

Abstract
We present a systematic investigation of the structural and electronic changes that occur in an Fe(0)–N2 unit (Fe(depe)2(N2); depe = 1,2-bis(diethylphosphino)-ethane) upon the addition of exogenous Lewis acids. Addition of neutral boranes, alkali metal cations, and an Fe2+ complex increases the N–N bond activation (Δ νNN up to 172 cm−1), decreases the Fe(0)–N2 redox potential, polarizes the N–N bond, and enables −N protonation at uncommonly anodic potentials. These effects were rationalized using combined experimental and theoretical studies.
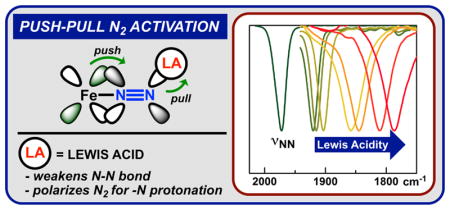
Graphical Abstract
INTRODUCTION
The reduction of dinitrogen to ammonia is one of the most important, yet challenging, chemical transformations. While the currently used industrial process for nitrogen fixation requires forcing temperature and pressure (~500 °C and >100 atm), nitrogen fixation by nitrogenases operates under ambient conditions.1 Nitrogenases use a multimetallic active site contained within a network of amino acid residues that are essential in orchestrating a series of multiproton, multielectron transfers to N2. Although this strategy may ultimately provide a low energy/carbon route to ammonia, the discrete mechanistic steps in the biological reduction sequence are largely unknown.2
A key step in the proposed mechanism of nitrogenase is the coordination and activation of a classically inert dinitrogen ligand at a reduced iron center (Figure 1).2a The high π-basicity (and highly cathodic reduction potentials) required of the metal center in synthetic systems has led to a primary coordination sphere-focused design strategy in the development of model complexes.3 In contrast, nitrogenases exhibit low overpotentials for N2 fixation, which suggests that additional design principles may be required.4 In particular, the role of acidic sites in the secondary-sphere of the nitrogenase active site (highly conserved α-195His, α-191Gln, arginine residues, and adjacent S–H and Fe centers)5 has been relatively unexamined in synthetic systems.6 These sites are required for nitrogenase activity, as demonstrated by mutation studies,7 and may play a crucial role in enabling nitrogenase’s low overpotential and high selectivity.
Figure 1.

FeMo–co active site with proposed interactions of acidic groups (left); conceptual framework of the current study illustrating the use of Lewis acids to test the push–pull hypothesis (right). Filled orbitals shown in green.
Hydrogen bonding groups and Lewis acidic sites are commonly used in metalloenzymes to modulate substrate binding and activation.8 In nitrogenase, these interactions have been used to formulate the so-called “push–pull” hypothesis in which electron density is “pushed” from a reduced iron center and “pulled” into the N2 unit by adjacent Lewis acidic sites.2a,b These interactions lead to polarization of N2 and enhanced charge transfer from the iron center; the two effects may contribute to the low overpotential and high protonation selectivity observed in the enzyme. The push–pull approach is an intriguing concept for understanding N2 activation in nitrogenase and may also provide a general strategy that could be used in the design of synthetic nitrogenases. However, this concept has not been experimentally tested, which may be due to the synthetic challenges inherent in combining highly acidic and reducing centers in a single molecule.6 Herein, we report an experimental test of the push–pull hypothesis through a systematic investigation of the ability of exogenous Lewis acids to activate a reduced Fe–N2 complex.
Fe(depe)2(N2) (1) was selected as a model Fe(0)–N2 unit for our investigation because it provides a sterically accessible, nucleophilic N2 ligand and was recently shown to catalyze N2 reduction.9 While Lewis acids (LAs) such as SiMe3+ and H+ have been reported to react with structural analogues of 1 to provide ill-defined mixtures,10 their excessive electrophilicity make them inadequate models for noncovalent interactions in the secondary-sphere of nitrogenase. In contrast, neutral boranes are a highly tunable class of LAs11 that are ideal for modeling these interactions. B(C6F5)3, a highly acidic borane, was initially selected to interrogate the push–pull hypothesis via adduct formation with 1.
RESULTS AND DISCUSSION
Synthesis and Characterization of 1–LA Complexes
When B(C6F5)3 was combined with 1, a LA/base adduct (2) was formed in quantitative yield as assessed by IR and NMR spectroscopy (Figure 2). The IR spectrum revealed that the N–N bond was significantly weakened by the addition of B(C6F5)3, noted by the 129 cm−1 shift of the νNN stretch (1: 1959 cm−1; 2: 1830 cm−1). The formation of 2 was general in a variety of nonpolar solvents (pentane, C6D6, toluene, PhF, methyl tert-butyl ether, THF), allowing analysis by 1H, 11B, 15N, 19F, and 31P NMR spectroscopy, which supported a persistent borane–N2 interaction in solution and a highly activated N2 ligand.
Figure 2.

(A) Synthesis of 2. (B) Generality of N2 activation using a variety of Lewis acids (Li+, Na+, K+, Rb+, Cs+; BR3 (R = 2,6-F2Ph, 2,4,6-F3Ph, C6F5, OC6F5, F)). (C) Crystal structure of 2. Fe1–N1: 1.717(2) Å; N1–N2: 1.186(3) Å; N1–N2–B1: 137.0(3)°. Non-essential carbon, fluorine, and hydrogen atoms omitted for clarity. Ellipsoids shown at 50% probability.
NMR spectra of 2 at room temperature contain a sharp 11B resonance at −6.35 ppm, consistent with a tetrahedral boron center. The 15N NMR spectrum of an 15N2-labeled derivative of 2 suggests significant polarization of the N2 unit induced by the LA: the Δδ for the iron-bound and terminal N atoms increases from 5 to 109 ppm (1: Nα −45.2, Nβ −40.5;12 2: Nα −10.5, Nβ −119.8 ppm). The significant upfield shift of Nβ (−74.6 ppm), as opposed to the downfield shift Nα (+30.0 ppm), suggests increased localization of electron density at the terminal nitrogen atom.13
A single crystal of 2 was grown from toluene/pentane and analyzed by an X-ray diffraction experiment. The N–N bond length is elongated by 0.04(1) Å (1: 1.142(7); 2: 1.186(3)), reflecting the significant N–N activation apparent in the IR and 15N NMR spectra.9b An important structural detail is the bent N1–N2–B1 geometry, with a bond angle of 137.0(3)°.14 This bond angle suggests a strong interaction between the empty boron p-orbital and the N2 π* orbital.15
To evaluate the generality of enhanced N2 activation by LAs, we selected a set of borane and alkali metal LAs of varying strength (BR3 (R = 2,6-F2Ph, 2,4,6-F3Ph, C6F5, OC6F5, F); Li [B(C6F5)4], Na+, K+, Rb+, Cs+ [BArF4 = B(3,5-(CF3)2C6H3)4]). Although select examples of alkali metal–N2 interactions have been reported, these complexes are often further supported by intramolecular cation–π interactions and charge pairing, making efforts to systematically evaluate the role of the alkali metal LAs in N2 activation highly challenging.1a,16
The availability of a broad set of LA–N2 adducts enabled an investigation of the relationship between Lewis acidity, N–N bond strength, and N2 binding affinity. The νNN undergoes a smooth bathochromic shift as a function of LA strength, as quantified by the acceptor number (AN).17 As the LA strength increases from Cs+ (AN: 26) to B(OC6F5)3 (AN: 89), the N–N bond is weakened as shown by a lowering of the νNN from 1920 to 1787 cm−1 (Figure 2). Within the series of alkali metal cations, the association constant (measured in Et2O) varies between 94(5) and 430(80) M−1, and stronger binding is correlated with increasing Lewis acidity. The same trend holds within the series of fluorinated triphenylboron LAs (measured in PhF, BR3; R = 2,6-F2 Ph, 2,4,6-F3Ph, C6F5), whose association constants increase from 4.6 × 103 to 7.9 × 104 M−1.18 The shift of the νNN and binding affinity clearly illustrates that the strength of the N–N and LA–N2 bonds can be dramatically tuned by the LA.
Electronic Structure and Predicted Electrochemical and Protonation Reactivity of 1–BR3 Adducts
The ability to finely tune the strength of boron-based LAs in 1-BR3 adducts presents a unique opportunity to explore the impact of exogenous LAs on the electronic structure of an Fe–N2 unit. Using DFT methods, we interrogated the molecular orbitals of 1 and 2 to provide quantitative orbital mixing coefficients between neutral Fe(depe)2 and LA–N2 fragments (Figure 3A) as well as natural bonding orbital (NBO) populations and net atomic charges.
Figure 3.

(A) π-Manifold for 1 and 2 highlighting the impact of LA interactions. (B) Orbital energies vs acceptor number (AN), terminal nitrogen atom NBO charge. (C) N2 π* population vs AN.
In complex 1, the combination of the empty, high-lying N2 π* orbital with the filled Fe(depe)2 dxy orbital leads to a high energy HOMO with a small contribution from unfilled N2 π* orbitals (Figure 3A). Upon introduction of B(C6F5)3 to the N2 fragment, the empty boron p orbital mixes with the N2 π* orbital, which lowers its energy from −1.18 to −4.60 eV. The resulting LA–N2 π* orbital can then better interact with the filled Fe(depe)2 dxy orbital, leading to stabilization of the HOMO (−3.73 to −4.60 eV) and an increase in its N2 π* character from 18% to 39%; these effects track closely with increasing Lewis acidity (Figure 3B). The increase in π* character in the HOMO equates to greater charge transfer (via π-backbonding) from Fe(depe)2 to the N2 unit. The relationship between the N–N–B angle and orbital energies of a N2–BF3 fragment was probed through a Walsh-type analysis. In contrast to the free N2–BF3 fragment, in which a linear geometry is favored by 21 kcal/mol, population of the N2 π* orbitals upon Fe(0) coordination leads to a net stabilization of the bent 120° geometry by 13 kcal/mol, compared to a linear (180°) geometry. These orbital considerations provide a rationale for the experimentally observed weakening of the N–N bond and bent activation in 1 upon N-coordination with a simple Lewis acid.
Crucially, the LA-induced increase in N2 activation is not provided at the expense of N2 polarization, even though a LA might be expected to quench the Lewis basic terminal nitrogen atom. The difference in NBO charge between the nitrogen atoms in 1 (Δ = 0.32) increases upon coordination of B(C6F5)3 (Δ = 0.39 in 2), indicating enhanced N2 polarization. Importantly, the negative charge of the terminal nitrogen atom increases from −0.22 in 1 to −0.30 in 2. Increasing Lewis acidity from B(C6F2H3)3 to B(OC6F5)3 further populates the N2 π* orbitals (0.217 to 0.228 e−) and the negative charge at the terminal nitrogen (−0.25 to −0.37), suggesting that the relative basicity of the terminal nitrogen atom is enhanced upon LA coordination.
Electrochemical Characterization of 1–BR3 Adducts
The addition of B(C6F5)3 to 1 lowers the HOMO energy by 0.9 V (Figure 3B), indicating that 2 and related LA-activated N2 adducts should exhibit significantly more anodic potentials than 1. Electrochemical experiments were used to test this prediction. The cyclic voltammogram of 1 featured a reversible redox couple with an E1/2 = −1.94 V (Epa = −1.72 V; 0.1 M [nBu4N][BArF4],19 PhF, vs Fc+/Fc, 100 mV s−1 at −45 °C).20 Upon addition of B(C6F5)3, the oxidative peak current undergoes a significant anodic shift (Epa = −0.89 V; 100 mV s−1) as assessed by cyclic voltammetry and becomes irreversible (Figure 4). The imparted irreversibility is consistent with the absence of reactivity between LAs and the corresponding Fe(I)–N2 complex, [Fe(depe)2(N2)]+; see the SI. The 0.8 V dif ference in the Epa values from 1 to 2 is consistent with the predicted 0.9 V shift from DFT calculations. The shift in the redox couple is a general effect and tracks closely with Lewis acidity (Figure 4 right). Overall, the significantly lower HOMO energies in the 1–LA adducts afford a significant anodic shift of the Fe–N2 unit and validate the conceptual foundation of the push–pull hypothesis.
Figure 4.

(A) Cyclic voltammograms for Fe0/FeI in 1 vs 2. (B) LA strength vs redox potential of 1–BR3 adducts. AN for 1 in B corresponds to that of fluorobenzene.
Selective −N Protonation of 2
An important prediction of our theoretical analysis is that Lewis acidic centers can bias the Fe–N2 unit toward −N protonation rather than −Fe protonation. The reactivity of H+ with 1 has been extensively investigated, but no protonated Fe–N2Hx adducts have been observed from these reactions.1b Furthermore, the electron yield for N2 reduction products can be low upon addition of acid, indicating preferential protonation at iron.9c In contrast to these systems, where a highly reducing iron center favors −Fe protonation, LA coordination to the terminal nitrogen atom in 2 shifts electron density from the metal center to the terminal nitrogen atom.21 We experimentally probed the impact of this effect by examining the selectivity of protonation reactions with 2.
Multinuclear NMR spectroscopy was used to assess protonation selectivity for 1 vs 2. When 2 was combined with HBArF4·(OEt2)2 (PhF, −45 °C), a new complex, 3, formed in 91% yield (31P integration) (Figure 5). The 15N NMR spectra revealed further separation between the iron-bound and terminal nitrogen resonances than either 1 or 2 and exhibited one-bond 15N–1H coupling (3: Nα −49.3, Nβ −140.7 ppm (1J15N-1H = 80.7 Hz)), consistent with −N protonation of the N–N unit. The protonation reaction is >95% selective; only a 4% yield of trans-Fe(H)(depe)2(N2)+ is observed by 31P and 15N NMR spectroscopy.12,22 In contrast, subjection of 1 to identical reaction conditions in the absence of B(C6F5)3 affords iron hydrides, indicating that protonation occurs exclusively at the iron center.23 The monoprotonated product (3) contains a structurally unique Fe–NN(B)H unit and was characterized by X-ray crystallography, NMR, and IR spectroscopy.
Figure 5.

Synthesis of 3 (Fe1–N1: 1.679(6) Å; N1–N2: 1.252(8) Å; N1–N2–B: 130.6(6)°); Synthesis of 4 (Fe1–N1: 1.742(3) Å; N1–N2: 1.177(5) Å; N2–Fe2: 1.884(3) Å; Fe1–N1–N2: 175.1(3)°; Fe2–N2–N1: 173.4(3)°). The bond metrics of 4 were of obtained from one of the two crystallographically unique units within the unit cell. See the SI for details. Nonessential atoms and counteranions omitted for clarity. Ellipsoids shown at 50% probability.
Comparison between 14N- and 15N-labeled 3 allowed identification of the 14N–H (3259 cm−1) and 14N–14N (1519 cm−1) stretches, the latter of which indicates a significant weakening of the N–N bond. The solid-state structure of 3 revealed an N1–N2 bond length that is elongated by 0.066(8) Å (vs 2) to 1.252(8) Å, consistent with further weakening of the N–N bond. H1 was located from the difference map and indicates protonation of the terminal nitrogen atom (N2), a conclusion reinforced by contraction of the N–N–B angle to 130.6(6)°. Furthermore, the Fe1–N1 bond length is shortened by 0.07(1) Å, consistent with an iron hydrazido formulation and similar to previously reported M–N–N(H)–LA species.24 The N–N bond length of 3 is longer than 2 and slightly shorter than the related iron hydrazido ([(SiPiPr3)Fe = N–N(Me)H]+: 1.284(4) Å) complex recently reported by Peters and Rittle.24a Progressive elongation of the N–N bond in 1, 2, and 3 is reflected in progressively lower Wiberg bond orders (2.5, 2.0, and 1.7), further highlighting the comparable impact of protons and neutral LAs on N–N bond strength. Collectively, these data demonstrate that −N coordination of a LA can simultaneously lower the redox potential of an Fe–N2 unit while enabling selective terminal −N protonation.
Fe(II) as a Lewis Acid Additive
In principal, the enhanced activation of N2 upon addition of main group LAs should also translate to transition metal LAs, affording M–(μ-N2)–M′ cores.25 Although such iron homo- and heterobimetallic units may be highly activated, their preparation commonly relies on combining two highly reduced metal fragments.23,26 As an alternative strategy to further activate the N2 unit, we investigated the effect of adding an oxidized, rather than a reduced, iron center to an Fe(0)–N2 unit. In this arrangement, the charge disparity between two iron sites may facilitate charge transfer to the N2 unit, similar to main-group LAs (vide supra). We selected an Fe(II)(iPr2Tp)Cl as a monomeric, yet sterically accessible, iron center.26b When 1 was combined with Fe(II)(iPr2Tp)Cl in the presence of NaBArF4, a new compound Fe(depe)2(μ-N2)Fe(iPr2Tp) (BArF4) (4) was obtained (Figure 5). This S = 2 (μeff = 4.77 μB) complex contains a high-energy, solvent-dependent IVCT band at 910 nm (ε = 1700 M−1 cm−1, Δν1/2 = 3398 cm−1) as well as structurally distinct environments for each Fe center in the X-ray structure (trigonal bipyramidal for Fe1 and tetrahedral for Fe2). These data are consistent with a Class II mixed-valence complex containing localized Fe(0)/Fe(II) centers.27 Importantly, the IR spectrum of 4 contains an activated N–N unit (νNN = 1825 cm−1), which is nearly identical to the shift found in 2 and more activated than the related homodimer (Fe(dmpe)2)2(μ-N2) (νNN = 1933 cm−1).23 This indicates that a high-spin Lewis acidic Fe(II) center induces greater activation of the N–N bond than the addition of a more reduced and Lewis basic Fe(0) center. The extent of N2 activation enabled by charge-disparate Fe centers may provide insights into how N2 is activated within the multimetallic core of nitrogenase.
CONCLUSIONS
In contrast to the mildly reducing conditions used for N2 reduction in nitrogenases, synthetic complexes that both bind and enable N2 protonation require powerful reducing equivalents and possess highly negative redox potentials. In this paper, we describe the application of secondary sphere Lewis acids based on main group elements, alkali metal cations, and an Fe(II) center to an Fe–N2 unit. Our study provides an alternative N2 activation strategy that weakens the N–N bond, enables mild redox potentials, and enhances protonation selectivity through the simple addition of Lewis acids. Theoretical analysis shows that this effect is caused by stabilization of the N2 π* orbital and polarization of the N2 unit between the reducing and acidic fragments, much like the frustrated Lewis pair systems widely used for small molecule activation.28 This work supports a mechanistic rationale for the conserved acidic residues in nitrogenase and may focus future efforts that exploit Lewis acids as a key design element.
Supplementary Material
Acknowledgments
This work was supported by the NIH (Grant No. 1R01GM111486-01A1) and the NSF (Grant No. CHE-0840456) for X-ray instrumentation. N.K.S. is a Camille Dreyfus Teacher–Scholar and an Alfred P. Sloan Research Fellow. We thank Dr. Jeff Kampf for crystallographic assistance, Prof. Charles McCrory for electrochemical assistance, and Prof. James Mayer and Dr. Oscar Tutusaus for helpful discussions.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b01982.
Synthetic details, characterization and crystallographic information for compounds 2–4 (PDF)
X-ray data for compounds 2–4 (CIF)
References
- 1.(a) Connor GP, Holland PL. Catal Today. 2017;286:21. doi: 10.1016/j.cattod.2016.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Crossland JL, Tyler DR. Coord Chem Rev. 2010;254:1883. [Google Scholar]; (c) Hazari N. Chem Soc Rev. 2010;39:4044. doi: 10.1039/b919680n. [DOI] [PubMed] [Google Scholar]
- 2.(a) Hoffman BM, Lukoyanov D, Yang ZY, Dean DR, Seefeldt LC. Chem Rev. 2014;114:4041. doi: 10.1021/cr400641x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hoffman BM, Lukoyanov D, Dean DR, Seefeldt LC. Acc Chem Res. 2013;46:587. doi: 10.1021/ar300267m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Howard JB, Rees DC. Proc Natl Acad Sci U S A. 2006;103:17088. doi: 10.1073/pnas.0603978103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Dance I. Chem Commun. 2013;49:10893. doi: 10.1039/c3cc46864j. [DOI] [PubMed] [Google Scholar]
- 3.(a) Coric I, Mercado BQ, Bill E, Vinyard DJ, Holland PL. Nature. 2015;526:96. doi: 10.1038/nature15246. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rittle J, Peters JC. Proc Natl Acad Sci U S A. 2013;110:15898. doi: 10.1073/pnas.1310153110. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Arashiba K, Miyake Y, Nishibayashi Y. Nat Chem. 2011;3:120. doi: 10.1038/nchem.906. [DOI] [PubMed] [Google Scholar]; (d) Yandulov DV, Schrock RR. Science. 2003;301:76. doi: 10.1126/science.1085326. [DOI] [PubMed] [Google Scholar]
- 4.Braaksma A, Haaker H, Grande HJ, Veeger C. Eur J Biochem. 1982;121:483. doi: 10.1111/j.1432-1033.1982.tb05813.x. [DOI] [PubMed] [Google Scholar]
- 5.(a) Spatzal T, Perez KA, Einsle O, Howard JB, Rees DC. Science. 2014;345:1620. doi: 10.1126/science.1256679. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dance I. Dalton Trans. 2012;41:7647. doi: 10.1039/c2dt30518f. [DOI] [PubMed] [Google Scholar]; (c) Dos Santos PC, Igarashi RY, Lee HI, Hoffman BM, Seefeldt LC, Dean DR. Acc Chem Res. 2005;38:208. doi: 10.1021/ar040050z. [DOI] [PubMed] [Google Scholar]
- 6.(a) Creutz SE, Peters JC. Chem Sci. 2017;8:2321. doi: 10.1039/c6sc04805f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bhattacharya P, Prokopchuk DE, Mock MT. Coord Chem Rev. 2017;334:67. [Google Scholar]
- 7.(a) Kim CH, Newton WE, Dean DR. Biochemistry. 1995;34:2798. doi: 10.1021/bi00009a008. [DOI] [PubMed] [Google Scholar]; (b) Yang ZY, Dean DR, Seefeldt LC. J Biol Chem. 2011;286:19417. doi: 10.1074/jbc.M111.229344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Borovik AS. Acc Chem Res. 2005;38:54. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]; (b) Neu HM, Baglia RA, Goldberg DP. Acc Chem Res. 2015;48:2754. doi: 10.1021/acs.accounts.5b00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Komiya S, Akita M, Yoza A, Kasuga N, Fukuoka A, Kai Y. J Chem Soc, Chem Commun. 1993:787. [Google Scholar]; (b) Perthuisot C, Jones WD. New J Chem. 1994;18:621. [Google Scholar]; (c) Hill PJ, Doyle LR, Crawford AD, Myers WK, Ashley AE. J Am Chem Soc. 2016;138:13521. doi: 10.1021/jacs.6b08802. [DOI] [PubMed] [Google Scholar]
- 10.Field LD, Hazari N, Li HL. Inorg Chem. 2015;54:4768. doi: 10.1021/acs.inorgchem.5b00211. [DOI] [PubMed] [Google Scholar]
- 11.Sivaev IB, Bregadze VI. Coord Chem Rev. 2014;270–271:75. [Google Scholar]
- 12.Field LD, Hazari N, Li HL, Luck IJ. Magn Reson Chem. 2003;41:709. [Google Scholar]
- 13.The increased Δδ for the N2 unit upon functionalization is similar to that observed for H+ (Δδ 176 Mo(HIPTN3N)(N2H)) or SiR3+ (Δδ 147.8–157.3 (SiPiPr3)Fe(N2SiR3)); see: Yandulov DV, Schrock RR. J Am Chem Soc. 2002;124:6252. doi: 10.1021/ja020186x.See also: Lee Y, Mankad NP, Peters JC. Nat Chem. 2010;2:558. doi: 10.1038/nchem.660.
- 14.W–NN–BR2 units have been reported; see: Ishino H, Ishii Y, Hidai M. Chem Lett. 1998;27:677.
- 15.Similarly bent angles in an M–NN(R) unit have been previously reported for cationic R groups. See refs 3b and 13. Klein HF, Ellrich K, Ackermann K. J Chem Soc, Chem Commun. 1983:888. and Fryzuk MD, MacKay BA, Johnson SA, Patrick BO. Angew Chem, Int Ed. 2002;41:3709. doi: 10.1002/1521-3773(20021004)41:19<3709::AID-ANIE3709>3.0.CO;2-U.In addition, AlR3 N2 adducts have been previously reported: Chatt J, Crabtree RH, Jeffery EA, Richards RLJ. J Chem Soc, Dalton Trans. 1973:1167.Broda H, Hinrichsen S, Krahmer J, Nather C, Tuczek F. Dalton Trans. 2014;43:2007. doi: 10.1039/c3dt52965g.
- 16.McWilliams SF, Rodgers KR, Lukat-Rodgers G, Mercado BQ, Grubel K, Holland PL. Inorg Chem. 2016;55:2960. doi: 10.1021/acs.inorgchem.5b02841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.The acceptor number was derived from the shift of the 31P resonance of O==P(Et)3 upon coordination of the LA. See: Beckett MA, Strickland GC, Holland JR, Varma KS. Polymer. 1996;37:4629.
- 18.Weaker boron LA, such as BPh3, do not coordinate even through the Lewis acidity is within the observed range. This is presumably due to the larger reorganization energy required of boron, vs alkali metal, LAs.
- 19.Compound 2 rapidly decomposes in the presence of most noncoordinating electrolyte anions (PF6, SbF6, ClO4, BF4, CB11H12). A variable-temperature NMR experiment showed that mixtures of 2 and [NBu4+] [B(3,5-(CF3)2C6H3)4−] in PhF are unstable at temperatures above −10 °C, but decomposition is prevented at −45 °C.
- 20.This is similar to the reported value of the Fe(0/I) couple in THF (0.1 M [NBu4+][OTf], −1.95 V); see ref 9c.
- 21.LA interactions have been proposed by theoretical calculations to facilitate hydride insertion into a Ru–N2 unit. See: Holscher M, Leitner W. Eur J Inorg Chem. 2014;2014:6126.
- 22.(a) Bancroft GM, Mays MJ, Prater BE. J Chem Soc D. 1969:585. [Google Scholar]; (b) Buys IE, Field LD, Hambley TW, Mcqueen AED. Acta Crystallogr, Sect C: Cryst Struct Commun. 1993;49:1056. [Google Scholar]
- 23.Doyle LR, Hill PJ, Wildgoose GG, Ashley AE. Dalton Trans. 2016;45:7550. doi: 10.1039/c6dt00884d. [DOI] [PubMed] [Google Scholar]
- 24.(a) Rittle J, Peters JC. J Am Chem Soc. 2017;139:3161. doi: 10.1021/jacs.6b12861. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rittle J, Peters JC. J Am Chem Soc. 2016;138:4243. doi: 10.1021/jacs.6b01230. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Takahashi T, Mizobe Y, Sato M, Uchida Y, Hidai M. J Am Chem Soc. 1980;102(25):7461. [Google Scholar]
- 25.(a) O’Donoghue MB, Zanetti NC, Davis WM, Schrock RR. J Am Chem Soc. 1997;119:2753. [Google Scholar]; (b) Field LD, Guest RW, Turner P. Inorg Chem. 2010;49:9086. doi: 10.1021/ic101646p. [DOI] [PubMed] [Google Scholar]; (c) Zhang QF, Chim JLC, Lai W, Wong WT, Leung WH. Inorg Chem. 2001;40:2470. doi: 10.1021/ic001341g. [DOI] [PubMed] [Google Scholar]
- 26.(a) Betley TA, Peters JC. J Am Chem Soc. 2004;126:6252. doi: 10.1021/ja048713v. [DOI] [PubMed] [Google Scholar]; (b) McSkimming A, Harman WH. J Am Chem Soc. 2015;137:8940. doi: 10.1021/jacs.5b06337. [DOI] [PubMed] [Google Scholar]; (c) Yu RP, Darmon JM, Hoyt JM, Margulieux GW, Turner ZR, Chirik PJ. ACS Catal. 2012;2:1760. doi: 10.1021/cs300358m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Smith JM, Lachicotte RJ, Pittard KA, Cundari TR, Lukat-Rodgers G, Rodgers KR, Holland PL. J Am Chem Soc. 2001;123:9222. doi: 10.1021/ja016094+. [DOI] [PubMed] [Google Scholar]; (e) McWilliams SF, Holland PL. Acc Chem Res. 2015;48:2059. doi: 10.1021/acs.accounts.5b00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D’Alessandro DM, Keene FR. Chem Soc Rev. 2006;35:424. doi: 10.1039/b514590m. [DOI] [PubMed] [Google Scholar]
- 28.Stephan DW. Science. 2016;354:1248. doi: 10.1126/science.aaf7229. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.