

Abstract
Glutamate is the major excitatory neurotransmitter in the brain, but excessive synaptic glutamate must be removed to prevent excitotoxic injury and death. Two astrocytic glutamate transporters, excitatory amino acid transporter (EAAT) 1 and 2, play a major role in eliminating excess glutamate from the synapse. Dysregulation of EAAT1 contributes to the pathogenesis of multiple neurological disorders, such as Alzheimer's disease (AD), ataxia, traumatic brain injuries and glaucoma. In the present study, we investigated the effect of arundic acid on EAAT1 to determine its efficacy in enhancing the expression and function of EAAT1, and its possible mechanisms of action. The studies were carried out in human astrocyte H4 cells as well as in human primary astrocytes. Our findings show that arundic acid upregulated EAAT1 expression at the transcriptional level by activating nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). Arundic acid increased astrocytic EAAT1 promoter activity, mRNA/protein levels and glutamate uptake, while pharmacological inhibition of NF-κB or mutation on NF-κB binding sites in the EAAT1 promoter region abrogated these effects. Arundic acid increased NF-κB reporter activity and induced NF-κB nuclear translocation as well as its bindings to the EAAT1 promoter. Furthermore, arundic acid activated the Akt and ERK signaling pathways to enhance EAAT1 mRNA/protein levels. Finally, arundic acid attenuated manganese-induced decrease in EAAT1 expression by inhibiting expression of the transcription factor Ying Yang 1 (YY1). These results demonstrate that arundic acid increases the expression and function of EAAT1 via the Akt, ERK and NF-κB signaling pathways, and reverses Mn-induced EAAT1 repression by inhibiting the Mn-induced YY1 activation.
Keywords: Astrocytes, EAAT1, arundic acid, NF-κB, manganese
Introduction
Glutamate is a major excitatory neurotransmitter in the central nervous system (CNS), playing a critical role in various brain functions such as cognition, memory and learning, as well as development of the CNS [1]. Synaptic glutamate concentrations are kept at low optimal levels to prevent excitotoxic neuronal death caused by excessive stimulation of glutamate receptors [2]. The rapid removal of excessive glutamate from the synaptic clefts is achieved by Na+-dependent excitatory amino acid transporters (EAATs). Among the five human sub-types of EAATs (EAAT1-5), EAAT1 and EAAT2 are also known as glutamate aspartate transporter (GLAST) and glutamate transporter-1 (GLT-1) in rodents, respectively. These transporters are predominantly expressed in astrocytes [3,4]. Consequentially, dysregulation of EAAT1 and EAAT2 causes pathophysiology inherent to multiple neurological disorders including stroke, epilepsy, amyotrophic lateral sclerosis (ALS), Alzheimer's disease (AD), Huntington's disease (HD), Parkinson's disease (PD), HIV-associated dementia and malignant glioma [5,6].
During neurodevelopment, EAAT1 is highly expressed in cerebellar Bergmann glia and retinal Muller cells [7-11]. In mice, deletion of GLAST induces neurological deficits and enhances susceptibility to cerebellar injury [4,12]. Glutamate neurotoxicity is inherent to GLAST knockout mice, leading to retinal ganglion cell degeneration and glaucoma [13]. In humans, reduced glutamate uptake and EAAT1 mRNA/protein levels are detected in the brain of the AD patients [14-16]. A heterozygous mutation in EAAT1 gene decreased glutamate uptake, leading to the development of seizures and ataxia [17]. Furthermore, a single missense C186S mutation in EAAT1 gene is associated with ataxia [18]. Reduction in EAAT1 protein expression is inherent to traumatic brain injury [19] and ophthalmic disorders [10], such as glaucoma [20].
Pharmacological agents targeting enhancement of expression and function of glutamate transporters have been explored as potential therapeutic candidates for the treatment of excitotoxic-related neurological diseases [21]. A recent study has reported that arundic acid [ONO-2506; (R)-(-)-2-propyloctanoic acid] prevented retinal ganglion cell death by increasing EAAT1 expression in a glaucoma mouse model [22]. Arundic acid is an astrocyte-modulating agent that was discovered during screening for synthesis inhibitors of S100β, a calcium-binding protein primarily produced in astrocytes [23]. S100β exerts neurotrophic effects at low concentrations, but is neurotoxic at high concentrations, and has served as a biochemical marker of several CNS disorders, such as traumatic brain injury and AD [24]. Arundic acid protected against deficits induced in cerebral ischemia and the loss of dopaminergic neurons in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD mouse model by suppressing S100β expression [25-28]. In addition, arundic acid reduced the levels of reactive nitrogen and oxygen species, further contributing to neuroprotection against the MPTP-induced dopaminergic neuronal damage in the mouse brain [29]. More importantly, these protective effects were persistent even with delayed treatment [30]. Arundic acid also decreased extracellular glutamate levels, leading to a decrease in infarct size in a cerebral ischemic rat model [31]. The protective effect was further established in a glaucoma mouse model where arundic acid prevented retinal ganglion cell death by increasing the expression of EAAT1 [22]. Therefore, understanding the molecular mechanisms of arundic acid-induced upregulation of EAAT1 at the transcriptional should impart new insight into the efficacy of arundic acid as a pharmacological therapeutics for neurodegenerative diseases associated with EAAT1-dysregulation.
Efforts to develop efficacious agents to treat neurological disorders that modulate glutamate transporters, led to the characterization of several compounds that act on EAAT2, including ceftriaxone that has been staged on the clinical trials [4,32,33]. There have been a few studies on potential endogenous and pharmacological agents that show efficacy in regulating EAAT1 expression. Dibutyryl-cyclic AMP (dbcAMP) and neuronal soluble factors increased EAAT1 mRNA and protein levels in astrocytes [34,35]. Various growth factors, including epidermal growth factor (EGF), transforming growth factor-α (TGF-α), fibroblast growth factor (FGF), insulin-like growth factor-1 (IGF-1) and glial cell line-derived neurotrophic factor (GDNF) also increased EAAT1 expression [36-38]. Estrogenic compounds such as 17β-estradiol and selective estrogen receptor modulators (SERMs) including tamoxifen and raloxifene increased EAAT1 expression by non-genomic estrogen receptors (ERs) of ER-α and ER-β as well as G-protein-coupled receptor 30 (GPR30) [39-41] via TGF-α as a mediator [40]. Moreover, our recent study on the transcriptional regulation of EAAT1 demonstrated that nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is a highly potent positive regulator of EAAT1 and mediates the stimulatory effects of positive modulators such as EGF on EAAT1 expression and function [42].
Astrocytic EAAT1 expression is decreased by tumor necrosis factor-α (TNF-α) and endothelins [43,44]. Neurotoxic heavy metals such as arsenic and manganese (Mn) also reduced EAAT1 expression and function [45,40]. As chronic exposure to Mn from occupational or environmental sources causes manganism, a neurodegenerative disorder with similar features to PD, understanding the mechanism of Mn-induced downregulation of EAAT1 is highly significant in the search for novel pharmacological therapeutics for manganism [46]. The transcription factor Yin Yang 1 (YY1) plays a critical role in the transcriptional repression of EAAT1 in chicken Bergman glia [47,48]. We have also recently reported that YY1 is a critical repressor of EAAT1 and its activation mediates the inhibitory effects of Mn on EAAT1 using epigenetic modifiers histone deacetylases (HDACs) as co-repressors in human astrocytes [42]. Several HDAC inhibitors such as valproic acid and trichostatin A have been reported to exert neuroprotective effects against glutamate excitotoxicity-induced neurological diseases, possibly by increasing EAAT1 expression [47,49-51].
Given arundic acid's efficacy as a neuroprotectant (see above) along with its ability to increase EAAT1 expression, the present study investigated the transcriptional regulation and intracellular signaling pathways by which arundic acid induces enhancement of EAAT1 expression and function in human astrocytes. Our results demonstrate that the NF-κB pathway plays a crucial role in arundic acid-induced upregulation of astrocytic EAAT1 at the transcriptional level by activating phosphoinositide 3-kinase/a serine/threonine kinase, also known as protein kinase B (PI3k/Akt) and extracellular signal-regulated kinase (ERK) signaling.
Materials and Methods
Materials
The human astrocyte H4 (HTB-148) cell line was purchased from ATCC (Manassas, VA) and human primary astrocytes were obtained from ScienCell Research Laboratories (Carlsbad, CA). The cell culture media, reagents and lipofectamine 2000 were purchased from Invitrogen (Carlsbad, CA). Growth media, supplements and transfection reagent astrofectagen for primary human astrocytes were from ScienCell. Luciferase reporter assay kit was obtained from Promega (Madison, WI). The plasmid DNA and RNA isolation kits were obtained from Qiagen (Valencia, CA). Arundic acid (ONO 2506) was purchased from Tocris Biosciences (Minneapoils, MN). Manganese chloride (MnCl2), N4-[2-(4-phenoxyphenyl) ethyl]-4,6-quinazolinediamine (QNZ), 1,4-diamino-2,3-dicyano-1,4-bis[2-aminophenylthio] butadiene (U0126) and protease inhibitor cocktail were obtained from Sigma-Aldrich (St. Louis, MO). Sulfo-NHS-SS-biotin was from Thermo Scientific (Rockford, IL) and 2-morpholino-8-phenyl-4H-chromen-4-one (LY294002) was from Selleck Chemicals (Houston, TX). L-[3H]-glutamate was purchased from Perkin Elmer (Boston, MA). EAAT1 (ab416) antibody was from Abcam (Cambridge, MA). Antibodies for YY1 (sc-281), NF-κB p65 (sc-372), NF-κB inhibitor-α (IκBα) (sc-1643), β-actin (sc-1616), histone H3 (sc-10809) and Na+/K+-ATPase (sc-48345) were from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies for phosphor-Akt (#4058S), total Akt (#2967), phosphor-ERK (#3510) and total ERK (#9102) were from Cell Signaling Technology (Danvers, MA).
Cell Culture
Human primary astrocytes (ScienCell) were grown in astrocyte growth medium for three weeks prior to the experiments, and H4 cells were cultured in DMEM with 10% fetal bovine serum and 1% penicillin and streptomycin (Gibco Life Tech, Grand island, NY). The cultures were maintained at 37°C in a 95% air, 5% CO2 incubator. For the experiments, astrocytes were plated in 6-well plates for protein/mRNA analysis and in 24-well plates for promoter activity and glutamate uptake assays.
Measurement of Promoter Activity
H4 astrocytes were transfected with the EAAT1 wild type or NF-κB mutant promoter plasmids with lipofectamine 2000. The plasmid transfection of human astrocytes was performed with astrofectagen. The mutations on NF-κB consensus binding sites (-116 and -538 position) in the EAAT1 promoter plasmid were previously described [42]. NF-κB reporter plasmid was obtained from Clontech Laboratories (Palo Alto, CA), and the YY1 promoter plasmid was a gift from Dr. Denis Guttridge (Ohio State University, OH). After overnight transfections, the effects of various compounds on promoter activities were determined with the Bright-Glo luciferase assay kit (Promega) according to the manufacturer's instructions.
Cell Viability Assay
At the end of the treatment with the designated compounds, 20 μl of 3,4,5-dimethyl thiazol-2,5-diphenyl tetrazolium bromide (MTT, 5 mg/ml) was added for 4 h to the cells in each of the 24-well plates. Next, dimethyl sulfoxide (100 μl) was added to dissolve the formazan crystals and the absorbance was read at 570 nm with a microplate spectrophotometer (Molecular Devices, Sunnyvale, CA).
Quantitative RT-PCR Analysis
The total RNA was extracted from the cells by using the RNA isolation kit (Qiagen) and 2 μg of RNA was transcribed to cDNA with the high capacity cDNA reverse transcription kit (Applied Biosystems, USA). For the quantitative real time PCR (qPCR), the reactions mixture of 1μg of cDNA template, 0.4 μM of the EAAT1, YY1 or GAPDH primers and RT2 SYBR Green qPCR master mix (Qiagen) was prepared in a total volume of 20 μl. The primers used were: for EAAT1, 5′- ACG GTC ACT GCT GTC ATT G-3′ (forward) and 5′-TGT GAC GAG ACT GGA GAT GA -3′ (reverse); for YY1, 5′- CTC CTG CAG CCC TGG GCG CAT C -3′ (forward) and 5′- GGT AAG CCC TTT AGC GCC TC -3′ (reverse); and for GAPDH, 5′-TCC CTC AAG ATT GTC AGC AA-3′ (forward) and 5′-AGA TCC ACA ACG GAT ACA TT-3′ (reverse). The qPCR was carried out in the CFX96 real time PCR detection system (Bio-Rad) with the following program: 1 cycle at 95°C for 10 min, 40 cycles at 95°C for 15 s and at 60 °C for 1 min. Data were analyzed using a PCR array data analysis program from Qiagen with GAPDH as an internal control.
Western Blot
Cells were lysed in radio immunoprecipitation assay (RIPA) [100 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.5% sodium deoxycholate and 0.1% sodium dodecyl sufate] buffer with a protease inhibitor cocktail (Sigma-Aldrich). The protein concentrations in the lysates were determined by bicinchoninic acid (BCA) assay and a total of 30 μg of protein samples were resolved on 10% SDS-PAGE. After the electrophoretic transfer of proteins onto the nitrocellulose membrane, the membranes were incubated overnight at 4°C with primary antibodies using the following dilutions: EAAT1 (1:1000), YY1 (1:500), NF-κB (1:500), IκBα (1:500), β-actin (1:1000) histone H3 (1:500) and Na+/K+-ATPase (1:500) phosphor-Akt (1:500) and total Akt (1:1000), phosphor-ERK (1:500) and total ERK (1:1000). Afterwards, incubation was performed with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:3000, Promega) at room temperature for 1 h. The blots were developed and detected with an enhanced chemiluminescence detection kit (Pierce).
Preparation of Cytoplasmic and Nuclear Fractions
Cells were lysed in hypnotic buffer (10 mM HEPES-KOH, pH 7.9, 10 mM KCl, 1.5 mM MgCl2) containing 0.5% NP-40 and centrifuged at 2500 rpm for 5 min at 4°C. The lysates containing cytoplasmic fractions were saved. The nuclei in the pellet were dissolved in hypertonic buffer (20 mM HEPES-KOH, pH 7.9, 0.4M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA and 25% glycerol) with periodic vortexing and incubation on ice for 30 min. The nuclear fractions were obtained after spinning at 20,000 × g for 10 min at 4°C.
Glutamate Uptake Assay
To determine EAAT1-mediated glutamate uptake, the assay was carried out in the presence of dihydrokainic acid (DHK), a specific inhibitor for EAAT2 [52]. Cells were first washed with the pre-warmed glutamate uptake buffer (122 mM NaCl, 3.3 mM KCl, 0.4 mM MgSO4, 1.3 mM CaCl2, 1.2 mM KH2PO4, 25 mM HEPES, and 10 mM D-(+)-glucose, pH 7.4), and 100 μM of DHK was added to cells and incubated at 37°C for 30 min. Then, the uptake buffer containing 0.25 μCi/ml l-[3H]-glutamate (specific activity, 49.0 Ci/mmol; PerkinElmer) and 100 nM unlabeled glutamate was added and further incubated for 10 min. After 3 washes with ice-cold phosphate buffered saline (PBS), cell lysis was achieved by adding 1 ml of 1 N NaOH. An aliquot of 750 μl of each sample was transferred into scintillation vials and neutralized with 75 μl of 10 N HCl. Finally, 5 ml of liquid scintillation fluid was added to each vial and the radioactivity was measured in LS 6500 liquid scintillation counter (Beckman Coulter, Fullerton, CA). Glutamate uptake was quantified as nmol of glutamate/mg of protein/min, and correction of protein levels were determined by the BCA assay.
Cell Surface Biotinylation
Cells grown in 100 mm dishes were washed with warm PBS containing 0.1 mM of calcium and magnesium each. Then, 2 ml of sulfo-NHS-SS-biotin solution was added to cells and incubated for 20 min with gentle rocking at 4°C. Cells were washed twice with ice-cold with quenching solution (100 mM glycine in PBS) and incubated for 45 min at 4°C. RIPA buffer was added to lyse cells, and lysates were obtained after centrifuging at 16,000 × g for 20 min. Then, 300 μl of cell lysate was mixed in 1:1 with avidin beads and incubated at room temperature with gentle shaking. The avidin beads were pelleted by spinning at 8,000 × g for 2 min and supernatant was collected as intracellular fractions. The beads were washed 4 times with RIPA buffer and resuspended in 300 μl of SDS loading buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 20% glycerol and 5% β-mercaptoethanol). After centrifuging at 8,000 × g for 5 min, the supernatant was collected as biotinylated membrane fractions. Equal amounts of intracellular and membrane fractions were analyzed by western blot.
Chromatin Immunoprecipitation (ChIP) Assay
The ChIP assay was performed using the EZ-ChIP kit (Millipore) according to the manufacturer's instructions. Briefly, the crosslinking was done by treating cells with formaldehyde for 10 min at room temperature. After washing with ice-cold PBS, cells were lysed in SDS lysis buffer containing a protease inhibitor cocktail. The cell lysates were sonicated and centrifuged at 15,000 × g for 10 min at 4°C. The supernatant was mixed with ChIP dilution buffer and 60 μl of protein G agarose was added. After 1 h incubation at 4°C, the agarose beads were pelleted by spinning at 3,000 × g for 1 min. Then, 1% of the supernatants were saved as inputs and NF-κB p65 or rabbit IgG (negative control) or anti-RNA polymerase (positive control) antibodies were added to the remainder and incubated overnight at 4°C. Protein G agarose beads (60 μl) were added and incubated at 4°C for 1 h. The agarose beads were pelleted and washed with low salt and high salt immune complex wash buffer. The free DNA obtained after reverse crosslinking of protein-DNA complex was purified and PCR was carried out with the primers- 5′- GCG TGA AAG TGG TCT AAG GAG -3′ (forward) and 5′- GCA AGT TAC TAT CAG GGC AAC -3′ (reverse).
Electrophoretic Mobility Shift Assay (EMSA)
EMSA was carried out using LightShift Chemiluminsescent Kit (Pierce) based on the manufacturer's instructions. Briefly, 5 μg of nuclear extract from control or arundic acid-treated cells was incubated for 20 min on ice with annealed biotin labeled oligos harboring the consensus sites (-116 and -538 positions) for NF-κB. The reaction mixtures were resolved in 6% DNA retardation gels (Life technologies) and transferred to nylon membrane. The DNA-protein complexes were detected with the Chemiluminescent Nucleic Acid Detection Module (Pierce). The primers used were: for EAAT1 -116 NF-κB, 5′- CAG AAA CCT CGG GGT TTC CCC CTC CTC CCT G -3′ (forward) and 5′- CAG GGA GGA GGG GGA AAC CCC GAG GTT TCT G -3′ (reverse); for EAAT1 -538 NF-κB, 5′- GAA ATA GAG GCA TGT CCC TAA CTT TAG AC -3′ (forward) and 5′- GTC TAA AGT TAG GGA CAT GCC TCT ATT TC -3′ (reverse).
DNA Affinity Purification Assay (DAPA)
DAPA was performed the μMACS FactorFinder kit (Miltenyi Biotec, Inc., Auburn, CA) following the supplier's protocol. Briefly, 1.5 μg of biotinylated oligos containing the consensus sites for NF-κB on the EAAT1 promoter was incubated with 50 μg of nuclear extract in binding buffer for 20 min. Then, 100 μl of streptavidin microbeads were added and the reaction mixture was further incubated for 10 min. The mixture was loaded onto the μ column and the bound proteins were eluted and analyzed by western blotting.
Statistical Analysis
The statistical analysis was performed using GraphPad Prism software (GraphPad Inc., La Jolla, CA) and described the data as mean ± standard error of mean (SEM). Statistical differences between control and various treated groups were determined by one-way analysis of variance (ANOVA) followed by Tukey's post hoc test with statistical significance set at p<0.05. Each experiment was carried out in three independent cell preparations with 3-6 replicates in each.
Results
Arundic acid increased EAAT1 promoter activity
Previous studies have reported that arundic acid increased EAAT1 (GLAST) mRNA levels in activated astrocytes [23] and retina of mice [22]. We have tested if AA modulates EAAT1, EAAT2 or both transporters with any preference on specific subtype in human astrocyte H4 cells. According to our preliminary data, AA (50 μM) increased glutamate uptake mainly via EAAT1 (64.5%) compared to those via EAAT2 (31.7%) (Suppl. Fig. 1), indicating that AA effects on EAAT1 is relevant to further study. Thus, we investigated the mechanism by which arundic acid (Fig. 1A) modulates EAAT1 gene expression at the transcriptional level. First, we tested the effect of arundic acid on EAAT1 promoter activity in H4 astrocytes. The results showed that arundic acid increased EAAT1 promoter activity in a concentration- and time-dependent manner (Fig. 1B and C). Arundic acid significantly increased EAAT1 promoter activity after 24 h treatments with concentrations at ≥ 25 μM (p<0.001). Accordingly, subsequent studies on EAAT1 promoter activity were carried out with 24 h treatments at 50 μM. The cell viability assay confirmed that arundic acid did not cause cytotoxicity at this concentration (Fig. 1D).
Fig. 1.
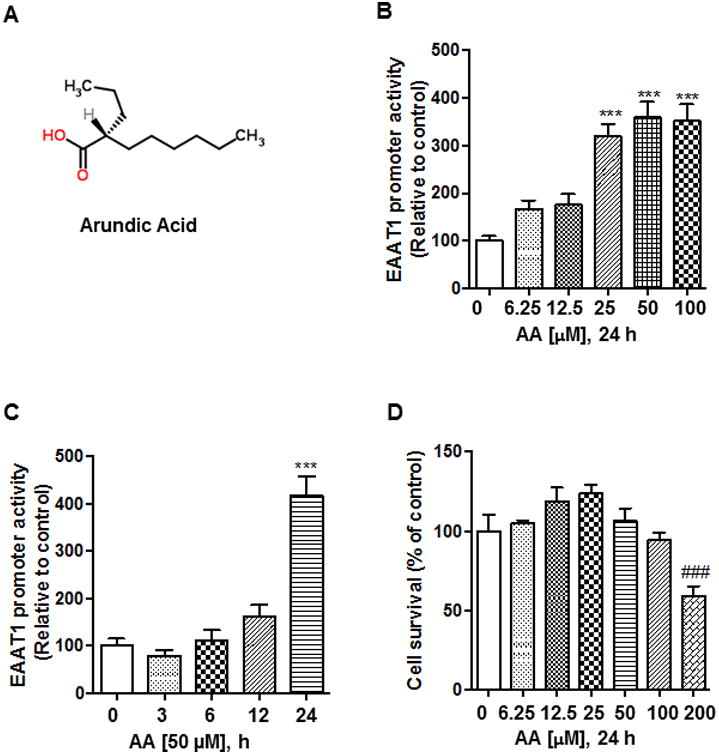
Arundic acid increases EAAT1 promoter activity. (A) After overnight transfection with 0.5 μg of EAAT1 promoter luciferase plasmid, human astrocyte H4 cells were treated with arundic acid for 24 h, followed by luciferase assay to measure EAAT1 promoter activity. (B) After overnight transfection with 0.5 μg of EAAT1 promoter plasmid, astrocytes were treated with 50 μM of arundic acid for 3, 6, 12 and 24 h, followed by measurement of EAAT1 promoter activity. (C) Cells were treated with the designated concentrations of arundic acid for 24 h and cell viability was determined by the MTT assay as described in the Methods section. (D) Chemical structure of arundic acid [ONO-2506; (R)-(-)-2-propyloctanoic acid]. ***p<0.001 (increase compared to the control); ###p<0.001 (decrease compared to the control); ANOVA followed by Tukey's post hoc test; N=6.
Arundic acid increased EAAT1 mRNA and protein levels as well as glutamate uptake
Next, we investigated whether arundic acid-increased promoter activity led to increased EAAT1 mRNA and protein expression in H4 astrocytes. Arundic acid increased both mRNA and protein levels of EAAT1 in a concentration-dependent manner (Fig. 2A, B). Fig. 2C shows that arundic acid increased glutamate uptake via EAAT1 in the presence of DHK, since DHK, a selective inhibitor of EAAT2, completely blocked glutamate uptake via EAAT2. Furthermore, cell surface biotinylation experiments established that arundic acid increased EAAT1 expression in the plasma membrane (Fig. 2D).
Fig. 2.

Arundic acid increases mRNA, protein levels and cell surface expression of EAAT1. (A) Astrocyte H4 cells were treated with designated concentrations of arundic acid for 24 h, followed by determination of mRNA levels by qPCR. (B) Cells were treated with designated concentrations of arundic acid for 24 h, followed by western blot to determine protein levels. The data showed as blots as well as in the bar graph for quantification. (C) After treatment with the designated concentrations of arundic acid, glutamate uptake assay was carried out in the presence of 100 μM of DHK to block EAAT2 transporter as described in the Materials and Methods section. (D) After H4 cells were treated with arundic acid (50 μM, 24 h), cell surface biotinylation experiment was conducted as described in the Methods section, followed by western blot with equal amounts of intracellular and membrane fractions to detect EAAT1 protein levels. β-Actin and Na+/K+-ATPase were used as loading controls for intracellular and membrane fractions, respectively. *p<0.05, **p<0.01, ***p<0.001 (increase compared to the control); ANOVA followed by Tukey's post hoc test; N=3-6.
NF-κB mediated arundic acid-induced EAAT1 expression at the transcriptional level
NF-κB is a critical positive regulator of EAAT1 [42]. To determine if NF-κB plays a role in arundic acid-induced EAAT1 expression, we first measured basal promoter activity in wild type and NF-κB mutants of the EAAT1 promoter in the absence of arundic acid in H4 human astrocytes. Mutation on the NF-κB consensus site (-116 or -538) significantly decreased EAAT1 promoter activity and a double mutation of both the NF-κB sites further decreased EAAT1 promoter activity (p<0.01, Fig. 3A).
Fig. 3.
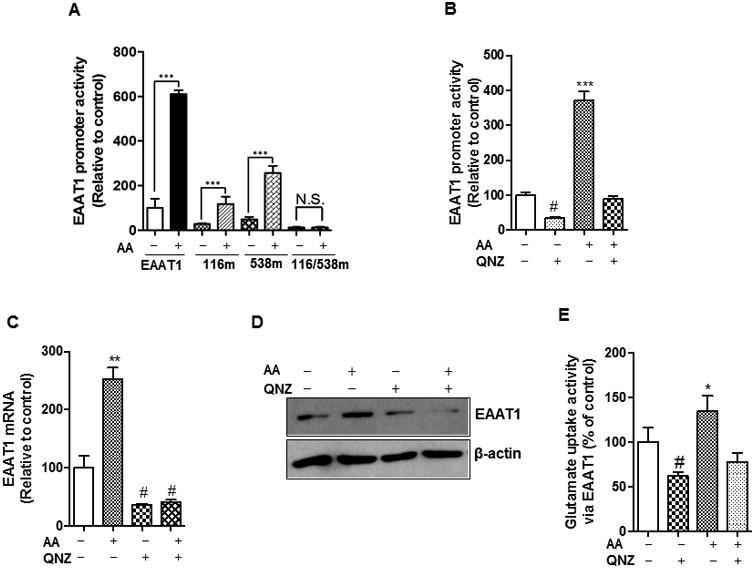
NF-κB mediates arundic acid-induced EAAT1 expression and function. (A) After overnight transfection with 0.5 μg of EAAT1 or single (-116m, -538m) or double (116/538m) NF-κB sites mutated EAAT1 promoter luciferase plasmids, H4 astrocytes were treated with 50 μM of arundic acid (AA) for 24 h, followed by luciferase assay to determine EAAT1 promoter activity. (B, C and D) After pre-treated with QNZ, an NF-κB inhibitor (50 μM, 30 min), H4 astrocytes were treated with arundic acid (50 μM, 24 h), followed by cell lysis preparations for luciferase assay, qPCR and western blot to determine EAAT1 promoter activity (B), mRNA levels (C) and protein levels (D) respectively. (E) At the end of arundic acid treatment (50 μM, 24 h), glutamate uptake was determined in H4 astrocytes in the presence of 100 μM of DHK to block EAAT2 transporter. *p<0.05, ** p<0.01, ***p<0.001 (increase compared to untreated group); #p<0.05, (decrease compared to untreated group); N.S., no significant change; ANOVA followed by Tukey's post hoc test; N=3-6.
Arundic acid increased EAAT1 promoter activity in both NF-κB single mutants, but not in a double mutant (Fig. 3A), suggesting that both NF-κB sites are critical for the arundic acid-induced enhancement of EAAT1 promoter activity. To further confirm the role of the NF-κB pathway, we assessed EAAT1 expression in the presence of QNZ, a pharmacological inhibitor of NF-κB. The arundic acid-induced increase in EAAT1 promoter activity, mRNA and protein level was completely abolished by QNZ (Fig. 3B, C and D). Furthermore, arundic acid failed to increase glutamate uptake via EAAT1 in the presence of QNZ (Fig. 3E), indicating that the NF-κB pathway mediates arundic acid-induced increase in EAAT1 expression and function, and that both NF-κB sites are crucial for arundic acid enhancement of EAAT1 promoter activity.
Arundic acid increased NF-κB reporter activity and induced NF-κB nuclear translocation
To gain further insight into the molecular mechanisms of arundic acid-induced activation of the NF-κB pathway, we tested if arundic acid directly activates NF-κB in H4 astrocytes. The results showed that after 12 h arundic acid increased NF-κB reporter activity (Fig. 4A), preceding the arundic acid-induced increase in EAAT1 promoter activity (seen at 24 h) (Fig. 4A and 1B, respectively).
Fig. 4.

Arundic acid activates the NF-κB pathway. (A) After overnight transfection with 0.5 μg of NF-κB reporter plasmid, H4 astrocytes were treated with 50 μM of arundic acid for the designated periods of time, followed by the luciferase assay to determine NF-κB reporter activity. (B) H4 cells were treated with 50 μM of arundic acid for the indicated time periods, followed by the preparation of cytoplasmic and nuclear fractions. Western blot was performed with equal protein amounts of cytoplasmic and nuclear fractions to detect IκBα and NF-κB p65, respectively. β-actin and histone H3 were used as loading controls for cytosolic and nuclear fractions, respectively. *p<0.05, ** p<0.01, ***p<0.001 (increase compared to the control); ##p<0.01 (decrease compared to the control); ANOVA followed by Tukey's post hoc test; N=3.
In quiescent cells, NF-κB dimers are sequestered by IκB-α or -β in cytosol, maintaining it in an inactive form, but phosphorylation of IκB-α or -β by IκB kinases (IKK) undertakes proteasomal degradation of IκB-α or -β, leading to the nuclear translocation of NF-κB. This is considered a canonical pathway for NF-κB activation [53]. To determine if arundic acid activated NF-κB via this pathway, we measured the levels of IκBα and NF-κB in cytoplasmic as well as nuclear fractions. Corroborating the NF-κB reporter activity data (Fig. 4A) 12 h of arundic acid treatment significantly increased the nuclear translocation of NF-κB. This effect occurred concomitantly with decreased IκBα levels in cytoplasmic fractions (Fig. 4B). We have also tested if AA modulates upstream of this IκBα pathway by assessing phosphorylation of IKK, but AA did not modulate IKK phosphorylation (Suppl. Fig. 2).
Arundic acid induced NF-κB binding to the EAAT1 promoter
Since arundic acid induced the nuclear translocation of NF-κB, next, we determined its efficacy in enhancing NF-κB binding to consensus sites on the EAAT1 promoter sequence. The in vivo binding of NF-κB to the EAAT1 promoter was determined with the ChIP assay, while the in vitro binding was tested with EMSA and DAPA experiments. The EMSA results showed that arundic acid increased NF-κB binding to both of its consensus sites -116 and -538 of the EAAT1 promoter (Fig. 5A). The specificity of the binding was verified by applying excess (100×) non-biotinylated oligos that can block binding of biotinylated oligos. The DAPA experiments also showed that arundic acid enhanced NF-κB binding to both of its consensus sites (-116 and -538) of the EAAT1 promoter, yet with higher binding to the -116 consensus site (Fig. 5C). The ChIP assay confirmed that arundic acid increased NF-κB binding to the EAAT1 promoter (Fig. 5B).
Fig. 5.

Arundic acid enhances NF-κB binding to the EAAT1 promoter. (A) H4 cells were treated with 50 μM of arundic acid (AA) for 24 h, followed by the preparation of nuclear extracts. EMSA was carried out with biotinylated oligonucleotides harboring -116 (left panel) and -538 (right panel) NF-κB binding sites of the EAAT1 promoter. Arrows show the shifts containing the DNA-protein complex. (B) H4 astrocytes were treated with arundic acid (50 μM, 24 h), followed by the ChIP assay to detect binding of NF-κB p65 to the EAAT1 promoter as described in the Methods section. Rabbit IgG was used as a negative control and input controls are also shown. (C) DAPA was carried out with the nuclear extracts prepared from arundic acid-treated astrocytes. The assay was conducted separately for two NF-κB binding sites (-116 and -538) containing biotinylated oligonucleotides. The eluted proteins along with the input controls were run for western blot to detect NF-κB p65. *p<0.05, ** p<0.01, ***p<0.001 (increase compared to the control); ANOVA followed by Tukey's post hoc test; N=3.
Arundic acid enhanced EAAT1 expression by activating the Akt and ERK pathways
Various intracellular signaling pathways are involved in regulating EAAT1 expression. Activation of protein kinase A (PKA), protein kinase C (PKC), ERK and Akt are required for the mediation of growth factors-induced expression of EAAT1 [37]. Estrogen and tamoxifen increase EAAT1 expression by activating the Akt and ERK pathways [40]. Results from the present study revealed that arundic acid activated astrocytic Akt as well as ERK by phosphorylation as early as 5 min after treatment (Fig. 6A). To further confirm the role of these pathways in arundic acid-induced EAAT1 expression, we used pharmacological inhibitors of these signaling proteins. Both LY294002 and UO126, specific inhibitors of Akt and ERK, respectively, significantly reduced arundic acid-induced EAAT1 promoter activity as well as EAAT1 mRNA and protein levels (Fig. 6B, C and D). These results indicate that both Akt and ERK pathways are critical for mediating arundic acid-induced upregulation of EAAT1.
Fig. 6.

Arundic acid (AA) activates Akt and ERK to enhance EAAT1 expression. (A) Astrocytes were treated with 50 μM of arundic acid, followed by western blot to detect phosphorylated ERK and Akt. The blots were re-probed with respective total ERK and Akt antibodies. β-actin was also used as a loading control. (B, C, D) Astrocytes were pre-treated with 10 μM of UO126 (UO) or 20 μM of LY294002 (LY) for 30 min to inhibit ERK and Akt, respectively, prior to treatment with arundic acid (50 μM, 24 h), followed by cell lysate preparation to determine EAAT1 promoter activity (B), mRNA levels (C) and protein levels (D). *p<0.05, **p<0.01, ***p<0.001 (increase compared to the control); ###p<0.001 (decrease compared to the indicated group); ANOVA followed by Tukey's post hoc test; N=3.
Arundic acid increased EAAT1 expression in human primary astrocytes via the Akt, ERK and NF-κB pathways
Consistent with the effects of arundic acid on EAAT1 in the astrocytic cell line, it increased EAAT1 promoter activity in human primary astrocytes. Double mutations of NF-κB binding sites of the EAAT1 promoter completely abolished this effect (Fig. 7A). These results confirm that NF-κB is critical for mediating the stimulatory effects of arundic acid on EAAT1 promoter activity in human primary astrocytes. Inhibitors of Akt, ERK and NF-κB, LY294002, UO126 and QNZ, respectively, blocked the effects of arundic acid on EAAT1 promoter activity and protein levels (Fig. 7B and C), indicating that the Akt, ERK and NF-κB pathways also mediate the stimulatory effects of arundic acid on EAAT1 expression in human primary astrocytes.
Fig. 7.
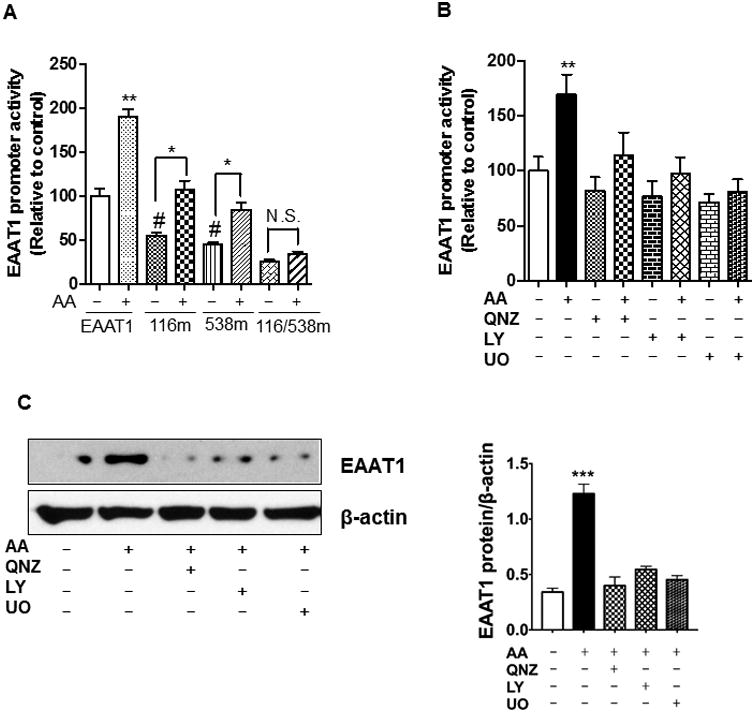
Arundic acid increases EAAT1 expression in human primary astrocytes. (A) After overnight transfection with 0.5 μg of EAAT1 or single (-116m, -538m) or double (116/538m) NF-κB mutants of EAAT1 promoter luciferase plasmids, human primary astrocytes were treated with 50 μM of arundic acid for 24 h, followed by luciferase assay to determine EAAT1 promoter activity. (B, C) Human primary astrocytes were pre-treated with 50 μM of QNZ, 10 μM of UO126 (UO) or 20 μM of LY294002 (LY) for 30 min to inhibit NF-κB, ERK and Akt, respectively, followed by treatment with arundic acid (50 μM, 24 h). Cells were lysed and EAAT1 promoter activity (B) and protein levels (C) were determined by luciferase assay and western blot, respectively. *p<0.05, ** p<0.01, ***p<0.001 (increase compared to the untreated or indicated group); #p<0.05 (decrease compared to the control); ANOVA followed by Tukey's post hoc test; N=3.
Arundic acid attenuated Mn-induced repression of EAAT1
Chronic overexposure to Mn may cause a PD-like neurological disorder, referred to as manganism. Mn-induced excitotoxicity has been considered as one of the main mechanisms involved in its neurotoxicity [46,54]. Mn decreases expression and function of GLAST (EAAT1) in rat primary astrocytes [40]. Since arundic acid afforded neuroprotection in a PD-animal model, we determined its efficacy in a Mn model associated with reduction in EAAT1 expression and function. Arundic acid attenuated Mn-induced decrease of EAAT1 promoter activity (Fig. 8A) as well as EAAT1 mRNA and protein levels (Fig. 8B and C). Similarly, the Mn-induced reduction in glutamate uptake activity via EAAT1 was attenuated by arundic acid (Fig. 8D). These data indicate that arundic acid protects against Mn-induced repression of EAAT1 expression and function in astrocytes.
Fig. 8.

Arundic acid attenuates Mn-induced EAAT1 repression. (A) After overnight transfection with 0.5 μg of EAAT1 promoter plasmid, H4 astrocytes were treated with arundic acid (50 μM, 24 h), followed by Mn exposure (250 μM, 6 h). The luciferase assay was carried out to determine EAAT1 promoter activity. (B, C) H4 astrocytes were treated with arundic acid (50 μM, 24 h), followed by Mn exposure (250 μM, 6 h). The mRNA (B) and protein (C) levels of EAAT1 were determined by qPCR and western blot, respectively. β-actin was used as a loading control. (D) After pre-treated with arundic acid (50 μM, 24 h), H4 astrocytes were exposed with Mn (250 μM, 6 h), followed by glutamate uptake assay as described in the Methods section. *p<0.05, **p<0.01, ***p<0.001 (increase compared to the control or indicated group); #p<0.05 (decrease compared to the control); ANOVA followed by Tukey's post hoc test; N=3.
Arundic acid inhibited Mn-induced YY1 activation
To further understand the mechanisms by which arundic acid attenuates Mn's effect on EAAT1 repression, we investigated if arundic acid modulates YY1, a transcription factor that is activated by Mn. YY1 acts as a repressor of EAAT1 [48], and mediates Mn-induced inhibition of EAAT1 with HDACs as co-repressors [42]. To determine whether the YY1 pathway plays a role in arundic acid-attenuation of Mn-induced repression of EAAT1, we tested its effects on YY1 expression. Arundic acid completely abolished Mn-activated YY1 promoter activity as well as YY1 mRNA and protein levels (Fig. 9A, B and C). These results indicate that arundic acid attenuates Mn-induced impairment in EAAT1 by modulating Mn activation of the YY1 pathway in addition to the other aforementioned mechanisms.
Fig. 9.

Arundic acid inhibits Mn-activated YY1 expression in H4 astrocytes. (A) After overnight transfection with 0.5 μg of YY1 promoter plasmid, H4 cells were pre-treated with of arundic acid (50 μM, 24 h) prior to Mn exposure (250 μM, 6 h), followed by measurement of YY1 promoter activity by luciferase assay. (B, C) Cells were pre-treated with arundic acid (50 μM, 24 h) prior to Mn exposure (250 μM, 6 h), followed by measurement of YY1 mRNA (B) and protein (C) levels by qPCR and western blot, respectively. β-actin was used as a loading control. *p<0.05, **p<0.01, (increase compared to the control); #p<0.05 (decrease compared to the indicated group); ANOVA followed by Tukey's post hoc test; N=3.
Arundic acid increased expression of EAAT2
We also tested if arundic acid modulates the function of the astrocytic glutamate transporter, EAAT2. EAAT2 is highly expressed in the frontal cortex of adult brain as well as other brain regions [9,55]. Arundic acid increased EAAT2 promoter activity in a concentration-dependent manner in H4 astrocytes (Fig. 10A). Since NF-κB serves as the major positive transcription factor of the EAAT2 promoter for various pharmacological stimulants [56,57,39,58], we tested if the NF-κB pathway mediates the effects of arundic acid on EAAT2. We measured promoter activities of the triple NF-κB mutant of EAAT2 in which all three critical NF-κB consensus sites (-251, -282 and -583) were mutated [57]. The results showed that the triple NF-κB mutant of EAAT2 did not completely abolish arundic acid-induced EAAT2 promoter activity (Fig. 10B). Inhibition of the NF-κB pathway with QNZ also only partially abolished the arundic acid-increase in EAAT2 promoter activity (Fig. 10C). These data indicate that NF-κB is not solely responsible for mediating the effects of arundic acid on EAAT2 upregulation, and that other transcription factors or pathways must be involved, warranting further investigation.
Fig. 10.

Arundic acid increases expression of EAAT2. (A) After overnight transfection with 0.5 μg of EAAT2 promoter luciferase plasmid, H4 astrocytes were treated with arundic acid (AA) for 24 h, followed by measurement of EAAT2 promoter activity by luciferase assay. (B) After overnight transfection with 0.5 μg of wild type EAAT2 or NF-κB mutant (NF-κBm) of EAAT2 promoter luciferase plasmids, H4 astrocytes were treated with arundic acid (50 μM) for 24 h, followed by measurement of EAAT2 promoter activity by luciferase assay. (C) H4 cells were pre-treated with QNZ (50 μM, 30 min) prior to treatment with arundic acid (50 μM, 24 h), followed by measurement of EAAT2 promoter activity by luciferase assay. *p<0.05, ** p<0.01, ***p<0.001 (increase compared to the control or untreated group); #p<0.05 (decrease compared to the control); ANOVA followed by Tukey's post hoc test; N=3.
Discussion
For the first time, we demonstrate that arundic acid enhances expression and function of EAAT1 by intracellularly activating the signaling pathways ERK and PI3K/Akt, and transcriptionally activating NF-κB. Moreover, arundic acid attenuates Mn-induced repression of EAAT1 by enhancing EAAT1 expression and inhibiting Mn-activated YY1 expression. These findings promote its potential as an efficacious treatment for neurological disorders associated with YY1 activation or excitotoxic dyshomeostasis, such as in Mn neurotoxicity.
Although the cellular and molecular mechanism(s) involved are not completely understood, the neuroprotective properties of arundic acid have been recognized, and is currently undergoing clinical trials for stroke, ALS, PD and AD [59]. Findings that the NF-κB pathway plays a critical role in arundic acid-induced upregulation of EAAT1 are noteworthy. The EAAT1 promoter sequence contains putative cis-elements for several transcription factors such as NF-κB, signal transducer of activated T cells (STAT), cAMP responsive element binding protein (CREB), activating protein 1 (AP1), nuclear factor of activated T cells (NFAT) and GC-box binding specificity protein (Sp) 1 and Sp3 [43]. It is well established that NF-κB mediates EGF as well as other growth factors-induced increase in EAAT1 expression [37]. We have recently demonstrated that two NF-κB binding sites at positions -116 and -538 on the EAAT1 promoter are critical for mediating the stimulatory effects of positive modulators [42]. Findings herein demonstrate that both NF-κB sites are critical for the increase in EAAT1 expression upon arundic acid treatment. Notably, single NF-κB mutants displayed a significant increase in EAAT1 promoter activity even upon treatment with arundic acid; yet in the double NF-κB mutant, arundic acid's-induced increase in EAAT1 promotor activity was fully abolished, corroborating that arundic acid increased NF-κB binding to both its consensus sites of the EAAT1 promoter (Fig. 5). There are several pathways for NF-κB signaling activation in cells [60], but arundic acid appears to activate the canonical NF-κB signaling as it increased nuclear translocation of NF-κB p65 with a decrease of IκBα in cytosol (Fig. 4) [53]. On a temporal axis, arundic acid enhancement of EAAT1 expression, i.e., NF-κB activation (12 h) prior to EAAT1 enhancement (24 h), supports the critical role of NF-κB in arundic acid-induced upregulation of EAAT1.
In addition to transcriptional modulation, intracellular signaling proteins ERK and Akt also appear to be involved in mediating the arundic acid-induced upregulation of EAAT1. This is consistent with earlier studies where ERK and Akt were shown to modulate the effects of growth factors and estrogen/SERMs on EAAT1 expression [37,40]. The ERK and Akt pathways may act in parallel and/or a coordinated fashion through cross-talk between these two pathways [61-63]. Furthermore, both the ERK and Akt pathways can also converge to activate NF-κB [64], as arundic acid induces the activation of both ERK and Akt, preceding that of NF-κB. Based on the results of EAAT1 promoter activity, the Akt pathway seems to be more prominent in arundic acid-induced EAAT1 stimulation, but further studies are required to determine the individual contribution and significance of each of these pathways.
Although Mn is an essential co-factor for numerous enzymes, including glutamine synthetase and Mn superoxide dismutase in its low or trace levels [65,66], chronic overexposure to Mn may cause manganism, a neurological disorder resembling PD symptoms. Glutamate excitotoxicity represents a critical mechanism in Mn-induced neurotoxicity, supported by observations that MK801, a pharmacological antagonist for the N-methyl-D-aspartate (NMDA) receptor, protected against Mn toxicity in the rat brain [54]. Mn has been also consistently shown to decrease glutamate uptake and expression of astrocytic glutamate transporters in in vitro as well as in vivo experimental settings [40,67,68,69]. Several pharmacological agents have been shown to attenuate Mn neurotoxicity by modulating astrocytic glutamate transporters. Estrogen (17β-estradiol), SERMs (tamoxifen and raloxifene), riluzole and HDAC inhibitors enhanced expression of GLAST and GLT-1, and attenuated Mn-induced repression of glutamate transporters [39,40,42,70]. Findings from the present study suggest that arundic acid might be a potential therapeutic candidate to treat Mn neurotoxicity as it fully reversed the Mn-induced decrease in EAAT1 promoter activity, mRNA/protein levels and glutamate uptake activity.
Interestingly, in addition to activation of NF-κB, arundic acid modulates YY1. Although YY1 can act as a positive or negative transcription factor depending on the cellular context, it serves as a repressor of EAAT1 since YY1 overexpression causes a decrease in EAAT1 mRNA and glutamate uptake activity in chicken Bergman glia [48]. We have previously reported that Mn increased YY1 promoter activity, mRNA and protein levels, and induced YY1 binding to the EAAT1 promoter [42,70]. These findings prompted us to test if arundic acid interferes with the YY1 pathway. Indeed, arundic acid attenuated the Mn-induced effects on EAAT1 repression via inhibition of YY1 promoter activity, mRNA and protein levels (Fig. 9), suggesting that modulation of YY1 might be another target for arundic acid in its efficacy in attenuating Mn-induced decrease in EAAT1 expression and function. Arundic acid attenuates Mn-induced repression of EAAT1 also by increasing EAAT1 trafficking to the cell surface [71,72]. Thus, EAAT1 function may be enhanced by increased expression of EAAT1 at the transcriptional/translational levels, and by increased functional EAAT1 localization in the plasma membrane by post-translational modifications or membrane trafficking. Arundic acid's ability to reverse the Mn-induced decrease of EAAT1 expression and cell surface localization requires further investigation to better understand the detailed molecular mechanisms involved.
Arundic acid also increases EAAT2 promoter activity (Fig. 10). EAAT2 is the other main astrocytic glutamate transporter and implicated in various neurological disorders such as ALS, AD and PD [73]. Earlier studies have reported that arundic acid protected against glutamate excitotoxicity by increasing the expression of EAAT1, EAAT2 and glutathione synthase in the rat model of transient middle cerebral artery occlusion (tMCAO) [31]. NF-κB is also a main positive transcription factor, playing a critical role in arundic acid-induced upregulation of EAAT2. It is well established that NF-κB mediates the stimulatory effects of various modulators including soluble neuronal factors, ceftriaxone, estrogen and SERMs on EAAT2 [39,56,57,58]. Findings from the present study that arundic acid significantly increased promoter activities of triple NF-κB mutant (-251, -282 and -583 positions) indicate that, unlike in EAAT1, inhibition of NF-κB does not completely block the stimulatory effects of arundic acid on EAAT2 promoter activity. The pharmacological inhibition of the NF-κB pathway also partially blocked the arundic acid-induced EAAT2 promoter activity, suggesting that other mechanisms may also play a critical role in mediating the effects of arundic acid on EAAT2 expression [57]. Our findings that mutations of both NF-κB and CRE binding sites of the EAAT2 promoter failed to inhibit arundic acid's stimulatory effect on EAAT2 completely (data not shown) suggest that multiple pathways might converge to mediate arundic acid's effect on EAAT2 expression.
In summary, we demonstrate that arundic acid upregulates astrocytic EAAT1 via activation of intracellular signaling proteins such as ERK and Akt, and the NF-κB pathway at the transcriptional level (Fig. 11). The findings from the present study open new venues for the development of therapeutics using arundic acid or its analogs to treat various neurological disorders associated with dysfunction of EAAT1. Furthermore, arundic acid-induced attenuation of Mn repression of EAAT1 by inhibiting YY1 activation suggests that it can also serve as a potential therapeutic against Mn-induced neurological disorders or pathological conditions associated with YY1 activation (Fig. 11).
Fig. 11.
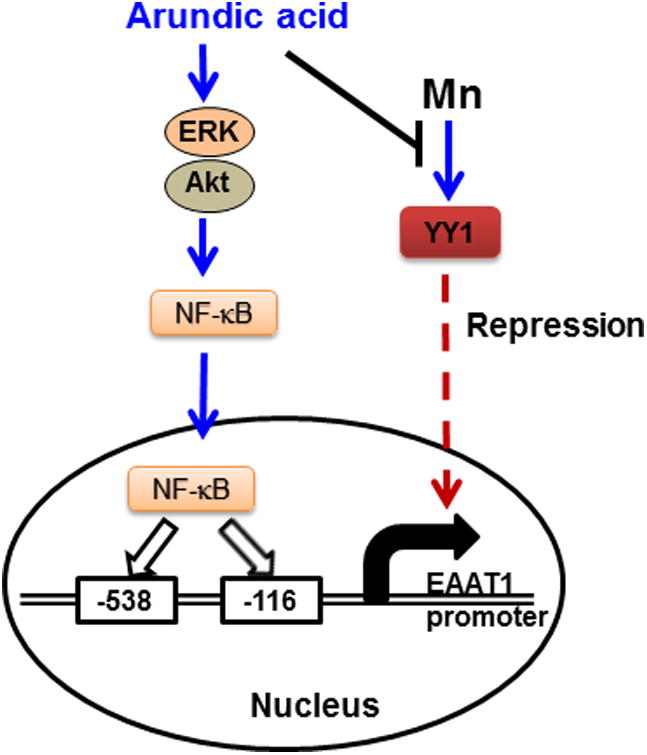
Proposed mechanism of arundic acid-induced EAAT1 expression. Arundic acid activates intracellular signaling proteins, ERK and Akt, as well as the NF-κB pathway, leading to nuclear translocation of NF-κB. In the nucleus, NF-κB binds to both consensus sites (-116 and -538) on the EAAT1 promoter, leading to the increased expression of EAAT1. Arundic acid also blocks Mn-induced repression of EAAT1 by inhibiting Mn-induced YY1 expression.
Supplementary Material
Acknowledgments
the present study has been supported in part by R01 ES024756 (EL), SC1 GM089630 (EL), SC1 CA200519 (DS), R01 ES10563 (MA), 1R03 ES024849 (MA) and 1R21 ES025415 (MA).
References
- 1.Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65(1):1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- 2.Platt SR. The role of glutamate in central nervous system health and disease--a review. Vet J. 2007;173(2):278–286. doi: 10.1016/j.tvjl.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 3.Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32(1):1–14. [PubMed] [Google Scholar]
- 4.Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16(3):675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 5.Maragakis NJ, Rothstein JD. Glutamate transporters: animal models to neurologic disease. Neurobiol Dis. 2004;15(3):461–473. doi: 10.1016/j.nbd.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 6.Shigeri Y, Seal RP, Shimamoto K. Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Res Brain Res Rev. 2004;45(3):250–265. doi: 10.1016/j.brainresrev.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 7.Bar-Peled O, Ben-Hur H, Biegon A, Groner Y, Dewhurst S, Furuta A, Rothstein JD. Distribution of glutamate transporter subtypes during human brain development. J Neurochem. 1997;69(6):2571–2580. doi: 10.1046/j.1471-4159.1997.69062571.x. [DOI] [PubMed] [Google Scholar]
- 8.Furuta A, Rothstein JD, Martin LJ. Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci. 1997;17(21):8363–8375. doi: 10.1523/JNEUROSCI.17-21-08363.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 1995;15(3 Pt 1):1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rauen T, Taylor WR, Kuhlbrodt K, Wiessner M. High-affinity glutamate transporters in the rat retina: a major role of the glial glutamate transporter GLAST-1 in transmitter clearance. Cell Tissue Res. 1998;291(1):19–31. doi: 10.1007/s004410050976. [DOI] [PubMed] [Google Scholar]
- 11.Harada T, Harada C, Watanabe M, Inoue Y, Sakagawa T, Nakayama N, Sasaki S, Okuyama S, Watase K, Wada K, Tanaka K. Functions of the two glutamate transporters GLAST and GLT-1 in the retina. Proc Natl Acad Sci U S A. 1998;95(8):4663–4666. doi: 10.1073/pnas.95.8.4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, Hori S, Takimoto M, Wada K, Tanaka K. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci. 1998;10(3):976–988. doi: 10.1046/j.1460-9568.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- 13.Harada T, Harada C, Nakamura K, Quah HM, Okumura A, Namekata K, Saeki T, Aihara M, Yoshida H, Mitani A, Tanaka K. The potential role of glutamate transporters in the pathogenesis of normal tension glaucoma. J Clin Invest. 2007;117(7):1763–1770. doi: 10.1172/JCI30178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen KH, Reese EA, Kim HW, Rapoport SI, Rao JS. Disturbed neurotransmitter transporter expression in Alzheimer's disease brain. J Alzheimers Dis. 2011;26(4):755–766. doi: 10.3233/JAD-2011-110002. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Liang Z, Valla J, Sefidvash-Hockley S, Rogers J, Li R. Effects of estrogen treatment on glutamate uptake in cultured human astrocytes derived from cortex of Alzheimer's disease patients. J Neurochem. 2002;80(5):807–814. doi: 10.1046/j.0022-3042.2002.00779.x. [DOI] [PubMed] [Google Scholar]
- 16.Masliah E, Alford M, DeTeresa R, Mallory M, Hansen L. Deficient glutamate transport is associated with neurodegeneration in Alzheimer's disease. Ann Neurol. 1996;40(5):759–766. doi: 10.1002/ana.410400512. [DOI] [PubMed] [Google Scholar]
- 17.Jen JC, Wan J, Palos TP, Howard BD, Baloh RW. Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures. Neurology. 2005;65(4):529–534. doi: 10.1212/01.wnl.0000172638.58172.5a. [DOI] [PubMed] [Google Scholar]
- 18.de Vries B, Mamsa H, Stam AH, Wan J, Bakker SL, Vanmolkot KR, Haan J, Terwindt GM, Boon EM, Howard BD, Frants RR, Baloh RW, Ferrari MD, Jen JC, van den Maagdenberg AM. Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake. Arch Neurol. 2009;66(1):97–101. doi: 10.1001/archneurol.2008.535. [DOI] [PubMed] [Google Scholar]
- 19.van Landeghem FK, Weiss T, Oehmichen M, von Deimling A. Decreased expression of glutamate transporters in astrocytes after human traumatic brain injury. J Neurotrauma. 2006;23(10):1518–1528. doi: 10.1089/neu.2006.23.1518. [DOI] [PubMed] [Google Scholar]
- 20.Naskar R, Vorwerk CK, Dreyer EB. Concurrent downregulation of a glutamate transporter and receptor in glaucoma. Invest Ophthalmol Vis Sci. 2000;41(7):1940–1944. [PubMed] [Google Scholar]
- 21.Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochemistry international. 2007;51(6-7):333–355. doi: 10.1016/j.neuint.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yanagisawa M, Aida T, Takeda T, Namekata K, Harada T, Shinagawa R, Tanaka K. Arundic acid attenuates retinal ganglion cell death by increasing glutamate/aspartate transporter expression in a model of normal tension glaucoma. Cell Death Dis. 2015;6:e1693. doi: 10.1038/cddis.2015.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asano T, Mori T, Shimoda T, Shinagawa R, Satoh S, Yada N, Katsumata S, Matsuda S, Kagamiishi Y, Tateishi N. Arundic acid (ONO-2506) ameliorates delayed ischemic brain damage by preventing astrocytic overproduction of S100B. Curr Drug Targets CNS Neurol Disord. 2005;4(2):127–142. doi: 10.2174/1568007053544084. [DOI] [PubMed] [Google Scholar]
- 24.Rothermundt M, Peters M, Prehn JH, Arolt V. S100B in brain damage and neurodegeneration. Microsc Res Tech. 2003;60(6):614–632. doi: 10.1002/jemt.10303. [DOI] [PubMed] [Google Scholar]
- 25.Matsui T, Mori T, Tateishi N, Kagamiishi Y, Satoh S, Katsube N, Morikawa E, Morimoto T, Ikuta F, Asano T. Astrocytic activation and delayed infarct expansion after permanent focal ischemia in rats. Part I: enhanced astrocytic synthesis of s-100beta in the periinfarct area precedes delayed infarct expansion. J Cereb Blood Flow Metab. 2002;22(6):711–722. doi: 10.1097/00004647-200206000-00010. [DOI] [PubMed] [Google Scholar]
- 26.Tateishi N, Mori T, Kagamiishi Y, Satoh S, Katsube N, Morikawa E, Morimoto T, Matsui T, Asano T. Astrocytic activation and delayed infarct expansion after permanent focal ischemia in rats. Part II: suppression of astrocytic activation by a novel agent (R)-(-)-2-propyloctanoic acid (ONO-2506) leads to mitigation of delayed infarct expansion and early improvement of neurologic deficits. J Cereb Blood Flow Metab. 2002;22(6):723–734. doi: 10.1097/00004647-200206000-00011. [DOI] [PubMed] [Google Scholar]
- 27.Kato H, Araki T, Imai Y, Takahashi A, Itoyama Y. Protection of dopaminergic neurons with a novel astrocyte modulating agent (R)-(-)-2-propyloctanoic acid (ONO-2506) in an MPTP-mouse model of Parkinson's disease. J Neurol Sci. 2003;208(1-2):9–15. doi: 10.1016/s0022-510x(02)00411-2. [DOI] [PubMed] [Google Scholar]
- 28.Kato H, Kurosaki R, Oki C, Araki T. Arundic acid, an astrocyte-modulating agent, protects dopaminergic neurons against MPTP neurotoxicity in mice. Brain Res. 2004;1030(1):66–73. doi: 10.1016/j.brainres.2004.09.046. [DOI] [PubMed] [Google Scholar]
- 29.Himeda T, Kadoguchi N, Kamiyama Y, Kato H, Maegawa H, Araki T. Neuroprotective effect of arundic acid, an astrocyte-modulating agent, in mouse brain against MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) neurotoxicity. Neuropharmacology. 2006;50(3):329–344. doi: 10.1016/j.neuropharm.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 30.Oki C, Watanabe Y, Yokoyama H, Shimoda T, Kato H, Araki T. Delayed treatment with arundic acid reduces the MPTP-induced neurotoxicity in mice. Cell Mol Neurobiol. 2008;28(3):417–430. doi: 10.1007/s10571-007-9241-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mori T, Tateishi N, Kagamiishi Y, Shimoda T, Satoh S, Ono S, Katsube N, Asano T. Attenuation of a delayed increase in the extracellular glutamate level in the peri-infarct area following focal cerebral ischemia by a novel agent ONO-2506. Neurochemistry international. 2004;45(2-3):381–387. doi: 10.1016/j.neuint.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 32.Carbone M, Duty S, Rattray M. Riluzole elevates GLT-1 activity and levels in striatal astrocytes. Neurochemistry international. 2012;60(1):31–38. doi: 10.1016/j.neuint.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433(7021):73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- 34.Gegelashvili G, Danbolt NC, Schousboe A. Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia. J Neurochem. 1997;69(6):2612–2615. doi: 10.1046/j.1471-4159.1997.69062612.x. [DOI] [PubMed] [Google Scholar]
- 35.Swanson RA, Liu J, Miller JW, Rothstein JD, Farrell K, Stein BA, Longuemare MC. Neuronal regulation of glutamate transporter subtype expression in astrocytes. J Neurosci. 1997;17(3):932–940. doi: 10.1523/JNEUROSCI.17-03-00932.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonde C, Sarup A, Schousboe A, Gegelashvili G, Noraberg J, Zimmer J. GDNF pre-treatment aggravates neuronal cell loss in oxygen-glucose deprived hippocampal slice cultures: a possible effect of glutamate transporter up-regulation. Neurochemistry international. 2003;43(4-5):381–388. doi: 10.1016/s0197-0186(03)00025-1. [DOI] [PubMed] [Google Scholar]
- 37.Figiel M, Maucher T, Rozyczka J, Bayatti N, Engele J. Regulation of glial glutamate transporter expression by growth factors. Exp Neurol. 2003;183(1):124–135. doi: 10.1016/s0014-4886(03)00134-1. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki K, Ikegaya Y, Matsuura S, Kanai Y, Endou H, Matsuki N. Transient upregulation of the glial glutamate transporter GLAST in response to fibroblast growth factor, insulin-like growth factor and epidermal growth factor in cultured astrocytes. Journal of cell science. 2001;114(Pt 20):3717–3725. doi: 10.1242/jcs.114.20.3717. [DOI] [PubMed] [Google Scholar]
- 39.Karki P, Webb A, Zerguine A, Choi J, Son DS, Lee E. Mechanism of raloxifene-induced upregulation of glutamate transporters in rat primary astrocytes. Glia. 2014;62(8):1270–1283. doi: 10.1002/glia.22679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee ES, Sidoryk M, Jiang H, Yin Z, Aschner M. Estrogen and tamoxifen reverse manganese-induced glutamate transporter impairment in astrocytes. J Neurochem. 2009;110(2):530–544. doi: 10.1111/j.1471-4159.2009.06105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pawlak J, Brito V, Kuppers E, Beyer C. Regulation of glutamate transporter GLAST and GLT-1 expression in astrocytes by estrogen. Brain Res Mol Brain Res. 2005;138(1):1–7. doi: 10.1016/j.molbrainres.2004.10.043. [DOI] [PubMed] [Google Scholar]
- 42.Karki P, Kim C, Smith K, Son DS, Aschner M, Lee E. Transcriptional Regulation of the Astrocytic Excitatory Amino Acid Transporter 1 (EAAT1) via NF-kappaB and Yin Yang 1 (YY1) J Biol Chem. 2015;290(39):23725–23737. doi: 10.1074/jbc.M115.649327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim SY, Choi SY, Chao W, Volsky DJ. Transcriptional regulation of human excitatory amino acid transporter 1 (EAAT1): cloning of the EAAT1 promoter and characterization of its basal and inducible activity in human astrocytes. J Neurochem. 2003;87(6):1485–1498. doi: 10.1046/j.1471-4159.2003.02128.x. [DOI] [PubMed] [Google Scholar]
- 44.Rozyczka J, Figiel M, Engele J. Endothelins negatively regulate glial glutamate transporter expression. Brain Pathol. 2004;14(4):406–414. doi: 10.1111/j.1750-3639.2004.tb00084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Castro-Coronel Y, Del Razo LM, Huerta M, Hernandez-Lopez A, Ortega A, Lopez-Bayghen E. Arsenite exposure downregulates EAAT1/GLAST transporter expression in glial cells. Toxicological sciences : an official journal of the Society of Toxicology. 2011;122(2):539–550. doi: 10.1093/toxsci/kfr126. [DOI] [PubMed] [Google Scholar]
- 46.Dobson AW, Erikson KM, Aschner M. Manganese neurotoxicity. Ann N Y Acad Sci. 2004;1012:115–128. doi: 10.1196/annals.1306.009. [DOI] [PubMed] [Google Scholar]
- 47.Aguirre G, Rosas S, Lopez-Bayghen E, Ortega A. Valproate-dependent transcriptional regulation of GLAST/EAAT1 expression: involvement of Ying-Yang 1. Neurochemistry international. 2008;52(7):1322–1331. doi: 10.1016/j.neuint.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 48.Rosas S, Vargas MA, Lopez-Bayghen E, Ortega A. Glutamate-dependent transcriptional regulation of GLAST/EAAT1: a role for YY1. J Neurochem. 2007;101(4):1134–1144. doi: 10.1111/j.1471-4159.2007.04517.x. [DOI] [PubMed] [Google Scholar]
- 49.Allritz C, Bette S, Figiel M, Engele J. Endothelin-1 reverses the histone deacetylase inhibitor-induced increase in glial glutamate transporter transcription without affecting histone acetylation levels. Neurochemistry international. 2009;55(1-3):22–27. doi: 10.1016/j.neuint.2008.12.020. [DOI] [PubMed] [Google Scholar]
- 50.Baltan S, Murphy SP, Danilov CA, Bachleda A, Morrison RS. Histone deacetylase inhibitors preserve white matter structure and function during ischemia by conserving ATP and reducing excitotoxicity. J Neurosci. 2011;31(11):3990–3999. doi: 10.1523/JNEUROSCI.5379-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kanai H, Sawa A, Chen RW, Leeds P, Chuang DM. Valproic acid inhibits histone deacetylase activity and suppresses excitotoxicity-induced GAPDH nuclear accumulation and apoptotic death in neurons. Pharmacogenomics J. 2004;4(5):336–344. doi: 10.1038/sj.tpj.6500269. [DOI] [PubMed] [Google Scholar]
- 52.Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci. 1994;14(9):5559–5569. doi: 10.1523/JNEUROSCI.14-09-05559.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1(4):a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brouillet EP, Shinobu L, McGarvey U, Hochberg F, Beal MF. Manganese injection into the rat striatum produces excitotoxic lesions by impairing energy metabolism. Exp Neurol. 1993;120(1):89–94. doi: 10.1006/exnr.1993.1042. [DOI] [PubMed] [Google Scholar]
- 55.Regan MR, Huang YH, Kim YS, Dykes-Hoberg MI, Jin L, Watkins AM, Bergles DE, Rothstein JD. Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the developing and mature CNS. J Neurosci. 2007;27(25):6607–6619. doi: 10.1523/JNEUROSCI.0790-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ghosh M, Yang Y, Rothstein JD, Robinson MB. Nuclear factor-kappaB contributes to neuron-dependent induction of glutamate transporter-1 expression in astrocytes. J Neurosci. 2011;31(25):9159–9169. doi: 10.1523/JNEUROSCI.0302-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Karki P, Webb A, Smith K, Lee K, Son DS, Aschner M, Lee E. cAMP response element-binding protein (CREB) and nuclear factor kappaB mediate the tamoxifen-induced up-regulation of glutamate transporter 1 (GLT-1) in rat astrocytes. J Biol Chem. 2013;288(40):28975–28986. doi: 10.1074/jbc.M113.483826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee SG, Su ZZ, Emdad L, Gupta P, Sarkar D, Borjabad A, Volsky DJ, Fisher PB. Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J Biol Chem. 2008;283(19):13116–13123. doi: 10.1074/jbc.M707697200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fernandes RA, Ingle AB. Arundic acid a potential neuroprotective agent: biological development and syntheses. Curr Med Chem. 2013;20(18):2315–2329. doi: 10.2174/0929867311320180003. [DOI] [PubMed] [Google Scholar]
- 60.Sun Y, Duan Y, Eisenstein AS, Hu W, Quintana A, Lam WK, Wang Y, Wu Z, Ravid K, Huang P. A novel mechanism of control of NFkappaB activation and inflammation involving A2B adenosine receptors. Journal of cell science. 2012;125(Pt 19):4507–4517. doi: 10.1242/jcs.105023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jarvis WD, Fornari FA, Jr, Auer KL, Freemerman AJ, Szabo E, Birrer MJ, Johnson CR, Barbour SE, Dent P, Grant S. Coordinate regulation of stress- and mitogen-activated protein kinases in the apoptotic actions of ceramide and sphingosine. Mol Pharmacol. 1997;52(6):935–947. doi: 10.1124/mol.52.6.935. [DOI] [PubMed] [Google Scholar]
- 62.Toulany M, Minjgee M, Saki M, Holler M, Meier F, Eicheler W, Rodemann HP. ERK2-dependent reactivation of Akt mediates the limited response of tumor cells with constitutive K-RAS activity to PI3K inhibition. Cancer Biol Ther. 2014;15(3):317–328. doi: 10.4161/cbt.27311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Winter JN, Jefferson LS, Kimball SR. ERK and Akt signaling pathways function through parallel mechanisms to promote mTORC1 signaling. Am J Physiol Cell Physiol. 2011;300(5):C1172–1180. doi: 10.1152/ajpcell.00504.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stice JP, Mbai FN, Chen L, Knowlton AA. Rapid activation of nuclear factor kappaB by 17beta-estradiol and selective estrogen receptor modulators: pathways mediating cellular protection. Shock. 2012;38(2):128–136. doi: 10.1097/SHK.0b013e31825da754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aschner JL, Aschner M. Nutritional aspects of manganese homeostasis. Mol Aspects Med. 2005;26(4-5):353–362. doi: 10.1016/j.mam.2005.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guilarte TR. Manganese and Parkinson's disease: a critical review and new findings. Environ Health Perspect. 2010;118(8):1071–1080. doi: 10.1289/ehp.0901748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hazell AS, Norenberg MD. Manganese decreases glutamate uptake in cultured astrocytes. Neurochem Res. 1997;22(12):1443–1447. doi: 10.1023/a:1021994126329. [DOI] [PubMed] [Google Scholar]
- 68.Mutkus L, Aschner JL, Fitsanakis V, Aschner M. The in vitro uptake of glutamate in GLAST and GLT-1 transfected mutant CHO-K1 cells is inhibited by manganese. Biol Trace Elem Res. 2005;107(3):221–230. doi: 10.1385/BTER:107:3:221. [DOI] [PubMed] [Google Scholar]
- 69.Erikson KM, Dorman DC, Lash LH, Aschner M. Manganese inhalation by rhesus monkeys is associated with brain regional changes in biomarkers of neurotoxicity. Toxicological sciences : an official journal of the Society of Toxicology. 2007;97(2):459–466. doi: 10.1093/toxsci/kfm044. [DOI] [PubMed] [Google Scholar]
- 70.Karki P, Webb A, Smith K, Johnson J, Jr, Lee K, Son DS, Aschner M, Lee E. Yin Yang 1 is a repressor of glutamate transporter EAAT2, and it mediates manganese-induced decrease of EAAT2 expression in astrocytes. Mol Cell Biol. 2014;34(7):1280–1289. doi: 10.1128/MCB.01176-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Duan S, Anderson CM, Stein BA, Swanson RA. Glutamate induces rapid upregulation of astrocyte glutamate transport and cell-surface expression of GLAST. J Neurosci. 1999;19(23):10193–10200. doi: 10.1523/JNEUROSCI.19-23-10193.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sims KD, Straff DJ, Robinson MB. Platelet-derived growth factor rapidly increases activity and cell surface expression of the EAAC1 subtype of glutamate transporter through activation of phosphatidylinositol 3-kinase. J Biol Chem. 2000;275(7):5228–5237. doi: 10.1074/jbc.275.7.5228. [DOI] [PubMed] [Google Scholar]
- 73.Takahashi K, Foster JB, Lin CL. Glutamate transporter EAAT2: regulation, function, and potential as a therapeutic target for neurological and psychiatric disease. Cell Mol Life Sci. 2015;72(18):3489–3506. doi: 10.1007/s00018-015-1937-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.