

 In this paper, we report an efficient strategy for the synthesis of Cu/Co double-doped CeO2 nanospheres (CuxCo1–x–CeO2–Pt, 0 ≤ x ≤ 1), which were fabricated via a simple water–glycol system.
In this paper, we report an efficient strategy for the synthesis of Cu/Co double-doped CeO2 nanospheres (CuxCo1–x–CeO2–Pt, 0 ≤ x ≤ 1), which were fabricated via a simple water–glycol system.
Abstract
In this paper, we report an efficient strategy for the synthesis of Cu/Co double-doped CeO2 nanospheres (CuxCo1–x–CeO2–Pt, 0 ≤ x ≤ 1), which were fabricated via a simple water–glycol system. The following in situ surface decoration of Pt nanoparticles make these nanospheres highly active for the catalytic reduction of nitrophenol and CO oxidation. Detailed tests show that their catalytic performance strongly depends on the doping components and ion concentration of Cu and Co ions. The best samples of Cu0.50Co0.50–CeO2–Pt and Cu0.34Co0.66–CeO2–Pt demonstrate an excellent turnover frequency (TOF) of more than 450 h–1 after five cycles and retains about 99% conversion by using NH3BH3 as a reductant to reduce nitrophenol. Moreover, Cu0.50Co0.50–CeO2–Pt possesses a much lower light-off and T100 (the temperature for 100% CO oxidation) temperature compared with the other catalysts.
Introductions
Among the metal oxides, CeO2 has attracted great attention due to its excellent physicochemical properties including its good optical properties, mechanical strength, oxygen ion conductivity, and high thermal stability. CeO2 has a fluorite-like cubic structure with each Ce4+ ion surrounded by eight O2– ions in a face-centered cubic (fcc) arrangement, whereas each O2– ion is tetrahedrally surrounded by four Ce4+ ions. Intrinsic oxygen vacancy defects can be rapidly formed and eliminated in the lattice of CeO2, which favors mediation of lattice expansion and strain, and hence contributes significantly towards stable grain boundary structures. Hence, CeO2 has been successfully employed in various applications such as in energy and magnetic data storage,1 photocatalytic applications,2–4 as sensors for CO,5–9 H2O2,10 NH3,11 and nitrophenol,12,13 as a UV blocker,14 in solar fuel synthesis,15 water oxidation,16,17 oxygen transfer,18 fuel cells,19,20 gates for metal-oxide semiconductor devices,21 as a promoter in three-way catalysts for the elimination of polluted auto-exhaust gases from vehicles,22–24 and so on.
The larger the amount of oxygen vacancies that CeO2 possesses, the more efficient it will be for storing oxygen. Thus, it seems meaningful to control the generation of oxygen vacancies to improve its physicochemical properties. Endeavours have been devoted to introduce dopants to improve the catalytic performance of CeO2. It has been reported that its catalytic activity can be considerably enhanced by tuning the surface and interface structure through doping with isovalent/aliovalent cations into the CeO2 lattice.25–30 The isovalent cations that are frequently used are Ti4+, Zr4+, Hf4+, and Sn4+, while aliovalent cations used include Mn2+, Ni2+, Zn2+, Ca2+, Mn3+, Sc3+, Y3+, Gd3+, Sm3+, Eu3+, La3+, etc. The substitution of isovalent dopants into the CeO2 lattice decreases the oxygen vacancy formation energy due to structural distortion, whereas in the case of aliovalent dopants, the decrease in the defect formation energy is due to structural distortion as well as electronic modification, resulting in the generation of extra oxygen vacancies.31,32 So once noble metals or metal oxides form hybrids with CeO2, they often exhibit greatly enhanced catalytic activity, stability and selectivity.
Noble metal nanoparticles have been extensively studied for decades due to their high performance in many kinds of catalytic reactions. Smaller sized noble metal nanoparticles often have a larger fraction of exposed atoms on the particle surface, demonstrating better catalytic activity. Moreover, the strong synergistic effect between noble metals and the support greatly favors improvement of the catalytic performance,33–38 especially for Pt–CeO2 systems.39–44 A recent report by Chowdhury et al., reveals that doping can influence the surface acid–base properties of mesoporous CeO2 and its catalytic behavior.43 It is highly expected that incorporation of the cations into a ceria lattice structure will influence the redox properties of ceria in favour of synergistic interactions with noble metal nanoparticles towards enhanced activity for catalytic nitrophenol reduction and CO oxidation.
In this paper, an efficient strategy was developed for the synthesis of CuxCo1–x–CeO2–Pt hybrids. First, double-doped CeO2 nanospheres were fabricated in a water–glycol mixed system, followed by a self-assembly process to in situ deposit Pt nanoparticles on the surface of the as-obtained double-doped CeO2 nanospheres to form the final hybrids. Then, the as-obtained catalysts with different doping components were studied in detail to find the optimal doping ratios with the best catalytic performance for the reduction of 4-NP (4-nitrophenol) by NH3BH3 and the oxidation of CO. Furthermore, a detailed discussion has been made to clarify the role of the doping elements in the reduction of 4-NP and oxidation of CO, according to the analytical results.
Experimental section
Synthesis of CeO2 nanospheres:
The synthesis of CeO2 nanospheres was carried out by a previously reported method.45 500 mg of Ce(NO3)3·6H2O and 200 mg of PVP were dissolved in 14 mL of ethylene glycol, and then 1 mL of deionized water was added to the above solution. After continuous stirring for 30 min, the clear solution was transferred into a Teflon-lined autoclave of 20 mL capacity and heated for 8 h at 160 °C. When the autoclave was cooled at room temperature, the products were collected and washed with deionized water and absolute alcohol several times. Finally, the products were dried at 60 °C overnight, and then calcined at 300 °C for 1 h at 1 °C min–1.
Synthesis of CuxCo1–x–CeO2 nanospheres
500 mg of Ce(NO3)3·6H2O and 200 mg of PVP were dissolved in 14 mL of ethylene glycol, and then 0.66 mL of 20 mg mL–1 CuCl2·2H2O and 0.34 mL of a 20 mg mL–1 CoCl2·2H2O solution were added to the above solution. The mixed solutions were transferred into a Teflon-lined autoclave of 20 mL capacity and heated for 8 h at 160 °C. When the autoclave was cooled to room temperature, the products were collected and washed with deionized water and absolute alcohol several times. Finally, the products were dried at 60 °C overnight, and then calcined at 300 °C at 1 °C min–1 for 1 h. The above products were labeled as Cu0.66Co0.34–CeO2. Cu–CeO2, Cu0.50Co0.50–CeO2, Cu0.34Co0.66–CeO2 and Co–CeO2 were prepared in a similar process, except for changing the CuCl2·2H2O/CoCl2·2H2O (v/v) to 1 mL CuCl2·2H2O and 0 mL CoCl2·2H2O, 0.5 mL CuCl2·2H2O and 0.5 mL CoCl2·2H2O, 0.34 mL CuCl2·2H2O and 0.66 mL CoCl2·2H2O, and 0 mL CuCl2·2H2O and 1 mL CoCl2·2H2O, respectively.
Synthesis of CuxCo1–x–CeO2–Pt hybrids
70 mg of CuxCo1–x–CeO2 nanospheres and 42 mg of PVP were first dissolved in 60 mL of ethylene glycol. After that, 0.84 mL of 0.02 M K2PtCl4 aqueous solution was added to the above solution. Then, the mixture was heated to 110 °C and was maintained at this temperature for 2 h. The product was collected by centrifugation and washed with deionized water several times and dried in an oven.
Characterization
The X-ray diffraction patterns of the products were collected on a Rigaku-D/max 2500 V X-ray diffractometer with CuKα radiation (λ = 1.5418 Å), with an operation voltage and current maintained at 40 kV and 40 mA. Transmission electron microscopic (TEM) images were obtained with a TECNAI G2 high-resolution transmission electron microscope operating at 200 kV. Inductively coupled plasma (ICP) analyses were performed with a Varian Liberty 200 spectrophotometer to determine the contents. X-ray photoelectron spectroscopy (XPS) measurements were taken on an ESCALAB-MKII 250 photoelectron spectrometer (VG Co.) with AlKα X-ray radiation as the X-ray source for excitation. Decreases in the concentration of 4-NP were analyzed by UV-vis-NIR (SHIMADZU, UV-3600) spectrophotometer. The catalytic performances of the catalysts for CO oxidation were monitored on-line by gas chromatography (GC9800).
Catalytic tests
Chemical reduction of nitrophenol by NH3BH3: aqueous solutions of 4-NP (0.01 M) and NH3BH3 (0.1 M) were freshly prepared. 20 μL of the 4-NP solution and 100 μL of the NH3BH3 solution were added to a quartz cuvette containing 2 mL of water. Then, 20 μL of 5 mg mL–1 catalysts were injected into the cuvette to start the reaction. Since the spectrophotometer has a function to display the instant absorbance of a fixed absorption peak such as 400 nm, we can easily monitor the intensity of the absorption peak at 400 nm as a function of time. After each round of reaction, another 20 μL of 4-NP solution and 100 μL NH3BH3 aqueous solution were added to the reaction solution. This step was repeated 10 times to study the stability of the catalysts. The reduction of 4-NP by NH3BH3 can be briefly expressed as follows: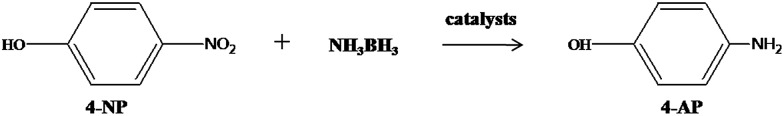
CO oxidation
20 mg of catalysts were put in a stainless steel reaction tube. The CO oxidation tests were performed under conditions in 1% CO and 20% O2 in N2 at a total flow rate of 30 mL min–1, and a space velocity (SV) of 90 000 mL h–1 gcat–1. The composition of the gas was monitored on-line by gas chromatography.
Results and discussion
The synthesis of the CuxCo1–x–CeO2–Pt hybrid nanocatalysts involved two steps. Uniform CuxCo1–x–CeO2 nanospheres were first acquired by tuning the doping concentration of Co2+ and Cu2+, and then they served as a support for the in situ deposition of Pt nanoparticles on their surface. Fig. 1 shows typical SEM images of the as-obtained CuxCo1–x–CeO2 samples, in which some detailed morphological and structural features can be found. All these samples are sphere-like with no obvious fragments. Their size distributions are shown in Fig. 1 (inset) and are listed in Table S1.† Compared with the pure CeO2 nanospheres of 174 nm, the Cu-rich ones are much smaller and are even less than 100 nm, while those Co-rich samples show a bigger size around 200 nm. Fig. 2 shows the SEM images of the as-obtained samples after the addition of K2PtCl4 aqueous solution and after being refluxed at 110 °C for 2 h. However, it is hard for us to distinguish the differences of the surface from the CuxCo1–x–CeO2 nanospheres, and no Pt particles can be found, indicating that the size of the deposited Pt nanoparticles should be very small in the CuxCo1–x–CeO2–Pt hybrids. The sizes of the as-obtained CuxCo1–x–CeO2–Pt hybrids are shown in Table S2.†
Fig. 1. SEM images of (A) Cu–CeO2; (B) Cu0.66Co0.34–CeO2; (C) Cu0.50Co0.50–CeO2; (D) Cu0.34Co0.66–CeO2; (E) Co–CeO2; (F) CeO2.

Fig. 2. SEM images of (A) Cu–CeO2–Pt; (B) Cu0.66Co0.34–CeO2–Pt; (C) Cu0.50Co0.50–CeO2–Pt; (D) Cu0.34Co0.66–CeO2–Pt; (E) Co–CeO2–Pt; (F) CeO2–Pt.

TEM characterization can tell us more information about the final CuxCo1–x–CeO2–Pt hybrids. As shown in Fig. 3, these CuxCo1–x–CeO2–Pt hybrids maintained their initial morphologies and inner nanostructures well. With a decrease in the amount of Cu2+ doping, the double-doped CeO2 nanospheres obviously underwent a morphology transformation from hollow to core–shell, and then to be solid. The high-resolution TEM image in Fig. 3 shows that each CuxCo1–x–CeO2–Pt nanosphere is decorated by hundreds of ultra-small Pt nanoparticles (less than 2 nm) and the CuxCo1–x–CeO2 nanospheres are composed of closely packed CeO2 nanoparticles with diameters of about 5 nm as primary building blocks. The clearly observed lattice spacing listed in Table S3† agrees well with that of Pt (200) (0.196 nm) and CeO2 (111) (0.312 nm). The high angle annular dark field scanning transmission electron microscopy (HAADF-STEM) images of Cu0.66Co0.34–CeO2–Pt demonstrate the evenly distributed Cu, Pt and Co elements, as shown in Fig. 4.
Fig. 3. TEM images of (A) and (B) Cu–CeO2–Pt; (C) and (D) Cu0.66Co0.34–CeO2–Pt; (E) and (F) Cu0.50Co0.50–CeO2–Pt; (G) and (H) Cu0.34Co0.66–CeO2–Pt; (I) and (J) Co–CeO2–Pt; (K) and (L) CeO2–Pt.

Fig. 4. HAADF-STEM images of Cu0.66Co0.34–CeO2–Pt nanospheres.

In the X-ray diffraction (XRD) patterns of the CuxCo1–x–CeO2–Pt and CeO2–Pt nanospheres (Fig. 5), the diffraction peaks of all the products can be indexed to a pure phase of fcc CeO2 structures (JCPDS no. 34-0394). The peaks at 2θ = 28.549°, 33.077°, 47.483°, 56.342°, 59.09°, 69.416°, 76.704° and 79.077° correspond to the characteristic (111), (200), (220), (311), (222), (400), (331) and (420) reflections, respectively. No signals related to impurities, such as cobalt oxide or copper oxide, can be found for all samples, indicating the homogeneous doping of Cu and Co ions in the CeO2 solid solutions. The contents of the elements Cu, Co, Ce and Pt for the CuxCo1–x–CeO2–Pt nanospheres were then determined by ICP-MS analysis and are listed in Table S4.† It can be seen that the loading amounts of Pt (mol%) are less than 1.4% if Cu2+ is introduced, however, in the absence of Cu2+, this percentage increased to 4.1% for Co–CeO2–Pt and 4.8% for CeO2–Pt, which were much higher than the others. On the contrary, Co2+ doping has little effect on the deposition amount of Pt.
Fig. 5. XRD patterns of (A) Cu–CeO2–Pt; (B) Cu0.66Co0.34–CeO2–Pt; (C) Cu0.50Co0.50–CeO2–Pt; (D) Cu0.34Co0.66–CeO2–Pt; (E) Co–CeO2–Pt; (F) CeO2–Pt.

XPS analysis was employed to determine the surface elements and their valence states of the CuxCo1–x–CeO2–Pt and CeO2–Pt nanospheres (Fig. 6). All of the samples show characteristic peaks at the binding energies of 71.3 eV (Pt 4f7/2) and 74.7 eV (Pt 4f5/2) of the element Pt, and the two peaks at 882.8 and 899.5 eV correspond well to the Ce 3d5/2 and Ce 3d3/2 spin–orbit peaks of CeO2. As previously reported, the XPS spectrum of Co 2p shows two major peaks at 795.5 and 780.4 eV, corresponding to Co 2p1/2 and Co 2p3/2 spin–orbit coupling, respectively.46 Two major peaks lying at 932.2 and 954.2 eV are characteristic signals of Cu2+ with Cu 2p3/2 and Cu 2p1/2 orbits, respectively.46 Unfortunately, due to the low doping content of Co and Cu in CuxCo1–x–CeO2–Pt, no Co and Cu signals can be detected in all the samples. However, in combination with the HAADF-STEM characterization and ICP results, the successful formation of CuxCo1–x–CeO2–Pt hybrids can be confirmed.
Fig. 6. (A) XPS spectra of (a) Cu–CeO2–Pt, (b) Cu0.66Co0.34–CeO2–Pt, (c) Cu0.50Co0.50–CeO2–Pt, (d) Cu0.34Co0.66–CeO2–Pt, (e) Co–CeO2–Pt, (f) CeO2–Pt. (B) XPS spectra of (a) Cu–CeO2–Pt; (b) Cu0.66Co0.34–CeO2–Pt; (c) Cu0.50Co0.50–CeO2–Pt; (d) Cu0.34Co0.66–CeO2–Pt; (e) Co–CeO2–Pt.

In the following, the reduction of 4-NP to 4-AP by NH3BH3 was selected to evaluate the catalytic performance of the as-obtained CuxCo1–x–CeO2–Pt and CeO2–Pt samples. As is known, 4-NP exhibits a strong characteristic absorption peak at 317 nm while at pH < 7, but it will be ionized as the alkalinity of the solution increases, resulting in a spectral shift to 400 nm.35 In our case, the absorption peak of 4-NP remained at 317 nm despite adding NH3BH3 solution, indicating that the NH3BH3 molecules were stable enough in water in the absence of the catalysts.34 However, after addition of the CuxCo1–x–CeO2–Pt hybrids, the absorption intensity at 400 nm gradually increased. The color of the reaction system changed from bright yellow to colorless. Since NH3BH3 was in a large excess relative to 4-NP, its concentration could be considered as constant during the reaction period. Thus, the reduction rate can be evaluated by pseudo-first-order kinetics with respect to 4-NP. Fig. 7A shows ln(C/C0) versus reaction time t, which was obtained from the relative intensity ratio of the absorbance (A/A0) at 400 nm. Here, C0 and C represent the initial and instantaneous concentrations of 4-NP, respectively; and k and t stand for the rate constant and the reaction time in turn. As all of these plots followed first-order reaction kinetics very well, the value k can be calculated from the equation ln(C/C0) = kt (Table 1). The catalytic activity of the six samples follows this sequence: Cu0.66Co0.34–CeO2–Pt > Cu0.50Co0.50–CeO2–Pt > Cu–CeO2–Pt > Cu0.34Co0.66–CeO2–Pt > Co–CeO2–Pt > CeO2–Pt, showing a strong dependence on the composition of Cu and Co. Furthermore, the turnover frequency (TOF), defined as moles of the reactant 4-NP converted per mole of active metal in the catalyst per hour was also calculated (Table 1). Though the most stable sample of Cu0.50Co0.50–CeO2–Pt shows a moderate catalytic activity compared to others for catalytic 4-NP conversion, its TOF of 480 h–1 is still at least five times higher than our previously reported Pt@CeO2 catalysts.34
Fig. 7. (A) ln(C/C0) versus t for the reduction of 4-NP catalyzed by (a) Cu–CeO2–Pt; (b) Cu0.66Co0.34–CeO2–Pt; (c) Cu0.50Co0.50–CeO2–Pt; (d) Cu0.34Co0.66–CeO2–Pt; (e) Co–CeO2–Pt; (f) CeO2–Pt. (B) Conversion in successive reaction cycles of CuxCo1–x–CeO2–Pt and CeO2–Pt.

Table 1. Comparison of the rate constant k, TOF and conversion for 4-NP reduction with CuxCo1–x–CeO2–Pt and CeO2–Pt catalysts a .
| Sample | k (s–1) | M Pt (mmol) | M 4-NP (mmol) | t (min) | TOF (h–1) | P (%) | Ref |
| Cu1.00–CeO2–Pt | 0.451 | 3.35 × 10–6 | 2.00 × 10–4 | 3.83 | 935 | 80 | This work |
| Cu0.66Co0.34–CeO2–Pt | 0.995 | 7.24 × 10–6 | 2.00 × 10–4 | 1.88 | 882 | 51.2 | This work |
| Cu0.50Co0.50–CeO2–Pt | 0.633 | 7.22 × 10–6 | 2.00 × 10–4 | 3.46 | 480 | 98.8 | This work |
| Cu0.34Co0.66–CeO2–Pt | 0.614 | 8.24 × 10–6 | 2.00 × 10–4 | 3.17 | 459 | 99.2 | This work |
| Co1.00–CeO2–Pt | 0.555 | 2.05 × 10–5 | 2.00 × 10–4 | 3.66 | 160 | 97.8 | This work |
| CeO2–Pt | 0.285 | 2.09 × 10–5 | 2.00 × 10–4 | 6.15 | 93 | 89.9 | This work |
| Pt@CeO2 | — | 1.00 × 10–4 | 1.00 × 10–3 | — | 46 | — | 34 |
| Pt@CeO2/RGO | — | 1.00 × 10–4 | 1.00 × 10–3 | — | 90 | — | 34 |
a M Pt: mole of noble metals; M4-NP: mole of 4-NP; t: conversion time; P: conversion percentage.
Good reproducibility and stability are also important for the evaluation of catalysts. In order to avoid the loss of the CuxCo1–x–CeO2–Pt catalysts caused by the separation process, cycling tests have been conducted in situ. Fig. 7B shows the conversion in successive reaction cycles of the CuxCo1–x–CeO2–Pt catalysts, and the conversions of 4-NP for the fifth cycle are listed in Table 1. It is found that the stability of the samples varied with the compositions of different doping ions, and that the Co rich samples show better stability among these catalysts.
Besides, catalytic CO oxidation was employed to evaluate these catalysts. In the catalytic process, the gas mixture of CO and O2 was introduced into the inner space of a stainless steel reaction tube filled with CuxCo1–x–CeO2–Pt catalysts. T100, the temperature for 100% CO oxidation, is used to compare the catalytic activity of these samples. Despite of the minor difference in the doped components, these samples show quite different catalytic performance. Fig. 8 presents their CO conversion curves, and the T100 values follow a sequence of: Cu0.50Co0.50–CeO2–Pt (90 °C) < Cu0.66Co0.34–CeO2–Pt (120 °C) < Co–CeO2–Pt (125 °C) < CeO2–Pt (135 °C) < Cu–CeO2–Pt (140 °C) < Cu0.34Co0.66–CeO2–Pt (155 °C). It can be seen that the catalytic performance of our samples improves with the increase in the doping amount of Co until the ratio of Cu2+/Co2+ = 0.5/0.5 is reached, and then this deteriorates quickly upon the doping of more Co2+ ions. This result indicates that Cu2+ and Co2+ ions can replace the tetravalent Ce4+ in the CeO2 fluorite lattice to produce more oxygen vacancies, but an appropriate doping ratio of Cu2+/Co2+ ions is needed in order to realize optimal catalytic performance in our case.
Fig. 8. CO conversion curves of CuxCo1–x–CeO2–Pt and CeO2–Pt.

Conclusions
We have successfully prepared a series of CuxCo1–x–CeO2–Pt hybrid nanospheres. Based on detailed characterization including SEM, TEM, XPS, ICP and XRD, it is found that (I) in the nucleation process, the introduction of Cu2+ accelerated the nucleation rate compared with CeO2, leading to the formation of the smaller sized CeO2. (II) The Cu2+ doping concentration can affect the amount of Pt nanoparticles deposited to a greater extent than Co2+. With the increase in Cu2+ doping concentration, the amount of Pt nanoparticles on the CuxCo1–x–CeO2 nanospheres decreases. However, the related effect of Cu2+ ions was quite small. (III) The high purity of all products indicates the formation of homogeneous Ce–Cu–Co–O, Ce–Cu–O or Ce–Co–O solid solutions.
Based on the catalytic tests, it is also found that the doping amount of Cu2+ greatly influences the catalytic performance of the CuxCo1–x–CeO2 nanospheres. Though the most stable sample of Cu0.50Co0.50–CeO2–Pt shows a moderate catalytic activity compared to others for catalytic 4-NP conversion, its TOF of 480 h–1 is still much higher than our previously reported Pt@CeO2 catalysts. Moreover, among the CuxCo1–x–CeO2–Pt samples, the Cu0.50Co0.50–CeO2–Pt nanospheres exhibited the best catalytic activity, attaining 100% CO conversion at 90 °C, which is also higher than previously reported Pt catalysts. It can be anticipated that this kind of double doped nanocatalysts will have great potential for application.
Supplementary Material
Acknowledgments
This work was financially supported by the fundamental research funds for the central universities, and the National Natural Science Foundation of China (Grant No. 51272249, 51372007 and 21301014). The work is also supported by the China Postdoctoral Science Foundation funded project.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc04069h
References
- Sen S., Edwards T., Kim S. K., Kim S. Chem. Mater. 2014;26:1918–1924. [Google Scholar]
- Zeng M., Li Y. Z., Mao M. Y., Bai J. L., Ren L., Zhao X. J. ACS Catal. 2015;5:3278–3286. [Google Scholar]
- Jiang D., Wang W. Z., Zhang L., Zheng Y. L., Wang Z. ACS Catal. 2015;5:4851–4858. [Google Scholar]
- Lei W. Y., Zhang T. T., Gu L., Liu P., Rodriguez J. A., Liu G., Liu M. H. ACS Catal. 2015;5:4385–4393. [Google Scholar]
- Liu Y. X., Sun X. C., Li B. K., Lei Y. J. Mater. Chem. A. 2014;2:11651–11659. [Google Scholar]
- Qi J., Chen J., Li G. D., Li S. X., Gao Y., Tang Z. Y. Energy Environ. Sci. 2012;5:8937–8941. [Google Scholar]
- Zhen J. M., Wang X., Liu D. P., Song S. Y., Wang Z., Wang Y. H., Li J. Q., Wang F., Zhang H. J. Chem.–Eur. J. 2014;20:1–6. [Google Scholar]
- Wang F., Wang X., Liu D. P., Zhen J. M., Li J. Q., Wang Y. H., Zhang H. J. ACS Appl. Mater. Interfaces. 2014;6:22216–22223. doi: 10.1021/am505853p. [DOI] [PubMed] [Google Scholar]
- Wang X., Liu D. P., Li J. Q., M Zhen J., Zhang H. J. NPG Asia Mater. doi: 10.1038/am.2014.128. [DOI] [Google Scholar]
- Edwards J. K., Pritchard J., Lu L., Piccinini M., Shaw G., Carley A. F., Morgan D. J., Kiely C. J., Hutchings G. J. Angew. Chem., Int. Ed. 2014;53:2381–2384. doi: 10.1002/anie.201308067. [DOI] [PubMed] [Google Scholar]
- Wang L. L., Huang H., Xiao S. H., Cai D. P., Liu Y., Liu B., Wang D. D., Wang C. X., Li H., Wang Y. R., Li Q. H., Wang T. H. ACS. Appl. Mater. Interfaces. 2014;6:14131–14140. doi: 10.1021/am503286h. [DOI] [PubMed] [Google Scholar]
- Xu P. F., Yu R. B., Ren H., Zong L. B., Chen J., Xing X. R. Chem. Sci. 2014;5:4221–4226. [Google Scholar]
- Zhao K., Qi J., Zhao S. L., Tang H. J., Yin H. J., Zong L. B., Chang L., Gao Y., Yu R. B., Tang Z. Y. Chin. J. Catal. 2015;36:261–267. [Google Scholar]
- Anupriya K., Vivek E., Subramanian B. J. Alloys Compd. 2014;590:406–410. [Google Scholar]
- Li P., Zhou Y., Zhao Z. Y., Xu Q. F., Wang X. Y., Xiao M., Zou Z. G. J. Am. Chem. Soc. 2015;137:9547–9550. doi: 10.1021/jacs.5b05926. [DOI] [PubMed] [Google Scholar]
- Zhao K., Qi J., Yin H. J., Wang Z. M., Zhao S. L., Ma X., Wan J. W., Chang L., Gao Y., Yu R. B., Tang Z. Y. J. Mater. Chem. A. 2015;3:20465–20470. [Google Scholar]
- Qi J., Zhao K., Li G. D., Gao Y., Zhao H. J., Yu R. B., Tang Z. Y. Nanoscale. 2014;6:4072–4077. doi: 10.1039/c3nr06822f. [DOI] [PubMed] [Google Scholar]
- Aneggi E., Rico-Perez V., Leitenburg C. D., Maschio S., Soler L., Llorca J., Trovarelli A. Angew. Chem., Int. Ed. 2015;54:1–5. doi: 10.1002/anie.201507839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snana S., Esposito V., Andreasen J. W., Hjelm J., Zhang W., Kasama T., Simonsen S. B., Christensen M., Linderoth S., Pryds N. Nat. Mater. 2015;14:500–504. doi: 10.1038/nmat4266. [DOI] [PubMed] [Google Scholar]
- Badwal S. P. S., Fini D., Ciacchi F. T., Munnings C., Kimptonb J. A., Drennan J. J. Mater. Chem. A. 2013;1:10768–10782. [Google Scholar]
- Rodriguez J. A., Ma S., Liu P., Hrbek J., Evans J., Pérez M. Science. 2007;318:1757–1759. doi: 10.1126/science.1150038. [DOI] [PubMed] [Google Scholar]
- Jiang D., Wang W. Z., Gao E., Sun S. M., Zhang L. Chem. Commun. 2014;50:2005–2007. doi: 10.1039/c3cc47806h. [DOI] [PubMed] [Google Scholar]
- Liang X., Wang X., Zhuang Y., Xu B., Kuang S., Li Y. D. J. Am. Chem. Soc. 2008;130:2736–2737. doi: 10.1021/ja7109629. [DOI] [PubMed] [Google Scholar]
- Mogensen M., Sammes N. M., Tompsett G. A. Solid State Ionics. 2000;129:63–94. [Google Scholar]
- Zhang D., Du X., Shi L., Gao R. Dalton Trans. 2012;41:14455–14475. doi: 10.1039/c2dt31759a. [DOI] [PubMed] [Google Scholar]
- Reddy G. K., Thrimurthulu G., Reddy B. M. Catal. Surv. Asia. 2009;13:237–255. [Google Scholar]
- Durgasri D. N., Vinodkumar T., Sudarsanam P., Reddy B. M. Catal. Lett. 2014;144:971–979. [Google Scholar]
- Zhao Y. X., Nie Z. W., Shi M. M., Zeng C. H., Li Y., Wang L., Zhong S. L. Inorg. Chem. Front. 2015;2:567–575. [Google Scholar]
- Lu B. W., Ju Y. W., Abe T., Kawamoto K. Inorg. Chem. Front. 2015;2:741–748. [Google Scholar]
- Trovarelli A. Comments Inorg. Chem. 1999;20:263–284. [Google Scholar]
- Tang Y., Zhang H., Cui L., Ouyang C., Shi S., Tang W., Li H., Lee J. S., Chen L. Phys. Rev. B: Condens. Matter Mater. Phys. 2010;82:125104–125112. [Google Scholar]
- Zhen J. M., Liu D. P., Wang X., Li J. Q., Wang F., Wang Y. H., Zhang H. J. Dalton Trans. 2015;44:2425–2430. doi: 10.1039/c4dt03141e. [DOI] [PubMed] [Google Scholar]
- Wang X., Liu D. P., Song S. Y., Zhang H. J. J. Am. Chem. Soc. 2013;135:15864–15872. doi: 10.1021/ja4069134. [DOI] [PubMed] [Google Scholar]
- Zheng Y. R., Gao M. R., Li H. H., Gao Q., Arshad M. N., Albar H. A., Sobahi T. R., Yu S. H. Science China Materials. 2015;58:179–185. [Google Scholar]
- Hu C. G., Han Q., Zhao F., Yuan Z. Y., Chen N., Qu L. T. Science China Materials. 2015;58:21–27. [Google Scholar]
- Wang X., Liu D. P., Li J. Q., Zhen J. M., Wang F., Zhang H. J. Chem. Sci. 2015;6:2877–2884. doi: 10.1039/c4sc03854a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D. P., Li W., Feng X. L., Zhang Y. Chem. Sci. 2015;6:7015–7019. doi: 10.1039/c5sc02774h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T., Zeng J., Lim B., Xia Y. N. Adv. Mater. 2010;22:5188–5192. doi: 10.1002/adma.201002763. [DOI] [PubMed] [Google Scholar]
- Zhou H. P., Wu H. S., Shen J., Yin A. X., Sun L. D., Yan C. H. J. Am. Chem. Soc. 2010;132:4998–4999. doi: 10.1021/ja101110m. [DOI] [PubMed] [Google Scholar]
- Yoon K., Yang K., Lu P., Wan D., Peng H., Masias K., Fanson P., Campbell C., Xia Y. N. Angew. Chem., Int. Ed. 2012;51:9543–9546. doi: 10.1002/anie.201203755. [DOI] [PubMed] [Google Scholar]
- Chu Y. Y., Wang Z. B., Jiang Z. Z., Gu D. M., Yin G. P. Adv. Mater. 2011;23:3100–3104. doi: 10.1002/adma.201100040. [DOI] [PubMed] [Google Scholar]
- Schweitzer N. M., Schaidle J. A., Ezekoye O. K., Pan X. Q., Linic S., Thompson L. T. J. Am. Chem. Soc. 2011;133:2378–2381. doi: 10.1021/ja110705a. [DOI] [PubMed] [Google Scholar]
- Postole G., Chowdhury B., Karmakar B., Pinki K., Banerji J., Auroux A. J. Catal. 2010;269:110–121. [Google Scholar]
- Liu W., Liu X. F., Feng L. J., Guo J. X., Xie A. R., Wang S. P., Zhang J. C., Yang Y. Z. Nanoscale. 2014;6:10693–10700. doi: 10.1039/c4nr02485k. [DOI] [PubMed] [Google Scholar]
- El-Sheikh S. M., Ismail A. A., Al-Sharab J. F. New J. Chem. 2013;37:2399–2407. [Google Scholar]
- Huang M., Zhang Y. X., Li F., Wang Z. C., Alamusi, Hu N., Wen Z. Y., Liu Q. Sci. Rep. doi: 10.1038/srep04518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.