

Abstract
Memory CD8 T cells that circulate in the blood and are present in lymphoid organs are an essential component of long-lived T cell immunity. These resting memory CD8 T cells remain poised to rapidly elaborate effector functions upon re-exposure to pathogen, but also have many properties in common with naïve cells, including the ability to migrate to lymph nodes and spleen, and their pluri-potency. Thus, memory cells embody features of both naïve and effector cells, fueling a long-standing debate centered on whether memory T cells develop from effector cells or directly from naïve cells1–4. To better define the developmental path of memory CD8 T cells we investigated changes in DNA methylation programming at naïve and effector genes in virus specific CD8 T cells during acute LCMV infection of mice. Methylation profiling of effector CD8 T cell subsets at day 4 and 8 after infection showed that, rather than retaining a naïve epigenetic state, the subset of cells that gives rise to memory cells acquired de novo DNA methylation programs at naïve-associated genes and became demethylated at loci of classically defined effector molecules. Conditional deletion of the de novo methyltransferase, Dnmt3a, at an early stage of effector differentiation strikingly reduced methylation of naïve-associated genes and resulted in faster re-expression of these naïve genes, accelerating memory cell development. Longitudinal phenotypic and epigenetic characterization of virus-specific memory-precursor CD8 T cells transferred into antigen-free mice revealed that their differentiation into memory cells was coupled to cell-division independent erasure of de novo methylation programs and re-expression of naïve-associated genes. These data provide evidence that epigenetic repression of naïve-associated genes in effector CD8 T cells can be reversed in cells that develop into long-lived memory CD8 T cells supporting a differentiation model where memory T cells arise from a subset of fate-permissive effector T cells.
We used the mouse model of acute LCMV infection to examine the transcriptional and epigenetic changes that occur as naïve CD8 T cells differentiate into effector and memory cells. It is well established that many effector genes are turned on when naïve CD8 T cells are stimulated by antigen but it is less well appreciated that several genes expressed by naïve t cells are also turned off upon T cell activation5,6. Interestingly, several of these naïve genes that are downregulated in effector CD8 T cells are expressed by central memory cells. This on-off-on pattern of gene expression is shown for LCMV-specific effector and memory CD8 T cells in Fig. 1a. Among the genes that show this pattern are L-selectin (CD62L) (Fig. 1b) and CCR7 that are needed for homing to lymphoid organs and Bcl-2 and CD127 that are important for long-term survival of memory T cells6,7. To examine epigenetic changes associated with this on-off-on pattern we analyzed DNA methylation profile of the CD62L promoter. Previous studies have defined CpG sites in the CD62L promoter region proximal to the binding sites for Klf2 and Ets1, the two transcription factors known to regulate CD62L expression (Extended data 1a)8,9. To determine if methylation status of these CpG sites has a direct impact on gene expression, we used a reporter construct to show that these CpG sites indeed regulate L-selectin expression (Extended data 1b,1c). Having established that methylation of these CpG sites decreases CD62L expression in vitro, we next examined the methylation status of these sites in LCMV-specific naïve, effector, and memory P14 CD8 T cells during acute LCMV infection in vivo (Fig.1c). Consistent with the high level of CD62L transcription in naïve CD8 T cells, the CpG sites proximal to the CD62L promoter were completely unmethylated in naïve P14 cells whereas the CD62L promoter was significantly methylated in both day 4 and day 8 LCMV specific effector CD8 T cells that did not express L-selectin (Extended data 1d). Interestingly, memory P14 cells showed minimal methylation at this promoter site and in accordance with this permissive epigenetic state there was expression of CD62L message (Fig.1c, Extended data 1d,1e). However, since >95% of the effector CD8 T cells undergo apoptosis it is possible that these surviving CD62L positive memory P14 cells may have never gotten methylated during the effector phase of the T cell response. The pool of effector CD8 T cells consists of two subsets; the majority (95%) are terminal effectors (TE) that are destined to die and the minority (5%) subset of effector cells, termed memory precursors (MP), survive to give rise to the pool of long-lived memory T cells5. These two subsets can be distinguished on the basis of their expression of cell surface markers Klrg1 and CD12710–12. So, we analyzed the TE and MP effector subsets at day 8 and quite strikingly both subsets were equally methylated at the CD62L promoter region and they also expressed low levels of CD62L message (Fig.1d, Extended data 1f,1g). We next analyzed memory cells at day 37 and found that the CD62L high population was significantly demethylated and expressed high levels of message (Fig.1e, Extended data 1f,1h). Taken together these results document that the MP effector CD8 T cell subset, which gives rise to memory cells, also gets methylated at the CD62L promoter during the acute phase of infection.
Figure 1. Dynamic changes in DNA methylation during effector and memory CD8 T cell differentiation.
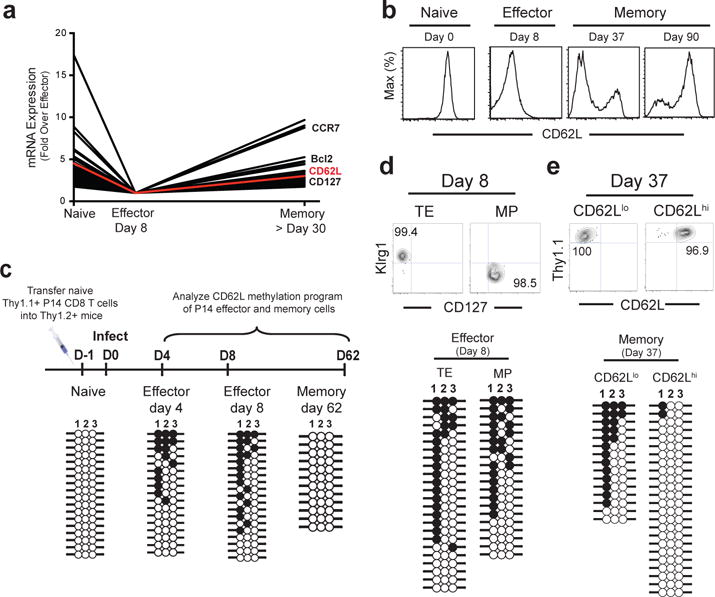
A) Analysis of on-off-on gene expression from published naïve, and LCMV-specific effector, and memory CD8 T cell microarray data sets6. B) Histogram analysis of CD62L protein expression on naïve, effector and memory P14 CD8 T cells following acute LCMV infection. C) Experimental setup and bisulfite sequencing DNA methylation analysis of the CD62L promoter from P14 CD8 T cell purified (>95% purity) at the indicated stages of differentiation. Each horizontal line represents an individual sequenced clone. Filled circles = methylated cytosine. Open circles = non-methylated cytosine. Representative post-sort purity and DNA methylation analysis of the CD62L proximal promoter CpG sites among D) terminal effector (TE) and memory precursor (MP) P14 CD8 T cells FACS purified 8 days after acute LCMV infection, or E) effector-memory (Tem) and central memory (Tcm) P14 CD8 T cells FACS purified 37 days after acute LCMV infection.
To get a more comprehensive assessment of methylation changes during naïve to effector differentiation we performed whole-genome bisulfite sequencing (WGBS) of antigen-specific MP and TE CD8 T cell subsets at day 4.5 and day 8.15 (Extended data Fig.2a, 2b). Both MP and TE effector subsets showed an increase in DNA methylation at about 1000 regions relative to naïve cells with a striking enrichment of these methylation events near or within genes (Fig.2a, Extended data 3a). The majority of these gain-of-methylation events occurred within the first 4 days of the effector response with > 50% of these newly methylated regions being similarly acquired in both TE and MP effector cells. These DMRs included the previously defined sites in the CD62L promoter as well as several other naïve-associated genes such as Ccr7 and Tcf7 (Fig.2b). Thus, the MP effector CD8 T cell subset, which is the precursor to memory cells, acquires repressive DNA methylation marks at many genes expressed by naïve cells. Interestingly, the MP subset not only methylated naïve genes but also demethylated several effector genes such as perforin, granzymes and interferon-gamma (Fig.2c,2d, Extended data 4a,4b,4c). These results are consistent with a model where MP cells transition through an effector phase during their differentiation into memory cells. It is important to note that while there is a high degree of overlap between TE and MP subsets in effector associated programming there were also some interesting differences in the level of demethylation (Extended data 4d). Of particular interest is a DMR in the Blimp1 (Prdm 1) locus that remains methylated in the MP subset. This is consistent with prior reports showing that MP cells have reduced Blimp1 expression relative to the TE subset and that deletion of Blimp 1 increases the number of effector cells with memory potential13. We also detected differential methylation of Runx2 and Runx3 further suggesting that MP and TE fates are coupled to epigenetic programming of transcriptional regulators.
Figure 2. Memory-precursor CD8 T cells acquire genome-wide effector-associated DNA methylation programs.
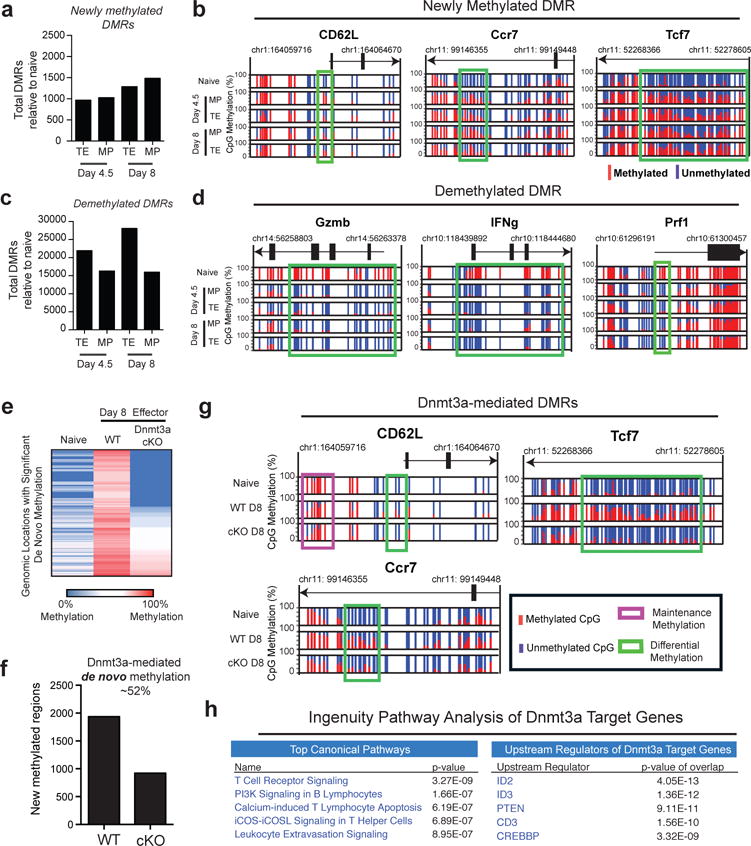
A) Summary graph of the number of newly methylated DMRs in TE and MP subsets relative to naïve cells identified from WGBS analyses. B) Normalized graph of CpG methylation in the Ccr7, Tcf7, and CD62L loci from WGBS data sets. Each vertical line indicates a CpG site and the ratio of red to blue indicate the % of methylated versus unmethylated CpGs at these sites. C) Summary graph of the number of demethylation DMRs between the effector subsets and naïve cells. D) Normalized graph of methylation at CpG sites Gzmb, IFNg, and Prf1 loci from TE and MP WGBS data sets. E) Heat-map representation of top 3000 newly methylated regions (relative to naïve CD8 T cell methylation) from WGBS analysis of tetramer+ WT and Dnmt3a cKO effector CD8 T cell. F) Summary graph of de novo methylated regions in WT and Dnmt3a cKO effector. G) Normalized graph of Dnmt3a-mediated de novo methylation at CpG sites in the CD62L, Ccr7, and Tcf7 loci. H) Top Canonical Pathways and Upstream Regulators from Ingenuity Pathway Analysis of gene-associated Dnmt3a-mediated DMRs.
Having determined that epigenetic repression of naïve-associated genes is a shared feature of both MP and TE CD8 T cell subsets we next asked which enzyme(s) are involved in this DNA methylation. We hypothesized that this would be a de novo process based on the rapid methylation and because this methylation was retained even while the CD8 T cells were rapidly dividing (10-15 divisions) during the first week after infection14. We have previously shown increased expression of the de novo methyltransferase Dnmt3a upon CD8 T cell activation15 and therefore tested whether it was required for methylation of the CD62L promoter. To do this we used Dnmt3a-granzyme b–cre mice to conditionally knock out Dnmt3a in virus-specific CD8 T cells following T cell activation in vivo (Extended data 5a)16,17. These Dnmt3a cKO mice were infected with LCMV and day 8 virus-specific effector CD8 T cells were sorted using the gp33 tetramer (Extended data 5b). PCR analysis of the Dnmt3a locus confirmed successful deletion of the enzyme (Extended data 5c). We then assessed the methylation status of WT and cKO gp33 tetramer positive effector CD8 T cells and found that in the absence of Dnmt3a there was no de novo methylation of the CD62L promoter (Extended data 5d). However, loss of Dnmt3a did not have any effect on maintenance methylation of a set of CpG sites distal to the CD62L promoter (Extended data 5e and 5f).
To more extensively profile loci targeted for de novo DNA methylation in effector cells, we performed WGBS with DNA from WT and cKO antigen-specific effector CD8 T cells (Fig.2e, 2f, 2g). Comparison of naïve and WT effector WGBS datasets revealed that the total pool of effector cells acquired ~2000 newly methylated regions (Fig.2e). Of these newly methylated regions, ~1000 were verified as targets of Dnmt3a as they were not acquired in the cKO effector cells (Fig.2f). Many of these Dnmt3a targets were naïve genes such as CD62L, CCR7, and Tcf7 loci (Fig.2g). Additionally, several genes associated with regulating T cell lineage commitment and differentiation, including Lef1 and IL6ST, were also targets for effector-stage de novo methylation (Extended data 6a). Unlike the striking effect on de novo methylation the deletion of Dnmt3a did not have any effect on maintenance DNA methylation (Extended data 6b). Given the selective nature of the Dnmt3a targeted loci we next assessed these genes for potential connections using Ingenuity Pathway Analysis (Fig.2h). Among the top canonical pathways identified by IPA were gene networks that were related to T cell receptor signaling and differentiation, further documenting the relationship between Dnmt3a-mediated programming and effector differentiation. Interestingly, our analysis revealed that loci targeted for de novo methylation were significantly associated with the ID2 and ID3 transcriptional axis, two well-established regulators of effector and memory T cell differentiation (Extended data 6c)18,19. Collectively, these results demonstrate that downregulation of naïve-associated genes is coupled to acquisition of Dnmt3a mediated de novo DNA methylation programs during development of both MP and TE cells.
We next asked how does Dnmt3a mediated de novo methylation regulate the generation and differentiation of effector and memory CD8 T cells in vivo. To address this question we performed longitudinal analysis of the magnitude and phenotype of virus specific CD8 T cells following LCMV infection of WT and Dnmt3a cKO mice. We found that the magnitude of the virus specific CD8 T cell response was similar in WT and cKO mice; there were equivalent numbers of LCMV specific CD8 T cells in multiple tissues at both effector and memory time points and both groups of mice controlled the viral infection (Fig.3a, Extended data 7a,7b,7c). We confirmed that the memory cells present in the cKO mice were indeed deficient in Dnmt3a and did not represent an outgrowth of WT cells (Extended data 7d). Thus, the magnitude of the virus specific CD8 T cell response was not impacted by Dnmt3a mediated de novo methylation. Longitudinal analysis of phenotypic markers showed that CD62L and CD127 were downregulated by both WT and cKO day 8 effector cells (Fig.3a, 3b, Extended data 7e). However, following viral clearance, re-expression of CD62L occurred at a significantly earlier time point in the Dnmt3a deficient virus-specific CD8 T cells compared to WT cells (Fig.3b). Dnmt3a cKO cells also showed enhanced conversion to central memory phenotype CD8 T cells compared to WT CD8 T cells (Fig.3c). Similar to the Dnmt3a deficient cells in the PBMC, cKO cells in the spleen and lymph nodes had significantly higher levels of CD62L expression and central memory cells compared to WT mice (Fig.3d, Extended data 7f). Not only did Dnmt3a cKO mice have significantly more CD62L+ memory CD8 T cells in lymphoid tissues, but they also had a greater number of CD62L+ memory CD8 T cells in nonlymphoid tissues (lung and liver) (Fig.3d). Additionally, the enhanced expression of CD62L was also observed with LCMV-specific CD8 T cells recognizing the gp276 and np396 epitopes of the virus (Extended data 7g). We next proceeded to determine if the faster re-expression of naïve-associated genes in the Dnmt3a cKO cells was due to a cell intrinsic mechanism. To address this issue Dnmt3a cKO mice were crossed with the P14 transgenic mouse to generate Dnmt3a cKO CD8 T cells with a TCR specific to the LCMV GP33 epitope. These Dnmt3a cKO naïve P14 cells, along with congenically distinct WT naïve P14 cells, were co-transferred into WT mice and these recipient mice were then acutely infected with LCMV (Fig.3e). Dnmt3a cKO P14 cells downregulated CD62L expression similar to WT P14 cells at 8 days post infection but they re-expressed CD62L much faster than WT P14 cells (Fig.3e). In addition, the Dnmt3a cKO P14 cells also exhibited an increased rate of conversion to central memory cells (CD127+ CD62L+ CD27+ Klrg1lo) compared to WT P14 cells (Fig.3f). Thus, these results show that Dnmt3a intrinsically regulates the execution of the memory CD8 T cell program.
Figure 3. Dnmt3a mediated de novo DNA methylation regulates the kinetics of gene re-expression during the effector to memory CD8 T cell transition.
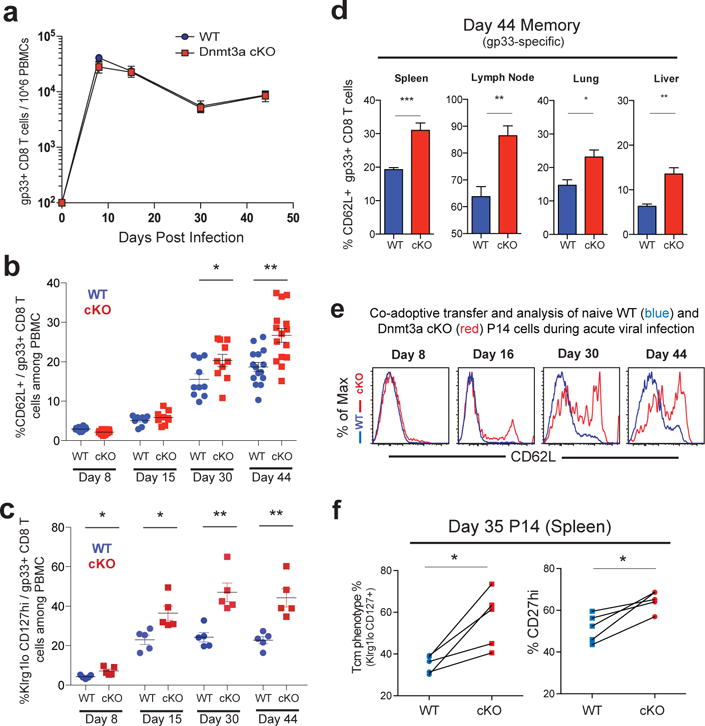
A) Longitudinal measurement of WT and Dnmt3a cKO gp33-specific CD8 T cell numbers among PBMC during acute LCMV infection. B) Longitudinal analysis of central-memory (CD62Lhi) and C) memory-precursor (Klrg1lo CD127hi) phenotypes of gp33-specific WT and Dnmt3 cKO CD8 T cells in PBMCs (n ≥ 5). D) Graphs of the %CD62L positive WT and Dnmt3a cKO gp33-specific memory CD8 T cells in the spleen and lymph node at 44 dpi. E) Representative histogram analysis of CD62L expression on WT (blue line) and Dnmt3a cKO (red line) P14 cells from the PBMC acutely infected chimeric animals at 8, 16, 30, and 44 days post infection. F) Paired analysis of Klrg1lo CD127hi and CD27hi phenotypes on WT and Dnmt3a cKO P14 splenocytes at 35 days post infection from co-transfer experiments (N= 5).
Since Dnmt3a cKO cells showed faster conversion to memory cells we asked to what extent did the cKO CD8 T cells undergo effector differentiation. We first assessed our whole-genome methylation analysis of day 8 cKO antigen-specific CD8 T cells for DNA demethyaltion events at effector-associated loci, including IFNg, Perforin, and GzmK. Similar to the level of demethylation we observed in WT MP and TE cells, these loci were fully demethylated in the day 8 cKO CD8 T cells demonstrating that they HAD acquired effector-associated epigenetic programs (Extended data 8a,8b). The day 8 cKO virus specific CD8 T cells ALSO expressed WT levels of Tbet and Ki67 providing further evidence that the cKO cells had mounted an effector response (Extended data 8c). Furthermore, the cKO effector and memory cells were able to rapidly express effector cytokines upon ex vivo peptide stimulation (Extended data 8d). These results explain the viral clearance data shown in Supplemental Figure 6b where we observed similar viral control in WT and Dnmt3a cKO mice.
Our results so far document that MP CD8 T cells not only demethylate effector genes, but they also methylate naïve genes. So a key question then is whether virus-specific MP CD8 T cells that had downregulated naïve genes like CD62L actually, re-express these genes and undergo associated epigenetic changes. To address this critical question we isolated purified populations of CD62L negative day 8 P14 TE and MP cells, labeled them with CFSE and adoptively transferred these cells into congenically distinct naïve B6 mice (Fig. 4a). We then longitudinally tracked cell division, CD62L expression, and promoter methylation of the transferred TE and MP populations. We took special care to avoid transferring any CD62L positive cells by sorting only CD62L negative TE and MP P14 CD8 T cells (Fig.4a). The take of the adoptively transferred TE and MP populations was determined by sacrificing a group of recipient mice one day after cell transfer and quantitating the number of P14 cells in several lymphoid and non-lymphoid tissues. The CFSE labeled transferred cells were present in all tissues analyzed, and it is important to note that all of these cells were CD62L negative. No CD62L positive P14 cells were detected in any of the tissues examined (Extended data 9a,9b). Thus, on day 1 we were starting with a pure population of CD62L negative P14 TE and MP cells. Longitudinal tracking showed that 28 days after transfer the TE cell population mostly remained CD62L negative and did not divide (Fig.4b). In striking contrast, many of the transferred MP cells now expressed CD62L and were undergoing homeostatic proliferation (Fig.4b, Extended data 9c).
Figure 4. Effector CD8 T cells erase Dnmt3a-mediated DNA methylation programs during their development into memory CD8 T cells.
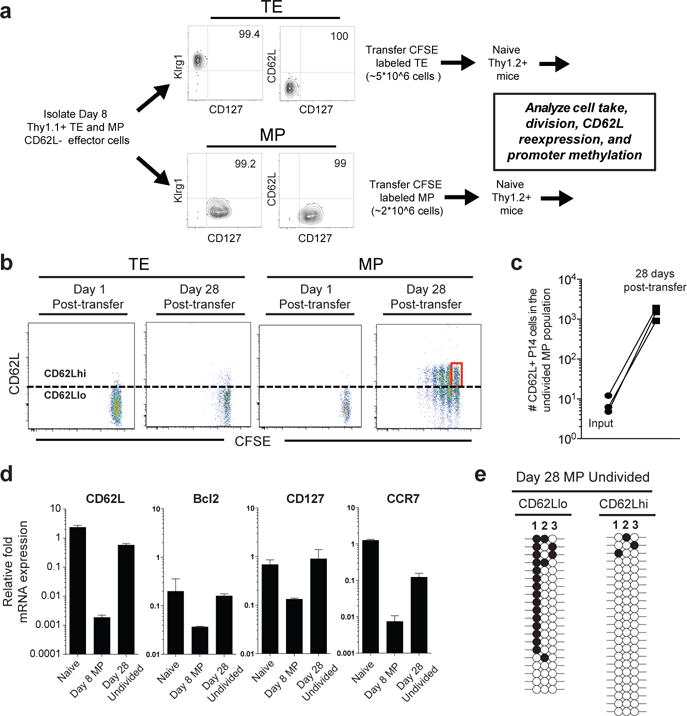
A) Experimental setup for obtaining CFSE labeled TE and MP CD8 T cells at day 1 and 28 post-transfer into naïve mice. B) Representative FACS analysis of CFSE label and CD62L expression of transferred TE and MP populations. C) Graph of the absolute number of transferred MP cells that were undivided and CD62L positive detected in the spleen on day 1 (input) and 28 days post-transfer. D) Real-time PCR analysis of the CD62L transcript from the undivided CD62hi versus CD62Llo memory populations. E) Bisulfite sequencing DNA methylation analysis of the CD62L promoter from the undivided CD62hi versus CD62Llo memory populations.
The most important finding from the above experiment was that, even the undivided MP cells expressed CD62L (Fig.4b, red box), and there were > 1000 of these CD62L positive undivided P14 cells in the spleens of individual mice (Fig.4c). Since there were no detectable (<10) CD62L positive P14 cells at day 1 and we now had >1000 CD62L positive P14 cells that had not undergone any division (i.e., their presence could not be accounted for by any cell proliferation), the only possible explanation for the existence of these cells would be re-expression of CD62L by previously CD62L negative MP effector cells. However, the one caveat is that CD62L protein can be cleaved from the surface of T cells so we wanted to be sure that the increase in CD62L protein was indeed due to an increase in CD62L transcription and not simply due to lack of proteolytic cleavage of L-selectin. To examine this we sorted the undivided day 28 antigen-specific CD8 T cells and quantitated mRNA expression levels compared to the input population of virus-specific MP cells. As shown in Fig.4D there was a 500-fold increase in the level of CD62L message in the undivided day 28 population compared to the input cells. In addition, these undivided day 28 cells had also upregulated several other naïve cell genes (Bcl-2, Ccr7, CD127) that had been downregulated in the input population (Fig.4d). Thus, these results provide unequivocal evidence that antigen-specific day 8 MP CD8 T cells can re-express naïve genes that had been downregulated during the effector phase.
We next sought to determine if re-expression of CD62L in the undivided CFSE+ memory CD8 T cells was coupled to demethylation of the promoter. Genomic DNA was isolated from FACS purified undivided CD62L positive and CD62L negative day 28 memory CD8 T cells (Extended data 9d) and the CD62L promoter methylation status was analyzed. Quite strikingly, the undivided CD62L positive memory population acquired a demethylated promoter, while the CD62L negative memory cells retained a level of promoter methylation equivalent to input effector cells (Fig.4e). These data demonstrate that erasure of previously acquired de novo DNA methylation program at the CD62L promoter in CD8 T cells occurs concordantly with re-expression of CD62L during the effector to memory transition. Taken together, these results support the idea that memory CD8 T cells are generated through a process of cellular de-differentiation that allows for the re-expression of naïve-associated genes.
As shown in Fig.2 and Extended data 4 the MP CD8 T cells had demethylated many canonical effector genes including perforin and granzyme b. It was of interest to determine the methylation status of these effector genes in memory cells. To do this virus-specific effector and memory CD8 T cells were isolated from mice 8 and 40 days after LCMV infection and the methylation status of the GzmB and Prf1 DMRs were measured. Quite strikingly, both GzmB and Prf1 loci remained demethylated in memory CD8 T cells (Extended data 10a, 10b) even though these memory cells did not express high levels of perforin or granzyme b. This finding shows that these memory CD8 T cells retain an epigenetic memory of their effector history.
The formation of memory CD8 T cells has been a topic of much interest and debate. During the past several years two broad and contrasting models have emerged to explain how memory CD8 T cells retain both naïve and effector features. One model describes effector and memory differentiation as distinct lineages arising from asymmetric cell division of the original naïve T cell, allowing memory T cells to retain gene expression programs from the naïve parental cell2,20,21. A contrasting model describes memory T cell differentiation as a process whereby memory CD8 T cells arise from a subset of effector cells (MP cells) that have the ability to re-acquire key naïve-like properties such as homing to lymphoid tissues but still retain the ability to rapidly elaborate effector functions2,22. In the current study, our results reveal that repression of a naïve transcriptional program in memory-precursor effector cells is coupled to de novo DNA methylation of the gene, which can then be erased in a cell-division independent process as the cells re-acquire specific aspects of the naïve-like gene expression program. Furthermore, we show that effector stage conditional deletion of the de novo DNA methyltransferase Dnmt3a results in enhanced kinetics for development of memory cells. A recent study examining the role of Dnmt3a during T cell differentiation also reports that de novo DNA methylation programs regulate the development of memory CD8 T cells. However, the authors of this study conclude that TE cells acquire de novo programs at critical loci, including Tcf7, but the MP cells lack these de novo programs23. In striking contrast, we show here that in fact MP cells also acquire Dnmt3a-mediated methylation programs but have the capacity to erase their newly acquired methylation programs and re-express naïve genes as they develop into memory CD8 T cells. It should be noted that our findings do not argue against a model of memory T cell differentiation whereby MP and TE cells undergo fate specification early in their development24. However, our results do provide evidence that the MP subset of effector CD8 T cells can dedifferentiate into memory T cells. In the accompanying manuscript by Akondy et al. we show that human virus-specific CD8 T cells also follow a similar program of memory differentiation and that both mouse and human memory CD8 T cells retain an epigenetic signature of their past effector history.
MATERIALS AND METHODS
Generation of antigen-specific T cells
Wild type male or female C57BL/6 mice (Jackson Laboratory), aged 6-8 weeks, were acutely infected with the Armstrong strain of LCMV (2*105 pfu i.p.). Effector and memory antigen-specific CD8 T cells were identified and purified by Fluorescence Activated Cell Sorting (FACS) using H-2Db tetramers bound to LCMV peptide GP33-41 conjugated to a fluorophore, along with CD8, CD44, CD127, Klrg1 and CD62L-fluorophore conjugated antibodies as previously described.11,14,25,26. To generate LCMV-specific CD8 T cell chimeras, transgenic P14 CD8 T cells with an engineered TCR that recognize the epitope GP33-41 of LCMV were harvested from naïve P14 TCR transgenic mice and adoptively transferred intravenously to C57BL/6 mice (cell# / mouse is specified in the figure legend)11,27–29. Congenically marked LCMV-specific CD8 T cells were sorted using fluorescently labeled CD90.1 (Thy1.1) and CD8 antibodies as previously described14. Naïve antigen-specific cells obtained from transgenic P14 mice30 were used as an antigen-specific naïve control. Dnmt3a conditional knockout mice were generated by breeding previously characterized floxed Dnmt3a mice with mice that contain a granzyme b driven recombinase transgene17,31. Genotyping and recombination of the Dnmt3a locus was performed using primers listed in Supplemental Table 1. All mouse experiments were approved by Emory University IACUC.
Bisulfite sequencing methylation analysis
DNA was isolated from FACS purified antigen-specific CD8 T cells using using the Qiagen DNeasy kit. Genomic DNA was bisulfite treated using the Zymo Research EZ DNA methylation kit. Bisulfite induced deamination of cytosine was used to determine the allelic frequency of cytosine methylation of the target genomic region32. The bisulfite modified DNA was PCR amplified with locus specific primers (Supplemental Table 1). The PCR amplicon was cloned into the pGEM-T TA cloning vector (Promega) then transformed into XL10-Gold ultracompetent bacteria (Stratagene). Individual bacterial colonies were grown and the cloning vector was isolated and sequenced.
Genome-wide methylation analysis
Genomic DNA from FACS purified WT naïve P14, D4.5 TE and MP effector P14s, Day 8 TE and MP effector P14s, Day 8 total tetramer+ WT and Dnmt3a cKO effector, and Day 35 tetramer+ WT memory CD8 T cells was isolated using the Qiagen DNeasy kit. Genomic DNA was bisulfite treated using the Zymo Research EZ DNA methylation kit. The bisulfite modified DNA sequencing library was generated using the Epicentre kit per the manufacturer’s instructions. Bisulfite modified DNA libraries were sequenced using an Illumina Hiseq. Sequencing was performed to yield >5x average coverage across the genome. Sequencing data were aligned to the mm10 mouse genome using BSMAP2.7433. Differentially methylated regions (DMRs) were identified using Bioconductor package DSS34. We first performed statistical test of differentially methylated loci (DML) using DMLtest function (smoothing=TRUE) in DSS, the results were then used to detect differentially methylated regions using CallDMR function in DSS, with a p value threshold for calling DMR set at 0.01. The minimum length for a DMR was set to 50 bps with a minimum number of 3 CpG sites. Genomic location classification was defined based on RefSeq annotation as follows: promoter from −2k to 1kb of transcriptional start site (TSS), TSS proximal enhancer from −10 kb to −2kb of TSS, distal from TTS from 1 bp to 2kb of transcriptional termination site (TTS), intron and exon were defined in RefSeq annotation, and any other genomic regions were defined as intergenic. The distance was defined based on strand of the gene. Each DMR was assigned in the order listed above and each DMR was assigned to one category only. The ShuffleBed function in bedtools35 was used to generate a set of random regions of same number and size distribution as the original DMRs to assess enrichment of DMR distribution among relative genomic locations. Specifically, each DMR was repositioned on a random chromosome at a random position. The number of DMRs and the size of DMRs were preserved. The random set of regions was then annotated the same way as the original DMRs. Differential methylation analysis of CpG methylation among the datasets were further assessed using a Bayesian hierarchical model to detect differences among methylation at 3 CpG sites36.
Real-time PCR analysis of mRNA
Splenocytes were harvested from mice at the described time points post infection with LCMV Armstrong. Antigen-specific CD8 T cells were purified by FACS based sorting. RNA was extracted from cells using the Qiagen RNeasy kit per the instructions of the manufacturer. Quantitative real-time PCR of CD62L was performed using primers that were previously described37.
In Vivo Cellular Proliferation
Virus-specific CD8 T cells were FACS purified and resuspended in PBS at a concentration of 2*107/ml. 1 volume of 5μM CFSE was mixed with 1 volume of cells for 7 minutes at room temperature. 100% Fetal Calf Serum was added to the cell suspension at final volume of 20% to quench the labeling. Cells were washed with PBS once and then adoptively transferred into naïve B6 mice.
In vitro promoter methylation and expression assay
The pCpGfree-basic reporter construct was purchased from InVivogen and the CD62L promoter was cloned into the MCS of the reporter construct. Proper orientation of the promoter was confirmed by sequencing. In vitro methylation of the plasmid was performed using MSsI purchased from New England Biolabs. The plasmid was incubated with the enzyme and cofactor for 2 hours at 37C and then additional cofactor was spiked into the reaction and incubated for another 2 hours. Methylation was confirmed using the established CD62L bisulfite sequencing assay. 0.5ug of unmethylated and in vitro methylated plasmids were then transfected into EL4 cells (purchased from ATCC) using Lipofectamine 2000 per the manufacture’s instructions. Supernatant was isolated from the cell culture at various times and the Lucia activity was measured using Quantiluc reagent on a Veritas luminometer.
Statistical significance for all in vivo and vitro studies, excluding the WGBS analyses, was determined on 3 or more biological replicate samples using Prism software.
Extended Data
Extended Data Figure 1. CD62L (L-selectin) gene expression changes during effector and memory CD8 T cell differentiation are coupled to epigenetic reprogramming of the CD62L promoter.
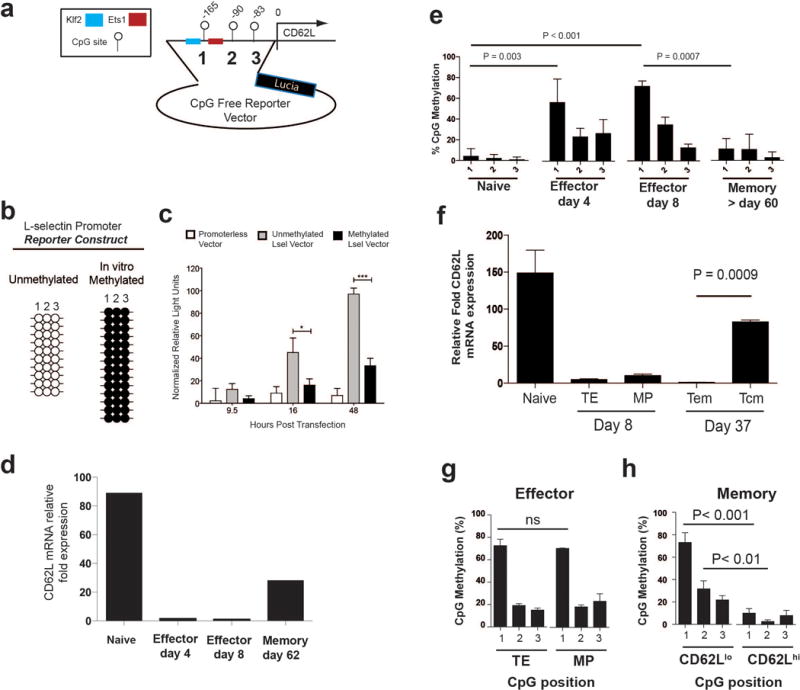
A) Real-time PCR analysis of CD62L mRNA in virus-specific naïve, effector, and memory P14 CD8 T cells. B) Cartoon diagram of CpG positions within the CD62L promoter region cloned into the CpG free Lucia Promoter reporter construct. Putative transcription factor binding sites are indicated by colored boxes. C) Representative methylation profiling of in vitro methylation efficiency of the reporter construct. D) Longitudinal measurement of Relative light units from EL4 cells transfected with unmethylated and in vitro methylated reporter constructs. E) Summary graph of CD62L proximal promoter methylation of naïve, day 4 effector, day 8 effector, >day 60 memory P14 CD8 T cells. Each horizontal line represents an individual sequenced clone. Filled circles = methylated cytosine. Open circles = non-methylated cytosine. F) Real-time PCR analysis of CD62L mRNA expression from day 8 terminal-effector and memory-precursor P14 CD8 T cells, and day 37 effector-memory and central-memory P14 CD8 T cells. Transcript data correspond to cell sorts used for DNA methylation measurements in figure 1f and 1h. G & H) Summary graph of the CD62L proximal promoter DNA methylation in TE and MP effector CD8 T cells and CD62Llo and CD62Lhi memory CD8 T cells.
Extended Data Figure 2. Isolation of memory precursor (MP) and terminal effector (TE) CD8 T cells for whole-genome methylation profiling.
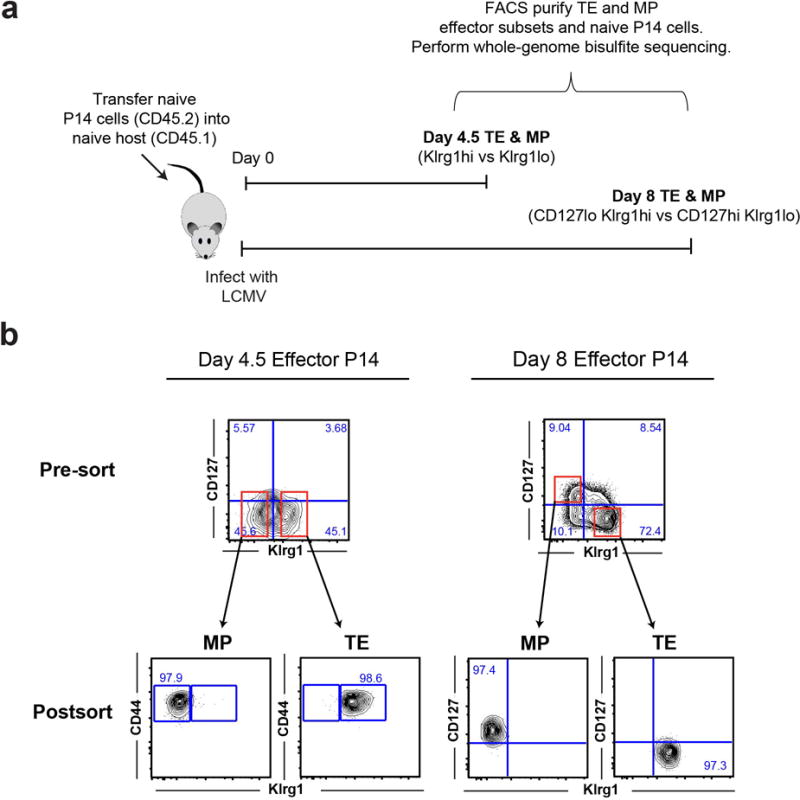
A) Experimental setup for isolating day 4.5 and day 8 MP and TE LCMV-specific CD8 T cells. B) Representative post-sort purity and phenotypic analysis of Day 4.5 and Day 8 MP and TE P14 CD8 T cells isolated from acutely infected mice used for whole-genome bisulfite sequencing (WGBS) methylation profiling.
Extended Data Figure 3. Effector-associated changes in DNA methylation occur predominantly at or near genes and are highly similar between MP and TE CD8 T cell subsets.
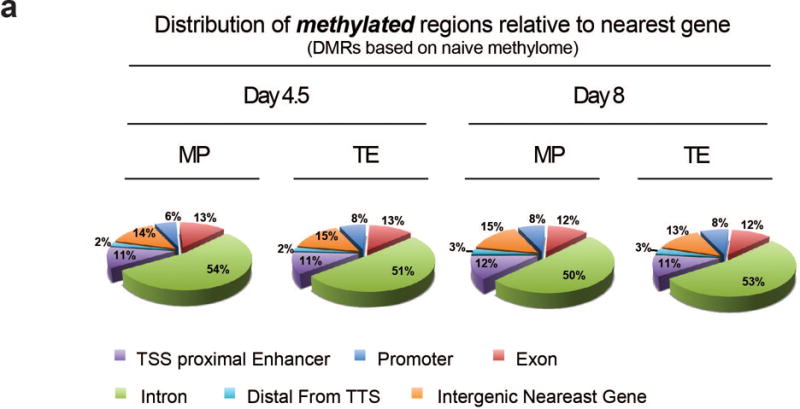
A) Pie chart representation of newly methylated DMR genomic distribution relative to the transcriptional start site of the nearest gene.
Extended Data Figure 4. Both MP and TE CD8 T cells acquire demethylated effector loci.
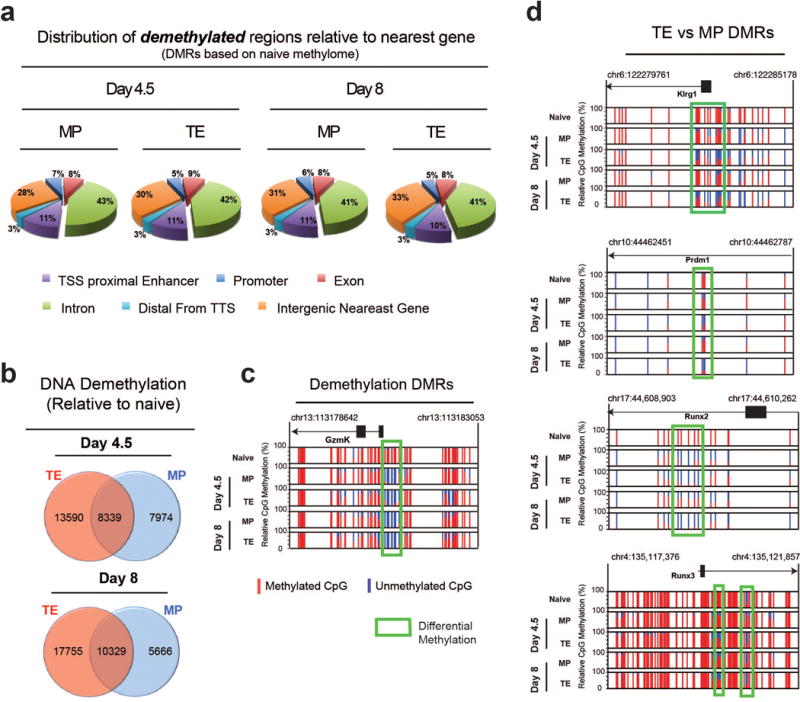
A) Pie chart representation demethylated DMR genomic distribution relative to the transcriptional start site of the nearest gene. B) Venn diagram of regions that undergo demethylation during differentiation of naïve CD8 T cells into TE and MP subsets. C) Normalized graph of methylation at CpG sites in the Gzmk locus of TE and MP WGBS data sets. D) Normalized graph of differentially methylated CpG sites in the Klrg1, Prdm1 (Blimp1), Runx2, and Runx3 loci from TE and MP WGBS data sets.
Extended Data Figure 5. Conditional deletion of Dnmt3a in activated CD8 T cells inhibits effector-associated de novo DNA methylation but does not impair maintenance methylation.
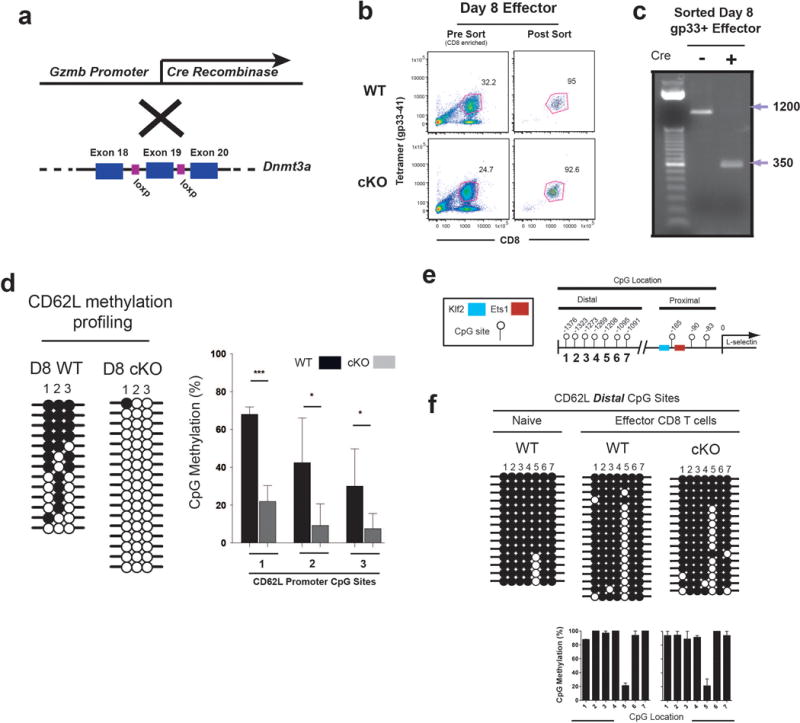
A) Cre recombinase expression is driven by the granzyme b promoter to initiate recombination of Dnmt3a exon 19 following T cell activation. B) Representative FACS analysis of virus-specific CD8 T cells sorted at 8 days post acute viral infection of WT and Dnmt3a cKO mice. C) Recombination of genomic DNA from FACS purified Dnmt3a cKO cells virus-specific CD8 T cells was assessed by PCR using primers that anneal to DNA outside of the floxed target region. The larger PCR amplicon corresponds to the intact locus and the smaller PCR product is the amplicon of the recombined locus. D) Representative and graphical summary of CD62L promoter methylation in WT and Dnmt3a cKO cells. Average and standard deviation were calculated from bisulfite sequencing analysis of 6 individually sorted populations. E) Cartoon diagram of CD62L promoter CpG location proximal and distal to the transcriptional start site. F) Representative DNA methylation analysis of CpG sites distal to the CD62L promoter regions in Day 8 WT and Dnmt3a cKO antigen-specific effector CD8 T cells. Graphical summary of the average CD62L distal CpG methylation in WT and Dnmt3a cKO cells calculated from bisulfite sequencing analysis of 4 individually sorted populations.
Extended Data Figure 6. Effector-stage de novo DNA methylation is enriched at genes that regulate effector and memory T cell differentiation.
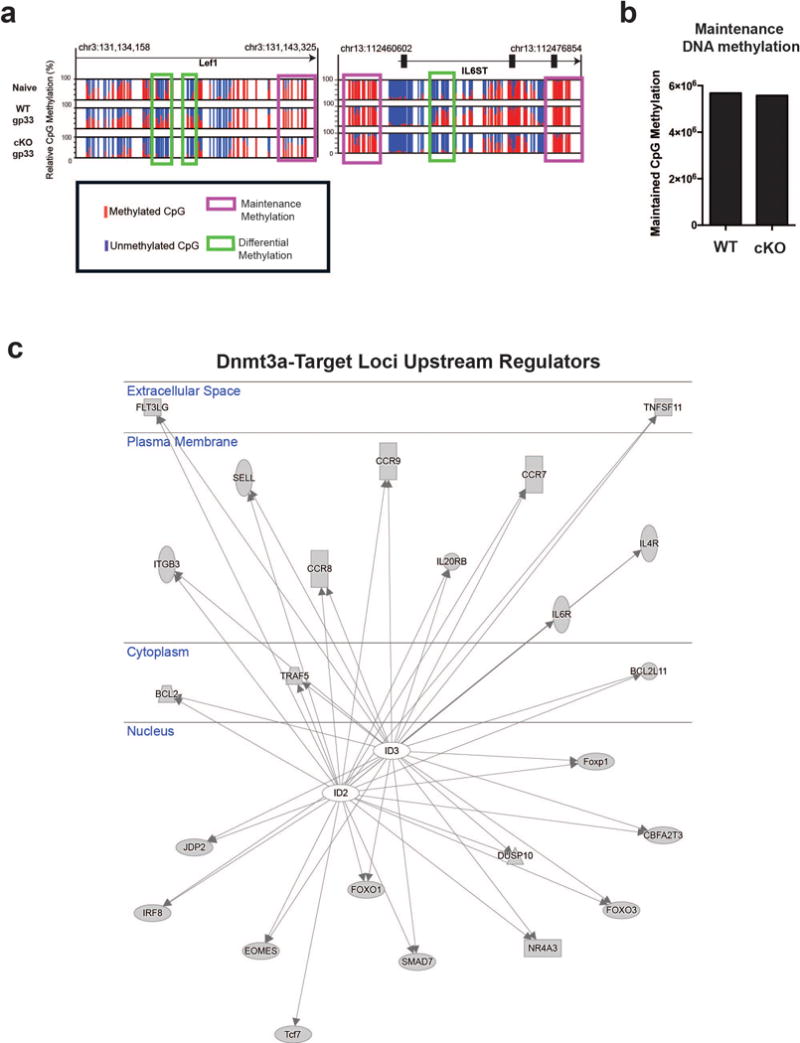
A) Normalized graph of Dnmt3a-mediated de novo methylation at CpG sites in the Lef1 and IL6ST loci from WGBS data sets. B) Summary graph of maintenance methylated regions in WT and Dnmt3a cKO effector WGBS datasets. C) Connectivity plot showing IPA predicted interactions of ID2 and ID3 with Dnmt3a-targeted loci.
Extended Data Figure 7. Dnmt3a deficient CD8 T cells undergo effector differentiation.
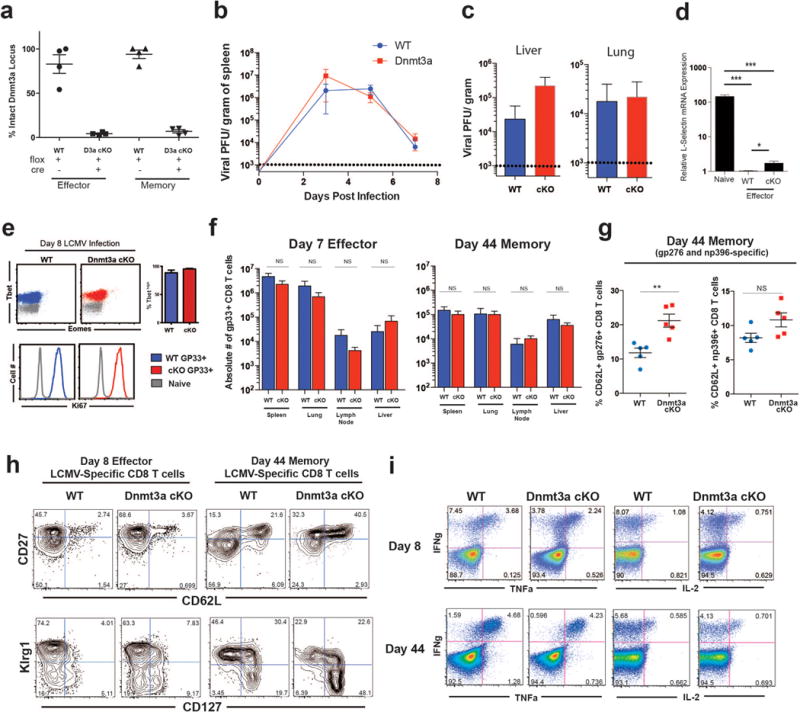
A) Summary graphs of gp33-specific CD8 T cell quantity at effector and memory time points in lymphoid and nonlymphoid tissues. Summary graphs of viral titers in spleens (B) and day 5 lung and liver (C) of acutely infected WT and Dnmt3a cKO mice. D) Quantitative-PCR analysis of Dnmt3a exon 19 recombination using a primer set that binds to DNA internal of the floxed target region. The average and standard deviation of intact (non-recombined) floxed Dnmt3a alleles were determined by quantitative PCR from 4 individually sorted gp33-specific effector and memory CD8 T cell populations. E) Real-time PCR analysis of CD62L mRNA expression of naïve and tetramer+ WT and Dnmt3a cKO effector CD8 T cells. F) Representative FACS analysis of Klrg1, CD127, CD27, and CD62L expression on WT and Dnmt3a cKO effector and memory gp33-specific CD8 T cell splenocytes. G) Summary graph showing the percent of CD62L positive LCMV-specific CD8 T cell splenocytes recognizing the LCMV dominate epitopes gp276, and np296.
Extended Data Figure 8. Effector molecule loci are demethylated during differentiation of virus-specific Dnmt3a cKO CD8 T cells.
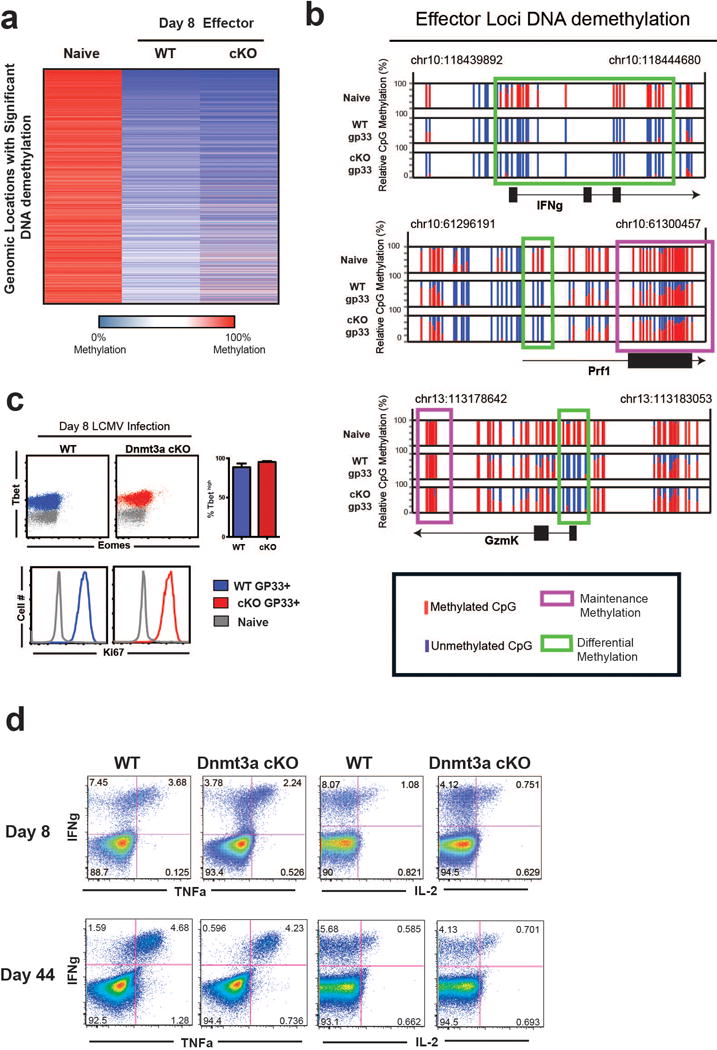
A) Heat-map representation of top 3000 demethylated regions among WT and cKO effector CD8 T cell WGBS data sets relative to the naïve WGBS data set. B) Normalized graph of effector loci methylation at CpG sites in the IFNg, Prf1, and GzmK loci from WT and cKO WGBS data sets. C) Representative FACS analysis of Tbet, Eomes, and Ki67 expression of gp33-specific effector CD8 T cells. D) Representative FACS analysis of cytokine production from virus-specific memory CD8 T cells following 5 hours of ex vivo gp33 peptide stimulation.
Extended Data Figure 9. CD62L low MP effector CD8 T cells develop into Tcm CD8 T cells.
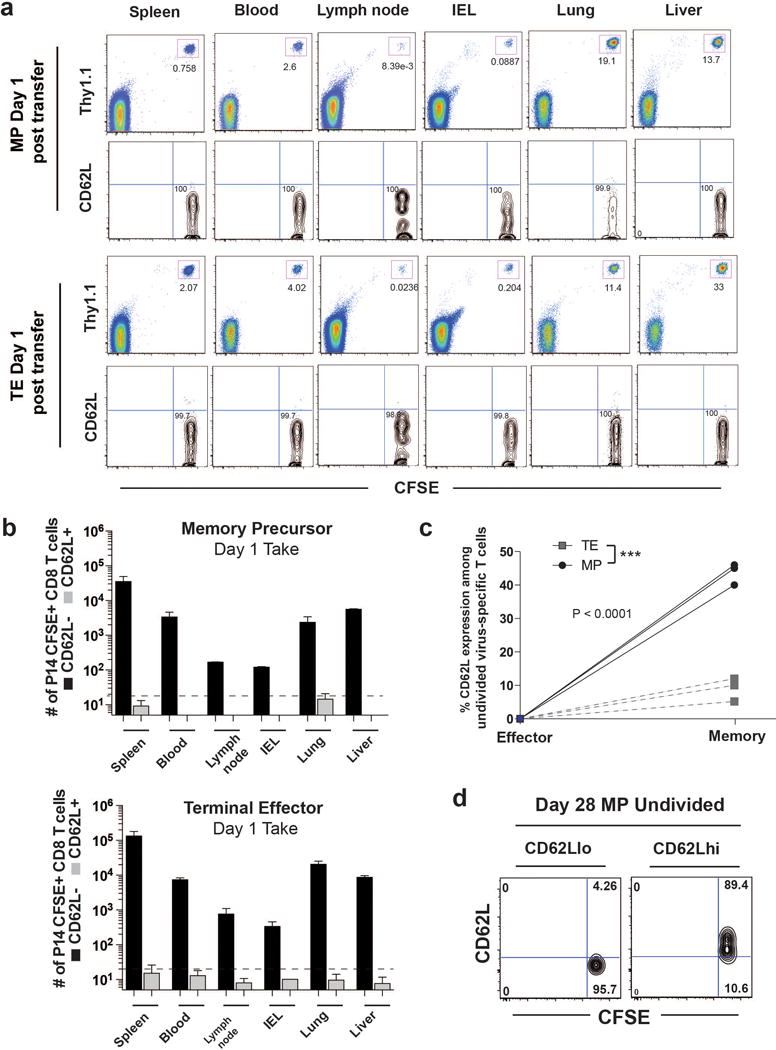
A) Representative FACS analysis of CD62L expression on Thy1.1+ CFSE+ MP and TE CD8 T cells 1 day post transfer into naïve recipient mice. The limit of our detection was ~10-20 CD62L+ cells in each of the lymphoid and nonlymphoid tissues at 1 day post-transfer. B) Summary graph of number of transferred TE and MP CD8 T cells in the spleen, blood, lymph node, IEL, lung, and liver of the recipient mice 1 day post-transfer. C) Summary graph of percent undivided (undiluted CFSE) CD62L positive virus-specific memory CD8 T cells arising from adoptively transferred MP vs. TE cells. Data are from 3 independent experiments. D) Representative post-sort purity FACS analysis of undivided CD62Lhi and CD62Llo MP P14 cells 28 days after adoptive transfer.
Extended Data Figure 10. Memory CD8 T cells retain demethylated effector loci.
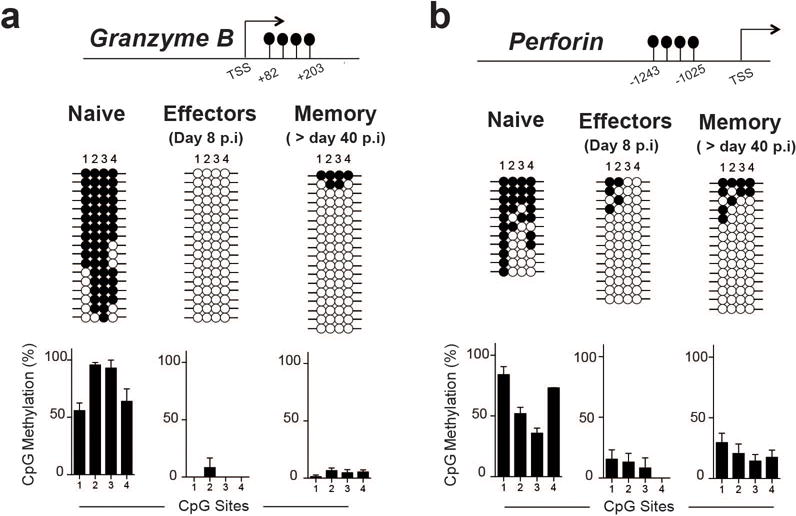
Representative analysis and summary graph for loci-specific methylation profiling of (A) Gzmb and (B) Perforin DMRs in naïve, effector (day 8 gp33 tetramer+) and memory (>day 40 gp33 tetramer+) CD8 T cells (Standard deviation is calculated from 3 independently sorted samples).
Supplementary Material
Supplemental Table. Primers for Bisulfite Sequencing and Analysis of Dnmt3a Genomic Deletion.
Acknowledgments
We thank R. Karaffa and S. Durham at the Emory University School of Medicine Flow Cytometry Core Facility Richard Cross and Greig Lennon in the St Jude Flow Cytometry Core Facility for FACS sorting. Whole-genome sequencing was performed by the St Jude Hartwell Sequencing facility. This work was supported by the National Institutes of Health (NIH) grant 1 P01 AI080192-01 (to R.A.), grant 2 R37 AI30048-17 (to R.A.), grant AHMED05GCGH0 (to R.A.), and funds from American Lebanese Syrian Associated Charities (ALSAC) (to B.A.Y.).
The data that support the findings of this study are available from the corresponding author upon request.
Footnotes
Supplementary Information is available in the online version of this paper.
AUTHOR CONTRIBUTIONS
BY & JSH designed experiments, collected data, analyzed and interpreted results.
HTK analyzed and interpreted results.
EEW, EA, XX, & AW collected data, analyzed and interpreted results.
YF, KA, XC, & RA interpreted results.
HG, PD, CWD & BTK collected data.
RA designed experiments and supervised the study.
All authors contributed in the preparation of the manuscript.
The authors report no competing financial interests.
References
- 1.Obar JJ, Lefrancois L. Memory CD8+ T cell differentiation. Annals of the New York Academy of Sciences. 2010;1183:251–266. doi: 10.1111/j.1749-6632.2009.05126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahmed R, Bevan MJ, Reiner SL, Fearon DT. The precursors of memory: models and controversies. Nature reviews Immunology. 2009;9:662–668. doi: 10.1038/nri2619. [DOI] [PubMed] [Google Scholar]
- 3.Buchholz VR, et al. Disparate individual fates compose robust CD8+ T cell immunity. Science. 2013;340:630–635. doi: 10.1126/science.1235454. [DOI] [PubMed] [Google Scholar]
- 4.Gerlach C, et al. Heterogeneous differentiation patterns of individual CD8+ T cells. Science. 2013;340:635–639. doi: 10.1126/science.1235487. [DOI] [PubMed] [Google Scholar]
- 5.Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 2002;111:837–851. doi: 10.1016/s0092-8674(02)01139-x. [DOI] [PubMed] [Google Scholar]
- 6.Wherry EJ, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27:670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 7.Wirth TC, et al. Repetitive antigen stimulation induces stepwise transcriptome diversification but preserves a core signature of memory CD8(+) T cell differentiation. Immunity. 2010;33:128–140. doi: 10.1016/j.immuni.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dang X, Raffler NA, Ley K. Transcriptional regulation of mouse L-selectin. Biochimica et biophysica acta. 2009;1789:146–152. doi: 10.1016/j.bbagrm.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carlson CM, et al. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature. 2006;442:299–302. doi: 10.1038/nature04882. [DOI] [PubMed] [Google Scholar]
- 10.Kaech SM, et al. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat Immunol. 2003;4:1191–1198. doi: 10.1038/ni1009. [DOI] [PubMed] [Google Scholar]
- 11.Sarkar S, et al. Functional and genomic profiling of effector CD8 T cell subsets with distinct memory fates. The Journal of experimental medicine. 2008;205:625–640. doi: 10.1084/jem.20071641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joshi NS, et al. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity. 2007;27:281–295. doi: 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rutishauser RL, et al. Transcriptional repressor Blimp-1 promotes CD8(+) T cell terminal differentiation and represses the acquisition of central memory T cell properties. Immunity. 2009;31:296–308. doi: 10.1016/j.immuni.2009.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Murali-Krishna K, et al. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- 15.Youngblood B, et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8+ T cells. Immunity. 2011;35:13. doi: 10.1016/j.immuni.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 17.Jacob J, Baltimore D. Modelling T-cell memory by genetic marking of memory T cells in vivo. Nature. 1999;399:593–597. doi: 10.1038/21208. [DOI] [PubMed] [Google Scholar]
- 18.Cannarile MA, et al. Transcriptional regulator Id2 mediates CD8+ T cell immunity. Nat Immunol. 2006;7:1317–1325. doi: 10.1038/ni1403. [DOI] [PubMed] [Google Scholar]
- 19.Yang CY, et al. The transcriptional regulators Id2 and Id3 control the formation of distinct memory CD8+ T cell subsets. Nat Immunol. 2011;12:1221–1229. doi: 10.1038/ni.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang JT, et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. 2007;315:1687–1691. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- 21.Arsenio J, et al. Early specification of CD8+ T lymphocyte fates during adaptive immunity revealed by single-cell gene-expression analyses. Nat Immunol. 2014;15:365–372. doi: 10.1038/ni.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Youngblood B, Hale JS, Ahmed R. T-cell memory differentiation: insights from transcriptional signatures and epigenetics. Immunology. 2013;139:277–284. doi: 10.1111/imm.12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ladle B, L K, Phillips M, Pucsek A, Haile A, Powell J, Jaffee E, Hildeman D, Gamper C. De novo DNA methylation by DNA methyltransferase 3a controls early effector CD8+ T -cell fate decisions following activation. Proceedings of the National Academy of Sciences of the United States of America. 2016 doi: 10.1073/pnas.1524490113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang JT, et al. Asymmetric proteasome segregation as a mechanism for unequal partitioning of the transcription factor T-bet during T lymphocyte division. Immunity. 2011;34:492–504. doi: 10.1016/j.immuni.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. Journal of virology. 2003;77:4911–4927. doi: 10.1128/JVI.77.8.4911-4927.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matloubian M, Somasundaram T, Kolhekar SR, Selvakumar R, Ahmed R. Genetic basis of viral persistence: single amino acid change in the viral glycoprotein affects ability of lymphocytic choriomeningitis virus to persist in adult mice. The Journal of experimental medicine. 1990;172:1043–1048. doi: 10.1084/jem.172.4.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blattman JN, et al. Estimating the precursor frequency of naive antigen-specific CD8 T cells. The Journal of experimental medicine. 2002;195:657–664. doi: 10.1084/jem.20001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kersh EN. Impaired memory CD8 T cell development in the absence of methyl-CpG-binding domain protein 2. Journal of immunology. 2006;177:3821–3826. doi: 10.4049/jimmunol.177.6.3821. [DOI] [PubMed] [Google Scholar]
- 29.Kersh EN, et al. Rapid demethylation of the IFN-gamma gene occurs in memory but not naive CD8 T cells. J Immunol. 2006;176:4083–4093. doi: 10.4049/jimmunol.176.7.4083. [DOI] [PubMed] [Google Scholar]
- 30.Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559–561. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]
- 31.Kaneda M, et al. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature. 2004;429:900–903. doi: 10.1038/nature02633. [DOI] [PubMed] [Google Scholar]
- 32.Trinh BN, Long TI, Laird PW. DNA methylation analysis by MethyLight technology. Methods. 2001;25:456–462. doi: 10.1006/meth.2001.1268. [DOI] [PubMed] [Google Scholar]
- 33.Xi Y, Li W. BSMAP: whole genome bisulfite sequence MAPping program. BMC bioinformatics. 2009;10:232. doi: 10.1186/1471-2105-10-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu H, et al. Detection of differentially methylated regions from whole-genome bisulfite sequencing data without replicates. Nucleic acids research. 2015;43:e141. doi: 10.1093/nar/gkv715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feng H, Conneely KN, Wu H. A Bayesian hierarchical model to detect differentially methylated loci from single nucleotide resolution sequencing data. Nucleic acids research. 2014;42:e69. doi: 10.1093/nar/gku154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Furukawa Y, et al. Identification of novel isoforms of mouse L-selectin with different carboxyl-terminal tails. The Journal of biological chemistry. 2008;283:12112–12119. doi: 10.1074/jbc.M801745200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table. Primers for Bisulfite Sequencing and Analysis of Dnmt3a Genomic Deletion.