

Abstract
Oxygen (O2) acts as a potent upstream regulator of cell function. In both physiological and pathophysiological microenvironments, the O2 concentration is not uniformly distributed but instead follows a gradient that depends on distance from oxygen-carrying blood vessels. Such gradients have a particularly important role in development, tissue regeneration, and tumor growth. In this protocol, we describe how to use our previously reported gelatin-based O2-controllable hydrogels that can provide hypoxic microenvironments in vitro. The hydrogel polymeric network is formed via a laccase-mediated cross-linking reaction. In this reaction, laccase catalyzes diferulic acid (diFA) formation to form hydrogels with an O2-consuming reaction. Cells, such as cancer or endothelial cells, as well as tumor/tissue grafts, can be encapsulated in the hydrogels during hydrogel formation and then analyzed for cellular responses to 3D hypoxic gradients and to elucidate the underlying mechanisms governing these responses. Importantly, oxygen gradients can be precisely controlled in standard cell/tissue culture conditions and in vivo. This platform has been applied to study vascular morphogenesis in response to hypoxia and to understand how oxygen gradients mediate cancer cell behavior. Herein, we describe the means to validate the assay from polymer synthesis and characterization—which take 1–2 weeks and include verification of ferulic acid (FA) conjugation, rheological measurements, and O2 monitoring—to the study of cellular responses and use in rodent models. Time courses for biological experiments using this hydrogel are variable, and thus they may range from hours to weeks, depending on the application and user end goal.
INTRODUCTION
Oxygen (dioxygen, O2) is a key molecule vital for the existence of all multicellular organisms. Variations in O2 are known to control signaling cascades that lead to metabolic and phenotypic changes. As partial pressure of O2 falls below 5%, herein defined as hypoxia (Table 1), a myriad of cellular and systemic adaptations commence.
TABLE 1.
Definitions of oxygen concentration and usual in vivo oxygen distribution.
| Term | %O2 |
|---|---|
| Normoxia (nonhypoxia) | 21 |
| Hypoxia | <5 |
| Anoxia | <0.1 |
| Organ | %O2 |
| Arterial blood | 13.2 |
| Brain | 4.4 |
| Liver | 5.4 |
| Kidney | 9.5 |
| Muscle | 3.8 |
| Bone marrow | 6.4 |
As shown in ref. 57.
During tumor development and progression, a low-O2 environment manifests when a tumor outgrows the existing O2 supply, leading to the upregulation of numerous important factors, perhaps most notably hypoxia-inducible factor 1-α (HIF-1α). HIF-1α upregulation has been correlated with negative patient outcomes in many cancers, such as sarcoma1, prostate cancer2, and squamous cell head and neck cancer3. Exacerbating such negative outcomes, tumors with high levels of HIFs are resistant to chemotherapy and radiation therapy because of limited perfusion4. At the cellular level, hypoxia has been shown to induce cancer cell migration5,6, extracellular matrix (ECM) modification7, and recruitment of blood vessels8. In sarcoma, low O2 leads to faster cell migration via HIF-1α (ref. 5), which upregulates collagen modifiers such as lysl oxidase, procollagenlysine, and 2-oxoglutarate 5-dioxygenase 2 (PLOD2)5,7. Hypoxic conditions in the tumor environment have also been shown to upregulate the secretion of angiogenic factors such as vascular endothelial growth factor (VEGF)9. Overall, the hypoxic microenvironment regulates multifaceted pathways that are critical to the development and progression of cancer.
A similar mechanism by which hypoxia upregulates VEGF and other effectors of cancer cell function in the tumor microenvironment has also been shown to affect various vascular diseases and to modulate wound healing and tissue regeneration. For example, hypoxic upregulation of VEGF secretion has been documented in macular degeneration, wherein high levels of VEGF cause uncontrolled blood vessel growth, resulting in poorly formed or leaky vessels, which can lead to blindness10–12. In wound healing, hypoxia plays a critical role in the enhancement of blood vessel recruitment and inflammatory responses. Here, a reduced blood flow at the injury site leads to a locally low-O2 environment, which in turn initiates angiogenesis and wound healing. Further, the hypoxic environment in wounded blood vessels increases the expression of intercellular adhesion molecule 1, a key factor in recruiting endothelial progenitor cells13,14 and immune cells15 to the injury site. Diseases with underlying HIF-1 overexpression can lead to chronic wounds and fibro-proliferative disorders, as well as to hypertrophic scars16. Conversely, low levels of HIF-1 are commonly seen in diabetic ulcers and in elderly patients, which may lead to impaired wound healing17.
Responses to hypoxia are required for the normal development of the vascular system18,19. In early embryonic development, fast-growing tissues must coordinate growth with O2 delivery. O2 in this environment is critically low and acts as a signal to stimulate vascular growth20 and endothelial cell differentiation21. It has also been suggested that, during embryonic vessel development, regions of relative hypoxia attract growing blood vessels22. During the early stages of embryogenesis, a primitive vascular pathway is formed through vasculogenesis, in which progenitor cells form an immature vascular plexus20. Once this primitive vascular network is formed, endothelial cells form new capillaries via angiogenesis. This pathway stimulates VEGF and other angiogenic factors via low O2, which facilitates blood vessel formation23. Overall, our current understanding suggests that vasculogenesis and angiogenesis in mammalian tissues occur in hypoxic conditions24–26. The vital roles of hypoxia and O2 gradients are made clear through their critical role in development, tissue regeneration, and cancer/ tumor growth and progression.
Methods of culture under hypoxic conditions
Variations in O2 tension in healthy or diseased tissues occur as gradients, ranging from anoxic (0%) to normoxic (~13%) depending on blood supply and tissue O2 demands (Table 1). Nonetheless, although hypoxia plays a pivotal role during various cellular processes, the design and utilization of a 3D hypoxic-gradient microenvironment to mimic the in vivo niche has only recently been realized.
To control O2, alternative in vitro research platforms use specialty equipment such as hypoxic chambers, hoods, and incubators, which do not allow for precise control over dissolved oxygen (DO) concentration in the cell’s local microenvironment, thus limiting the ability to study cell responses to O2 gradients. Numerous methods exist in the literature to combat this issue. These include many 2D systems with precision spatial and temporal control of O2 (refs. 27,28) and even O2 gradients29,30. In addition, methods for control of O2 tension and O2 gradients compatible with 3D cell encapsulations have been developed31–34. Although these platforms may be preferred for some experimental setups and should be considered in an application-dependent manner, several general limitations exist. For example, many of the methods describe complex platforms requiring microfabrication techniques32 and intricate pump and flow setups31, the use of hypoxic incubators and supplemental gas tanks35, and high cell concentrations (107–108 cells per ml)36, and prohibit real-time monitoring and imaging37. To address the above-mentioned limitations and the increasing interest in the study of cell responses to O2 gradients, we developed O2-controllable hydrogels that can serve as 3D hypoxic-gradient microenvironments.
Our hydrogel polymeric network is formed by diFA formation in a laccase-mediated cross-linking reaction with O2 consumption (Fig. 1a). After completion of the reaction, the low DO level is maintained through O2 consumption by the cells or tissue38. The cross-linking reaction allows the cells to be rapidly exposed to a hypoxic environment, without the need to wait for a spontaneous (cell-mediated) gradient to form. This is beneficial because it takes time for these cell-mediated gradients to form spontaneously, and the construct cannot be guaranteed to remain mechanically stable. A biomimetic extracellular environment is critical for research into vascular differentiation and development, in which 3D networks are formed through cell–matrix interactions, including adhesion and degradation39–41. In the case of cancer cells, O2 gradients can form in sufficiently thick 3D tissue, but the gradients are transient and very difficult to control. Finally, it should also be noted that rapid, predictable O2 gradients cannot form when encapsulating tissue grafts or when transplanting non-O2-controllable materials in vivo. Our platform circumvents these pitfalls by rapid O2 consumption during hydrogel formation via a laccase-mediated cross-linking reaction. When the chemical reaction is completed, the O2-consuming reaction is terminated and atmospheric O2 diffusion begins. In vitro, the O2 levels within the gel can be simply controlled by varying the thickness of the hydrogel construct. The gradients in the gel can last up to 1–2 weeks, allowing for long-term hypoxia experiments while minimizing the generation of reactive oxygen species that may be elicited through rapid reoxygenation. These gels can be fabricated in traditional polystyrene 96-well plates, and once cells are encapsulated or microtissues are embedded, the constructs are cultured with traditional cell culture media in a standard incubator. The DO levels and gradients are controlled by varying hydrogel thickness in a volume-dependent manner in wells of 96-well plates (e.g., nonhypoxic hydrogels, 45 μl; hypoxic hydrogels, 90 μl). In addition to using conventional cell culture equipment, these gels use a small volume of polymer solution, typically 45–100 μl, and therefore small volumes of reagents can be used for molecular biology assays such as PCR, histology, and immunofluorescence analysis. These gels are optically clear and therefore are ideal for the real-time measurement of O2 and cell migration, as well as in situ staining for immunofluorescence imaging. Furthermore, the platform can be used to screen small-molecular inhibitors5. In vivo experiments using hydrogels as therapeutics or to study the effects of O2 gradients in disease models may also be designed and performed (Box 1).
Figure 1.

Schematic representation of the Gel-HI hydrogels for in vitro and in vivo applications. (a) Synthesis of Gtn–FA, which can form a hydrogel network via a laccase-mediated cross-linking reaction with O2 consumption. (b) The Gtn–FA precursor solution is dissolved in DPBS at 37 °C. It can be mixed with either cells or tissues to provide artificial hypoxic microenvironments with oxygen gradients (see inserted computer simulation of oxygen tension and gradients). The precursor solution can also be directly injected into the animal as a hypoxia-inducible acellular matrix that induces temporal hypoxia and an oxygen gradient in the body (see inserted computer simulation of oxygen tension and gradients). Adapted with permission from Park and Gerecht38, Nature Publishing Group.
Box 1. In vivo procedures ● TIMING 1–2 weeks.
-
1
If you wish to test acellular hydrogels in vivo, follow option A. If you wish to inject cellularized hydrogels, follow option B.
(A) In vivo injection
! CAUTION Any experiments involving live rodents must conform to relevant institutional and national guidelines and regulations.
▲ CRITICAL The animal study used to generate the results shown was performed using one of the protocols (RA15A152, MO15A154, and MO16A284) approved by the Johns Hopkins University Institutional Animal Care and Use Committee.
Inject hydrogels, either acellular or containing cells of interest, s.c. into mice (we use n = 4, 6- to 10-week-old C57/6 female mice for acellular applications, or 6- to 10-week-old NU/NU female mice for cellular applications).
Sterilize polymer and laccase solutions by filtering with a syringe filter (pore size, 0.2 μm).
-
Prepare Gel-HI hydrogel solution by mixing Gtn–FA and laccase stock solutions at a 3:1 (150 μl of Gtn–FA:50 μl of laccase) ratio.
▲ CRITICAL STEP Mix the solutions by pipetting and mild shaking to prevent bubble formation.
-
To minimize the differences between the sample groups, also encapsulate 0.02% (wt/vol) Ca(OH)2, a by-product of CaO2 decomposition, within the hypoxic hydrogels.
▲ CRITICAL STEP To control O2 levels in vivo, mix a small amount of calcium peroxide (CaO2; Sigma-Aldrich) and calcium hydroxide (Ca(OH)2; Sigma-Aldrich) with the polymer solution to obtain 0.02% (wt/vol) CaO2.
Pre-incubate at 37 °C for the time determined in Step 22A(vii).
Inject the polymer precursor solution into the back of each mouse using 26-gauge needles.
(B) Hydrogel s.c. injection (with cells)
Sterilize the polymer solutions by filtering with a syringe filter (pore size, 0.2 μm).
Pellet the cells by centrifugation at room temperature for 5 min at 89g. to obtain a cell suspension in hydrogel precursor solution at desired concentration.
Resuspend the cell pellet in Gtn–FA polymer stock solution.
Mix laccase stock solution with the cell+polymer solution.
Pre-incubate at 37 °C for the time determined in Step 22A(vii).
-
Inject the polymer precursor solution into the back of each mouse using 26-gauge needles.
▲ CRITICAL Maintain the polymer solution at 35–40 °C before injection to avoid thermogelation.
▲ CRITICAL Shave the mouse’s hair before injection, and mark the injection sites to ensure that the injection region can be identified after in vivo implantation.
-
2
Following predetermined time points, analysis of the effects of either step 1A or B can be completed by assessing blood vessel perfusion (option A), measuring O2 gradient (option B), or performing histochemistry (option C).
(A) Blood vessel perfusion
To confirm the vascular functionality of blood vessels infiltrated into the hydrogel matrix, inject Alexa-Fluor-568-conjugated isolectin GS-IB4 from Griffonia simplicifolia (Invitrogen) through the tail veins of the mice.
After 30 min, harvest the mice.
Remove the hydrogels with surrounding tissues.
Stain the explants with Hoechst dye for 30 min.
-
Proceed to visualization using confocal microscopy (LSM 780 Meta, Carl Zeiss).
▲ CRITICAL STEP The stained tissues are light-sensitive. Keep the explants in the dark by wrapping the samples in aluminum foil.
(B) Gradient measurement in vivo (following subcutaneous injection)
! CAUTION Needle-type O2 microsensors are extremely fragile. Take extra care when handling microsensors. Although they are protected by the needles, the microsensor portion is <50 μm in diameter, so it may break if subjected to impact.
▲ CRITICAL Needle-type O2 microsensors are an invasive tool for measuring O2 gradients. As such, these measurements may affect the integrity of the hydrogel. Thus, we suggest that gradient measurements serve as the end point of a given experiment.
▲ CRITICAL Calibrate the microsensors to mouse body temperature. During readings, the body temperature will be slightly lower than normal because of the anesthesia. Keep the mice on the heating pad to maintain mouse comfort.
Attach a needle-type O2 microsensor to the manual micromanipulator according to the manufacturer’s instructions.
Place the temperature probe under the mouse’s body to monitor body temperature in real time.
Mark the proximal and distal ends of the hydrogel.
Puncture the mouse skin with the needle microsensor.
-
Using the fine-adjustment knob on the micromanipulator, move the microsensor into the distal end of the hydrogel.
▲ CRITICAL STEP In vivo, O2 measurements take time to equilibrate (~30–45 min). It is critical to achieve a stable reading before moving on to another measurement.
Using the fine-adjustment knob, move the sensor a predetermined distance to make another measurement. Continue until all measurements have been made.
(C) Histochemistry
At predetermined time points (e.g., 1, 3, and 5 d), harvest a subset of the animals, by cervical dislocation or CO2 asphyxiation.
Remove the hydrogels with the surrounding tissue.
Fix the explants by placing each in 30 ml of formalin-free Accustain fixative.
Using standard histological procedures, dehydrate the explants in biological-grade ethanol (80–100%).
-
Agar-embed the explants by encapsulating them within agar solution dissolved in PBS at 60 °C using custom-made molds (diameter = 8 mm; thickness = 10 mm).
▲ CRITICAL STEP Before paraffin-embedding, embed the constructs/explants within 0.1–0.5% agar gels to prevent sample destruction during sectioning.
Paraffin-embed following standard histological procedures and serially section using a microtome (5 mm).
Using standard immunohistochemical techniques, stain the slides with either H&E or immunolabeling for markers of interest, such as CD31 or α-SMA.
Cells encapsulated in hydrogels can be administered to mice. O2 gradients and severity of hypoxia in these cell grafts are dependent on two main factors in vivo: (i) hydrogel volume and (ii) surrounding tissue O2 concentration. Hydrogels with larger relative volumes will have a more severely hypoxic core, and thus a steeper O2 gradient. If hydrogels are injected or implanted in tissues with high relative O2, steep gradients may be obtained. Those tissues with lower relative O2 levels will exhibit shallower gradients38. We have demonstrated that insights into the role of varying O2 during tumor growth, vascular development42, and angiogenesis38,43 can be acquired using these hydrogels.
Development of the protocol
Gelatin hypoxia-inducible (Gel-HI) hydrogels are prepared by chemical functionalization of porcine gelatin. Gelatin has numerous inherent advantages for use as a tool for biological experimentation. Gelatin is produced through denaturation (partial hydrolysis) of collagen, a critical ECM protein found in nearly all tissues throughout the human body. Thus, gelatin possesses bioactive characteristics with broad implications for many biological applications of numerous cell and tissue types. In particular, the presence of integrin-mediated cell adhesion motifs and the amenability to cell-mediated proteolytic degradation highlight the underlying advantages of the use of a gelatin backbone. Such properties alleviate the need for user-defined optimization of the concentration of cell adhesion motifs and degradable add-ins, leading to a highly biomimetic culture system. Because gelatin is naturally derived, there is a certain degree of batch-to-batch variability, as with any natural polymer. However, widespread use and characterization of gelatin both in 2D culture systems44 and as 3D platforms45–47, along with a high level of bioactivity, motivate its use in our system.
Control over O2 tension in our Gel-HI hydrogel necessitates O2 consumption. In addition, in order to develop gelatin-based culture systems, chemical functionalization to achieve a cross-linking mechanism is necessary, as gelatin thermally cross-links only below physiological temperatures. These two needs define our design approach in developing this system. Importantly, a large number of molecules can be conjugated because a high number of chemically active functional groups is available, making chemical functionalization simple. In particular, for our application, primary amines are abundant. Less abundant are biocompatible reactions that include physiologically relevant O2 consumption. Laccase, an enzyme derived from mushrooms (among other select fungi and trees), has particular specificity in catalyzing reactions between phenol-containing molecules48. Specifically, laccase can catalyze the four-oxygen reduction of O2 to H2O, which results in oxidation of two phenol-containing molecules. Similar to laccase, other enzymes can be adopted to create hypoxia-inducible hydrogel matrices (e.g., tyrosinase and glucose oxidase, which consume molecular O2 during cross-linking of phenolic molecules). Our choice of phenolic moiety required the presence of a carboxyl group opposite the phenol to facilitate chemical conjugation to the gelatin backbone. FA satisfies our design criteria, and thus was conjugated to the gelatin backbone by using N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC)/N-hydroxysuccinimide (NHS) chemistry. The FA-conjugated gelatin polymers form hydrogels through diFA formation in a laccase-mediated reaction, resulting in bioactive and O2-controllable Gel-HI hydrogels.
It is critical to understand how O2 tension and O2 gradients are regulated in Gel-HI hydrogels. As discussed above, Gel-HI hydrogels are used in standard incubators and culture plates. In such conditions, passive diffusion of atmospheric (~21%) O2 is an important consideration. Such diffusion acts to increase the O2 tension in the culture microenvironment. With that, two elements of the system act to decrease O2 tension: (i) laccase-mediated O2 consumption and (ii) cell-mediated O2 consumption. An initial decrease is induced by the laccase-mediated cross-linking reaction, and in acellular experiments, following completion of the reaction, the hydrogel microenvironment returns to atmospheric conditions within hours. Hydrogels containing encapsulated cells maintain low-O2 (<5% is considered hypoxic) conditions for 3–7 d in culture. Maintenance of low-O2 conditions is highly dependent on cell concentration and cell type5,38. In general, higher cell concentrations lead to prolonged maintenance of hypoxia. In addition, cell types vary in rates of O2 consumption, and thus understanding cell O2 needs is critical to experimental design. These parameters should be considered as key factors in efforts to accurately mimic the native tumor (or ischemic) microenvironment, as cellular activities are highly variable within the dynamic O2 microenvironment. Growing evidence has demonstrated that transcription factors (e.g., HIFs), which regulate cellular activities, accumulate when the cells or tissues are exposed to hypoxia10. For example, cells may change their metabolism to reduce O2 consumption by upregulating GLUT gene expression in hypoxic environments. In addition, HIFs activate various cellular processes, including metabolism, proliferation, invasion and metastasis, and tumor angiogenesis. In our previous study, we also demonstrated that mouse sarcoma cultured within 3D hypoxic matrices upregulated numerous hypoxia-related genes including GLUT-1, MMP genes, and VEGF. Furthermore, there are variations in the cellular O2 consumption rate, depending on the cell type and number within artificial hypoxic microenvironments. Thus, we should consider the variations in the cellular metabolism within artificial hypoxic microenvironments as a critical design parameter throughout experiments in this hydrogel system. Last, because of the constant and delicate balance between active O2 decreases and passive O2 increases, the volume of the hydrogel has a substantial role in in vitro assays. Steepness of O2 gradients and maintenance of hypoxic conditions rely heavily on the volume and, perhaps more specifically, on the thickness of the hydrogel. In smaller-volume (decreased thickness) hydrogels, atmospheric concentrations of O2 diffuse quickly throughout the gel, resulting in higher O2 levels with a shallow gradient. Larger-volume (increased thickness) hydrogels diffuse O2 more slowly throughout the entire volume, resulting in hypoxic conditions with substantial O2 gradients.
In addition to O2 tension, other important ECM parameters, such as stiffness and degradability, can be controlled by simply adjusting polymer or enzyme concentration. Further adjustment through secondary cross-linking with an additional enzyme, microbial transglutaminase, has been described29,43. In this article, we describe the detailed protocol for synthesis, characterization, manipulation, and application of our versatile Gel-HI hydrogels. Limitations in 3D hypoxic cell culture and O2 gradient studies are alleviated by the use of Gel-HI hydrogels, with a wide range of use both in vitro as a culture platform and in vivo as a cell delivery vehicle and as an acellular therapeutic.
Applications of the protocol
Cancer
Hypoxia is a key regulator affecting cell migration, ECM modification, and blood vessel recruitment during tumor development, growth, and metastasis. Gel-HI hydrogels allow us to simultaneously study the effects of a wide range of O2 levels, whereas most studies focus on a fixed O2 level, typically drawing comparisons between experiments conducted within the severe hypoxic range (0.5–1%) and those conducted at atmospheric (21%) O2. One of the key benefits of this platform is the DO gradient along the hydrogel depth, which uniquely provides opportunities to study how gradients regulate cell behavior in vitro in three dimensions. Moreover, this platform not only allows for the use of conventional cell and molecular biology assays, but it also permits real-time imaging to track single cancer cell proliferation and study motility/migration. Conventional hypoxic platforms, such as chambers, do not allow for a controlled gradient to be formed, and are prohibitive for live-cell imaging because of the chamber’s size, as it cannot fit into conventional microscope setups. Tumor sections can also be encapsulated in the hydrogel, and the migration of cells out of the tumor can be documented. This allows the user to study cells in pathophysiologically relevant O2 gradients using real-time imaging to elucidate pathways that lead to cancer cell migration and potential metastasis.
Vascular applications
Not surprisingly, the vascular regeneration niche has been studied in great detail, owing to the high mortality and morbidity of cardiovascular disease in the developed world49. Complex mechanisms have been uncovered and described relating to the interplay between multiple cell types (including interactions between endothelial cells and mural cells50), growth factors51, ECM properties40, and O2 tension52. Specifically, as detailed above, hypoxic conditions have been shown to trigger vascular formation and growth during embryonic development, various vascular-related disorders, and cancer. However, studying the effects of O2 tension and O2 gradients in a 3D microenvironment was not possible until the development of Gel-HI hydrogels. Gel-HI hydrogels facilitate studies in a highly biomimetic experimental system by harnessing the endothelial cell’s own vasculogenic capacity. Many platforms that are used to study vascular network formation require the addition of hyperphysiological concentrations of proangiogenic growth factors or an engineered ‘angiogenic’ or ‘vasculogenic’ cocktail of factors39,53. However, encapsulation of vascular cells in Gel-HI leads to cell-mediated production of angiogenic growth factors and subsequent vascular network formation. Thus, Gel-HI hydrogels provide a system that more closely mimics the native regenerative niche than do other culture systems. For example, our laboratory has shown that early vascular cells derived from induced pluripotent stem cells of diabetic patients can generate lumenized networks in hypoxic Gel-HI hydrogels, indicating their responsiveness to hypoxic pathways42.
Limitations of the platform
No biomaterial is without limitations, whether in function or application. Several limitations have been briefly mentioned in the preceding text, which we will again describe here. First, batch-to-batch variability exists with these hydrogels, as with any naturally derived polymeric hydrogel. Even with a relatively simple polymer synthesis protocol, variability can be seen in an academic lab setting. As such, it is critical to maintain consistency in technique throughout every step of the process, including synthesis, purification, storage, and particularly cell encapsulation. To ensure reproducibility, we recommend careful analysis of polymer properties before cell/tissue encapsulation experiments. Enzymatic cross-linking is particularly sensitive to temperature and timing, both of which are critical to maintaining experimental reproducibility. Hydrogel precursor solutions, particularly laccase, may become cytotoxic outside of the concentration ranges suggested by this protocol. Laccase is sensitive to storage conditions and may lose activity with extended storage in aqueous solution (>2–3 months). In addition, because the matrix can be degraded in a cell-mediated manner, over the course of the experiment mechanical properties are subject to cell-mediated change. This property of hydrogels is critical to many applications but can serve as a limitation to culture time. If left in culture long enough (usually >5 d), the matrix may completely degrade. Furthermore, cell culture time may vary with cell type and encapsulation concentration.
Experimental design
A vast array of powerful experiments can be developed by following the steps described in this protocol. Examples of several end-point analyses are included, which can be used or omitted as researchers tailor the experimental design to fit their needs. Polymer synthesis, purification, and characterization are key first stages to any and all experiments following this protocol. Upon initial synthesis, we recommend that the gel components undergo a full array of characterization strategies, including NMR imaging, UV/visible measurement, mechanical/rheological testing, and O2 monitoring. O2 may be monitored using commercially available O2 sensors (PreSens), which measure local O2 levels in real time via a fluorescence-quenching methodology. Successful hydrogel synthesis requires several benchmarks. First, NMR will confirm conjugation of FA to the gelatin backbone and UV/visible measurement will determine the concentration of conjugated FA (40–60 μmol/g polymer). Rheological characterization provides verification of hydrogel formation and measurement of mechanical properties. Finally, acellular O2 measurements in 96-well plates are critical to ensuring that O2 is reduced to <5% (in 90- to 100-μl hydrogels), confirming successful establishment of a hypoxic microenvironment. Characterization may be streamlined and strategies omitted once the user has mastered the synthesis portion of the protocol. After successful synthesis, determination of gelation time is key to ensuring a homogeneous encapsulation. This may vary slightly with cell type and concentration, so gelation time measurements should be done on a per-experiment basis. Monitoring of cell morphology may be done via light microscopy or time-lapse microscopy and can be coupled with DO monitoring (either over the entire time course or as gradient measurements at specified time points) to gain an enhanced understanding of the microenvironmental properties over time. End-point analysis can involve RNA extraction or fixation and immunofluorescence staining for in vitro experiments. In vivo experiments can also incorporate cells and DO monitoring (although only at specified time points) and end-point experiments may include lectin tail-vein injection for EC applications or tissue excision and histology for numerous other applications. Indeed, Gel-HI hydrogels provide an impressive potential for breadth of study and may be adapted to many applications (Fig. 1).
Expertise in cell culture techniques is a necessity for performing experiments using this protocol. In addition, experience with microscopy, immunofluorescence staining, and microbiological techniques will aid in the range of experiments possible with this protocol. Background in materials science and polymer synthesis is suggested, but not required, as the synthesis protocol is relatively simple.
MATERIALS
REAGENTS
! CAUTION Wear suitable personal protective equipment, including a lab coat, chemical gloves, and safety goggles, when handling chemical reagents. Please refer to the appropriate material safety data sheet provided by the manufacturer.
▲ CRITICAL All reagents should be maintained at listed storage conditions and should not be used after manufacturer-defined expiration date.
Synthesis and characterization of Gtn–FA polymers
Gelatin, type A from porcine skin, <300 bloom (Sigma-Aldrich, cat. no. G2500; store at room temperature, 23–25 °C)
Laccase, lyophilized powder from mushroom, ≥4.0 units per mg (Sigma-Aldrich, cat. no. 75117; store in a dry place at −20 °C; Laccase from other sources such as Agaricus bisporus (Sigma-Aldrich, cat. no. 40452) or Trametes versicolor (Sigma-Aldrich, cat. no. 38429) may be used, but these must be checked for solubility, activity, and cytotoxicity before use.
3-Methoxy-4-hydroxycinnamic acid (ferulic acid (FA); Sigma-Aldrich, cat. no. 90034; store in a dry place at room temperature)
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC; Sigma-Aldrich, cat. no. E6383; store in a dry place at −20 °C)
N-hydroxysuccinimide (NHS; Sigma-Aldrich, cat. no. 130672; store in a dry place at room temperature)
DMSO (Sigma-Aldrich, cat. no. D8418; store in a dry place at room temperature) ! CAUTION DMSO is a toxic organic solvent that may induce skin irritation, serious eye damage, and respiratory irritation upon contact or inhalation. Always wear a face mask, goggles and gloves when handling DMSO. ▲ CRITICAL All chemicals can be purchased from other suppliers. If purchased from other suppliers, some properties of polymers and hydrogels may vary (e.g., conjugation efficiency of FA, solubility of polymer, and phase transition time and mechanical properties of hydrogels) because of the differences in the purity grade. It is advisable to characterize each polymer batch and the resultant hydrogels to ensure reproducibility of results. In particular, laccase is subject to manufacturer availability. Alternative laccase sources may be used. In such cases, ensure that the activity and cross-linking/O2 consumption properties match those of previously published results38.
Validation of polymer
Deuterium oxide (D2O; Sigma-Aldrich, cat. no. 151882; store in a dry place at room temperature)
Polymer and cross-linker preparation
Dulbecco’s PBS (DPBS; Gibco, cat. no. 14040133; store at room temperature)
Cytotoxicity of gelatin-g–ferulic acid (Gtn–FA) and laccase (XTT assay)
Newborn human foreskin fibroblasts (Global Stem, cat. no. GSC-3002) ! CAUTION The cell lines used in your research should be regularly checked to ensure that they are authentic and that they are not infected with mycoplasma.
Media base (DMEM; Thermo Fisher Scientific, cat. no. 11965092; store at 4 °C)
Heat-inactivated FBS (Thermo Fisher Scientific, cat. no. 16140071; store at −20 °C)
Cell and tumor encapsulation (endothelial cell culture)
Human endothelial cells such as endothelial colony-forming cells (Lonza, cat. no. 00189423) ! CAUTION The cell lines used in your research should be regularly checked to ensure that they are authentic and that they are not infected with mycoplasma.
Endothelial cell growth medium (EGM-2 BulletKit; Lonza, cat. no. CC-3162; store media at 4 °C; store kit at −20 °C)
Hyclone FBS (Hyclone, cat. no. SH30071.03; store at −20 °C)
Cell and tumor encapsulation(cancer cell or tumor graft culture)
KIA cells or any other cancer cell line of interest (Reagent Setup). KIA cells were derived from a genetic murine model of sarcoma kindly provided by T.S.K. Eisinger at the University of Pennsylvania. ! CAUTION The cell lines used in your research should be regularly checked to ensure that they are authentic and that they are not infected with mycoplasma.
Cancer media base (DMEM; Corning, cat. no. 10-013-CV; store it at 4 °C)
Penicillin–streptomycin (10,000 U/ml; Life Technologies, cat. no. 151; store at −20 °C)
Heat-inactivated FBS (Thermo Fisher Scientific, cat. no. 16140071; store at −20 °C)
Continuous O2 monitoring in vitro
Dow Corning high-vacuum silicone grease (Sigma Aldrich, cat. no. Z273554)
Silicone glue (3M, cat. no. 255967)
Protease activity
DQ gelatin (Thermo Fisher Scientific, cat. no. D-12054; store at −20 °C)
PBS (see above)
Cell proliferation activity
Cell proliferation reagent WST-1 (Sigma Aldrich, cat. no. 5015944001; store at −20 °C)
PBS (see above)
Triton X-100 (Sigma-Aldrich, cat. no. X100; store at room temperature) ! CAUTION Triton X-100 is hazardous and toxic, and it may induce skin irritation, serious eye damage, and respiratory irritation if contacted or inhaled. Always wear eye shields, face shields, full-face respirator (US), and gloves when handling Triton X-100 to avoid any direct contact.
Immunofluorescence staining
Paraformaldehyde (PFA; Sigma-Aldrich, cat. no. 158127; store the powder at 4 °C, and store the solution at 20 °C) ! CAUTION PFA is a potent fixative and it is hazardous. Avoid any direct contact and wear appropriate personal protective equipment.
Triton X-100 (Sigma-Aldrich, cat. no. X100; store it at room temperature)
PBS (see above)
BSA (Sigma-Aldrich, cat. no. A9418; store at 4 °C)
Secondary antibody such as Alexa Fluor 488 (Invitrogen Molecular Probes, cat. no. A11001)
DAPI (Life Technologies, cat. no. D1306; store the powder at room temperature, and store the solution at −20 °C) ! CAUTION PFA is hazardous and toxic, and it may induce skin irritation, serious eye damage, and respiratory irritation upon contact or inhalation. Always wear appropriate personal protective equipment.
Tween 20 (Fisher Scientific, cat. no. BP337; store at room temperature)
RNA extraction
TRIzol Reagent (Life Technologies, cat. no. 15596-026; store at 4 °C) ! CAUTION TRIzol Reagent contains phenol (toxic and corrosive) and guanidine isothiocyanate (an irritant), and it may be hazardous if not handled properly. Follow the manufacturer’s instructions and always work in a chemical safety fume hood and wear appropriate personal protective equipment.
In vivo subcutaneous injection
Mice, 6–10 weeks old, C57/6 female (Charles River, strain code 027; for acellular applications) or NU/NU (Charles River, strain code 088 (homozygous); for human cell applications) ! CAUTION Any experiments involving live rodents must conform to relevant institutional and national regulations. ▲ CRITICAL The animal study used to produce the example results shown was performed using one of the protocols (RA15A152, MO15A154, or MO16A284) as approved by the Johns Hopkins University Institutional Animal Care and Use Committee (RA, rat protocol; MO, mouse protocol).
Calcium peroxide (Sigma-Aldrich, cat. no. 466271; store in a dry place at room temperature)
Calcium hydroxide (Sigma-Aldrich, cat. no. 31219; store in a dry place at room temperature)
Accustain, formalin-free tissue fixative (Sigma-Aldrich, cat. no. A5472; store at room temperature) ! CAUTION Accustain is a toxic organic solvent that may induce skin irritation, serious eye damage, and respiratory irritation upon contact or inhalation. Work in a chemical safety fume hood and always wear appropriate personal protective equipment. Avoid any direct contact.
Blood vessel perfusion
Hoescht (Thermo Fisher, cat. no. 62249)
Alexa-Fluor-568-conjugated isolectin GS-IB4 (Molecular Probes, cat. no. I21412; store at −20 °C) ! CAUTION Alexa Fluor 568 is a harmful and toxic reagent. Always wear appropriate personal protective equipment. Avoid any direct contact.
Histology
Antibody α-SMA (Abcam, cat. no. ab5694; store at −20 °C)
Antibody CD31 (Abcam, cat. no. ab28364; store at −20 °C)
Agar (Sigma-Aldrich, cat. no. 1296; store at room temperature)
EQUIPMENT
Synthesis and characterization of Gtn–FA polymers
Round-bottom flask with a magnetic stir bar
Dialysis membrane, molecular weight cutoff = 3,500 Da, (Spectrum Laboratories, cat. no. 132724; store at 4 °C)
Freeze-dryer
Syringe filter (Millipore, cat. no. SLGV013SL)
Validation of polymer
1H NMR spectrometer (Bruker, model no. AMX-300)
UV/visible spectrometer (SpectraMax; Molecular Devices)
Polymer and cross-linker preparation (sterilization)
Syringe filter unit, such as EMD Millipore Millex sterile syringe filters with PES membrane, green (Millipore, cat. no. SLGP033RS)
Rheological analysis of Gtn-HI hydrogels
Rheometer (TA Instruments, model no. RFS3)
25-mm plate geometry (TA Instruments)
Temperature-controlled water bath
Cell and tumor encapsulation
96-well tissue culture plate (BD Bioscience, cat. no. 353072)
500-ml filter (Corning, cat. no. 431475)
Continuous O2 monitoring in vitro
▲ CRITICAL Calibrate O2 sensors by either using manufacturer-provided values (PreSens) or calibrating in-house. Calibration of sensors should be performed at the same temperature at which experiments will be carried out (37 °C for in vitro experiments and animal body temperature for in vivo experiments).
Oxy-4 micro fiber-optic oxygen transmitter (PreSens)
Sensor spots (PreSens, model no. PSt1)
Implantable oxygen microsensor (PreSens)
O2 gradient measurement (in vitro or in vivo)
▲ CRITICAL Calibrate O2 sensors by either using manufacturer-provided values (PreSens) or calibrating in-house. Calibration of sensors should be performed at the same temperature at which experiments are carried out (37 °C for in vitro experiments and animal body temperature for in vivo experiments). ▲ CRITICAL Other O2 sensors are available. Ensure compatibility of O2-sensing with your specific application.
Microx TX3 microfiber-optic oxygen transmitter; temperature-compensated system (PreSens)
Needle-type oxygen microsensors (PreSens)
Manual micromanipulator (PreSens)
3D real-time imaging
Confocal microscope (e.g., Carl Zeiss, model no. LSM 780)
Cell tracking analysis
Imaris (Bitplane, http://www.bitplane.com/download)
MATLAB (MathWorks, https://www.mathworks.com/products/matlab.html)
Prism (GraphPad, https://www.graphpad.com/scientific-software/prism/)
Protease activity
POLARstar OPTIMA plate reader (BMG Labtech) or similar fluorescent plate reader for wavelength measurements of fluorescent signals
Cell proliferation assay
UV/visible spectrometer (SpectraMax; Molecular Devices)
RNA extraction
NanoDrop spectrophotometer (Thermo Fisher Scientific, model no. ND-1000)
Blood vessel perfusion
Confocal microscope (Carl Zeiss, model no. LSM 780 Meta)
General laboratory equipment
Cell culture incubator
Sterile 96-well plates for 3D cell culture
Hemocytometer or cell counter
Positive-displacement pipette and sterile capillary piston tips
Water bath
Heat block
Sterile 1.5-, 2-, 15-, and 50-ml conical (micro)centrifuge tubes
Glass beaker, 50 ml
Spatula
Forceps
REAGENT SETUP
! CAUTION Wear suitable personal protective equipment, including a lab coat, chemical gloves, and safety goggles when handling chemical reagents. Please refer to the appropriate material safety data sheet provided by the manufacturer.
Cancer cell isolation Isolate cells from murine soft-tissue sarcomas, as described in Yoon et al.54 and Kirsch et al.55.
Cancer cell culture medium Filter cancer media base supplemented with 50 μg/ml penicillin–streptomycin and 10% (vol/vol) heat-inactivated FBS using a 0.1-μm-pore-size filter such as a Corning 500-ml filter (cat. no. 431475). This medium can be stored at 4 °C for 4–6 weeks.
Endothelial cell culture medium Filter EGM medium with a BulletKit supplemented with 10% (vol/vol) Hyclone FBS using a 0.1-μm-pore-size filter such as a Corning 500-ml filter (cat. no. 431475). This medium can be stored at 4 °C for 4–6 weeks.
Solvent preparation Prepare a mixture of DMSO and dH2O with a 1:1 volume ratio as a solvent. The solution can be stored in the dark at room temperature until use. ! CAUTION This solution will heat up upon mixing
Triton X-100 solution Dilute Triton X-100 to 1.0% (vol/vol) Triton X-100 in sterile deionized water and store the solution at room temperature. It can be stored for up to 6 months if it is sealed and refrigerated at 4 °C.
BSA solution Dilute BSA to 10% (wt/vol) in PBS. It can be stored for up to 6 months if it is sealed and refrigerated at 4 °C.
Formaldehyde solution Dilute formaldehyde to 3.7% (wt/vol) in PBS and store it at room temperature in the dark. It can be stored for up to 1 year if it is sealed.
PROCEDURE
Synthesis and characterization of Gtn–FA polymers ● TIMING 1–2 weeks
! CAUTION Perform polymer synthesis in a chemical safety fume hood. The experimenter should wear appropriate personal protective equipment, including a lab coat, chemical gloves, and safety goggles.
▲ CRITICAL Maintain the polymer solution at 35–40 °C to dissolve the gelatin, as the polymer is thermosensitive during polymer synthesis and purification—for example, in the oil bath and an oven.
-
1|
Prepare a mixture of DMSO and dH2O with a 1:1 volume ratio as a solvent.
-
2|
Dissolve gelatin (Gtn, 1.0 g) in 50 ml of the cosolvent by stirring at 40 °C for >1 h. Keep the solution at 40 °C while stirring moderately in an oil bath until the solution is completely dissolved and becomes clear (without any polymer powder).
▲ CRITICAL STEP Ensure that the polymers are completely dissolved in the solvent.
? TROUBLESHOOTING
-
3|
Dissolve FA (0.78 g, 4.0 mmol) in 20 ml of the cosolvent.
-
4|
React the FA solution with EDC (0.92 g, 4.8 mmol, 1.2 equivalents for each carboxyl unit of FA) and NHS (0.64 g, 5.6 mmol, 1.4 equivalents for each carboxyl unit of FA) at room temperature for 15 min to activate the terminal carboxyl groups of FA.
-
5|
After activation of carboxyl groups, add the activated solution from Step 4 to the Gtn solution from Step 2.
-
6|
Conduct a conjugative reaction by stirring the combined solution at 40 °C for 24 h.
-
7|
After the reaction, transfer the solution to a dialysis membrane (molecular weight cutoff = 3,500 Da) and dialyze at 40 °C against 5 liters of deionized water for 3–5 d in a chemical safety fume hood. Change water at least twice daily.
▲ CRITICAL STEP FA should be removed completely, as residual FA (free FA molecules) may influence hydrogel properties because of disruption of diFA formation in the Gtn-FA polymer backbone by the free FA molecules; e.g., it might result in a slow phase transition or in hydrogels with poor mechanical properties. Dialysis is complete when the solution becomes colorless and transparent. If there is any free FA, the solution will be slightly yellow.
▲ CRITICAL STEP The solution should be purified by filtering with a 0.2-um-pore-size membrane to remove gelatin aggregates and other impurities before the freeze-drying step.
-
8|
After dialysis and purification (Step 7), freeze-dry to obtain Gtn–FA conjugates.
■PAUSE POINT Keep the product in a refrigerator at 4 °C until use. The product can be stored for up to 6 months.
Validation of Gtn–FA polymer ● TIMING 1–2 h
-
9|
Chemical structures (1H NMR). Dissolve the Gtn–FA polymer solution in deuterium oxide (10 mg/ml) and acquire a 1H NMR spectrum to confirm the structure (Supplementary Fig. 1).
-
10|
Degree of substitution (DS) of FA. Dissolve Gtn–FA polymer (10 mg) in 1 ml of a mixture of DMSO and dH2O with a volume ratio of 1:1.
-
11|
Measure absorbance at the 320-nm wavelength using a UV/visible spectrometer.
▲ CRITICAL STEP For UV/visible measurements, the polymers and standard FA molecules should be dissolved in the cosolvent.
▲ CRITICAL STEP Ensure that the polymer and standard FA molecules are completely dissolved in the solvent.
-
12|
Calculate the concentration of the conjugated FA molecules from a calibration curve given by monitoring the absorbance of a known concentration of FA; standardize with the baseline measured using Gtn solution (10 mg/ml).
Assessment of cytotoxicity of Gtn–FA and laccase (XTT assay; control experiment) ● TIMING 3–5 d
-
13|
Seed newborn human foreskin fibroblasts in a well of a 96-well plate, with cell density at 2.8 × 104 cells per cm2 in 200 μl of culture medium; incubate for 24 h at 37 °C.
-
14|
Remove the culture medium and add fresh medium including polymers (0.63–10.0 mg/ml) and laccase (1.6–25 U/ml).
-
15|
After 24 h, remove the medium and culture the cells in the presence of the XTT solution (20% (vol/vol)) dissolved in medium for 4 h.
▲ CRITICAL STEP The XTT solutions are light-sensitive. Keep the solutions in the dark by wrapping the samples in aluminum foil.
-
16|
For quantitative analysis, place the medium in a 96-well plate and read it in a microplate reader at the 450-nm wavelength.
-
17|
Determine cell viability as a percentage relative to the viability of control cells (nontreated cells; defined as 100% viable).
Polymer and cross-linker preparation ● TIMING 1–2 h
-
18|
Prepare Gtn–FA (4.0–6.7 wt%) and laccase (25–100 U/ml) stocks by dissolving in DPBS (pH 7.4).
▲ CRITICAL STEP Keep stock solutions in a freezer at −20 °C and incubate the solutions at 37 °C for 15 min before use. Incubate the laccase stock solution at 37 °C for 5 min before use.
▲ CRITICAL STEP Maintain small-volume aliquots of laccase to avoid repeated freeze–thaw cycles.
■PAUSE POINT Stock solutions can be prepared for later use. Laccase stock solutions should be used within 2–3 months.
Rheological analysis of Gel-HI hydrogels ● TIMING 2–3 h
-
19|
Prepare the Gel-HI hydrogel solution by mixing Gtn–FA and laccase stock solutions at a 3:1 (Gtn–FA/laccase) ratio.
▲ CRITICAL STEP Mix the solutions by pipetting and mild shaking to prevent bubble formation.
-
20|
Pipette 200 μl of Gel-HI hydrogel solution onto 25-mm plate geometry in the instrument (TA Instruments, model no. RFS3).
? TROUBLESHOOTING
-
21|
To monitor elastic modulus (G′) and viscous modules (G″), perform dynamic time sweeps on hydrogels at 10% of strain and a frequency of 0.1 Hz at 37 °C.
▲ CRITICAL STEP Maintain the polymer solution at 37 °C to avoid thermogelation.
▲ CRITICAL STEP Ensure that the hydrogels are flat and parallel to the plate in the instruments.
▲ CRITICAL STEP To prevent water evaporation in the hydrogels, keep the hydrogels with a solvent trap (purchased from TA Instruments).
-
22|
Follow option A to encapsulate cells and option B to encapsulate tumors. If you wish to continually monitor oxygen in vitro, follow option C. To continually monitor the oxygen gradient, follow option D.
(A) Cell encapsulation ● TIMING 1–2 h
▲ CRITICAL STEP Prepare the polymer solutions using DPBS, and filter the solutions using a syringe filter with a pore size of 0.2 μm for sterilization.
Detach and collect cells (endothelial or cancer cells), using your standard laboratory protocol.
Determine the required cell number (n) based on the final cell concentration (c) and the total volume of gel solution (V) that must be prepared. n = c*V.
Spin down the desired number of cells into a pellet (5 min at 89g at room temperature).
-
Aspirate all of the medium from the pellet.
▲ CRITICAL STEP Any liquid that is left can interfere with the gelation.
-
To make 500 μl of polymer solution, first add 375 μl of polymer solution to the cell pellet and pipette up and down 20 times.
▲ CRITICAL STEP Minimize air bubbles in gel precursor solutions by careful pipetting.
Add laccase to yield a 3:1 polymer/laccase ratio. Thus, to 375 μl of polymer solution, add 125 μl of laccase to obtain a final volume of 500 μl. Pipette up and down 20 times to mix. Following mixing, immediately move the solution to a 37 °C water bath, continuously swirl it, and then start the timer.
-
Swirl the solution in the water bath and then check the solution every 10 s to look for gelation. When the polymer gels in the tube, subtract a minute from this time and that will be the ‘preincubation time’ for the polymer. This varies by batch and should take between 2 and 8 min.
▲ CRITICAL STEP Determination of this time is critical to estimating the gelation time of the polymer and laccase solution. Proper preincubation time will ensure a homogeneous distribution of cells throughout the volume of the hydrogel.
Repeat Step 22A(i–vi).
Swirl the solution in the water bath for the preincubation time.
-
Immediately take the solution to a 96-well plate and pipette the desired volume into the plate: 45–50 μl for nonhypoxic (control) gels and 90–100 μl for hypoxic gels.
▲ CRITICAL STEP Ensure the use of an experimental control. In general, nonhypoxic hydrogels may be used as controls for variations in O2 tension. However, other experiments may require additional controls.
▲ CRITICAL STEP This step must be done quickly so that gels can be fabricated before gelation occurs.
Place the 96-well plate with gels in the incubator for 20 min.
Add media to the gels: 100 μl for nonhypoxic gels and 200 μl for hypoxic gels.
-
Check the gels to make sure that cells did not sink to the bottom.
? TROUBLESHOOTING
Place the gels back into a 37 °C incubator or alternatively implant the hydrogels in vivo (Box 1, option B).
Change the medium daily for the length of the experiment.
(B) Tumor encapsulation ● TIMING 1–2 h
-
(i)
Generate the desired tumors using your standard laboratory protocol (Fig. 2).
-
(ii)
Slice a tumor to 1-mm thickness using standard laboratory technique.
-
(iii)
Use a 3-mm biopsy punch to create a uniform-sized disk.
-
(iv)
Pipette 5 ml of Gel-HI solution into the 96-well plate to create a hydrogel disk/pad. This is used to keep the tumor off the bottom of the 96-well plate (see polymer and cross-linking preparation).
-
(v)
Place the plate in the incubator at 37 °C for 5 min.
-
(vi)
Place the tissue disk on top of the hydrogel disk/pad bottom.
-
(vii)
Make the desired amount of polymer solution in a separate tube. The solution has a ratio of three parts polymer solution to one part laccase. For 500 μl of Gel-HI solution, add 375 μl of polymer solution and then 125 μl of laccase (as in Step 19 above). Pipette the solution to mix it.
-
(iii)
Add 45 μl of Gel-HI solution to the tumor disks in the 96-well plate to make nonhypoxic gels and 90 μl to make hypoxic gels.
-
(ix)
Place the 96-well plate in a 37 °C incubator for 20 min.
-
(x)
Add medium to the gels: 100 μl for nonhypoxic gels and 200 μl for hypoxic gels.
-
(xi)
Change the medium daily for the length of the experiment.
Figure 2.
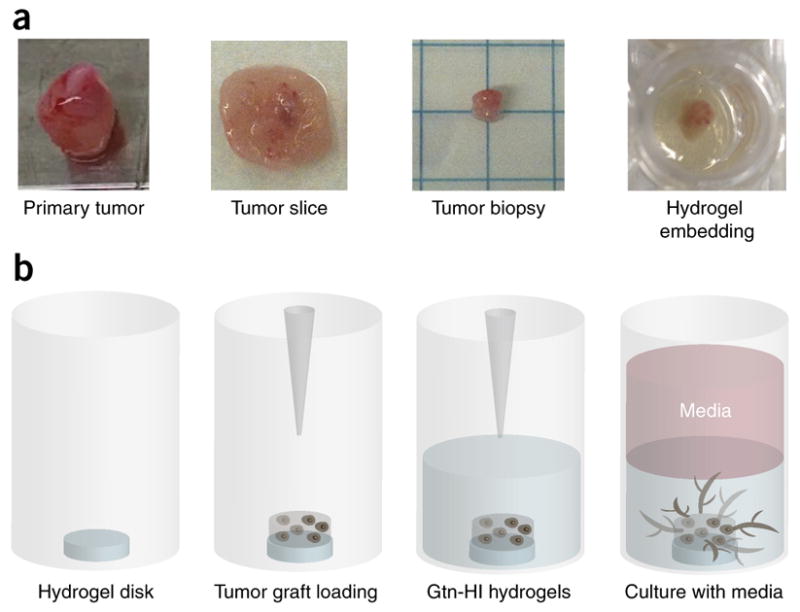
Gtn-HI hydrogel preparation for in vitro 3D tissue/biopsy culture. (a) For 3D tumor encapsulation, the primary tumor is first sectioned by using a custom-made and sterilized Teflon mold (thickness 3 mm) to obtain tumor slices. Then, the slices are stamped by using a biopsy punch (diameter, 3 mm) to prepare small tumor grafts to encapsulate within hydrogels. (b) To encapsulate the tumor biopsy, a Gtn-HI hydrogel disk is prepared, which prevents the attachment and growth of tumors on the bottom of the culture plates. The small tumor graft is placed on the top of the hydrogel disk and the precursor solution is added. The 3D tumor-encapsulated hydrogel construct can be cultured with conditioned medium for up to 14 d. Drawing is not to scale. Adapted with permission from Lewis et al.5, National Academy of Sciences.
(C) Continuous oxygen monitoring in vitro ● TIMING ~2 weeks
▲ CRITICAL STEP All experiments should be conducted in a controlled environment at 37 °C and 5% CO2 in incubators.
-
Immobilize the sensor spot on the cell culture well-plate using either vacuum grease or silicone glue.
▲ CRITICAL STEP Ensure that the sensor spot is immobilized on the center of the well-plate bottom. If the sensor spot is not properly placed, sensor readings will fail to accurately reflect microenvironmental O2 because of the weak optical signal.
Follow option A, pipetting the polymer solution on top of the O2 sensor.
-
Place the cell culture well-plate so that the sensor spot is directly above the O2 microsensor.
▲ CRITICAL STEP Ensure that the sensor spot is fully covered with the polymer solution.
▲ CRITICAL STEP Ensure that the optical signal is >80%. The microsensor and sensor spot should be as close as possible to obtain the best signal.
After 20-min incubation time, add appropriate media volume (100–200 μl).
-
Monitor O2 continuously throughout the experimental time course (up to 2 weeks; (Fig. 3)).
▲ CRITICAL STEP Media changes may be performed as normal. Removing the cell culture well-plate for a brief time will not affect the integrity of the data.
Following the experimental end-point, export data to the analysis software (Microsoft Excel, MATLAB, GraphPad, and so on).
Figure 3.
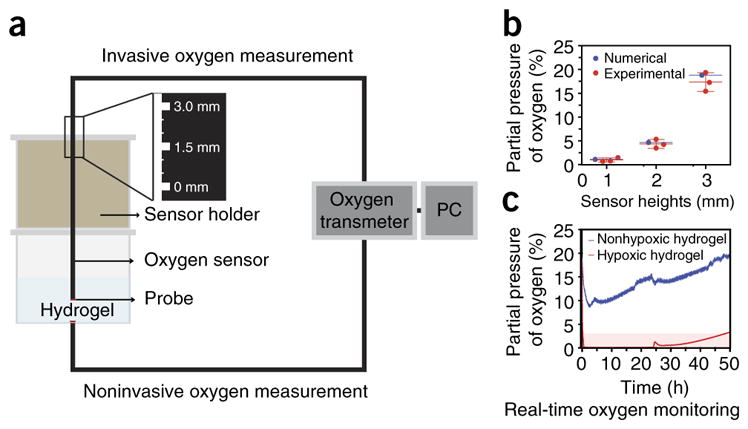
Real-time oxygen monitoring system. (a) To measure oxygen tension and gradients, we use commercially available invasive and noninvasive oxygen sensors. (b) Using invasive oxygen sensors (e.g., needle-type microsensors), we can measure oxygen tension and gradients throughout the hydrogel matrices by controlling the height of the sensors (0–3 mm; mean ± s.d.; n = 3). (c) Using noninvasive oxygen sensors, we can monitor the oxygen tension at the bottom of the hydrogels during culture periods. PC, personal computer. Adapted from with permission from Park and Gerecht38, Nature Publishing Group.
(D) O2 gradient measurement (in vitro) ● TIMING 2–3 h
! CAUTION Needle-type O2 microsensors are extremely fragile. Take extra care when handling microsensors. Although they are protected by needles, the microsensor portion is <50 μm diameter, and thus it may break if subjected to impact.
▲ CRITICAL STEP Needle-type O2 microsensors are an invasive tool for measuring O2 gradients. As such, these measurements may affect the integrity of the hydrogel. Thus, we suggest that gradient measurements serve as the end point of a given experiment.
Fabricate the hydrogel as described (Step 22A or B)
Attach a needle-type O2 microsensor to the manual micromanipulator according to the manufacturer’s instructions for calibration.
Fully extend the LED fiber-optic cable (housed inside the needle) so that is it still protected by the needle bevel, but comes into direct contact with the hydrogel.
Insert the needle through the top of the gel until it makes contact with the bottom of the plate.
-
Beginning at the bottom of the hydrogel, measure O2.
▲ CRITICAL STEP In hydrogels, O2 measurements take time to equilibrate (~15 min). It is critical to achieve a stable reading before moving on to another measurement.
Using the fine-adjustment knob, move the sensor up to a predetermined height to make another measurement. Continue until all measurements have been made.
Further analyses
-
23|
To carry out 3D real-time imaging, follow option A. To measure protease activity, follow option B. To measure cell proliferation and viability, follow option C. For immunofluorescence staining, follow option D, and for RNA extraction follow option E.
(A) 3D real-time imaging ● TIMING 24 h for experiment; 1–2 h for tracking analysis
-
Experimental component. Place the 96-well plate insert on the microscope stage.
▲ CRITICAL STEP For real-time cell imaging experiments, cells that are fluorescently tagged must have been encapsulated in the Gel-HI.
? TROUBLESHOOTING
Warm the incubation chamber to 37 °C and 5% CO2.
Let the microscope warm up for ~1 h.
-
Place the sample on the microscope and let it warm for an additional half hour.
▲ CRITICAL STEP This allows thermal drift on the microscope to be minimized.
Determine the height of z-stack that should be imaged.
-
In each experiment, pick the number of positions that are desired to be imaged. For Zeiss microscopes, pick the center position of the z-stack. The height step of each confocal slice in the z-stack should be 1.5 μm.
▲ CRITICAL STEP This minimizes vertical stretching during tracking analysis.
Set the microscope to image for the desired length of the experiment.
Cell tracking analysis. Using confocal images, track cell trajectories using software such as Imaris spot analysis (Bitplane).
Export cell trajectories data from the cell tracking software into an Excel file.
Analyze with software, such as that created by Wu et al.56.
Plot the data and run statistical analysis using software such as GraphPad Prism.
(B) Assessment of protease activity ● TIMING 1–2 h
▲ CRITICAL STEP When measuring protease activity, cells should not be used if they will interfere with the wavelength of the assay, 495 nm. Turn off lights in the hood while encapsulating cells, as DQ gelatin is light-sensitive.
▲ CRITICAL STEP Add the desired volume of DQ gelatin stock to the polymer/cell mixture at Step 23A(v–vi) so that the final concentration of DQ gelatin in the gel is 10 μg/ml.
-
Check the gels to make sure that cells did not sink to the bottom
? TROUBLESHOOTING
Take the 96-well plate to the fluorescence spectrophotometer and take a baseline read at day 0 with excitation/emission of 495/515 nm.
Put the plate back into the incubator and culture for the desired length of time of the experiment.
On the final day of the experiment, repeat Step 23B(ii).
Subtract day 0 data from the data generated in Step 23B(iv) to get a normalized data set.
Remove the culture medium and add 200 μl of 3.7% (vol/vol) paraformaldehyde (PFA) to each gel.
Image the gels using confocal microscopy to observe protease localization.
(C) Cell proliferation and viability assay ● TIMING 1–2 h
▲ CRITICAL STEP The following steps comprise a modified standard cell proliferation procedure that also assesses the viability of cells encapsulated in the hydrogel. Some modifications may be required, depending on the experimental conditions.
Prepare WST-1 solution as stated in the manufacturer’s protocol.
Aspirate the medium from the 96-well plate.
Add 100 μl of WST-1 solution to nonhypoxic gels and 200 μl to hypoxic gels.
Place gels in the incubator.
Let the solution incubate at 37 °C for 1 h for nonhypoxic gels and 2 h for hypoxic gels.
-
Transfer the WST-1 solution from atop the gels to another 96-well plate after the desired incubation time.
▲ CRITICAL STEP Avoid bubbles when transferring, as they may obscure UV spectroscopy readings.
Stop the reaction with the addition of 1% (vol/vol) Triton X-100 and incubate for 5 min at room temperature.
Add medium to the gels and place them back in the incubator; continue culturing for the desired length of the experiment.
Read the samples on a UV spectrometer according to the manufacturer’s protocol.
Repeat Steps 23C(i–ix) at the desired end point and subtract Day 0 data to obtain final values.
(D) Immunofluorescence staining ● TIMING 1–2 d
▲ CRITICAL STEP Whenever a volume of solution must be removed, use a pipettor rather than a vacuum, as the Gel-HI structure may be altered by the powerful suction of a vacuum.
▲ CRITICAL STEP The following protocol is a modified standard immunofluorescence staining of fixed cell cultures. Some optimization of incubation times for blocking and antibodies may be required, depending on experimental conditions. The following is a suggested starting point, with which we have had success across a range of antibodies and experiments.
-
Fix the hydrogels for 30 min in 3.7% PFA at room temperature. Use the same volume of PFA as medium used in maintaining cells in the hydrogel (e.g., if 200 μl of medium was used, fix the hydrogels in 200 μl of PFA).
? TROUBLESHOOTING
Remove the PFA (and dispose of it in accordance with the relevant local and national guidelines and regulations). Wash the hydrogels with PBS for 10 min at room temperature.
-
Repeat for a total of three washes with PBS.
■ PAUSE POINT Store at 4 °C if you cannot complete the remainder of the procedure immediately. Samples can be stored for up to a week at 4 °C.
-
Add 1% (vol/vol) Triton X-100, and incubate the hydrogels at room temperature for 30 min.
? TROUBLESHOOTING
Remove Triton X-100. Wash the hydrogels with PBS for 10 min at room temperature.
Repeat PBS wash for a total of three washes.
Add 10% (wt/vol) BSA, and incubate the hydrogels at room temperature for 60 min.
Remove the BSA, and wash with 0.05% (vol/vol) Tween/PBS for 10 min at room temperature.
Remove Tween/PBS. Wash the hydrogels with PBS for 10 min at room temperature.
-
Repeat for a total of two washes with PBS.
■ PAUSE POINT Store at 4 °C if you cannot complete the remainder of the procedure immediately. Samples may be stored at 4 °C for ~1–2 weeks.
-
Remove the PBS. Add primary antibody in antibody diluent and incubate overnight at 4 °C.
▲ CRITICAL STEP Primary antibody concentrations on the high end of the manufacturer’s suggestions are recommended for staining hydrogels.
Remove the primary antibody. Wash the hydrogels with PBS for 10 min at room temperature.
Repeat PBS wash for a total of three washes.
-
Remove the PBS. Add secondary antibody in antibody diluent and incubate for 2 h at room temperature.
▲ CRITICAL STEP Protect fluorophores from light.
Remove the secondary antibody. Wash the hydrogels with PBS for 10 min at room temperature.
Repeat PBS wash for a total of three washes.
Remove the PBS. Add nuclear stain (e.g., DAPI). Incubate for 15 min at room temperature.
Remove the nuclear stain. Wash the hydrogels with PBS for 10 min at room temperature.
-
Repeat PBS wash for a total of three washes.
▲ CRITICAL STEP Image quality is greatly improved by transferring the hydrogels to glass-bottom microscopy dishes.
■ PAUSE POINT Samples may be stored for ~1–2 weeks in PBS at 4 °C.
Take images with an available microscope. Confocal or light-sheet microscopy yields the best results for 3D imaging.
(E) RNA extraction ● TIMING 1–2 h
▲ CRITICAL STEP The following protocol is a modified standard RNA extraction of cell cultures, to enable its use in the Gtn-HI hydrogel.
Add 200 μl of TRIzol Reagent to the gel.
Incubate for 5 min at room temperature
Using a pipette tip, mash up the gel.
Transfer the gel and solution to 300 μl of TRIzol Reagent in an Eppendorf tube.
Using a homogenizer, continue to crush the gel and mix the solution. ▲ CRITICAL STEP Make sure to homogenize thoroughly to get the maximum yield of RNA.
Centrifuge the Eppendorf tubes at 12,000g for 10 min at 4 °C.
Continue with the manufacturer’s protocols for RNA extraction.
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 2.
TABLE 2.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 2 | Weight loss of the polymer (incorrect balancing) | Static electricity | Use a static gun (e.g., Milty Zerostat 3 Anti-Static Gun) |
| 20 | Air bubble in the gels | Unsuitable pipette was used | Use a positive-displacement pipette |
| 22A(xiii), 23B(i) | Cells sink to the bottom | Preincubation time is too short | Increase the preincubation time |
| 23A(i) | Inability to obtain clear images of the 96-well plate | A long-working-distance objective was not used | Use a long-working-distance objective or switch to a glass-bottom 96-well plate |
| 23D(i) | Cell structures collapse | Fixation conditions are too harsh | Decrease PFA concentration (from 3.7 to 2%) or decrease the PFA incubation time |
| 23D(iv) | Poor image quality | Permeabilization conditions are too harsh | Decrease Triton X-100 concentration (to between 0.1 and 1% ((vol/vol))) or decrease Triton X-100 incubation time (to between 10 and 20 min) |
● TIMING
Steps 1–8, synthesis and characterization of Gtn–FA polymers: 1–2 weeks
Steps 9–12, validation of polymer: 1–2 h
Steps 13–17, assessment of cytotoxicity of Gtn–FA and laccase (XTT assay): 3–5 d
Step 18, polymer and cross-linker preparation: 1–2 h
Steps 19–22, rheological analysis of Gel-HI hydrogels: 2–3 h to ~2 weeks
Step 22A, cell encapsulation: 1–2 h
Step 22B, tumor encapsulation: 1–2 h
Step 22C, continuous oxygen monitoring in vitro: ~2 weeks
Step 22D, O2 gradient measurement (in vitro): 2–3 h
Step 23A, 3-D real-time imaging: 24 h for experiment; 1–2 h for tracking analysis
Step 23B, assessment of protease activity: 1–2 h
Step 23C, cell proliferation and viability assay: 1–2 h
Step 23D, immunofluorescence staining: 1–2 d
Step 23E, RNA extraction: 1–2 h
Box 1, In vivo procedures: 1–2 weeks
ANTICIPATED RESULTS
The 1H NMR spectra and chemical structure of Gtn–FA are shown in Supplementary Figure 1.
Gel-HI hydrogels provide a uniquely biomimetic microenvironment that can be used for a variety of applications. In particular, the ability to control hypoxic gradients and overall O2 tension may be applied to vascular applications38,43 (Fig. 4). Vasculogenesis and angiogenesis often rely on the hypoxic microenvironment to facilitate the formation or invasion of new blood vessels. We anticipate that Gel-HI hydrogels may provide novel insights into such vascular development and regenerative processes. This knowledge may be used to inform targets for drug discovery, as well as aid in the design of vascularized tissue constructs. Overall, the ability to mimic the physiological, or pathophysiological, microenvironments in vitro will probably provide a platform for continued innovation.
Figure 4.

Tubulogenesis in Gel-HI. (a) Light microscopy images of early vascular cells (EVCs) derived from induced pluripotent stem cells in the Gel-HI hydrogels (hypoxic versus nonhypoxic) encapsulated at 2 million cells per ml. Scale bars, 100 μm. (b) Dissolved oxygen levels measured in EVC-Gtn-HI constructs encapsulated at 2 million cells per ml. Measurements were taken immediately after encapsulation. (c) Fluorescence microscopy images of EVCs after 6 d of culture. Phalloidin (red) merged with DAPI (blue). Scale bars, 500 μm. (d) Quantitative analysis of vascular tube formation. Results are shown as means ± s.d. n = 8 for hypoxic hydrogels; n = 10 for nonhypoxic hydrogels. *P < 0.05; **P < 0.01.
For cancer research, Gel-HI hydrogels provide a powerful platform for studying single-cell responses to hypoxic gradients, such as matrix remodeling and cell migration5 (Fig. 5). As a display of the potential breadth of study, cellular mechanisms can be analyzed with standard cell and molecular biology techniques. The Gel-HI hydrogels can also provide a platform for analyzing tumor biopsies that can be grafted into the gel (Fig. 2) to analyze cell migration out of the graft into the surrounding hydrogel, as an in vitro corollary to tumor growth, migration, and potential metastasis (Fig. 6). As a tool in cancer biology, Gel-HI hydrogels may identify novel pathways for tumor growth and migration induced only in hypoxic conditions, as well as provide a method of screening cancer therapeutics and determining potential pitfalls or unintended consequences that cannot be studied with standard in vitro platforms.
Figure 5.

Tumor encapsulation and tracking. (a) (i) Sarcoma cells encapsulated in Gel-HI on day 3. (ii) Images of GFP-tagged sarcoma cells encapsulated in the hydrogel on day 3. Scale bars, 25 μm. (iii) Invasive oxygen measurements of KIA cells encapsulated in the Gel-HI hydrogel. (b) 3D projection of cell trajectories to determine migration of GFP cells. (c–e) Overall speed (c), velocity (d), and mean squared displacement (MSD; e) were calculated (in the x, y, and z directions (d,e)). Results are shown as the average of all cells tracked ± s.e.m. Significance levels were evaluated using a t-test: *P < 0.05; ^P < 0.01; #P < 0.001. Adapted with permission from Lewis et al.5, National Academy of Sciences.
Figure 6.

Tumor encapsulation and tracking. (a) Noninvasive oxygen readings of engrafted tumors in the Gel-HI hydrogel. (b) Light microscopy (left) and confocal microscopy (right) images of GFP-tagged sarcoma tumors encapsulated in the hydrogel on day 3. Scale bars, 25 μm. (c) 3D projection of cell trajectories to determine migration of GFP cells. (d,e) Overall speed (d) and mean squared displacement (e) were calculated in the x, y, and z directions (e). Results are shown as the average of all cells tracked ± s.e.m. Significance levels were evaluated using a t-test: *P < 0.05; ^P < 0.01; #P < 0.001. Adapted with permission from Lewis et al.5, National Academy of Sciences.
During in vivo implantation of the hydrogel, real-time O2 measurements can be made using the needle-type O2 sensor (Fig. 7). After implantation of the hydrogel, an analysis of the vasculature can be performed to look at vascular recruitment to the wound, as well as the number of vessels (Fig. 8)27. If proper O2 tension is maintained, recruitment should be clear under histological analysis and fluorescence staining. The translational potential of Gel-HI hydrogels is immense, as hypoxia governs production of many signaling molecules that may influence cell behavior.
Figure 7.
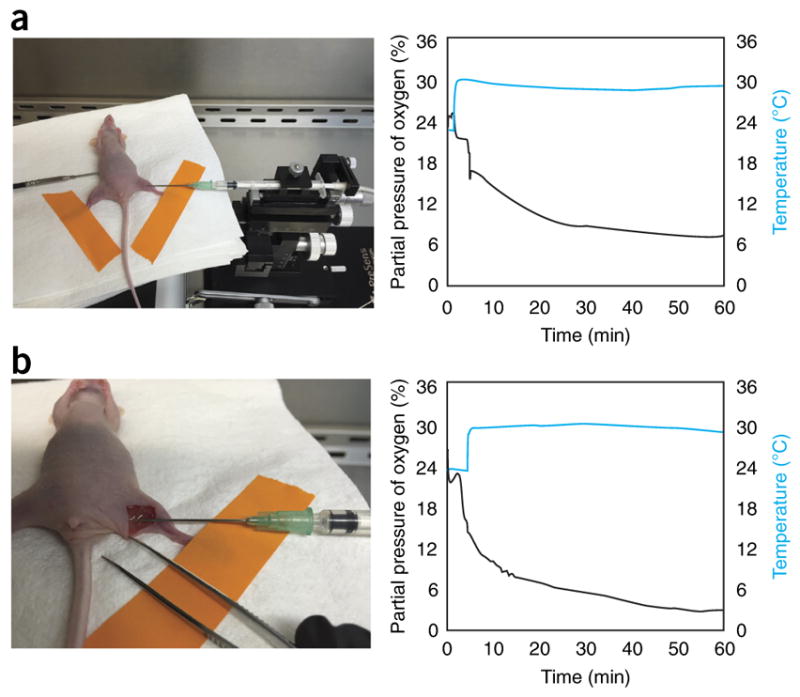
Subcutaneous and intramuscular in vivo oxygen sensing. Commercially available needle-type oxygen sensors (PreSens) were used to measure partial pressure of oxygen over time both (a) subcutaneously and (b) intramuscularly in 10- to 12-week-old nude mice. Sensors remained stationary and readings equilibrated after ~60 min to 7.4 and 3.0% in the subcutaneous and intramuscular regions, respectively. A temperature probe was used concomitantly to ensure the accuracy of the oxygen readings. The animal study was performed using a protocol (MO16A284) approved by The Johns Hopkins University Institutional Animal Care and Use Committee.
Figure 8.

Application of Gtn-HI hydrogels for in vivo vascular tissue recruitment. The Gtn-HI hydrogels showed in vivo angiogenic effects. (a) Histological sections of hydrogels 1, 3, and 5 d after injection stained with CD31. Right-hand images are close-ups of areas outlined by the boxes in the left-hand images. (b) Blood vessel perfusion within the Gtn-HI hydrogels. Perfused blood vessels infiltrated the hydrogel matrices 5 d after injection. GS-IB4 lectin in red and nuclei in blue. Scale bars, 100 μm. Adapted with permission from Park and Gerecht38, Nature Publishing Group. The animal study was performed using a protocol (RA15A152 or MO15A154) approved by The Johns Hopkins University Institutional Animal Care and Use Committee. MO, mouse protocol; RA, rat protocol.
Overall, the Gel-HI hydrogels have a great potential either as 3D artificial hypoxic cellular microenvironments or as hypoxia-inducible acellular matrices for the treatment of vascular disorders. The HI hydrogels also provide an innovative platform as an engineered tumor microenvironment in which to study basic cancer biology and to screen drug candidates for better clinical outcomes in cancer treatment. In addition to the applications discussed, looking forward, we anticipate that the use of Gel-HI hydrogels will provide a new platform for innovation in the stem cell and developmental biology fields. We anticipate that studying the effects of developmentally relevant O2 gradients will lead to conclusions previously unattainable with available research tools. Such revelations may lead to advances in tissue engineering and regenerative medicine, as well as accelerate the understanding of the role of hypoxic gradients in development and tissue regeneration.
Supplementary Material
Acknowledgments
This work was supported by a fellowship from the National Cancer Institute (NCI; T-32 2T32CA153952-06), a Nanotechnology Cancer Research training grant (to D.M.L.), the American Heart Association (15EIA22530000), a President’s Frontier Award from Johns Hopkins University, and Project 3 of the NCI Physical Sciences-Oncology Center (U54CA210173 to S.G.).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS D.M.L., M.R.B., and K.M.P. developed, tested, applied, and validated the Gel-HI hydrogel system; D.M.L., M.R.B., and K.M.P. performed the experiments; D.M.L., M.R.B., K.M.P., and S.G. designed the experiments, analyzed the results, and wrote the paper.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Brizel DM, et al. Tumor oxygenation predicts for the likelihood of distant metastases in human soft tissue sarcoma. Cancer Res. 1996;56:941–943. [PubMed] [Google Scholar]
- 2.Mori R, et al. The relationship between proangiogenic gene expression levels in prostate cancer and their prognostic value for clinical outcomes. Prostate. 2010;70:1692–1700. doi: 10.1002/pros.21204. [DOI] [PubMed] [Google Scholar]
- 3.Koukourakis MI, et al. Hypoxia-inducible factor (HIF1A and HIF2A), angiogenesis, and chemoradiotherapy outcome of squamous cell head-and-neck cancer. Int J Radiat Oncol Biol Phys. 2002;53:1192–1202. doi: 10.1016/s0360-3016(02)02848-1. [DOI] [PubMed] [Google Scholar]
- 4.Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8:967–975. doi: 10.1038/nrc2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lewis DM, et al. Intratumoral oxygen gradients mediate sarcoma cell invasion. Proc Natl Acad Sci. 2016;113:9292–9297. doi: 10.1073/pnas.1605317113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chaturvedi P, et al. Hypoxia-inducible factor-dependent breast cancer-mesenchymal stem cell bidirectional signaling promotes metastasis. J Clin Invest. 2013;123:189–205. doi: 10.1172/JCI64993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisinger-Mathason TK, et al. Hypoxia-dependent modification of collagen networks promotes sarcoma metastasis. Cancer Discov. 2013;3:1190–1205. doi: 10.1158/2159-8290.CD-13-0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carmeliet P, et al. Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- 9.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 10.Kvanta A, Algvere P, Berglin L, Seregard S. Subfoveal fibrovascular membranes in age-related macular degeneration express vascular endothelial growth factor. Invest Ophthalmol Vis Sci. 1996;37:1929–1934. [PubMed] [Google Scholar]
- 11.Lin CH, et al. Silibinin inhibits VEGF secretion and age-related macular degeneration in a hypoxia-dependent manner through the PI-3 kinase/Akt/mTOR pathway. Br J Pharmacol. 2013;168:920–931. doi: 10.1111/j.1476-5381.2012.02227.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stefánsson E, Geirsdóttir Á, Sigurdsson H. Metabolic physiology in age related macular degeneration. Prog Ret Eye Res. 2011;30:72–80. doi: 10.1016/j.preteyeres.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 13.Lewis DM, Abaci HE, Xu Y, Gerecht S. Endothelial progenitor cell recruitment in a microfluidic vascular model. Biofabrication. 2015;7:045010. doi: 10.1088/1758-5090/7/4/045010. [DOI] [PubMed] [Google Scholar]
- 14.Arnould T, Michiels C, Remacle J. Increased PMN adherence on endothelial cells after hypoxia: involvement of PAF, CD18/CD11b, and ICAM-1. Am J Physiol. 1993;264:C1102–1110. doi: 10.1152/ajpcell.1993.264.5.C1102. [DOI] [PubMed] [Google Scholar]
- 15.Luster AD, Alon R, von Andrian UH. Immune cell migration in inflammation: present and future therapeutic targets. Nat Immunol. 2005;6:1182–1190. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- 16.Craig R, Schofield J, Jackson S. Collagen biosynthesis in normal human skin, normal and hypertrophic scar and keloid. Eur J Clin Invest. 1975;5:69–74. doi: 10.1111/j.1365-2362.1975.tb00430.x. [DOI] [PubMed] [Google Scholar]
- 17.Botusan IR, et al. Stabilization of HIF-1α is critical to improve wound healing in diabetic mice. Proc Natl Acad Sci. 2008;105:19426–19431. doi: 10.1073/pnas.0805230105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramirez-Bergeron DL, Simon MC. Hypoxia-inducible factor and the development of stem cells of the cardiovascular system. Stem Cells. 2001;19:279–286. doi: 10.1634/stemcells.19-4-279. [DOI] [PubMed] [Google Scholar]
- 19.Licht AH, Muller-Holtkamp F, Flamme I, Breier G. Inhibition of hypoxia-inducible factor activity in endothelial cells disrupts embryonic cardiovascular development. Blood. 2006;107:584–590. doi: 10.1182/blood-2005-07-3033. [DOI] [PubMed] [Google Scholar]
- 20.Krock BL, Skuli N, Simon MC. Hypoxia-induced angiogenesis good and evil. Genes Cancer. 2011;2:1117–1133. doi: 10.1177/1947601911423654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kusuma S, Peijnenburg E, Patel P, Gerecht S. Low oxygen tension enhances endothelial fate of human pluripotent stem cells. Arterioscler, Thromb, Vasc Biol. 2014;34:913–920. doi: 10.1161/ATVBAHA.114.303274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee YM, et al. Determination of hypoxic region by hypoxia marker in developing mouse embryos in vivo: a possible signal for vessel development. Dev Dyn. 2001;220:175–186. doi: 10.1002/1097-0177(20010201)220:2<175::AID-DVDY1101>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 23.Liu Y, Cox SR, Morita T, Kourembanas S. Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells identification of a 5′ enhancer. Circ Res. 1995;77:638–643. doi: 10.1161/01.res.77.3.638. [DOI] [PubMed] [Google Scholar]
- 24.Covello KL, Simon MC. HIFs, hypoxia, and vascular development. Curr Top Dev Biol. 2004;62:37–54. doi: 10.1016/S0070-2153(04)62002-3. [DOI] [PubMed] [Google Scholar]
- 25.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 26.Marti HJ, et al. Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am J Pathol. 2000;156:965–976. doi: 10.1016/S0002-9440(10)64964-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oppegard SC, Nam KH, Carr JR, Skaalure SC, Eddington DT. Modulating temporal and spatial oxygenation over adherent cellular cultures. PLoS One. 2009;4:8. doi: 10.1371/journal.pone.0006891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rexius-Hall ML, Mauleon G, Malik AB, Rehman J, Eddington DT. Microfluidic platform generates oxygen landscapes for localized hypoxic activation. Lab Chip. 2014;14:4688–4695. doi: 10.1039/c4lc01168f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adler M, Polinkovsky M, Gutierrez E, Groisman A. Generation of oxygen gradients with arbitrary shapes in a microfluidic device. Lab Chip. 2010;10:388–391. doi: 10.1039/b920401f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen YA, et al. Generation of oxygen gradients in microfluidic devices for cell culture using spatially confined chemical reactions. Lab Chip. 2011;11:3626–3633. doi: 10.1039/c1lc20325h. [DOI] [PubMed] [Google Scholar]
- 31.Funamoto K, et al. A novel microfluidic platform for high-resolution imaging of a three-dimensional cell culture under a controlled hypoxic environment. Lab Chip. 2012;12:4855–4863. doi: 10.1039/c2lc40306d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oppegard SC, Eddington DT. A microfabricated platform for establishing oxygen gradients in 3-D constructs. Biomed Microdevices. 2013;15:407–414. doi: 10.1007/s10544-013-9737-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DelNero P, et al. 3D culture broadly regulates tumor cell hypoxia response and angiogenesis via pro-inflammatory pathways. Biomaterials. 2015;55:110–118. doi: 10.1016/j.biomaterials.2015.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodenhizer D, et al. A three-dimensional engineered tumour for spatial snapshot analysis of cell metabolism and phenotype in hypoxic gradients. Nat Mater. 2016;15:227–234. doi: 10.1038/nmat4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu D, Yotnda P. Induction and testing of hypoxia in cell culture. J Vis Exp. 2011;(54):2899. doi: 10.3791/2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin X, et al. Oxygen-induced cell migration and on-line monitoring biomarkers modulation of cervical cancers on a microfluidic system. Sci Rep. 2015;5:9643. doi: 10.1038/srep09643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allen CB, Schneider BK, White CW. Limitations to oxygen diffusion and equilibration in in vitro cell exposure systems in hyperoxia and hypoxia. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1021–L1027. doi: 10.1152/ajplung.2001.281.4.L1021. [DOI] [PubMed] [Google Scholar]
- 38.Park KM, Gerecht S. Hypoxia-inducible hydrogels. Nat Commun. 2014;5:4075. doi: 10.1038/ncomms5075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanjaya-Putra D, et al. Controlled activation of morphogenesis to generate a functional human microvasculature in a synthetic matrix. Blood. 2011;118:804–815. doi: 10.1182/blood-2010-12-327338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davis GE, Senger DR. Endothelial extracellular matrix - biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ Res. 2005;97:1093–1107. doi: 10.1161/01.RES.0000191547.64391.e3. [DOI] [PubMed] [Google Scholar]
- 41.Davis GE, Bayless KJ, Mavila A. Molecular basis of endothelial cell morphogenesis in three-dimensional extracellular matrices. Anat Rec. 2002;268:252–275. doi: 10.1002/ar.10159. [DOI] [PubMed] [Google Scholar]
- 42.Chan XY, et al. Three-dimensional vascular network assembly from diabetic patient-derived induced pluripotent stem cells. Arterioscler, Thromb, Vasc Biol. 2015;35:2677–2685. doi: 10.1161/ATVBAHA.115.306362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blatchley M, Park KM, Gerecht S. Designer hydrogels for precision control of oxygen tension and mechanical properties. J Mater Chem B. 2015;3:7939–7949. doi: 10.1039/C5TB01038A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Higuchi A, Ling QD, Hsu ST, Umezawa A. Biomimetic cell culture proteins as extracellular matrices for stem cell differentiation. Chem Rev. 2012;112:4507–4540. doi: 10.1021/cr3000169. [DOI] [PubMed] [Google Scholar]
- 45.Paguirigan AL, Beebe DJ. Protocol for the fabrication of enzymatically crosslinked gelatin microchannels for microfluidic cell culture. Nat Protoc. 2007;2:1782–1788. doi: 10.1038/nprot.2007.256. [DOI] [PubMed] [Google Scholar]
- 46.Yung CW, et al. Transglutaminase crosslinked gelatin as a tissue engineering scaffold. J Biomed Mater Res Part A. 2007;83A:1039–1046. doi: 10.1002/jbm.a.31431. [DOI] [PubMed] [Google Scholar]
- 47.Loessner D, et al. Functionalization, preparation and use of cell-laden gelatin methacryloyl-based hydrogels as modular tissue culture platforms. Nat Protoc. 2016;11:727–746. doi: 10.1038/nprot.2016.037. [DOI] [PubMed] [Google Scholar]
- 48.Minussi RC, Pastore GM, Duran N. Potential applications of laccase in the food industry. Trends Food Sci Technol. 2002;13:205–216. [Google Scholar]
- 49.Mozaffarian D, et al. Heart disease and stroke statistics-2016 update: a report from the American Heart Association. Circulation. 2016;133:e38. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 50.Gaengel K, Genove G, Armulik A, Betsholtz C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol. 2009;29:630–638. doi: 10.1161/ATVBAHA.107.161521. [DOI] [PubMed] [Google Scholar]
- 51.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Semenza GL. Vascular responses to hypoxia and ischemia. Arterioscler Thromb Vasc Biol. 2010;30:648–652. doi: 10.1161/ATVBAHA.108.181644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bayless KJ, Kwak HI, Su SC. Investigating endothelial invasion and sprouting behavior in three-dimensional collagen matrices. Nat Protoc. 2009;4:1888–1898. doi: 10.1038/nprot.2009.221. [DOI] [PubMed] [Google Scholar]
- 54.Yoon SS, et al. Efficacy of sunitinib and radiotherapy in genetically engineered mouse model of soft-tissue sarcoma. Int J Radiat Oncol Biol Phys. 2009;74:1207–1216. doi: 10.1016/j.ijrobp.2009.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 55.Kirsch DG, et al. A spatially and temporally restricted mouse model of soft tissue sarcoma. Nat Med. 2007;13:992–997. doi: 10.1038/nm1602. [DOI] [PubMed] [Google Scholar]
- 56.Wu PH, Giri A, Wirtz D. Statistical analysis of cell migration in 3D using the anisotropic persistent random walk model. Nat Protoc. 2015;10:517–527. doi: 10.1038/nprot.2015.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koh MY, Powis G. Passing the baton: the HIF switch. Trends Biochem Sci. 2012;37:364–372. doi: 10.1016/j.tibs.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.