these activities in VSMCs is relatively sparse and the mechanisms not well defined (Wang, Prince, Mou, & Pollman, 2002). However, based on the ubiquity of noncanonical signaling in other contexts, it seems likely that some of the effects of Notch signaling in VSMCs are due to undiscovered noncanonical interactions.
Most of the observed noncanonical signaling involves interactions of Notch components with members of other signaling pathways, but the overlap of Notch signaling and other pathways is not limited to the non-canonical pathway. Signals from outside of the Notch pathway can regulate Notch expression and activity as well as alter the transcription factors recruited to Notch target genes. Notch signaling can also in turn regulate the expression and activity of other signaling pathways. Much of the diversity in Notch signaling outcomes can be attributed to its interaction with other signaling pathways. Here we will discuss some of the known interactions with other signaling pathways in VSMCs.
2.2.1 Platelet-Derived Growth Factor B
Platelet-derived growth factor B (PDGF-B) is a major mitogen and chemotractant in VSMCs that acts through its receptor, PDGFRβ (Donovan, Abraham, & Norman, 2013). PDGF signaling has been shown to interact with Notch signaling in VSMCs in two major capacities. First, Notch signaling has been shown to directly promote the transcription of PDGFRβ and enhance the signaling effects of PDGF-B (Donovan et al., 2013; Jin et al., 2008). This relationship is especially apparent in pericytes, as both PDGFRβ and Notch3 are considered markers of pericytes and their dysfunction in animal models and humans can produce similar phenotypes (Lee, 2013; Martignetti et al., 2013; Wang et al., 2014). Second, it has been shown that PDGF-B signaling can regulate the expression of Notch components (Baeten & Lilly, 2015; Campos, Wang, Pollman, & Gibbons, 2002). Overall, it is apparent that the PDGF and Notch signaling pathways are strongly intertwined, with significant functional overlap in pericytes and coregulation in VSMCs.
2.2.2 Transforming Growth Factor β
The transforming growth factor β (TGFβ) pathway is a vital contributor to the differentiation of VSMCs (Guo & Chen, 2012). The Notch and TGFβ signaling pathways cooperatively regulate the differentiation of VSMCs through direct interaction of NICD and SMAD2/3 (a transcription factor within the TGFβ pathway) and transcription of smooth muscle contractile genes (Tang et al., 2010). This cooperative induction of VSMC differentiation has also been shown in progenitor cell types (Grieskamp, Rudat, Ludtke, Norden, & Kispert, 2011; Kurpinski et al., 2010), and the direct interaction of NICD and SMAD3 as transcriptional activators has been shown in other systems (Blokzijl et al., 2003). Interestingly, it has also been shown that these two pathways can negatively regulate each other’s expression. This is seen in fibroblasts where TGFβ signaling decreases expression of Notch3 (Kennard, Liu, & Lilly, 2008) and in VSMCs where Notch signaling promotes the transcription of miR145 which inhibits the TGFβ pathway by targeting the TGFβ receptor 2 (Boucher, Peterson, Urs, Zhang, & Liaw, 2011; Zhao et al., 2015). Taken together, the Notch and TGFβ pathways are able to both cooperatively promote transcription of their downstream genes and inhibit each other’s expression, suggesting a very complex relationship and potentially a mechanism for negative feedback within each pathway.
2.2.3 Mitogen-Activated Protein Kinase
The mitogen-activated protein kinase (MAPK) pathway is one of the major signaling pathways controlling cell growth, survival, and many other processes and is a main transducer of growth factor signals (Pearson et al., 2001). In cultured VSMCs, Notch3 is capable of activating this pathway by phosphorylation of the MAPK component ERK (Baeten & Lilly, 2015; Wang, Campos, Prince, Mou, & Pollman, 2002), through a mechanism that is not yet known. Physical interaction of Notch and MAPK components has previously been shown in other models (Hodkinson et al., 2007). However, it may simply be a result of upregulation of growth factor ligands or receptors by Notch signaling, such as PDGFRβ. Regardless of how it is enacting this effect, it is clear that at least in some contexts Notch signaling is capable of promoting MAPK pathway activity.
2.2.4 Wingless-Related Integration Site
The Wnt (wingless-related integration site) signaling pathway is an important developmental pathway that regulates smooth muscle proliferation, migration, and survival (Mill & George, 2012). The Wnt and Notch pathways have significant cross talk in many systems, by both direct effect on each other’s pathway components and shared regulation of developmental processes (Andersen et al., 2012; Duncan et al., 2005; Espinosa, Ingles-Esteve, Aguilera, & Bigas, 2003; Fre et al., 2009; Hayward et al., 2005). These interactions have been shown to extend to VSMCs as well, as early mesoderm cells in chick embryos are pushed toward a VSMC lineage through cooperation of the Notch and Wnt pathway (Shin, Nagai, & Sheng, 2009). While evidence for Wnt/Notch pathway interaction in VSMCs is limited, the results in mesodermal smooth muscle progenitors coupled with the known cross talk between Notch and Wnt signaling in other systems suggest that these pathways cooperate to drive early VSMC differentiation.
3. NOTCH SIGNALING IN VSMC DEVELOPMENT
Notch signaling plays an essential role in the development of the cardiovascular system, from hematopoiesis to cardiac development to endothelial cell sprouting and vasculogenesis. In the development and differentiation of VSMCs, the functions of Notch signaling can be categorized into two main roles: construction of the vessel wall and arterial–venous specification.
3.1 Constructing a Vessel Wall
Following the initial stages of vasculogenesis, nascent vessels are composed of an endothelial tube surrounded by perivascular cells. For a vessel to mature into a conduit capable of withstanding increasing pressure and volume demands, it must recruit mural cells to encompass it and differentiate into one or more layers of vascular smooth muscle. This recruitment is largely driven by a PDGF-B gradient secreted by the endothelial cells (Hellstrom, Kalen, Lindahl, Abramsson, & Betsholtz, 1999). Once recruited to the nascent vessel, endothelial cell-derived Notch ligands can activate Notch signaling in these mural cells, which induces integrin adhesion to the endothelial basement membrane and initiates maturation and differentiation (Scheppke et al., 2012). We have learned a great deal about how endothelial-derived Notch signals affect VSMCs and mural cells from mutant mouse models and in vitro systems. In the great vessels of the outflow tract, cardiac neural crest progenitors give rise to the VSMCs within the vessel walls. With the complete abrogation of Notch signaling by “knocking in” of a dominant-negative MAML1 in cardiac neural crest cells, mice display various defects of the outflow tract and aortic arch associated with decreased differentiation and expression of smooth muscle markers (High et al., 2007). Mice with an endothelial deletion of Jagged1 also display decreased VSMC differentiation and expression of smooth muscle markers (High et al., 2008). Through a combination of in vitro coculture experiments (Liu, Kennard, & Lilly, 2009) and the cardiac neural crest-specific Jagged1 knockout mice (Manderfield et al., 2012), it was demonstrated that induction of Notch signaling in VSMCs by endothelial-expressed Jagged1 leads to the increased expression of both Notch3 and Jagged1. This allows for lateral induction of Notch signaling by homotypic VSMC–VSMC interactions through multiple layers of smooth muscle to promote VSMC differentiation after the initial signal is received from endothelial-derived Jagged1 (Hoglund & Majesky, 2012). This model has been further supported by smooth muscle deletion of Jagged1, RBPJ, and a smooth muscle deletion of Notch2 coupled with heterozygous deletion of Notch3 (Baeten et al., 2015; Feng, Krebs, & Gridley, 2010; Krebs, Norton, & Gridley, 2016). All three of these models develop patent ductus arteriosus (PDA) due to contractile deficits of the VSMCs and present decreased expression of smooth muscle markers. An interesting parallel exists between this model and one proposed from experiments performed by the Krasnow lab (Greif et al., 2012), where they demonstrate that the expression of PDGFRβ and ACTA2 (alpha smooth muscle actin) moves through layers of smooth muscle like a wave. They describe a major role for endothelial-derived PDGF-B in the migration, orientation, and proliferation of these smooth muscle cell layers and suggest that there may be another signaling pathway contributing to the radial pattern of construction. Indeed, Notch signaling would elegantly fit into this proposed model, as it is known to induce PDGFRβ and ACTA2, and could be passed through the layers of smooth muscle via cell–cell contact (Jin et al., 2008; Noseda et al., 2006).
An interesting finding from comparing the knockout phenotypes of mice deficient in Notch2 and Notch3 is their unique roles in different vascular beds. Notch2 is mainly expressed in the outflow tract and other large-caliber vessels, while Notch3 is expressed in all mural cells (High et al., 2007; Joutel et al., 2000). Likewise, in large-caliber vessels, Notch2 signaling is indispensable, with loss of Notch3 only acting to amplify defects in Notch2-null mice (McCright et al., 2001; Wang et al., 2012). This demonstrates that while there may be some functional redundancy of Notch2/3 in large-caliber vessels, Notch3 cannot completely replace Notch2 function. However, in pericytes and VSMCs in smaller caliber vessels where Notch2 expression is significantly diminished, Notch3 plays a major role, especially in the cerebral vasculature (Henshall et al., 2015; Liu et al., 2010; Volz et al., 2015; Wang et al., 2014).
3.2 Arterial–Venous Specification
Another important role for Notch signaling in VSMCs in the developing vasculature is the specialization of vessels into arteries and veins. Arteries and veins have distinct structural properties and express distinct protein markers that contribute to their unique functions in the cardiovascular system (Wang, Chen, & Anderson, 1998). In endothelial cells, Notch signaling regulates arterial specification in cooperation with VEGF (Hirashima, 2009; Kim et al., 2008). Similarly, Notch3 regulates arterial specification in VSMCs. In a mouse model with global knockout of Notch3, arteries acquired a more venous phenotype, with enlarged vessels, thinner and disorganized smooth muscle layers, and less festooned elastic lamina (Domenga et al., 2004). They did not see these changes in the outflow tract or aorta where Notch2 is highly expressed, likely suggesting a functional redundancy for Notch2 and Notch3 in this context. The expression of the arterial marker smoothelin and a LacZ reporter driven by an arterial-specific promoter element was also reduced in these Notch3-null mice, showing both structural and morphological defects in arterial specification. In zebrafish, Notch3 expression was found to be restricted to arteries, and inhibition of Notch signaling in zebrafish leads to decreased arterial specification and similar phenotypes compared to the mouse model (Lawson et al., 2001).
4. NOTCH SIGNALING AND VSMC PHENOTYPE
The ability of VSMCs to display various phenotypes and enact disparate functions depending on their cellular context, known as phenotypic switching or plasticity, is central to their changing roles from early developmental vasculogenesis to vascular homeostasis and remodeling in adult animals. Notch signaling has been closely tied to the many possible phenotypic endpoints of VSMCs. While there are still disparities in the literature about some of the phenotypic effects of Notch signaling in VSMCs, we are starting to obtain a clearer picture of how Notch activation drives VSMCs to different endpoints and the contributions from the different Notch receptors.
4.1 Differentiation
A “differentiated” VSMC is generally described as a quiescent cell expressing contractile proteins (such as ACTA2, smooth muscle myosin heavy chain [MYH11], transgelin [SM22α], and calponin [CNN1]) and primed to provide contractile force to constrict (or dilate) the vessel in which it resides in response to external cues. This is the state necessary for mature vessels to maintain physiological homeostasis. Notch signaling has been tied to regulation of VSMC differentiation and expression of contractile proteins for some time. While initial studies in vitro and in injury models showed that Notch-activated genes of the HRT (hairy-related transcription factors) family could repress myocardin-induced contractile protein expression (Doi et al., 2005; Proweller, Pear, & Parmacek, 2005; Yoshida et al., 2003), there were also several studies, showing that Notch activity promoted VSMC differentiation and contractile marker expression (Doi et al., 2006; Domenga et al., 2004; High et al., 2007). Although the discrepancy has not been conclusively explained, it is likely that the observed repression of contractile genes by the HRT family is negative feedback, as this family of repressors interferes with the transcription of many Notch target genes, including the HRT genes themselves (King et al., 2006; Nakagawa et al., 2000; Takebayashi et al., 1994). Notch activity has since been shown to directly promote the transcription of ACTA2 (Noseda et al., 2006), and Notch-induced transcription of ACTA2 was repressed by HRT transcription factors (Tang, Urs, & Liaw, 2008). Overall, the preponderance of data supports the hypothesis that Notch signaling in VSMCs promotes contractile differentiation (Baeten et al., 2015; Boscolo et al., 2011; Boucher, Gridley, & Liaw, 2012; Doi et al., 2006; Domenga et al., 2004; High et al., 2008, 2007; Lin & Lilly, 2014b; Liu et al., 2010; Meng, Zhang, Lee, & Wang, 2013; Noseda et al., 2006; Tang et al., 2010; Wang et al., 2012), which fits with our understanding of its role in development of the vessel wall and response to contact-induced signaling with endothelial cells (Baeten et al., 2015; Gaengel, Genove, Armulik, & Betsholtz, 2009; High et al., 2008; Hoglund & Majesky, 2012; Lilly & Kennard, 2009; Lin & Lilly, 2014a, 2014b; Liu et al., 2009; Manderfield et al., 2012; Regan & Majesky, 2009; Wang et al., 2012).
4.2 Proliferation
The proliferation of VSMCs is negatively tied to their differentiation status, so much so that they are often discussed as polar opposites, where mitotically active cells are considered “dedifferentiated” or “synthetic” and differentiated VSMCs are assumed to be quiescent, though there is likely a range of intermediate phenotypes between these extremes (Owens et al., 2004). Since Notch signaling in VSMCs promotes contractile differentiation, one would assume cells with active Notch signaling would not be actively proliferating. However, the initial data suggested the opposite, with several papers showing that Notch signaling promoted proliferation in vitro (Campos et al., 2002; Havrda, Johnson, O’Neill, & Liaw, 2006; Sweeney et al., 2004) and in the context of vascular injury in vivo (Li, Takeshita, et al., 2009; Wang, Campos, et al., 2002). In 2013, findings from Lucy Liaw’s lab demonstrated that the Notch2 receptor specifically inhibited proliferation via cell-cycle arrest and Notch2 was localized in non-proliferating regions of injured vessels, suggesting a receptor-specific regulation of VSMC proliferation (Boucher et al., 2013). Indeed, the previous data demonstrating promotion of proliferation were largely focused on the activity of Notch1 and/or Notch3 (Havrda et al., 2006; Li, Takeshita, et al., 2009; Sweeney et al., 2004; Wang, Campos, et al., 2002). Delving further into receptor-specific roles, our own lab demonstrated that manipulation of Notch2 and Notch3 had opposite effects on PDGF-B-dependent proliferation, and that PDGF-B treatment decreased expression of Notch2, but increased expression of Notch3 (Baeten & Lilly, 2015). Interestingly, while Notch2 and Notch3 have disparate roles in regulating VSMC proliferation, they both promote contractile differentiation (Baeten et al., 2015). Together these results indicate that regulation of the expression of the different Notch receptors in VSMCs could have a large impact on phenotypic outcome.
4.3 Survival
The role of Notch signaling in VSMC survival and apoptosis has drawn significant attention due to the human disease CADASIL (cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy), which is involves the loss of VSMCs and pericytes from cerebral vessels and is caused by mutation of the Notch3 receptor (which will be discussed further in the next section). Notch signaling has been implicated in suppression of apoptosis in a number of cell types (Dror et al., 2007; Jehn, Bielke, Pear, & Osborne, 1999; Meurette et al., 2009; Rosati et al., 2009), especially in many forms of cancer (Dang, 2012). This role as an inhibitor or apoptosis and promoter of cell survival appears to be expressed in VSMCs and pericytes as well (Arboleda-Velasquez et al., 2014; Baeten & Lilly, 2015; Henshall et al., 2015; Li, Takeshita, et al., 2009; Liu et al., 2010; Sweeney et al., 2004; Wang, Campos, et al., 2002; Wang et al., 2014; Wang, Prince, et al., 2002). Interestingly, the Notch receptors responsible for promotion of cell survival in mural cells are also those implicated in the promotion of proliferation, with Notch1 and Notch3 promoting survival, but with no known role for Notch2 (Baeten & Lilly, 2015; Henshall et al., 2015; Li, Takeshita, et al., 2009; Liu et al., 2010; Sweeney et al., 2004; Wang, Campos, et al., 2002; Wang et al., 2014; Wang, Prince, et al., 2002). Direct comparison of the effects of Notch2 and Notch3 manipulation in vitro on VSMC survival revealed that Notch3 promoted survival, but Notch2 had no effect (Baeten & Lilly, 2015). Notch3 activity correlates with MAPK/ERK pathway activation and with survival gene expression, which could be abrogated by inhibition of the MAPK/ERK pathway. This suggests that Notch3 may be promoting VSMC survival through a noncanonical association with the MAPK pathway, which had previously been suggested (Wang, Prince, et al., 2002). Overall, Notch activity promotes survival in VSMCs, but this effect is receptor specific and involves a noncanonical interaction with the MAPK pathway.
4.4 Extracellular Matrix Synthesis
VSMCs and their precursors are predominant contributors to the production of the extracellular matrix (ECM) that comprises the basement membrane and supports tensile strength and elasticity in the vessel (Armulik, Genove, & Betsholtz, 2011; Underwood, Bean, & Whitelock, 1998; Wagenseil & Mecham, 2009). The ability of Notch signaling to promote ECM synthesis has been demonstrated in fibroblasts, where it induces collagen production (Dees et al., 2011; Lilly & Kennard, 2009), and in renal podocytes, where inhibition of Notch signaling reduced expression of collagen IV and laminin, but increased expression of MMP-2 and MMP-9 (Yao, Wang, Zhang, Chi, & Gao, 2015). This relationship is maintained in VSMCs, where coculture studies identified that endothelial-derived Notch signaling in VSMCs promotes collagen synthesis and other markers of a synthetic phenotype (Lilly & Kennard, 2009; Lin & Lilly, 2014b). This may be a recapitulation of the developmental program in early vessel development, wherein Notch and other synthetic signaling pathways (such as TGFβ) induce ECM synthesis in perivascular cells to produce the framework of the vessel wall.
4.5 Migration
The migration of VSMCs is primarily important in two situations: recruitment of mural cells to the developing vessel and in response to vessel injury (Hellstrom et al., 1999). The main chemotractant for VSMCs is PDGF-B secreted by endothelial cells as well as platelets and macrophages (Hellstrom et al., 1999; Shimokado et al., 1985). Because Notch signaling has been shown to increase expression of the PDGF receptor β (Jin et al., 2008), it would follow that active Notch signaling in VSMCs would increase migration in response to PDGF-B. Indeed, much of the work done on Notch and VSMC migration has shown an induction of migratory ability. Similar to their roles in proliferation, Notch1 and Notch3 were shown to promote VSMC migration in vitro (Sweeney et al., 2004) and in a mouse injury model Notch1 was shown to be important for migration, proliferation, and formation of the neointimal layer (Li, Takeshita, et al., 2009). The Notch-responsive gene HEY2 was also shown to promote proliferation, migration, and neointimal formation (Sakata et al., 2004), and Notch signaling has been shown to regulate expression of MMP-2 and MMP-9, which are important in the matrix remodeling necessary for migration through the vessel wall rich in ECM (Delbosc et al., 2008). Notch signaling has also been shown to regulate cellular adhesions between cells via N-cadherin (Li et al., 2011; Lindner et al., 2001) and to the surrounding matrix via integrins (Scheppke et al., 2012). These results support the idea that Notch signaling is important for recruitment of mural cells to the vessel wall in response to a PDGF-B gradient, and maintaining their position via cellular adhesions.
5. NOTCH SIGNALING IN VASCULAR DISEASE
The various roles of Notch signaling in regulating smooth muscle development and phenotypic modulation make it an obvious player in many vascular diseases associated with VSMCs. These include both inherited genetic diseases and ailments brought on by defective physiological homeostasis (Table 1).
Table 1.
Vascular Diseases Associated with Mutations of Notch Pathway Components
| Notch Component | Mutation Site | Notch Signaling Effect | Disease Outcome |
|---|---|---|---|
| Jagged1 | Identified in every exon and several splice sites | Loss of function | Alagille syndrome1 |
| EGF-like repeat 2 | Not known | Tetralogy of Fallot2 | |
| DLL4 | DSL domain, N-terminal domain, and EGF-like repeats | Predicted loss of function | Adams–Oliver syndrome3 |
| Notch1 | Haploinsufficiency, EGF-like repeat 11, LNR 2, ANK domain 3 | Predicted loss of function | Adams–Oliver syndrome4 |
| Haploinsufficiency, EGF-like repeat 29 | Predicted loss of function | Bicuspid aortic valve5 | |
| Notch2 | Truncated NICD | Predicted loss of function | Alagille syndrome6 |
| PEST domain | Predicted gain of function | Hajdu–Cheney7 | |
| Notch3 | EGF-like repeats 1–32 | Likely loss of function | CADASIL8–11 |
| PEST domain | Predicted gain of function | Lehman12 | |
| Heterodimerization domain | Predicted gain of function | Infantile myofibromatosis13 | |
| EGF-like repeats 21 and 23 | Not known | Childhood PAH14 | |
| RBPJ | DNA-binding domain | Loss of Function | Adams–Oliver syndrome15 |
| EOGT | Domains not defined | Predicted loss of function | Adams–Oliver syndrome16 |
References:
5.1 Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy
Perhaps the most studied Notch-related disease in VSMCs, CADASIL, is caused by mutations in the Notch3 gene through a mechanism we still do not fully understand (Joutel et al., 1996). This disorder presents in the patient as migraines, mood disorders, ischemic attacks, cognitive impairment, and stroke and is marked by lesions within the cerebral and systemic vasculature characterized by accumulation of granular osmiophilic material (GOM) at smooth muscle basement membrane and progressive loss of VSMCs (Chabriat et al., 2009; Joutel et al., 2000, 1997). These GOM deposits appear to be comprised of oligomerized Notch3 ECD, and the mutations found in human patients lead to a gain or loss of a cysteine residue within the EGF-like repeats of the ECD (Joutel et al., 1997). It is not clear what exactly causes the disease progression of CADASIL, whether it is impaired function of Notch3, aberrant effects of GOM deposition, or a combination of both. Mouse models expressing Notch3 receptors containing disease causing mutations have had mixed results in recreating CADASIL disease states. A knockin model of a CADASIL Notch3 mutation did not develop disease phenotypes or GOM deposits (Lundkvist et al., 2005), but another mouse with ectopically overexpressed mutant Notch3 protein did produce GOM deposits and mirrored many of the CADASIL disease symptoms (Joutel et al., 2010). Notch3-null mutant mice are viable and fertile and do not show any overt symptoms associated with CADASIL (Krebs et al., 2003), though they do exhibit some disruption of the VSMCs in cerebral arteries (Henshall et al., 2015). It has also been shown that hypomorphic mutations of Notch3 in humans do not cause CADASIL (Rutten et al., 2013). These results would seem to suggest that it is the gained aberrant function of GOM deposits that causes CADASIL phenotypes. However, a homozygous-null Notch3 mutation has been shown to produce a recessive early-onset arteriopathy and cavitating leukoencephalopathy very similar to CADASIL without the deposition of GOM (Pippucci et al., 2015), and a mouse model with combined deficiency of Notch1 and Notch3 causes pericyte dysfunction and models some phenotypes of CADASIL (Kofler et al., 2015). While it is still difficult at this point to definitively rule out any of the possible explanations, it seems likely that impaired Notch3 signaling plays at least some role in the CADASIL phenotypes, based on the known function of Notch3 in regulating cell survival in VSMCs and pericytes (Baeten & Lilly, 2015; Liu et al., 2010).
5.2 Patent Ductus Arteriosus
PDA results from the failure of the ductus arteriosus (DA), which diverts blood around the fetal lungs, to close shortly after the first breath and inflation of the lungs (Schneider & Moore, 2006). There are several signaling pathways involved in regulating the timing of this closure, but ultimately, it relies on the VSMCs of the DA to contract and functionally close the vessel. Because of this, defects in contractile proteins or VSMC differentiation often result in PDA (Guo et al., 2007; Huang et al., 2008; Zhu et al., 2006). Since Notch signaling promotes VSMC differentiation, it is not surprising that certain mouse models of Notch deficiency also present with the PDA phenotype (Baeten et al., 2015; Feng et al., 2010; Krebs et al., 2016). This connection is also seen in human disease, where a subset of patients afflicted with Alagille, Adams–Oliver, or Hajdu–Cheney syndromes, all caused by mutation of Notch family members, present with PDA (Battelino, Writzl, Bratanic, Irving, & Novljan, 2016; Li et al., 1997; Stittrich et al., 2014).
5.3 Alagille Syndrome
Alagille syndrome is an autosomal dominant disorder caused by loss of Notch2 or Jagged1 and is characterized by many developmental defects, including those of the liver, heart, skeleton, eye, face, and kidney (Li et al., 1997; McDaniell et al., 2006). While many of these defects are caused by the roles of Notch signaling in other cell types, there are some interesting vascular phenotypes that could be related to dysfunctional Notch signaling in VSMCs. In addition to the PDA previously discussed, Alagille syndrome patients often present with defects of the cardiac outflow tract and great vessels (McElhinney et al., 2002). These defects are similarly modeled in mice with abrogated Notch signaling in the cardiac neural crest lineage that gives rise to the VSMCs of the outflow tract and great vessels (High et al., 2007).
5.4 Pulmonary Arterial Hypertension
Pulmonary arterial hypertension (PAH) is characterized by a persistent elevation of pulmonary arterial pressure, pulmonary arterial remodeling, and right ventricular hypertrophy (Crosswhite & Sun, 2014). This pulmonary arterial remodeling presents as increased VSMC proliferation, survival, and expression of contractile proteins such as ACTA2. Notch signaling has been proposed to regulate the pathogenesis of PAH, as elevated Notch3 expression is one of the hallmarks of the disease (Crosswhite & Sun, 2014; Geraci et al., 2001), and this would fit with the proliferation, survival, and differentiation roles that have been shown for Notch3. Two Notch3 mutations were identified in cases of childhood PAH, and in vitro experiments using the mutant proteins demonstrated increased proliferation (Chida et al., 2014). This role for Notch3 in PAH is further confirmed by the discovery that Notch3-null mice do not develop PAH in a hypoxia-induced PAH mouse model and that treatment with the Notch inhibitor DAPT (Li, Zhang, et al., 2009), or with a soluble Jagged1 ligand, which functions as a Notch antagonist, could prevent disease progression (Xiao, Gong, & Wang, 2013).
5.5 Infantile Myofibromatosis
Infantile myofibromatosis is an unusual mesenchymal malignancy that is seen at or near birth and typically spontaneously regresses in early childhood (Hatzidaki et al., 2001). Although the tumors rarely present any significant harm to the patient, they are of interest to us because of their causative mutations: PDGFRβ and Notch3 (Lee, 2013; Martignetti et al., 2013). These mutations are predicted to create constitutively active forms of the proteins (Lee, 2013), which fits our current understanding of the relationship between Notch3 and PDGFRβ in promotion of VSMC proliferation (Baeten & Lilly, 2015; Jin et al., 2008).
5.6 Vascular Injury
In situations of vascular injury, many signaling pathways are activated to repair and remodel the vessel. However, these changes can become pathological if not properly regulated and can lead to neointimal hyperplasia, an aggressive migration, and proliferation of VSMCs into the lumen of the vessel (Wu & Zhang, 2009). Expression of members of the Notch signaling pathway, including Notch1, Notch3, HEY1, HEY2, and JAG1, is regulated in response to vascular injury, with an initial downregulation, followed by upregulation after several days (Gridley, 2010; Li, Takeshita, et al., 2009; Lindner et al., 2001). Several of these members have been specifically shown to regulate migration and proliferation of the neointima in injury models, including Notch1, Notch3, and HEY2 (Li, Takeshita, et al., 2009; Sakata et al., 2004; Wang, Campos, et al., 2002). Overall, the general consensus is that the Notch signaling pathway drives neointimal formation after vascular injury and is therefore a potential pharmacological target in situations of restenosis or other vascular injuries.
6. CONCLUSION
Our understanding of the molecular mechanisms of Notch signaling in VSMCs is constantly expanding, and in the last few years we have made great strides in uncovering some of the unique functions of the different Notch family members. There is still much we do not know, however, both in what roles each Notch component has in VSMC development and disease, and the precise mechanisms that allow for functional divergence of the Notch signal. How do the different receptors enact different functional outcomes while utilizing common cofactors? Work from the Gridley lab recently showed that the NICDs of Notch1 and Notch2 are functionally equivalent in development by creating Notch1/2 chimeric proteins (Liu et al., 2015), despite the known differences in Notch1 and Notch2 discussed in this review, especially in regard to VSMC proliferation. Can these differences be explained by different expression profiles and ligand affinities alone? Or are there noncanonical interactions yet to be discovered? The Kitajewski lab has developed decoys that specifically target DLL/Notch or JAG/Notch ligand–receptor interactions, and demonstrated their different functions in tumor angiogenesis (Kangsamaksin et al., 2015). Further use of these tools in VSMCs will go a long way toward understanding the differing functionalities of Notch ligand–receptor pairs. Additionally, the advent of CRISPR technology will greatly expedite our ability to make compound and chimeric Notch receptor mouse models to pinpoint the precise source of the unique functions of the different Notch receptors. These are some of the most pressing questions for the Notch field to answer in the coming years as we strive to uncover the intricacies of Notch signaling in vascular development and disease.
Acknowledgments
We would like to acknowledge the Center for Cardiovascular Research and the Heart Center at Nationwide Children’s hospital for their support.
ABBREVIATIONS
- ACTA2
alpha smooth muscle actin
- CADASIL
cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
- ECD
extracellular domain
- ECM
extracellular matrix
- EGF
epidermal growth factor-like
- GOM
granular osmiophilic material
- HDACs
histone deacetylases
- HES
hairy/enhancer of split
- HRT
hairy-related transcription factors
- MAML
mastermind-like
- MAPK
mitogen-activated protein kinase
- NICD
Notch intracellular domain
- NRR
negative regulatory region
- PAH
pulmonary arterial hypertension
- PDA
patent ductus arteriosus
- PDGF-B
platelet-derived growth factor B
- PEST
domain rich in proline [P]glutamic acid [E], serine [S], and threonine [T]
- RBPJ
recombination signal-binding protein for immunoglobulin kappa J region
- TGFβ
transforming growth factor β
- VSMCs
vascular smooth muscle cells
- Wnt
wingless-related integration site
Footnotes
CONFLICT OF INTERESTS
We have no conflicts of interest to report.
References
- Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annual Review of Physiology. 2012;74:13–40. doi: 10.1146/annurev-physiol-012110-142315. http://dx.doi.org/10.1146/annurev-physiol-012110-142315. [DOI] [PubMed] [Google Scholar]
- Andersen P, Uosaki H, Shenje LT, Kwon C. Non-canonical Notch signaling: Emerging role and mechanism. Trends in Cell Biology. 2012;22(5):257–265. doi: 10.1016/j.tcb.2012.02.003. http://dx.doi.org/10.1016/j.tcb.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arboleda-Velasquez JF, Primo V, Graham M, James A, Manent J, D’Amore PA. Notch signaling functions in retinal pericyte survival. Investigative Ophthalmology & Visual Science. 2014;55(8):5191–5199. doi: 10.1167/iovs.14-14046. http://dx.doi.org/10.1167/iovs.14-14046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armulik A, Genove G, Betsholtz C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Developmental Cell. 2011;21(2):193–215. doi: 10.1016/j.devcel.2011.07.001. http://dx.doi.org/10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: Cell fate control and signal integration in development. Science. 1999;284(5415):770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- Ayaz F, Osborne BA. Non-canonical notch signaling in cancer and immunity. Frontiers in Oncology. 2014;4:345. doi: 10.3389/fonc.2014.00345. http://dx.doi.org/10.3389/fonc.2014.00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeten JT, Jackson AR, McHugh KM, Lilly B. Loss of Notch2 and Notch3 in vascular smooth muscle causes patent ductus arteriosus. Genesis. 2015;53(12):738–748. doi: 10.1002/dvg.22904. http://dx.doi.org/10.1002/dvg.22904. [DOI] [PubMed] [Google Scholar]
- Baeten JT, Lilly B. Differential regulation of NOTCH2 and NOTCH3 contribute to their unique functions in vascular smooth muscle cells. The Journal of Biological Chemistry. 2015;290:16226–16237. doi: 10.1074/jbc.M115.655548. http://dx.doi.org/10.1074/jbc.M115.655548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battelino N, Writzl K, Bratanic N, Irving MD, Novljan G. End-stage renal disease in an infant with Hajdu-Cheney syndrome. Therapeutic Apheresis and Dialysis. 2016;20(3):318–321. doi: 10.1111/1744-9987.12444. http://dx.doi.org/10.1111/1744-9987.12444. [DOI] [PubMed] [Google Scholar]
- Beckers J, Clark A, Wunsch K, Hrabe De Angelis M, Gossler A. Expression of the mouse Delta1 gene during organogenesis and fetal development. Mechanisms of Development. 1999;84(1–2):165–168. doi: 10.1016/s0925-4773(99)00065-9. [DOI] [PubMed] [Google Scholar]
- Benedito R, Roca C, Sorensen I, Adams S, Gossler A, Fruttiger M, Adams RH. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137(6):1124–1135. doi: 10.1016/j.cell.2009.03.025. http://dx.doi.org/10.1016/j.cell.2009.03.025. [DOI] [PubMed] [Google Scholar]
- Blokzijl A, Dahlqvist C, Reissmann E, Falk A, Moliner A, Lendahl U, Ibanez CF. Cross-talk between the Notch and TGF-beta signaling pathways mediated by interaction of the Notch intracellular domain with Smad3. The Journal of Cell Biology. 2003;163(4):723–728. doi: 10.1083/jcb.200305112. http://dx.doi.org/10.1083/jcb.200305112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borggrefe T, Oswald F. The Notch signaling pathway: Transcriptional regulation at Notch target genes. Cellular and Molecular Life Sciences. 2009;66(10):1631–1646. doi: 10.1007/s00018-009-8668-7. http://dx.doi.org/10.1007/s00018-009-8668-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boscolo E, Stewart CL, Greenberger S, Wu JK, Durham JT, Herman IM, Bischoff J. JAGGED1 signaling regulates hemangioma stem cell-to-pericyte/vascular smooth muscle cell differentiation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2011;31(10):2181–2192. doi: 10.1161/ATVBAHA.111.232934. http://dx.doi.org/10.1161/ATVBAHA.111.232934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher J, Gridley T, Liaw L. Molecular pathways of notch signaling in vascular smooth muscle cells. Frontiers in Physiology. 2012;3:81. doi: 10.3389/fphys.2012.00081. http://dx.doi.org/10.3389/fphys.2012.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher JM, Harrington A, Rostama B, Lindner V, Liaw L. A receptor-specific function for Notch2 in mediating vascular smooth muscle cell growth arrest through cyclin-dependent kinase inhibitor 1B. Circulation Research. 2013;113(8):975–985. doi: 10.1161/CIRCRESAHA.113.301272. http://dx.doi.org/10.1161/CIRCRESAHA.113.301272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher JM, Peterson SM, Urs S, Zhang C, Liaw L. The miR-143/145 cluster is a novel transcriptional target of Jagged-1/Notch signaling in vascular smooth muscle cells. The Journal of Biological Chemistry. 2011;286(32):28312–28321. doi: 10.1074/jbc.M111.221945. http://dx.doi.org/10.1074/jbc.M111.221945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos AH, Wang W, Pollman MJ, Gibbons GH. Determinants of Notch-3 receptor expression and signaling in vascular smooth muscle cells: Implications in cell-cycle regulation. Circulation Research. 2002;91(11):999–1006. doi: 10.1161/01.res.0000044944.99984.25. [DOI] [PubMed] [Google Scholar]
- Cappellari O, Benedetti S, Innocenzi A, Tedesco FS, Moreno-Fortuny A, Ugarte G, Cossu G. Dll4 and PDGF-BB convert committed skeletal myoblasts to pericytes without erasing their myogenic memory. Developmental Cell. 2013;24(6):586–599. doi: 10.1016/j.devcel.2013.01.022. http://dx.doi.org/10.1016/j.devcel.2013.01.022. [DOI] [PubMed] [Google Scholar]
- Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG. Cadasil. Lancet Neurology. 2009;8(7):643–653. doi: 10.1016/S1474-4422(09)70127-9. http://dx.doi.org/10.1016/S1474-4422(09)70127-9. [DOI] [PubMed] [Google Scholar]
- Chida A, Shintani M, Matsushita Y, Sato H, Eitoku T, Nakayama T, Nakanishi T. Mutations of NOTCH3 in childhood pulmonary arterial hypertension. Molecular Genetics & Genomic Medicine. 2014;2(3):229–239. doi: 10.1002/mgg3.58. http://dx.doi.org/10.1002/mgg3.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosswhite P, Sun Z. Molecular mechanisms of pulmonary arterial remodeling. Molecular Medicine. 2014;20:191–201. doi: 10.2119/molmed.2013.00165. http://dx.doi.org/10.2119/molmed.2013.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang TP. Notch, apoptosis and cancer. Advances in Experimental Medicine and Biology. 2012;727:199–209. doi: 10.1007/978-1-4614-0899-4_15. http://dx.doi.org/10.1007/978-1-4614-0899-4_15. [DOI] [PubMed] [Google Scholar]
- Dees C, Tomcik M, Zerr P, Akhmetshina A, Horn A, Palumbo K, Distler JH. Notch signalling regulates fibroblast activation and collagen release in systemic sclerosis. Annals of the Rheumatic Diseases. 2011;70(7):1304–1310. doi: 10.1136/ard.2010.134742. http://dx.doi.org/10.1136/ard.2010.134742. [DOI] [PubMed] [Google Scholar]
- Delbosc S, Glorian M, Le Port AS, Bereziat G, Andreani M, Limon I. The benefit of docosahexaenoic acid on the migration of vascular smooth muscle cells is partially dependent on Notch regulation of MMP-2/-9. The American Journal of Pathology. 2008;172(5):1430–1440. doi: 10.2353/ajpath.2008.070951. http://dx.doi.org/10.2353/ajpath.2008.070951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi H, Iso T, Sato H, Yamazaki M, Matsui H, Tanaka T, Kurabayashi M. Jagged1-selective notch signaling induces smooth muscle differentiation via a RBP-Jkappa-dependent pathway. The Journal of Biological Chemistry. 2006;281(39):28555–28564. doi: 10.1074/jbc.M602749200. http://dx.doi.org/10.1074/jbc.M602749200. [DOI] [PubMed] [Google Scholar]
- Doi H, Iso T, Yamazaki M, Akiyama H, Kanai H, Sato H, Kurabayashi M. HERP1 inhibits myocardin-induced vascular smooth muscle cell differentiation by interfering with SRF binding to CArG box. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(11):2328–2334. doi: 10.1161/01.ATV.0000185829.47163.32. http://dx.doi.org/10.1161/01.ATV.0000185829.47163.32. [DOI] [PubMed] [Google Scholar]
- Domenga V, Fardoux P, Lacombe P, Monet M, Maciazek J, Krebs LT, Joutel A. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes and Development. 2004;18(22):2730–2735. doi: 10.1101/gad.308904. http://dx.doi.org/10.1101/gad.308904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan J, Abraham D, Norman J. Platelet-derived growth factor signaling in mesenchymal cells. Frontiers in Bioscience (Landmark Edition) 2013;18:106–119. doi: 10.2741/4090. [DOI] [PubMed] [Google Scholar]
- Dror V, Nguyen V, Walia P, Kalynyak TB, Hill JA, Johnson JD. Notch signalling suppresses apoptosis in adult human and mouse pancreatic islet cells. Diabetologia. 2007;50(12):2504–2515. doi: 10.1007/s00125-007-0835-5. http://dx.doi.org/10.1007/s00125-007-0835-5. [DOI] [PubMed] [Google Scholar]
- Duncan AW, Rattis FM, DiMascio LN, Congdon KL, Pazianos G, Zhao C, Reya T. Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nature Immunology. 2005;6(3):314–322. doi: 10.1038/ni1164. http://dx.doi.org/10.1038/ni1164. [DOI] [PubMed] [Google Scholar]
- Ehebauer M, Hayward P, Arias AM. Notch, a universal arbiter of cell fate decisions. Science. 2006;314(5804):1414–1415. doi: 10.1126/science.1134042. http://dx.doi.org/10.1126/science.1134042. [DOI] [PubMed] [Google Scholar]
- Eldadah ZA, Hamosh A, Biery NJ, Montgomery RA, Duke M, Elkins R, Dietz HC. Familial tetralogy of Fallot caused by mutation in the jagged1 gene. Human Molecular Genetics. 2001;10(2):163–169. doi: 10.1093/hmg/10.2.163. [DOI] [PubMed] [Google Scholar]
- Espinosa L, Ingles-Esteve J, Aguilera C, Bigas A. Phosphorylation by glycogen synthase kinase-3 beta down-regulates Notch activity, a link for Notch and Wnt pathways. The Journal of Biological Chemistry. 2003;278(34):32227–32235. doi: 10.1074/jbc.M304001200. http://dx.doi.org/10.1074/jbc.M304001200. [DOI] [PubMed] [Google Scholar]
- Feng X, Krebs LT, Gridley T. Patent ductus arteriosus in mice with smooth muscle-specific Jag1 deletion. Development. 2010;137(24):4191–4199. doi: 10.1242/dev.052043. http://dx.doi.org/10.1242/dev.052043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouillade C, Monet-Lepretre M, Baron-Menguy C, Joutel A. Notch signalling in smooth muscle cells during development and disease. Cardiovascular Research. 2012;95(2):138–146. doi: 10.1093/cvr/cvs019. http://dx.doi.org/10.1093/cvr/cvs019. [DOI] [PubMed] [Google Scholar]
- Fre S, Pallavi SK, Huyghe M, Lae M, Janssen KP, Robine S, Louvard D. Notch and Wnt signals cooperatively control cell proliferation and tumorigenesis in the intestine. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(15):6309–6314. doi: 10.1073/pnas.0900427106. http://dx.doi.org/10.1073/pnas.0900427106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaengel K, Genove G, Armulik A, Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29(5):630–638. doi: 10.1161/ATVBAHA.107.161521. http://dx.doi.org/10.1161/ATVBAHA.107.161521. [DOI] [PubMed] [Google Scholar]
- Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437(7056):270–274. doi: 10.1038/nature03940. http://dx.doi.org/10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- Geraci MW, Moore M, Gesell T, Yeager ME, Alger L, Golpon H, Voelkel NF. Gene expression patterns in the lungs of patients with primary pulmonary hypertension: A gene microarray analysis. Circulation Research. 2001;88(6):555–562. doi: 10.1161/01.res.88.6.555. [DOI] [PubMed] [Google Scholar]
- Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovascular Research. 2012;95(2):156–164. doi: 10.1093/cvr/cvs115. http://dx.doi.org/10.1093/cvr/cvs115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greif DM, Kumar M, Lighthouse JK, Hum J, An A, Ding L, Krasnow MA. Radial construction of an arterial wall. Developmental Cell. 2012;23(3):482–493. doi: 10.1016/j.devcel.2012.07.009. http://dx.doi.org/10.1016/j.devcel.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gridley T. Notch signaling in vascular development and physiology. Development. 2007;134(15):2709–2718. doi: 10.1242/dev.004184. http://dx.doi.org/10.1242/dev.004184. [DOI] [PubMed] [Google Scholar]
- Gridley T. Notch signaling in the vasculature. Current Topics in Developmental Biology. 2010;92:277–309. doi: 10.1016/S0070-2153(10)92009-7. http://dx.doi.org/10.1016/S0070-2153(10)92009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieskamp T, Rudat C, Ludtke TH, Norden J, Kispert A. Notch signaling regulates smooth muscle differentiation of epicardium-derived cells. Circulation Research. 2011;108(7):813–823. doi: 10.1161/CIRCRESAHA.110.228809. http://dx.doi.org/10.1161/CIRCRESAHA.110.228809. [DOI] [PubMed] [Google Scholar]
- Gripp KW, Robbins KM, Sobreira NL, Witmer PD, Bird LM, Avela K, Sol-Church K. Truncating mutations in the last exon of NOTCH3 cause lateral meningocele syndrome. American Journal of Medical Genetics. Part A. 2015;167A(2):271–281. doi: 10.1002/ajmg.a.36863. http://dx.doi.org/10.1002/ajmg.a.36863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Chen SY. Transforming growth factor-beta and smooth muscle differentiation. World Journal of Biological Chemistry. 2012;3(3):41–52. doi: 10.4331/wjbc.v3.i3.41. http://dx.doi.org/10.4331/wjbc.v3.i3.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Milewicz DM. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nature Genetics. 2007;39(12):1488–1493. doi: 10.1038/ng.2007.6. http://dx.doi.org/10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- Hassed SJ, Wiley GB, Wang S, Lee JY, Li S, Xu W, Gaffney PM. RBPJ mutations identified in two families affected by Adams-Oliver syndrome. The American Journal of Human Genetics. 2012;91(2):391–395. doi: 10.1016/j.ajhg.2012.07.005. http://dx.doi.org/10.1016/j.ajhg.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzidaki E, Korakaki E, Voloudaki A, Daskaloyannaki M, Manoura A, Giannakopoulou C. Infantile myofibromatosis with visceral involvement and complete spontaneous regression. The Journal of Dermatology. 2001;28(7):379–382. doi: 10.1111/j.1346-8138.2001.tb00153.x. [DOI] [PubMed] [Google Scholar]
- Havrda MC, Johnson MJ, O’Neill CF, Liaw L. A novel mechanism of transcriptional repression of p27kip1 through Notch/HRT2 signaling in vascular smooth muscle cells. Thrombosis and Haemostasis. 2006;96(3):361–370. doi: 10.1160/TH06-04-0224. http://dx.doi.org/10.1160/TH06-04-0224. [DOI] [PubMed] [Google Scholar]
- Hayward P, Brennan K, Sanders P, Balayo T, DasGupta R, Perrimon N, Martinez Arias A. Notch modulates Wnt signalling by associating with Armadillo/beta-catenin and regulating its transcriptional activity. Development. 2005;132(8):1819–1830. doi: 10.1242/dev.01724. http://dx.doi.org/10.1242/dev.01724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzler P, Simpson P. The choice of cell fate in the epidermis of Drosophila. Cell. 1991;64(6):1083–1092. doi: 10.1016/0092-8674(91)90263-x. [DOI] [PubMed] [Google Scholar]
- Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126(14):3047–3055. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- Henshall TL, Keller A, He L, Johansson BR, Wallgard E, Raschperger E, Lendahl U. Notch3 is necessary for blood vessel integrity in the central nervous system. Arteriosclerosis, Thrombosis, and Vascular Biology. 2015;35(2):409–420. doi: 10.1161/ATVBAHA.114.304849. http://dx.doi.org/10.1161/ATVBAHA.114.304849. [DOI] [PubMed] [Google Scholar]
- Hicks C, Johnston SH, diSibio G, Collazo A, Vogt TF, Weinmaster G. Fringe differentially modulates Jagged1 and Delta1 signalling through Notch1 and Notch2. Nature Cell Biology. 2000;2(8):515–520. doi: 10.1038/35019553. http://dx.doi.org/10.1038/35019553. [DOI] [PubMed] [Google Scholar]
- High FA, Lu MM, Pear WS, Loomes KM, Kaestner KH, Epstein JA. Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(6):1955–1959. doi: 10.1073/pnas.0709663105. http://dx.doi.org/10.1073/pnas.0709663105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- High FA, Zhang M, Proweller A, Tu L, Parmacek MS, Pear WS, Epstein JA. An essential role for Notch in neural crest during cardiovascular development and smooth muscle differentiation. The Journal of Clinical Investigation. 2007;117(2):353–363. doi: 10.1172/JCI30070. http://dx.doi.org/10.1172/JCI30070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirashima M. Regulation of endothelial cell differentiation and arterial specification by VEGF and Notch signaling. Anatomical Science International. 2009;84(3):95–101. doi: 10.1007/s12565-009-0026-1. http://dx.doi.org/10.1007/s12565-009-0026-1. [DOI] [PubMed] [Google Scholar]
- Hodkinson PS, Elliott PA, Lad Y, McHugh BJ, MacKinnon AC, Haslett C, Sethi T. Mammalian NOTCH-1 activates beta1 integrins via the small GTPase R-Ras. The Journal of Biological Chemistry. 2007;282(39):28991–29001. doi: 10.1074/jbc.M703601200. http://dx.doi.org/10.1074/jbc.M703601200. [DOI] [PubMed] [Google Scholar]
- Hoglund VJ, Majesky MW. Patterning the artery wall by lateral induction of Notch signaling. Circulation. 2012;125(2):212–215. doi: 10.1161/CIRCULATIONAHA.111.075937. http://dx.doi.org/10.1161/CIRCULATIONAHA.111.075937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Cheng L, Li J, Chen M, Zhou D, Lu MM, Parmacek MS. Myocardin regulates expression of contractile genes in smooth muscle cells and is required for closure of the ductus arteriosus in mice. The Journal of Clinical Investigation. 2008;118(2):515–525. doi: 10.1172/JCI33304. http://dx.doi.org/10.1172/JCI33304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iso T, Hamamori Y, Kedes L. Notch signaling in vascular development. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23(4):543–553. doi: 10.1161/01.ATV.0000060892.81529.8F. http://dx.doi.org/10.1161/01.ATV.0000060892.81529.8F. [DOI] [PubMed] [Google Scholar]
- Jehn BM, Bielke W, Pear WS, Osborne BA. Cutting edge: Protective effects of notch-1 on TCR-induced apoptosis. The Journal of Immunology. 1999;162(2):635–638. [PubMed] [Google Scholar]
- Jin S, Hansson EM, Tikka S, Lanner F, Sahlgren C, Farnebo F, Lendahl U. Notch signaling regulates platelet-derived growth factor receptor-beta expression in vascular smooth muscle cells. Circulation Research. 2008;102(12):1483–1491. doi: 10.1161/CIRCRESAHA.107.167965. http://dx.doi.org/10.1161/CIRCRESAHA.107.167965. [DOI] [PubMed] [Google Scholar]
- Joutel A, Andreux F, Gaulis S, Domenga V, Cecillon M, Battail N, Tournier-Lasserve E. The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. The Journal of Clinical Investigation. 2000;105(5):597–605. doi: 10.1172/JCI8047. http://dx.doi.org/10.1172/JCI8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Tournier-Lasserve E. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383(6602):707–710. doi: 10.1038/383707a0. http://dx.doi.org/10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Tournier-Lasserve E. Notch3 mutations in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), a mendelian condition causing stroke and vascular dementia. The Annals of the New York Academy of Sciences. 1997;826:213–217. doi: 10.1111/j.1749-6632.1997.tb48472.x. [DOI] [PubMed] [Google Scholar]
- Joutel A, Monet-Lepretre M, Gosele C, Baron-Menguy C, Hammes A, Schmidt S, Hubner N. Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. The Journal of Clinical Investigation. 2010;120(2):433–445. doi: 10.1172/JCI39733. http://dx.doi.org/10.1172/JCI39733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kangsamaksin T, Murtomaki A, Kofler NM, Cuervo H, Chaudhri RA, Tattersall IW, Kitajewski J. NOTCH decoys that selectively block DLL/NOTCH or JAG/NOTCH disrupt angiogenesis by unique mechanisms to inhibit tumor growth. Cancer Discovery. 2015;5(2):182–197. doi: 10.1158/2159-8290.CD-14-0650. http://dx.doi.org/10.1158/2159-8290.CD-14-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennard S, Liu H, Lilly B. Transforming growth factor-beta (TGF-1) down-regulates Notch3 in fibroblasts to promote smooth muscle gene expression. The Journal of Biological Chemistry. 2008;283(3):1324–1333. doi: 10.1074/jbc.M706651200. http://dx.doi.org/10.1074/jbc.M706651200. [DOI] [PubMed] [Google Scholar]
- Kim YH, Hu H, Guevara-Gallardo S, Lam MT, Fong SY, Wang RA. Artery and vein size is balanced by Notch and ephrin B2/EphB4 during angiogenesis. Development. 2008;135(22):3755–3764. doi: 10.1242/dev.022475. http://dx.doi.org/10.1242/dev.022475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King IN, Kathiriya IS, Murakami M, Nakagawa M, Gardner KA, Srivastava D, Nakagawa O. Hrt and Hes negatively regulate Notch signaling through interactions with RBP-Jkappa. Biochemical and Biophysical Research Communications. 2006;345(1):446–452. doi: 10.1016/j.bbrc.2006.04.097. http://dx.doi.org/10.1016/j.bbrc.2006.04.097. [DOI] [PubMed] [Google Scholar]
- Kofler NM, Cuervo H, Uh MK, Murtomaki A, Kitajewski J. Combined deficiency of Notch1 and Notch3 causes pericyte dysfunction, models CADASIL, and results in arteriovenous malformations. Scientific Reports. 2015;5:16449. doi: 10.1038/srep16449. http://dx.doi.org/10.1038/srep16449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs LT, Norton CR, Gridley T. Notch signal reception is required in vascular smooth muscle cells for ductus arteriosus closure. Genesis. 2016;54:86–90. doi: 10.1002/dvg.22916. http://dx.doi.org/10.1002/dvg.22916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs LT, Xue Y, Norton CR, Sundberg JP, Beatus P, Lendahl U, Gridley T. Characterization of Notch3-deficient mice: Normal embryonic development and absence of genetic interactions with a Notch1 mutation. Genesis. 2003;37(3):139–143. doi: 10.1002/gene.10241. http://dx.doi.org/10.1002/gene.10241. [DOI] [PubMed] [Google Scholar]
- Kurpinski K, Lam H, Chu J, Wang A, Kim A, Tsay E, Li S. Transforming growth factor-beta and notch signaling mediate stem cell differentiation into smooth muscle cells. Stem Cells. 2010;28(4):734–742. doi: 10.1002/stem.319. http://dx.doi.org/10.1002/stem.319. [DOI] [PubMed] [Google Scholar]
- Kwon C, Cheng P, King IN, Andersen P, Shenje L, Nigam V, Srivastava D. Notch post-translationally regulates beta-catenin protein in stem and progenitor cells. Nature Cell Biology. 2011;13(10):1244–1251. doi: 10.1038/ncb2313. http://dx.doi.org/10.1038/ncb2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai EC. Protein degradation: Four E3s for the notch pathway. Current Biology. 2002;12(2):R74–R78. doi: 10.1016/s0960-9822(01)00679-0. [DOI] [PubMed] [Google Scholar]
- Lawson ND, Scheer N, Pham VN, Kim CH, Chitnis AB, Campos-Ortega JA, Weinstein BM. Notch signaling is required for arterial-venous differentiation during embryonic vascular development. Development. 2001;128(19):3675–3683. doi: 10.1242/dev.128.19.3675. [DOI] [PubMed] [Google Scholar]
- LeBon L, Lee TV, Sprinzak D, Jafar-Nejad H, Elowitz MB. Fringe proteins modulate Notch-ligand cis and trans interactions to specify signaling states. eLife. 2014;3:e02950. doi: 10.7554/eLife.02950. http://dx.doi.org/10.7554/eLife.02950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW. Mutations in PDGFRB and NOTCH3 are the first genetic causes identified for autosomal dominant infantile myofibromatosis. Clinical Genetics. 2013;84(4):340–341. doi: 10.1111/cge.12238. http://dx.doi.org/10.1111/cge.12238. [DOI] [PubMed] [Google Scholar]
- Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Spinner NB. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nature Genetics. 1997;16(3):243–251. doi: 10.1038/ng0797-243. http://dx.doi.org/10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- Li F, Lan Y, Wang Y, Wang J, Yang G, Meng F, Yang X. Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Developmental Cell. 2011;20(3):291–302. doi: 10.1016/j.devcel.2011.01.011. http://dx.doi.org/10.1016/j.devcel.2011.01.011. [DOI] [PubMed] [Google Scholar]
- Li Y, Takeshita K, Liu PY, Satoh M, Oyama N, Mukai Y, Liao JK. Smooth muscle Notch1 mediates neointimal formation after vascular injury. Circulation. 2009;119(20):2686–2692. doi: 10.1161/CIRCULATIONAHA.108.790485. http://dx.doi.org/10.1161/CIRCULATIONAHA.108.790485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhang X, Leathers R, Makino A, Huang C, Parsa P, Thistlethwaite PA. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nature Medicine (New York) 2009;15(11):1289–1297. doi: 10.1038/nm.2021. http://dx.doi.org/10.1038/nm.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilly B, Kennard S. Differential gene expression in a coculture model of angio-genesis reveals modulation of select pathways and a role for Notch signaling. Physiological Genomics. 2009;36(2):69–78. doi: 10.1152/physiolgenomics.90318.2008. http://dx.doi.org/10.1152/physiolgenomics.90318.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Lilly B. Endothelial cells direct mesenchymal stem cells toward a smooth muscle cell fate. Stem Cells and Development. 2014a;23(21):2581–2590. doi: 10.1089/scd.2014.0163. http://dx.doi.org/10.1089/scd.2014.0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Lilly B. Notch signaling governs phenotypic modulation of smooth muscle cells. Vascular Pharmacology. 2014b;63(2):88–96. doi: 10.1016/j.vph.2014.09.004. http://dx.doi.org/10.1016/j.vph.2014.09.004. [DOI] [PubMed] [Google Scholar]
- Lindner V, Booth C, Prudovsky I, Small D, Maciag T, Liaw L. Members of the Jagged/Notch gene families are expressed in injured arteries and regulate cell phenotype via alterations in cell matrix and cell-cell interaction. The American Journal of Pathology. 2001;159(3):875–883. doi: 10.1016/S0002-9440(10)61763-4. http://dx.doi.org/10.1016/S0002-9440(10)61763-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Brunskill E, Varnum-Finney B, Zhang C, Zhang A, Jay PY, Kopan R. The intracellular domains of Notch1 and Notch2 are functionally equivalent during development and carcinogenesis. Development. 2015;142(14):2452–2463. doi: 10.1242/dev.125492. http://dx.doi.org/10.1242/dev.125492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Kennard S, Lilly B. NOTCH3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed JAGGED1. Circulation Research. 2009;104(4):466–475. doi: 10.1161/CIRCRESAHA.108.184846. http://dx.doi.org/10.1161/CIRCRESAHA.108.184846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Zhang W, Kennard S, Caldwell RB, Lilly B. Notch3 is critical for proper angiogenesis and mural cell investment. Circulation Research. 2010;107(7):860–870. doi: 10.1161/CIRCRESAHA.110.218271. http://dx.doi.org/10.1161/CIRCRESAHA.110.218271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundkvist J, Zhu S, Hansson EM, Schweinhardt P, Miao Q, Beatus P, Lendahl U. Mice carrying a R142C Notch 3 knock-in mutation do not develop a CADASIL-like phenotype. Genesis. 2005;41(1):13–22. doi: 10.1002/gene.20091. http://dx.doi.org/10.1002/gene.20091. [DOI] [PubMed] [Google Scholar]
- Luo B, Aster JC, Hasserjian RP, Kuo F, Sklar J. Isolation and functional analysis of a cDNA for human Jagged2, a gene encoding a ligand for the Notch1 receptor. Molecular and Cellular Biology. 1997;17(10):6057–6067. doi: 10.1128/mcb.17.10.6057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manderfield LJ, High FA, Engleka KA, Liu F, Li L, Rentschler S, Epstein JA. Notch activation of Jagged1 contributes to the assembly of the arterial wall. Circulation. 2012;125(2):314–323. doi: 10.1161/CIRCULATIONAHA.111.047159. http://dx.doi.org/10.1161/CIRCULATIONAHA.111.047159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martignetti JA, Tian L, Li D, Ramirez MC, Camacho-Vanegas O, Camacho SC, Hakonarson H. Mutations in PDGFRB cause autosomal-dominant infantile myofibromatosis. The American Journal of Human Genetics. 2013;92(6):1001–1007. doi: 10.1016/j.ajhg.2013.04.024. http://dx.doi.org/10.1016/j.ajhg.2013.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Arias A, Zecchini V, Brennan K. CSL-independent Notch signalling: A checkpoint in cell fate decisions during development? Current Opinion in Genetics and Development. 2002;12(5):524–533. doi: 10.1016/s0959-437x(02)00336-2. [DOI] [PubMed] [Google Scholar]
- McCright B, Gao X, Shen L, Lozier J, Lan Y, Maguire M, Gridley T. Defects in development of the kidney, heart and eye vasculature in mice homozygous for a hypomorphic Notch2 mutation. Development. 2001;128(4):491–502. doi: 10.1242/dev.128.4.491. [DOI] [PubMed] [Google Scholar]
- McDaniell R, Warthen DM, Sanchez-Lara PA, Pai A, Krantz ID, Piccoli DA, Spinner NB. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. The American Journal of Human Genetics. 2006;79(1):169–173. doi: 10.1086/505332. http://dx.doi.org/10.1086/505332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElhinney DB, Krantz ID, Bason L, Piccoli DA, Emerick KM, Spinner NB, Goldmuntz E. Analysis of cardiovascular phenotype and genotype-phenotype correlation in individuals with a JAG1 mutation and/or Alagille syndrome. Circulation. 2002;106(20):2567–2574. doi: 10.1161/01.cir.0000037221.45902.69. [DOI] [PubMed] [Google Scholar]
- Meester JA, Southgate L, Stittrich AB, Venselaar H, Beekmans SJ, den Hollander N, Wuyts W. Heterozygous loss-of-function mutations in DLL4 cause Adams-Oliver syndrome. The American Journal of Human Genetics. 2015;97(3):475–482. doi: 10.1016/j.ajhg.2015.07.015. http://dx.doi.org/10.1016/j.ajhg.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng H, Zhang X, Lee SJ, Wang MM. Von Willebrand factor inhibits mature smooth muscle gene expression through impairment of Notch signaling. PLoS One. 2013;8(9):e75808. doi: 10.1371/journal.pone.0075808. http://dx.doi.org/10.1371/journal.pone.0075808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurette O, Stylianou S, Rock R, Collu GM, Gilmore AP, Brennan K. Notch activation induces Akt signaling via an autocrine loop to prevent apoptosis in breast epithelial cells. Cancer Research. 2009;69(12):5015–5022. doi: 10.1158/0008-5472.CAN-08-3478. http://dx.doi.org/10.1158/0008-5472.CAN-08-3478. [DOI] [PubMed] [Google Scholar]
- Mill C, George SJ. Wnt signalling in smooth muscle cells and its role in cardiovascular disorders. Cardiovascular Research. 2012;95(2):233–240. doi: 10.1093/cvr/cvs141. http://dx.doi.org/10.1093/cvr/cvs141. [DOI] [PubMed] [Google Scholar]
- Moloney DJ, Panin VM, Johnston SH, Chen J, Shao L, Wilson R, Vogt TF. Fringe is a glycosyltransferase that modifies Notch. Nature. 2000;406(6794):369–375. doi: 10.1038/35019000. http://dx.doi.org/10.1038/35019000. [DOI] [PubMed] [Google Scholar]
- Morgan TH. The theory of the gene. The American Naturalist. 1917;51(609):513–544. http://dx.doi.org/10.1086/279629. [Google Scholar]
- Nakagawa O, McFadden DG, Nakagawa M, Yanagisawa H, Hu T, Srivastava D, Olson EN. Members of the HRT family of basic helix-loop-helix proteins act as transcriptional repressors downstream of Notch signaling. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(25):13655–13660. doi: 10.1073/pnas.250485597. http://dx.doi.org/10.1073/pnas.250485597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noseda M, Fu Y, Niessen K, Wong F, Chang L, McLean G, Karsan A. Smooth muscle alpha-actin is a direct target of Notch/CSL. Circulation Research. 2006;98(12):1468–1470. doi: 10.1161/01.RES.0000229683.81357.26. http://dx.doi.org/10.1161/01.RES.0000229683.81357.26. [DOI] [PubMed] [Google Scholar]
- Ong CT, Cheng HT, Chang LW, Ohtsuka T, Kageyama R, Stormo GD, Kopan R. Target selectivity of vertebrate notch proteins. Collaboration between discrete domains and CSL-binding site architecture determines activation probability. The Journal of Biological Chemistry. 2006;281(8):5106–5119. doi: 10.1074/jbc.M506108200. http://dx.doi.org/10.1074/jbc.M506108200. [DOI] [PubMed] [Google Scholar]
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiological Reviews. 2004;84(3):767–801. doi: 10.1152/physrev.00041.2003. http://dx.doi.org/10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- Patel-Hett S, D’Amore PA. Signal transduction in vasculogenesis and developmental angiogenesis. The International Journal of Developmental Biology. 2011;55(4–5):353–363. doi: 10.1387/ijdb.103213sp. http://dx.doi.org/10.1387/ijdb.103213sp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: Regulation and physiological functions. Endocrine Reviews. 2001;22(2):153–183. doi: 10.1210/edrv.22.2.0428. http://dx.doi.org/10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- Phng LK, Gerhardt H. Angiogenesis: A team effort coordinated by notch. Developmental Cell. 2009;16(2):196–208. doi: 10.1016/j.devcel.2009.01.015. http://dx.doi.org/10.1016/j.devcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- Pippucci T, Maresca A, Magini P, Cenacchi G, Donadio V, Palombo F, Seri M. Homozygous NOTCH3 null mutation and impaired NOTCH3 signaling in recessive early-onset arteriopathy and cavitating leukoencephalopathy. EMBO Molecular Medicine. 2015;7(6):848–858. doi: 10.15252/emmm.201404399. http://dx.doi.org/10.15252/emmm.201404399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proweller A, Pear WS, Parmacek MS. Notch signaling represses myocardin-induced smooth muscle cell differentiation. The Journal of Biological Chemistry. 2005;280(10):8994–9004. doi: 10.1074/jbc.M413316200. http://dx.doi.org/10.1074/jbc.M413316200. [DOI] [PubMed] [Google Scholar]
- Regan JN, Majesky MW. Building a vessel wall with notch signaling. Circulation Research. 2009;104(4):419–421. doi: 10.1161/CIRCRESAHA.109.194233. http://dx.doi.org/10.1161/CIRCRESAHA.109.194233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca C, Adams RH. Regulation of vascular morphogenesis by Notch signaling. Genes and Development. 2007;21(20):2511–2524. doi: 10.1101/gad.1589207. http://dx.doi.org/10.1101/gad.1589207. [DOI] [PubMed] [Google Scholar]
- Rosati E, Sabatini R, Rampino G, Tabilio A, Di Ianni M, Fettucciari K, Marconi P. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood. 2009;113(4):856–865. doi: 10.1182/blood-2008-02-139725. http://dx.doi.org/10.1182/blood-2008-02-139725. [DOI] [PubMed] [Google Scholar]
- Rostama B, Peterson SM, Vary CP, Liaw L. Notch signal integration in the vasculature during remodeling. Vascular Pharmacology. 2014;63(2):97–104. doi: 10.1016/j.vph.2014.10.003. http://dx.doi.org/10.1016/j.vph.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutten JW, Boon EM, Liem MK, Dauwerse JG, Pont MJ, Vollebregt E, Lesnik Oberstein SA. Hypomorphic NOTCH3 alleles do not cause CADASIL in humans. Human Mutation. 2013;34(11):1486–1489. doi: 10.1002/humu.22432. http://dx.doi.org/10.1002/humu.22432. [DOI] [PubMed] [Google Scholar]
- Sakata Y, Xiang F, Chen Z, Kiriyama Y, Kamei CN, Simon DI, Chin MT. Transcription factor CHF1/Hey2 regulates neointimal formation in vivo and vascular smooth muscle proliferation and migration in vitro. Arteriosclerosis, Thrombosis, and Vascular Biology. 2004;24(11):2069–2074. doi: 10.1161/01.ATV.0000143936.77094.a4. http://dx.doi.org/10.1161/01.ATV.0000143936.77094.a4. [DOI] [PubMed] [Google Scholar]
- Sanalkumar R, Dhanesh SB, James J. Non-canonical activation of Notch signaling/target genes in vertebrates. Cellular and Molecular Life Sciences. 2010;67(17):2957–2968. doi: 10.1007/s00018-010-0391-x. http://dx.doi.org/10.1007/s00018-010-0391-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheppke L, Murphy EA, Zarpellon A, Hofmann JJ, Merkulova A, Shields DJ, Cheresh DA. Notch promotes vascular maturation by inducing integrin-mediated smooth muscle cell adhesion to the endothelial basement membrane. Blood. 2012;119(9):2149–2158. doi: 10.1182/blood-2011-04-348706. http://dx.doi.org/10.1182/blood-2011-04-348706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider DJ, Moore JW. Patent ductus arteriosus. Circulation. 2006;114(17):1873–1882. doi: 10.1161/CIRCULATIONAHA.105.592063. http://dx.doi.org/10.1161/CIRCULATIONAHA.105.592063. [DOI] [PubMed] [Google Scholar]
- Shaheen R, Aglan M, Keppler-Noreuil K, Faqeih E, Ansari S, Horton K, Alkuraya FS. Mutations in EOGT confirm the genetic heterogeneity of autosomal-recessive Adams-Oliver syndrome. The American Journal of Human Genetics. 2013;92(4):598–604. doi: 10.1016/j.ajhg.2013.02.012. http://dx.doi.org/10.1016/j.ajhg.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimokado K, Raines EW, Madtes DK, Barrett TB, Benditt EP, Ross R. A significant part of macrophage-derived growth factor consists of at least two forms of PDGF. Cell. 1985;43(1):277–286. doi: 10.1016/0092-8674(85)90033-9. [DOI] [PubMed] [Google Scholar]
- Shin M, Nagai H, Sheng G. Notch mediates Wnt and BMP signals in the early separation of smooth muscle progenitors and blood/endothelial common progenitors. Development. 2009;136(4):595–603. doi: 10.1242/dev.026906. http://dx.doi.org/10.1242/dev.026906. [DOI] [PubMed] [Google Scholar]
- Shutter JR, Scully S, Fan W, Richards WG, Kitajewski J, Deblandre GA, Stark KL. Dll4, a novel Notch ligand expressed in arterial endothelium. Genes and Development. 2000;14(11):1313–1318. [PMC free article] [PubMed] [Google Scholar]
- Simpson MA, Irving MD, Asilmaz E, Gray MJ, Dafou D, Elmslie FV, Trembath RC. Mutations in NOTCH2 cause Hajdu-Cheney syndrome, a disorder of severe and progressive bone loss. Nature Genetics. 2011;43(4):303–305. doi: 10.1038/ng.779. http://dx.doi.org/10.1038/ng.779. [DOI] [PubMed] [Google Scholar]
- Spinner NB, Colliton RP, Crosnier C, Krantz ID, Hadchouel M, Meunier-Rotival M. Jagged1 mutations in Alagille syndrome. Human Mutation. 2001;17(1):18–33. doi: 10.1002/1098-1004(2001)17:1<18::AID-HUMU3>3.0.CO;2-T. http://dx.doi.org/10.1002/1098-1004(2001)17:1<18::AID-HUMU3>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Stanley P, Okajima T. Roles of glycosylation in Notch signaling. Current Topics in Developmental Biology. 2010;92:131–164. doi: 10.1016/S0070-2153(10)92004-8. http://dx.doi.org/10.1016/S0070-2153(10)92004-8. [DOI] [PubMed] [Google Scholar]
- Stittrich AB, Lehman A, Bodian DL, Ashworth J, Zong Z, Li H, Patel MS. Mutations in NOTCH1 cause Adams-Oliver syndrome. The American Journal of Human Genetics. 2014;95(3):275–284. doi: 10.1016/j.ajhg.2014.07.011. http://dx.doi.org/10.1016/j.ajhg.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney C, Morrow D, Birney YA, Coyle S, Hennessy C, Scheller A, Cahill PA. Notch 1 and 3 receptor signaling modulates vascular smooth muscle cell growth, apoptosis, and migration via a CBF-1/RBP-Jk dependent pathway. The FASEB Journal. 2004;18(12):1421–1423. doi: 10.1096/fj.04-1700fje. http://dx.doi.org/10.1096/fj.04-1700fje. [DOI] [PubMed] [Google Scholar]
- Takebayashi K, Sasai Y, Sakai Y, Watanabe T, Nakanishi S, Kageyama R. Structure, chromosomal locus, and promoter analysis of the gene encoding the mouse helix-loop-helix factor HES-1. Negative autoregulation through the multiple N box elements. The Journal of Biological Chemistry. 1994;269(7):5150–5156. [PubMed] [Google Scholar]
- Tang Y, Urs S, Boucher J, Bernaiche T, Venkatesh D, Spicer DB, Liaw L. Notch and transforming growth factor-beta (TGFbeta) signaling pathways cooperatively regulate vascular smooth muscle cell differentiation. The Journal of Biological Chemistry. 2010;285(23):17556–17563. doi: 10.1074/jbc.M109.076414. http://dx.doi.org/10.1074/jbc.M109.076414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Urs S, Liaw L. Hairy-related transcription factors inhibit Notch-induced smooth muscle alpha-actin expression by interfering with Notch intracellular domain/CBF-1 complex interaction with the CBF-1-binding site. Circulation Research. 2008;102(6):661–668. doi: 10.1161/CIRCRESAHA.107.165134. http://dx.doi.org/10.1161/CIRCRESAHA.107.165134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood PA, Bean PA, Whitelock JM. Inhibition of endothelial cell adhesion and proliferation by extracellular matrix from vascular smooth muscle cells: Role of type V collagen. Atherosclerosis. 1998;141(1):141–152. doi: 10.1016/s0021-9150(98)00164-6. [DOI] [PubMed] [Google Scholar]
- Varadkar P, Kraman M, Despres D, Ma G, Lozier J, McCright B. Notch2 is required for the proliferation of cardiac neural crest-derived smooth muscle cells. Developmental Dynamics. 2008;237(4):1144–1152. doi: 10.1002/dvdy.21502. http://dx.doi.org/10.1002/dvdy.21502. [DOI] [PubMed] [Google Scholar]
- Villa N, Walker L, Lindsell CE, Gasson J, Iruela-Arispe ML, Weinmaster G. Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mechanisms of Development. 2001;108(1–2):161–164. doi: 10.1016/s0925-4773(01)00469-5. [DOI] [PubMed] [Google Scholar]
- Volz KS, Jacobs AH, Chen HI, Poduri A, McKay AS, Riordan DP, Red-Horse K. Pericytes are progenitors for coronary artery smooth muscle. eLife. 2015;4:e10036. doi: 10.7554/eLife.10036. http://dx.doi.org/10.7554/eLife.10036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiological Reviews. 2009;89(3):957–989. doi: 10.1152/physrev.00041.2008. http://dx.doi.org/10.1152/physrev.00041.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Campos AH, Prince CZ, Mou Y, Pollman MJ. Coordinate Notch3-hairy-related transcription factor pathway regulation in response to arterial injury. Mediator role of platelet-derived growth factor and ERK. The Journal of Biological Chemistry. 2002;277(26):23165–23171. doi: 10.1074/jbc.M201409200. http://dx.doi.org/10.1074/jbc.M201409200. [DOI] [PubMed] [Google Scholar]
- Wang HU, Chen ZF, Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell. 1998;93(5):741–753. doi: 10.1016/s0092-8674(00)81436-1. [DOI] [PubMed] [Google Scholar]
- Wang Y, Pan L, Moens CB, Appel B. Notch3 establishes brain vascular integrity by regulating pericyte number. Development. 2014;141(2):307–317. doi: 10.1242/dev.096107. http://dx.doi.org/10.1242/dev.096107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Prince CZ, Mou Y, Pollman MJ. Notch3 signaling in vascular smooth muscle cells induces c-FLIP expression via ERK/MAPK activation. Resistance to Fas ligand-induced apoptosis. The Journal of Biological Chemistry. 2002;277(24):21723–21729. doi: 10.1074/jbc.M202224200. http://dx.doi.org/10.1074/jbc.M202224200. [DOI] [PubMed] [Google Scholar]
- Wang Q, Zhao N, Kennard S, Lilly B. Notch2 and Notch3 function together to regulate vascular smooth muscle development. PLoS One. 2012;7(5):e37365. doi: 10.1371/journal.pone.0037365. http://dx.doi.org/10.1371/journal.pone.0037365. [DOI] [PMC free article] [PubMed] [Google Scholar]